

Abstract
While traditional approaches for disease management in the era of modern medicine have saved countless lives and enhanced patient well-being, it is clear that there is significant room to improve upon the current status quo. For infectious diseases, the steady rise of antibiotic-resistance has resulted in super pathogens that do not respond to most approved drugs. In the field of cancer treatment, the idea of a cure-all silver bullet has long been abandoned. As a result of the challenges facing current treatment and prevention paradigms in the clinic, there has been an increasing push for personalized therapeutics, where plans for medical care are established on a patient-by-patient basis. Along these lines, vaccines, both against bacteria and tumors, are a clinical modality that could benefit significantly from personalization. Effective vaccination strategies could help to address many challenging disease conditions, but current vaccines have been limited by factors such as a lack of potency and antigenic breadth. Recently, researchers have turned towards the use of biomimetic nanotechnology as a means of addressing these hurdles. Here, we discuss recent progress in the development of biomimetic nanovaccines for antibacterial and anticancer applications, with an emphasis on their potential for personalized medicine.
Keywords: nanomedicine, biomimetic nanoparticle, personalized medicine, anticancer vaccination, bacteria vaccination
Graphical Abstract
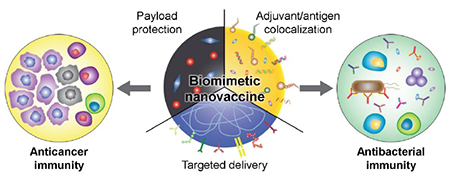
Table of content entry:
Personalized medicine is transforming how diseases are managed in the clinic. At the same time, biomimetic nanotechnology offers many advantages that can be leveraged towards the design of more effective medical interventions. In this progress report, we discuss recent developments in biomimetic nanovaccines against cancer and bacterial infections, with a specific emphasis on potential avenues for personalization.
1. Introduction
A marked increase in human longevity over the past century has engendered new challenges in healthcare.[1] While traditional medications have been able to significantly improve patient outcomes, it has become increasingly evident that a one-size-fits-all approach to disease management has major limitations.[2–4] An important point to consider is that clinical responses to the same therapy can be highly variable. For instance, while some cancer patients treated with a monoclonal antibody can experience complete tumor eradication, others receiving the same treatment often see little improvement.[5–7] An underlying reason for this discrepancy is the complexity of cancer pathogenesis, in which an accumulation of mutations ultimately leads to uncontrolled cell growth. As the pathways involved in tumorigenesis can vary greatly,[8–12] the origin, development, and severity of cancer is generally different for each individual patient. Beyond variability in treatment outcomes, a cure-all mentality in medical practice can lead to the over-prescription and misuse of pharmaceutical drugs by doctors and uninformed patients.[13] The classical example is when patients with mild symptoms such as a sore throat and fever are indiscriminately prescribed antibiotics.[14–16] These prescriptions often do not address the root cause of illness, particularly in the case of non-bacterial infections. Overuse of antibiotics is a major driver of drug resistance in probiotic bacteria, and the associated genes can subsequently be acquired by harmful pathogens through horizontal transfer mechanisms.[17, 18] Once these pathogens are drug-resistant, treatment options become significantly limited in clinical settings, which is why more selective and appropriate usage of antibiotics has been widely advocated. Overall, there has been a strong drive towards more individualized forms of therapy for both cancer[19–21] and infectious diseases.[22, 23]
Personalized medicine is a concept that has been garnering attention due to our enhanced ability to characterize and classify diseases, particularly through the use of genomic technologies.[24] Over the past decade, the term has been used to encompass medical decisions that are tailored to individual patients or specific groups of patients. This type of approach has the potential to maximize efficacy by providing each patient with optimal care while avoiding ineffective remedies.[25] One recent example of personalization is chimeric antigen receptor (CAR) T cell therapy, where the relevant effector cell populations are isolated from each individual patient for processing.[26–30] The cells are then genetically engineered to target the patient’s own cancer cells, expanded ex vivo, and then reinfused for treatment. In this manner, CAR T cells are manufactured on a case-by-case basis, which helps to ensure that the treatment provokes minimal unwanted immune responses. Despite significant developments, personalized medicine is still a field in its infancy, and continued research along these lines will undoubtedly lead to better treatment options for patients in the clinic.
Recent advances in vaccine nanotechnology have led to the development of new prophylactic and therapeutic modalities that can improve upon current clinical standards. Vaccines focus on training a patient’s own immune system to recognize and combat deadly diseases. By activating the inherent ability of the immune system to potentiate highly specific responses against pathogens or tumors, vaccine-based immunotherapies can be leveraged to facilitate disease eradication in a safe and reliable manner.[31, 32] In this progress report, we begin with a basic immunology overview and discuss the principles governing effective vaccine design. The advantages of incorporating nanotechnology in the engineering of vaccines and the current state of nanovaccines will be discussed in detail. Then, we will review recent developments in nanovaccines for antibacterial and anticancer therapy, with a special focus on biomimetic platforms that have future implications for personalized medicine.
2. Vaccine Immunology and Design Principles
The human body has evolved so that it can address a variety of threats quickly and efficiently. The innate branch of the immune system is tasked with rapid and nonspecific clearance of any foreign bodies that enter the host. Over time, key components of the innate immune system will activate and prime adaptive immune cells against the threat. The adaptive branch of immunity is then responsible for generating focused responses against specific antigenic targets, including pathogens, toxins, or infected cells. The main mediators between the innate and adaptive immune systems are antigen-presenting cells (APCs), which include macrophages and dendritic cells.[33] These specialized cells can take up and digest protein-based antigens and present epitopic peptide fragments on their surfaces in the context of major histocompatibility complexes (MHCs).[34–36] Exogenous antigens from pathogens like viruses, parasites, or bacteria are generally taken up by APCs and digested, followed by peptide loading onto MHC class II (MHC-II) before being presented on the cell surface. Endogenous antigens, such as mutated proteins or parts of intracellular viruses and bacteria, are processed in the cytosol and presented onto MHC class I (MHC-I). In some subsets of APCs, exogenous antigens can also be loaded onto MHC-I through a process called cross-presentation, and this is facilitated by the shuttling of antigens into the cytosolic compartment after uptake.[37, 38] Important to immune activation is the expression of costimulatory markers by APCs on their surface, which can be triggered through the engagement of pattern recognition receptors (PRRs) by pathogen-associated molecular patterns (PAMPs).[39] Once activated, APCs often migrate to secondary lymphoid organs such as lymph nodes or the spleen. There, the APCs can interact with cells from the adaptive immune system, consisting mostly of T cells and B cells, in order to elicit cellular or humoral immunity.
Cytotoxic T cells (CTLs), or CD8+ T cells, are one of the main drivers of cellular immunity. Activation of these T cells require two signals from APCs.[40] First, the CD8 coreceptor and T cell receptor (TCR) on T cells must interact with a peptide-loaded MHC-I complex on the APC. The second signal consists of costimulatory markers, such as CD80 or CD86, which interact with T cell surface proteins like CD28. When CD8+ T cells successfully interact with both a costimulatory marker and their cognate antigen, they will undergo clonal expansion to increase the number of antigen-specific CTLs that can survey the body for antigen-expressing target cells.[41] Upon interaction with a cell expressing its cognate antigen, the cytotoxic T cells will release various cytotoxins that eventually lead to apoptosis of the infected or mutated cells.
APCs can also interact with helper T cells, or CD4+ T cells, to support CTLs. Similar to CTLs, helper T cells also need two signals to become activated. The first signal is mediated by the interaction of antigen-loaded MHC-II with its cognate TCR as well as CD4, while the second signal results from costimulatory interactions between markers such as CD80 or CD86 and CD28. After activation, the helper T cells will proliferate and differentiate into different subclasses.[42] Among those subclasses, T helper type 1 (Th1) cells can release cytokines such as interferon-γ (IFN-γ) and interleukin-2 (IL-2) to assist CTL activation and support the development of cellular immunity. Importantly, Th1 cells are essential to the formation of memory T cells that provide long-term immunity against specific antigens. Activated Th1 cells can return to interact with dendritic cells through interactions between CD40 and CD40L in order to “license” APCs and allow them to more robustly interact with CTLs in ways that can cause differentiation into memory T cells.[43] These memory T cells can rapidly respond to subsequent antigen exposure and have superior self-renewal capacity ideal for long-term protection.
Helper T cells can alternatively differentiate into T helper type 2 (Th2) cells, which work to support humoral immunity. Humoral immunity is driven by B cells that secrete antibodies against specific antigens that bind mostly to extracellular pathogens such bacteria or parasites, as well as their virulence factors. The binding of an antibody to its target results in neutralization or destruction via processes such as complement activation and phagocytosis.[44] B cells need to directly interact with their cognate antigen in order to secrete antibodies, and this interaction alone can fully activate B cells specific for some antigens called thymus-independent antigens.[45] However, many important antigens, including most of those that are protein-derived, can be considered thymus-dependent, and help is required from Th2 cells in order to fully activate the corresponding B cells.[46] In these cases, Th2 cells bind to B cells presenting their cognate antigen loaded onto MHC-II. The T cells then present CD40L, which binds to CD40 on B cells and fully activates them.[47] Additionally, Th2 cells are essential for clonal proliferation and isotype switching; both processes help to generate B cells that produce antibodies with high affinity for their targets.[48]
Traditional immunizations are designed to take advantage of these innate and adaptive immune pathways to generate cellular or humoral immunity in the absence of the actual threat. Most vaccines consist of an antigen and an immunological adjuvant that work together to fully stimulate antigen-specific immunity. Antigens are protein material that serve as a means of training the immune system to recognize a specific target of interest. They are generally comprised of attenuated pathogens or proteins isolated from the pathogen being vaccinated against. Adjuvants help to induce APC maturation, leading to the presentation of costimulatory markers. Historically, they have been based on aluminum salts (alum) or oil emulsions, but can also include natural or synthetic PRR agonists for toll-like receptors (TLRs) or nucleotide-binding oligomerization domain (NOD)-like receptors.[49–51] When the complete vaccine is administered to a patient, the components are mostly taken up by APCs, particularly dendritic cells. These APCs are then stimulated by the adjuvant and present the antigen on their surface via MHCs, after which they migrate to nearby lymph nodes to interact with CTLs, T helper cells, and B cells in order to elicit specific immune responses.
3. Current Vaccine Nanotechnology
Nanotechnology offers many benefits that can be leveraged to increase the potency of vaccine formulations (Figure 1). One advantage of nanoparticle-based formulations is the ability to co-deliver antigenic materials with an immunostimulatory adjuvant. This is highly important for proper immune stimulation, because the spatial colocalization of the two components ensures that a prompt immune response against the antigen of interest is generated.[52] To achieve co-delivery, the adjuvant can either be encapsulated into the nanoparticle core, functionalized onto the surface, or the nanoparticle material itself can serve as the stimulus. In one example of the design of an inherently immunostimulatory nanoformulation, alpha-alumina nanoparticles were conjugated with the model antigen ovalbumin (OVA).[53] The nanoparticle-based vaccine was able to significantly enhance cross-presentation and activation of CD8+ T cells by at least 500-fold compared to vaccination with the soluble antigen alone. Animals treated therapeutically with the nanovaccine were able to completely reject tumor growth and remained tumor-free for more than 40 days, whereas none of the other control formulations showed any efficacy. It should be noted that when using a mixture of OVA along with alum, a traditional adjuvant, there was minimal improvement in survival and no observable reduction in tumor growth. Other examples of stimulatory nanomaterials include calcium phosphate,[54] a mineral-based adjuvant, and small-sized proteoliposomes.[55]
Figure 1.
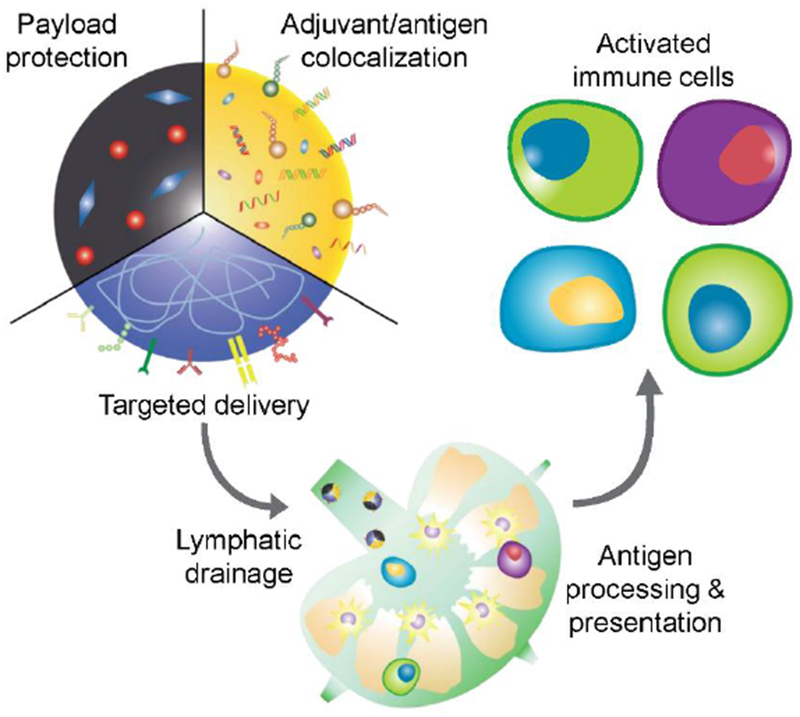
Overview of vaccine nanotechnology. Nanoparticles offer several advantages that can aid in the design of more effective vaccine formulations, including the ability to protect the bioactivity of encapsulated payloads, colocalize antigen and adjuvant for unified delivery to immune cells, and target specific cell subsets through the introduction of functional surface ligands. Their small size also enables efficient lymphatic transport, which can facilitate processes such as antigen presentation and lead to more potent immune activation.
Adjuvant molecules can be directly incorporated within a nanoparticulate matrix. In one example, interbilayer-crosslinked multilamellar vesicles (ICMVs) were formed by crosslinking the headgroups of adjacent lipid bilayers.[56] The complex structure of the ICMVs could incorporate high levels of OVA proteins as well as monophosphoryl lipid A (MPLA), a lipid-like immunostimulatory adjuvant. The ICMVs coloaded with antigen and adjuvant could ultimately be engulfed and degraded by intracellular lipases for direct delivery of the payloads into cells. Immunization with ICMVs elicited a strong humoral response with a substantial increase in antibody titer levels, about 1000-fold compared with immunizations using the soluble OVA antigen. ICMVs could also generate potent cell-mediated immunity by eliciting a strong CD8+ T cell response against OVA, the magnitude of which was 14 times stronger than when using soluble antigen. ICMV-based nanovaccines have also been utilized in conjunction with the adjuvant polyIC, a TLR3 agonist, to produce antigen-specific effector memory T cells at the mucosal surface,[57] which is important given that many pathogens infiltrate their hosts through these barriers. Another means of incorporating adjuvants into nanoformulations is to directly functionalize them onto nanoparticle surfaces. Along these lines, synthetic high-density lipoprotein-mimicking nanodiscs have been used for immunotherapeutic applications.[58] In one instance, the nanodisc surface was functionalized with the adjuvant CpG, a TLR9 agonist, as well as with tumor antigens via reduction-sensitive linkages. This nanodisc platform, when administered to mice, was able to produce a 41-fold increase in antigen-specific CD8+ T cells compared with antigen-linked nanodiscs mixed with free CpG as a control. When the vaccine was used in conjunction with checkpoint inhibitors to generate responses against a mutated neoantigen in an MC38 tumor model, complete regression in 87.5% of the mice was achieved. As a comparison, only 25% survival was achieved with a soluble neoantigen and adjuvant formulation. The nanodisc platform has also been combined with chemotherapeutics to achieve impressive therapeutic results against tumors.[59]
Another advantage of nanoparticle-based vaccines is the enhanced bioavailability of the payloads. By encapsulating or conjugating antigen and adjuvant to a nanocarrier, the materials can more effectively be protected from host interactions during transport. Within the circulatory system, there is a wide range of enzymes that can cause the degradation of biomolecules and bioactive cargoes.[60] In addition, nanoparticulate delivery can also prevent systemic toxicities that are often associated with the administration of adjuvants in their free form. Thus, by shielding their payloads from the surrounding environment, nanoparticles can concurrently protect the host from nonspecific biologicals interactions, which can cause unintended side effects.[61] The protection imparted by nanocarriers can effectively prolong in vivo residence, which increases the probability of successful delivery to APCs. Furthermore, targeting moieties can be introduced onto the nanoparticle surface to enhance delivery towards desired cell subsets. Some ligands, such as mannose, have a natural affinity for dendritic cells and macrophages.[62] Lastly, nanoparticle-based formulations can greatly prolong immune stimulation due to sustained and controlled release of the encapsulated molecules. A careful selection of the materials used for constructing nanoformulations can ensure that the encapsulated materials are slowly released over time in a controlled manner,[63] which has been shown to prolong the elevation of antibody titers and results in the production of more effector memory T cells.[64]
The unique size range of nanoparticles is another factor that can enable improved delivery of vaccine components. The nanoscale dimensions of nanocarriers allow for more efficient lymphatic drainage into the lymphoid organs where antigen uptake and processing can occur.[65–67] In an example application of this phenomenon, iron oxide–zinc oxide nanoparticles with a core–shell structure were used to deliver carcinoembryonic antigen.[68] These nanoparticles had an average size of around 15 nm, enabling them to effectively travel to the lymph nodes. Once at their destination, the particles could be taken up by the resident dendritic cells for processing in order to elicit a specific immune response against the antigen. When administered to mice, there was a tenfold increase in splenic CD8+ T cells secreting IFN-γ, which is a proinflammatory cytokine that is commonly correlated with the activation of cell-mediated immunity. Although the formulation was not able to completely eradicate tumors, vaccination with the iron oxide–zinc oxide nanoparticles did delay tumor growth and extended the mean survival from 10.5 days to 19.5 days. An added advantage of these nanoparticles was their inherent ability to be used for magnetic resonance imaging. It is important to note that effective lymphatic drainage of nanoparticles is greatly dependent on size. Research has found that, after intradermal administration, 100 nm nanoparticles were only 10% as efficient in accumulating at the draining lymph nodes when compared to 25 nm nanoparticles.[67]
Nanoparticles can also be designed for efficient cytosolic delivery, which has major implications for improving vaccine performance. Antigen delivery to the cytosolic compartment of cells is a unique way of activating CD8+ T cells through the MHC-I pathway. Under normal circumstances, internalized antigens need to be shuttled from the endosomal compartment to the cytosol in order to be presented in the context of MHC-I.[69] In contrast, targeted delivery of antigenic material directly into the cytosol can allow CD8+ T cell stimulation while bypassing endogenous cross-presentation mechanisms. For cancer vaccine applications, effective cytosolic delivery can benefit both antigen and adjuvant, leading to simultaneous enhancement of antigen presentation and improvement of immune stimulation.[70] In one example, a synthetic polymeric nanoparticle was used to deliver OVA to the cytosol while simultaneously triggering the stimulator of interferon gene (STING) pathway.[71] The nanoparticles were designed to be pH-sensitive, enabling them to disrupt the endosomal membrane and release their payloads into the cytosol prior to being degraded. Compared with a soluble OVA control, the nanoparticle formulation was able to increase the frequency of OVA-specific CD8+ T cells by 29-fold. Ultimately, when used under a therapeutic setting, the STING agonistic nanoformulation was capable of significantly controlling tumor growth. Other strategies for cytosolic delivery include employing hydrophobic nanoparticles and cationic nanoparticles to facilitate direct uptake.[72, 73]
Advantages of nanovaccines have been similarly utilized for managing bacterial infections.[74] In one case, inherently stimulatory gold nanoparticles were used to vaccinate against the flagellin of Pseudomonas aeruginosa, producing a highly specific humoral response.[75] In another example, deactivated Chlamydia trachomatis bacterium were conjugated onto a polymeric nanoparticle loaded with R848, a potent TLR7/8 agonist, for mucosal vaccination.[76] After intrauterine immunization, only the bacteria-conjugated nanoparticle formulation was able to significantly protect mice from C. trachomatis challenge. A mixture of the inactivated bacteria with the adjuvant-loaded nanoparticles had a negligible effect, supporting the need for antigen–adjuvant colocalization to achieve strong antibacterial immunity.
As described in this section, nanotechnology can confer several unique advantages when it comes to the engineering of vaccine formulations. Careful manipulation of nanoparticle parameters and conscious design choices can significantly improve potency compared with traditional vaccine delivery systems.
4. Biomimetic Nanoparticle Technology
The design of vaccines using nanotechnology offers several key advantages that can be used to help improve upon what is currently available in the clinic. To further enhance the utility of nanoscale platforms, researchers have more recently looked towards nature for inspiration. Through millions of years of evolution and refinement, living systems have evolved the ability to perform complex functions in highly efficient ways. Many technologies that have been adopted in the modern world were adapted from nature, and many of these are employed on a daily basis.[77] Likewise, biomimetic design principles have become increasingly prevalent within the field of nanomedicine, where the result is a streamlined approach for introducing and fabricating multifunctional nanoparticle platforms that can effectively interface with biological systems.[78–80]
One way in which biomimetic design can enhance the utility of nanoparticle technology is by enabling targeted delivery through the use of natural ligands.[81] A prominent example is arginylglycylaspartic acid (RGD), which is a binding peptide that can be found in fibronectin.[82] Composed of only three amino acids, RGD is the minimal motif required for some cellular adhesion processes. In cancer, integrin adhesion molecules are overexpressed on angiogenic endothelial cells and serve as a prime target for the peptide.[83] In an example, nanoparticulate delivery of paclitaxel to tumors was enhanced using RGD as the targeting ligand.[84] Compared with their nontargeted counterparts, RGD-functionalized nanoparticles had a fivefold higher accumulation at the tumor vasculature, which prolonged the survival of tumor-bearing mice from 13 days to 21 days. Another biomimetic targeting ligand is the CDX peptide, which was synthesized with inspiration from candoxin, a snake neurotoxin that binds to nicotinic acetylcholine receptors on brain endothelial cells with high affinity and selectivity.[85–87] Once bound, candoxin can subsequently be transported to brain cells through receptor-mediated transcytosis.[88] Leveraging this property, CDX-conjugated nanoparticles have been shown to cross the blood–brain barrier, a major obstacle that generally prevents delivery to the central nervous system. When used to treat glioblastoma, micelles functionalized with CDX significantly extended survival in mice, whereas micelles lacking the ligand did not have any noticeable effect.[87] When combined with RGD, a dual-targeted formulation achieved even better therapeutic efficacy in an intracranial U87 glioma mouse model compared to those functionalized with either ligand alone.[89] Some carbohydrates such as mannose have strong binding affinity to immune cells, and thus can be used for vaccine delivery.[90] On the other hand, mannose-specific lectins have been utilized for antibacterial applications. In one example, gliadin nanoparticles were conjugated with lectins that selectively bind to carbohydrate receptors on Helicobacter pylori.[91] The enhanced binding allowed the drug-containing nanoparticles to inhibit bacterial growth by more than twofold as compared to nontargeted nanoparticles. Many other naturally derived moieties have been used for targeting, since this process bypasses the need to synthetically replicate complex receptor–ligand interactions.[81, 92]
In addition to targeting interactions, biomimetic functionalization of nanoparticles can also be used to modulate the activity of biological targets. This can be particularly useful for biodetoxification, where therapies are designed to neutralize the activity of toxic molecules that pose a threat to human health. An innovative biomimetic approach along these lines is the use of molecularly imprinted polymers, which mimic the physical specificity of antibodies to achieve neutralization.[93] Target molecules are used as the template and mixed with a solution of polymerizable monomers with various functional groups. After complexation of the monomers with the template, polymerization is carried out to effectively freeze the positioning of each component. Removal of the template produces polymeric nanoparticles that are highly specific to the original target molecule. Molecularly imprinted nanoparticles produced with melittin bee toxin as the template were able to neutralize the hemolytic activity of the toxin.[94] In an in vivo model, the imprinted nanoparticles were able to save 50% of mice from a lethal dose of melittin, whereas a 100% mortality rate was observed for untreated mice.[95] Other biomimetic platforms can exert their effects directly on cells, and one way to achieve this is through the use of natural particulates.[96] This includes the engineering of viruses or virus-like particles (VLPs) to take advantage of their ability to invade and manipulate target cells.[97] Due to their unique advantages, viral nanoparticles have been strategically employed for gene therapy.[98–101] In a different type of application, oncolytic viruses have been designed to specifically infect and kill cancerous cells.[102–104] Other strategies for biomimetic nanoparticle design have taken advantage of the biological activity of individual ligands. For example, membrane vesicles have been engineered to express programmed cell death protein 1 (PD-1) in order to block the biological function of its corresponding ligand to prevent T cell exhaustion.[105] Along these lines, there are countless ligands that could be leveraged for the design of nanoparticles capable of executing a wide range of specific biological functions.
The decoration of nanoparticles with natural ligands represents a streamlined approach for introducing desirable biomimetic functions. However, a challenge associated with the use of these individual ligands is the difficulty in incorporating multiple functionalities at the same time. In general, it is hard to replicate the multifaceted biological interactions found in nature using bottom-up synthetic strategies. To address this issue, an emerging biomimetic strategy for creating multifunctional nanoparticles has been employed to leverage the unique properties of cellular membranes.[106] As a fundamental unit of living organisms, cells are involved in countless biological interactions, and thus they represent a rich source of natural targeting ligands, functional modulators, and antigenic materials. Rather than recreating complex cellular functions, researchers have directly isolated plasma membrane and coated them onto the surface of nanoparticles to enable more effective biointerfacing.[107–110] The faithful transfer of cell membrane onto nanoparticles ensures that all of the associated surface proteins and receptors are preserved in their entirety. In contrast to bottom-up synthesis techniques, this top-down membrane coating approach is largely function-driven and does not require prior identification of individual ligands. Cell membrane-coated nanoparticles are capable of performing cell-like functions in a manner that is dependent on their membrane source, and they have been employed in a variety of ways, including for biodetoxification,[111, 112] targeted delivery,[113, 114] and bioimaging.[115, 116] In particular for vaccine development, cell membranes provide a rich source of multiantigenic material, which can enable the development of formulations that confer broader protection.
Nanoparticles functionalized with natural cell membrane have proven to be useful for a number of biomedical applications. For example, by camouflaging nanoparticle surfaces with red blood cell (RBC) membrane, it has been demonstrated that the immune system will recognize the resulting nanoparticles as self rather than foreign.[108] This is in large part due to the surface receptors and complement regulatory proteins found on RBC membrane that prevent the binding of opsonins and clearance by the mononuclear phagocytic system.[117] As a result, these nanoparticles have exhibited enhanced circulation, which can increase the bioavailability of encapsulated payloads and allow them to more effectively reach their intended targets. Different targeting moieties can also easily be introduced onto RBC membrane-coated nanoparticles (RBC-NPs) to enhance delivery. In addition to conjugation onto the membrane surface, other techniques such as lipid insertion have been used to introduce targeting functionality.[85, 118, 119] Rather than adding exogenous ligands onto the nanoparticle surface, targeted drug delivery can also be achieved through leveraging innate receptors found on the plasma membrane of certain cell types. For example, platelets have a multitude of surface moieties that naturally bind to different disease substrates, including damaged vasculature and certain pathogens.[120–122] Accordingly, nanoparticles camouflaged with platelet membrane have demonstrated the same types of binding affinities (Figure 2).[110] In this fashion, targeted delivery can be achieved with minimal disruption to the nanoparticle surface. In another example, cancer cells have been shown to naturally bind with one another through a homotypic aggregation phenomenon.[123] By taking advantage of this mechanism, cancer cell membrane-coated nanoparticles have been used to enable more preferential delivery of payloads to cancer cells.[109] This targeting phenomenon has been exploited for modes of cancer treatment such as photothermal therapy and photodynamic therapy.[124–126]
Figure 2.
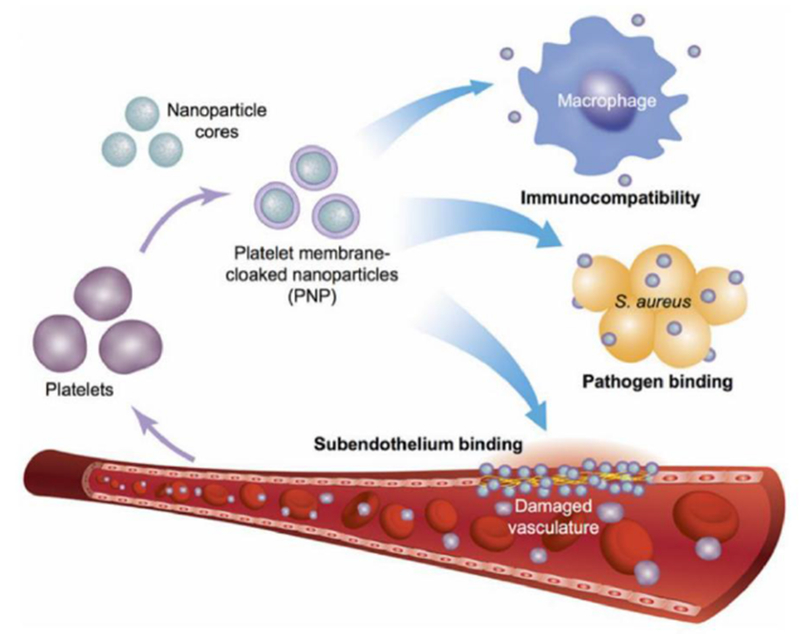
Platelet membrane-coated nanoparticles. Platelet membrane is derived from whole platelets by a repeated freeze-thaw process. The purified membranes can then be fused with a nanoparticulate core, enabling natural targeting affinity towards pathogens and damaged vasculature. Reproduced with permission.[110] Copyright 2015, Springer Nature.
Overall, biomimetic nanoparticle platforms, including those fabricated using cell membrane coating technology, are incredibly versatile and have a limitless number of potential applications. More recently, they have been increasingly used in the design of more effective vaccine formulations, and development along these lines will be discussed more in detail in the following sections.
5. Biomimetic Antibacterial Nanovaccines
The discovery of penicillin in 1928 revolutionized the way in which bacterial infections are treated, but the recent surge in “superbug” pathogens has shown that the evolution of drug resistance in pathogens far outpaces our ability to discover new antibiotics.[127] It thus appears that the strategy of continuously developing new classes of therapeutics for treating increasingly resistant bacteria will eventually prove futile. To overcome this, innovative strategies, including those focused on prevention rather than treatment, are needed to combat disease-causing pathogens. This is an area in which biomimetic nanotechnology can provide significant benefits, and increasing attention has been placed on the use of extracellular vesicles and nanotoxoids as antibacterial vaccines. Both of these biomimetic systems are amenable to personalization, where formulations can be facilely tailored to address any number of individual bacterial strains or fabricated on-demand for specific patient populations.
5.1. Outer Membrane Vesicles
Outer membrane vesicles (OMVs) are ~20-250 nm nanostructures generated from the blebbing of the cell envelope of bacteria.[128] Along with various outer membrane proteins, OMVs can also contain inner membrane and cytoplasmic proteins, making them a complex mixture of antigenic material. Many factors impact the biogenesis of OMVs, including the presence of envelope crosslinking proteins and the lipid composition of the outer membrane. The production rate of OMVs can be altered by environmental factors such as temperature and the availability of nutrients. Virulence factors are also an important component of OMVs, although the underlying mechanisms that determine their partitioning into OMVs are still being elucidated. Once secreted from the cell envelope, OMVs serve a variety of functions for bacteria, including mediating stress responses, acquiring nutrients, and acting as decoys for antibiotics and phages.
The multiantigenic nature of OMVs makes them attractive candidates for antibacterial vaccination. Their unique membrane protein profile activates both the innate and adaptive immune systems, and these responses are prompted by the presentation of PAMPs that bind to PRRs on APCs.[129] In addition, their small size increases their lymph node entry and APC uptake rates, further improving their immune activation capabilities. The concept of using OMVs for vaccination has already been successfully translated, with an OMV-derived meningococcal vaccine currently used in clinic.[130] This vaccine contains meningococcal group B OMVs and aluminum hydroxide, which acts as the adjuvant. Aside from the meningococcal vaccine, many others based on similar principles have been proposed.[131, 132] For example, an engineered Staphylococcus aureus strain has been used to generate nontoxic extracellular vesicles containing antigens such as magnesium transport system membrane proteins and ferrichrome-binding periplasmic proteins.[133] Immunization with these engineered vesicles provided significant protection at a rate of approximately 60% in a lethal mouse model of sepsis.
Bacteria have also been manipulated in other ways to produce OMVs that can be used for improved vaccination efficacy.[134] For example, lab strain Escherichia coli have been genetically engineered to express O-antigen polysaccharides from other pathogenic bacteria.[135] When vesiculation was induced, the altered E. coli secreted OMVs containing the engineered polysaccharides on their surfaces. OMVs derived in this fashion contained pathogen-mimetic glycotopes that could subsequently be used to confer protection against the pathogens from which the antigen originated. In another case, OMVs were genetically engineered to improve the efficacy of subunit vaccines without the need to utilize adjuvants.[136] Using green fluorescent protein as a model subunit antigen, mice vaccinated with the protein-fused OMVs were able to produce significant antibody titers, indicating that a potent humoral response could be elicited with this strategy. These novel bacterial membrane-based vaccines show significant promise, and more work along these lines will help to facilitate their clinical translation.
5.2. OMV Nanoparticles
Employing OMVs as antigenic material for use in conjunction with adjuvanting nanoparticulate delivery systems may further improve vaccine efficacy. Shigella flexneri releases OMVs whose membrane contains many components that can be targeted to confer protection against shigellosis, including lipopolysaccharides (LPS), outer membrane proteins, and Ipa proteins, making these vesicles an ideal antigen source for vaccine formulations. In one example, whole S. flexneri OMVs were loaded into poly(anhydride) nanoparticles by a solvent displacement method.[137] Here, the nanoparticulate material, a copolymer made up of methyl vinyl ether and maleic anhydride, was used as a potent adjuvant by enhancing bioadhesive interactions, and the vaccine formulation was able to induce TLR2 and TLR4-mediated innate immunity. While nasal vaccination using free OMVs provided only 40% protection against bacterial challenge, OMV-loaded nanoparticles administered by the same route and at the same concentration protected all mice. By incorporating the OMV material into the poly(anhydride) nanoparticles, a bioadhesive formulation with high antigen density that could release the material in a controlled manner over time could be fabricated.
While loading OMV material inside of a nanocarrier has been shown to greatly boost immunity, presenting intact OMV membrane on nanoparticle surfaces could more closely replicate the membrane protein profile encountered during an infection and may provide better training cues for the immune system. This can be achieved by coating OMVs onto a nanoparticulate core. In one case, gold nanoparticles were coated with E. coli OMVs (Figure 3), which resulted in precise control of the final formulation’s size distribution.[138] This in theory allowed for the enhanced unification of transport kinetics compared with crude OMV preparations, which exhibited high polydispersity. The coating process concurrently improved the stability of the gold nanoparticle cores, and significant lymph node accumulation was observed after administration of the OMV-coated nanoparticles. Further, the strong association of the membrane to the gold core, as demonstrated by a dye binding assay, likely aided in multivalent display of OMV membrane antigens. Vaccination with the bacterial OMV-coated nanoparticles, as compared to OMVs only, significantly increased dendritic cell activation, and E. coli-binding IgG antibody production was elicited in a sustained manner.
Figure 3.
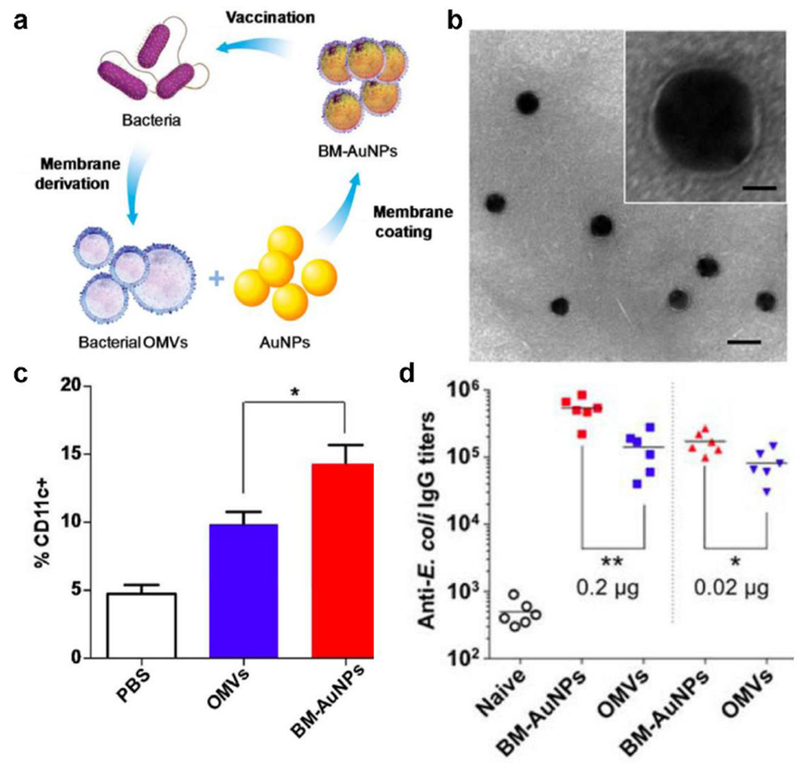
OMV-coated gold nanoparticles for antibacterial vaccination. a) OMVs are collected from bacteria and then coated onto gold nanoparticles (AuNPs), and the resulting bacterial membrane-coated AuNPs (BM-AuNPs) can be used to vaccinate against the source bacteria. b) Transmission electron microscopy (TEM) image of BM-AuNPs (scale bar, 50 nm). Inset: a single BM-AuNP (scale bar, 10 nm). c,d) When administered in vivo, BM-AuNPs can recruit more APCs in the lymph nodes (c) and generate stronger anti-E. coli IgG titers (d). Reproduced with permission.[138] Copyright 2015, American Chemistry Society.
5.3. Nanotoxoids
An emerging strategy to treat infectious diseases is antivirulence therapy.[139, 140] Infections often become lethal due to the toxins secreted by the associated pathogen. These compounds can inhibit protein synthesis, cause hemolysis and tissue damage, disrupt the immune system, and lead to sepsis. By targeting virulence factors, including the harmful toxins secreted by bacteria, toxoid vaccines have the potential to prevent infections while circumventing the risk of evolutionary resistance.[141] In toxoid vaccination, an inactivated form of the targeted toxin is used to elicit an immune response. By depending on toxin neutralization rather than the cytotoxic activity of antibiotics, this vaccination strategy can inhibit a pathogen’s ability to colonize a host without directly exerting pressure on individual bacterium. Although attractive, this approach often requires an in-depth understanding of the target antigen in order to design a vaccine that effectively generates the appropriate anti-toxin immunity.[142] To improve their safety profiles, toxoids are usually generated through harsh chemical or heat treatments, and their immunogenicity and antigenicity can be significantly impacted as a result of these processes. Recently, cell membrane coating nanotechnology has been used to address the aforementioned challenges, resulting in toxoid vaccines with improved efficacy and safety profiles.[143]
Most virulence factors must in some way interact with cells via the plasma membrane in order to exert their toxicity.[144] To leverage this fact, cell membrane-coated nanoparticles have been used as decoys that are capable of neutralizing toxins and preventing them from harming healthy cells.[80, 145] This concept was first demonstrated using red blood cell (RBC) membrane-coated nanoparticles (RBC-NPs) as a means of guarding against the activity of hemolytic toxins (Figure 4).[146] One of the most important pore-forming toxins released by Staphylococcus aureus is α-toxin, which embeds itself as an oligomer into RBCs and causes destruction through pore formation on the cellular membrane. When RBC-NPs were preincubated with α-toxin, toxicity and hemolytic activity of the toxin were completely abrogated. Accordingly, when mice challenged with α-toxin were treated using RBC-NPs, 89% of mice were rescued in a prophylactic setting and 44% of mice survived in a therapeutic scenario.
Figure 4.
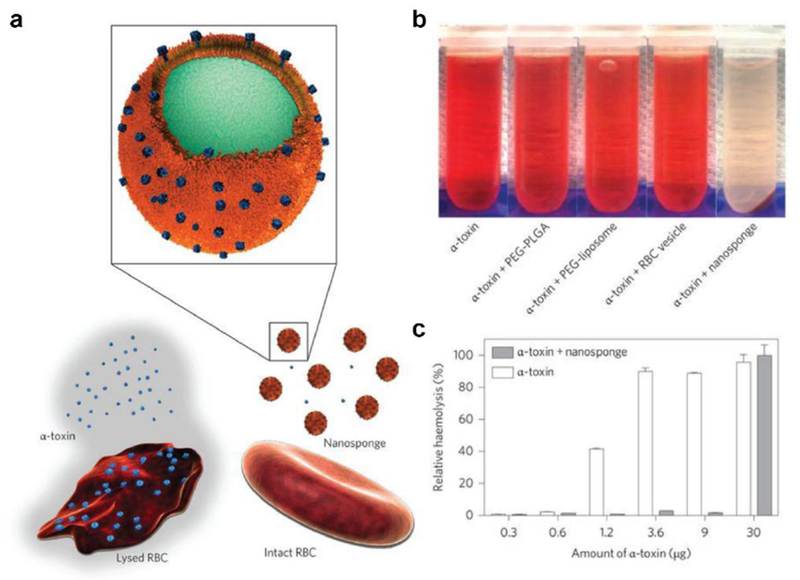
RBC nanosponges for biodetoxification. a) RBC nanosponges are fabricated by coating RBC membrane onto a polymeric core. The nanosponges can protect healthy RBCs from toxin-mediated hemolysis. b) Staphylococcal α-toxin preincubated with RBC nanosponges are unable to lyse native RBCs, whereas no protection is conferred after preincubation with various controls. c) RBC nanosponges negate the hemolytic activity of α-toxin in a concentration-dependent manner. Reproduced with permission.[146] Copyright 2013, Springer Nature.
Toxin-bound cell membrane-coated nanoparticle constructs have been used as nanoparticle-based toxoids, or nanotoxoids, for enhancing antivirulence vaccination efficacy.[147] Due to the ability of cell membrane-coated nanoparticles to neutralize pore-forming toxins in their native state, mice vaccinated with the nanotoxoids demonstrated superior protective immunity and higher antibody titer formation as compared to a conventional heat-denatured toxoid. When challenged with α-toxin systemically, 90% of the mice vaccinated with a single dose of the nanotoxoids survived, compared to only 10% survival using a control heat-treated toxoid. Importantly, nanotoxoids exhibited no observable toxicity when administered in vivo, indicating that they were able to offer superior protection without compromising safety. Vaccination using the same formulation was also able to help lower disease burden in an animal model of live methicillin-resistant S. aureus (MRSA) infection.[148]
Another advantage of using cell membrane-coated nanoparticles in the design of toxoid vaccines is that it bypasses the need to intimately understand the toxins involved in the pathogenesis of infections. Since biomimetic RBC-NPs are in essence miniaturized RBCs, any bacterial toxins that bind to and target the source cells will similarly bind to the nanoparticle surface. In addition to α-toxin, it was recently proven that RBC-NPs can neutralize the toxicity of melittin, listeriolysin O, and streptolysin O.[149] The broad-spectrum neutralization capability of RBC-NPs enables them to concurrently capture a multitude of different bacterial toxins for vaccination, which can be used to elicit immune responses against several targets at once (Figure 5).[150] Vaccinating against multiple virulence factors has the potential to offer superior protection against bacteria, thus reducing their chance of successfully colonizing a host. Animals vaccinated with nanotoxoids that were preincubated with crude hemolytic protein preparations derived from MRSA culture supernatants were able to generate antibody responses against α-toxin, γ-toxin, and Panton–Valentine leukocidin. Although this proof-of-concept study was limited to the use of previously characterized toxins, other works have demonstrated the ability of cell membrane-coated nanoparticles to bind novel virulence factors, thus aiding in their identification.[151]
Figure 5.

RBC-based nanotoxoids for multiantigenic antibacterial vaccination. a) Bacteria secrete numerous virulence factors that can cause cellular damage. When incubated with nanosponges, the toxicity of the secretions is neutralized, enabling the resulting toxin-inserted nanotoxoid formulations to be used as a vaccine for eliciting multiantigenic immunity. b) Vaccination using nanotoxoids loaded with a hemolytic supernatant fraction (hSP) from S. aureus concurrently potentiates humoral antibody responses against multiple known toxins. Reproduced with permission.[150] Copyright 2017, Wiley-VCH.
The nanotoxoid approach for generating antivirulence vaccine formulations is highly versatile. Different cocktails of bacterial toxins can be preincubated with cell membrane-coated nanoparticles in order to modulate the specificity of the resultant vaccine. Furthermore, the cell membrane on the nanoparticle surface can be easily interchanged with the membrane from other types of cells.[109, 110, 152, 153] For example, murine macrophage membrane-coated nanoparticles have demonstrated the ability to capture and neutralize the activity of endotoxins such as LPS.[152] The flexibility to easily modulate the membrane coating source and the captured toxins means that the nanotoxoid platform is potentially applicable across the whole span pathogenic bacteria, including both gram-positive and gram-negative strains. In the future, this biomimetic nanotechnology may also be exploited for personalized therapies where vaccine formulations can be generated against different bacterial strains and administered to patients based on their individual risk profiles.
6. Biomimetic Anticancer Nanovaccines
Anticancer therapy is another area in which there is a high demand for personalized medicine. The pathogenesis of cancer is extremely complex, and the disease is inherently hard to treat given that malignant cells are derived from mutated versions of one’s own healthy cells. With many disease-causing factors involved, cancer varies greatly from patient to patient.[154] Recently, researchers have engineered different biomimetic platforms, including cell-derived nanovesicles, VLPs, artificial antigen-presenting cells (aAPCs), and cell membrane-coated nanoparticles, for use as anticancer vaccines. Many of these have the potential to develop into personalized therapies that can ultimately help to overcome tumor heterogeneity.
6.1. Augmenting Preexisting Immunity
One commonly studied immunotherapeutic approach for cancer treatment is to enhance the power of preexisting immunity. Cancer cells utilize many immunosuppressive mechanisms in the tumor microenvironment to prevent cytotoxic activity and evade destruction. Since anticancer vaccination is mainly focused on producing new subsets of T cells with antitumor specificities, therapeutics that augment this process are often used to overcome immunosuppression. One evasion mechanism employed by tumors involves the upregulation of certain immune checkpoint markers that obstruct T cell function. Those such as the cognate ligands for PD-1 and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) can bind to T lymphocytes, thereby inhibiting their cytotoxic activity.[155] By disrupting these receptor–ligand interactions, the inhibitory signals on the suppressed T cells are removed, subsequently enabling CTLs to carry out the task of cancer elimination. Currently, monoclonal antibodies are used in the clinic as checkpoint blockade therapies, but their systemic administration can induce toxicities due to undesirable off-target effects.[156] A new means of employing checkpoint blockade therapy using genetically engineered nanovesicles was recently reported.[105] HEK293T, a human embryonic kidney cell line that can be easily manipulated to generate large amounts of recombinant proteins, was engineered to express PD-1 on its surface. The cells were then collected and lysed before the cell membrane was purified and extruded to form nanovesicles. It was shown that nanovesicles expressing PD-1 could bind to PD-1 ligand (PD-L1) on tumor cells to disrupt the corresponding T cell exhaustion pathway. While conventional anti-PD-L1 monoclonal antibody therapy was only able to save 10% of treated mice, use of the engineered vesicles brought the survival rate up to 20%. In addition to a better safety profile, PD-1 nanovesicles could be loaded with additional therapeutic payloads such as indoleamine 2,3-dioxygenase inhibitors to disrupt other immune tolerance pathways and enhance efficacy.
It has been shown that engineered nanovesicles could be especially useful within the context of surgery. Surgical removal of tumors remains one of the primary means of treating many cancers. However, the complete elimination of all malignant cells is highly challenging, and even small amounts of residual tumor can lead to relapse. Platelets serve as circulating sentinels for vascular damage and naturally bind to and accumulate at surgical sites.[110] Researchers took advantage of this natural targeting property and engineered platelet microparticles to express PD-1 for postsurgical cancer therapy.[157] Because platelets are terminally differentiated cells, megakaryocytes, a platelet progenitor, were engineered to express the protein instead. Treatment of mice with the engineered membrane particles after tumor resection delayed tumor recurrence and led to a survival rate of 25%. Besides genetic manipulation, checkpoint inhibitors have been conjugated directly onto the surface of platelets through a bifunctional maleimide linker.[158] It has been shown that anti-PD-L1 antibody-conjugated platelets can form microparticles after thrombin-induced activation, and these particles can be used to reverse tumor immunosuppression (Figure 6). In multiple murine tumor recurrence and metastasis models, the antibody-conjugated membrane particles were able to prolong survival. In a final example, targeted immune checkpoint blockade therapy was achieved by conjugating platelets functionalized with anti-PD-L1 onto another cellular carrier, such as hematopoietic stem cells, and this platform has demonstrated utility for treating acute myeloid leukemia.[159] In the future, these platelet-based therapies can be personalized by employing autologous cells, which would mitigate concerns about immune incompatibility. Their prospects for clinical use are also aided by the fact that platelet infusions are oftentimes indicated for patients recovering from surgery.[160]
Figure 6.
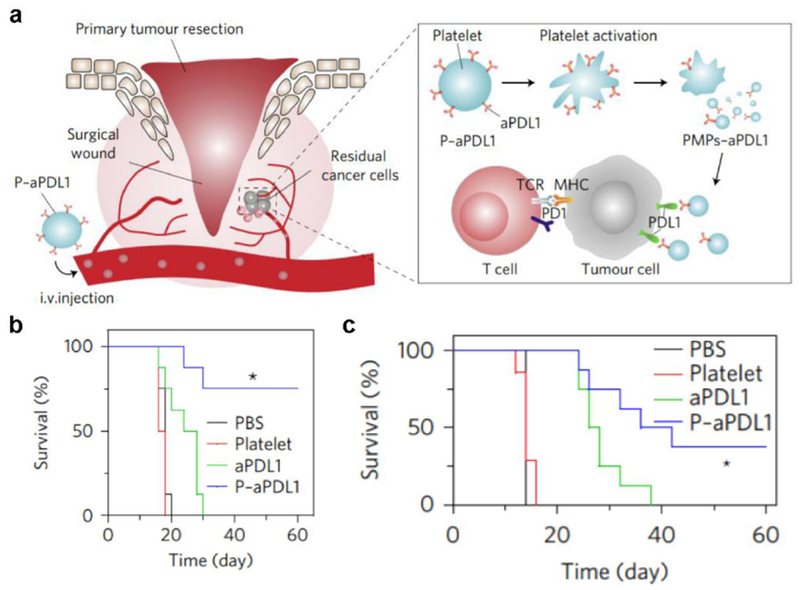
Engineered platelet microparticles for targeted checkpoint blockade therapy. a) Platelet-derived vesicles functionalized with anti-PD-L1 (aPDL1) can bind to surgical wounds after tumor resection, reducing tumor immunosuppression and enabling attack of tumor cells by T cells. b, c) When used to treat tumor-bearing mice, aPDL1-functionalized platelet vesicles significantly enhance survival in an incomplete surgery tumor model (b) and an incomplete resection and metastasis model (c). Reproduced with permission.[158] Copyright 2017, Springer Nature.
6.2. Nonspecific Immune Modulation
An effective strategy for overcoming tumor heterogeneity has been the use of immunostimulatory agents that promote innate immune system activity and nonspecifically recruit immune cells to the tumor site. These foreign agents can reduce immunosuppression by increasing the presence of activated immune cells within the tumor microenvironment. In this case, the patient’s own tumor cells can then serve as the antigenic material for generating tumor-specific T cells. A biomimetic example of this vaccination strategy used vesicles derived from bacteria. When CT26 tumor-bearing mice were systemically treated four times at three-day intervals with E. coli OMVs, all of the tumors were completely eradicated.[161] Furthermore, a second re-challenge with the tumor four weeks later and a third re-challenge seven weeks later did not result in any noticeable growth. This phenomenon was not limited to OMVs secreted by E. coli, as vesicles purified from Lactobacillus acidophilus, S. aureus, and Salmonella enterica were similarly capable of inhibiting tumor growth. For this study, the antitumor immunity likely resulted from the recruitment of immune cells at the tumor site caused by secretion of the proinflammatory cytokine IFN-γ. The recruited immune cells could then use nearby tumor cells for antigen processing to generate highly specific CTLs. IFN-γ knockout mice and mice injected with monoclonal anti-IFN-γ antibodies prior to treatment did not experience any tumor regression.
VLPs are another class of foreign agents that can elicit a strong immune reaction. The particles are generally produced from the spontaneous assembly of viral coat proteins into virus capsids.[162, 163] Plant-based VLPs derived from cowpea mosaic virus that are free of viral nucleic acids have been exploited to suppress tumor growth (Figure 7).[164, 165] Inhalation of the VLPs in tumor-bearing mice increased the presence of tumor-infiltrating neutrophils and elevated levels of cytokines and chemokines such as granulocyte-macrophage colony-stimulating factor, CXCL1, CCL5, and macrophage inflammatory protein 1α, among others. In a B16F10 lung metastasis model, weekly intratracheal administration of the cowpea mosaic virus VLPs significantly reduced tumor burden. The protective efficacy was not unique to the B16F10 model, as similar effects were observed in the context of 4T1 lung metastasis, CT26 intradermal colon tumor, and ID8-Defb29/Vegf-A ovarian tumor models. Anticancer immunity initiated by the VLPs was shown to be long-lasting, and mice with complete responses that were re-challenged four weeks later exhibited strong memory with a 75% tumor rejection rate. A slow-release version of this treatment has also been developed to overcome challenges posed by the frequent treatment schedule.[166] In this formulation, VLPs were combined with generation-4 polyamidoamine dendrimers to form aggregates based on electrostatic interactions. A single dose of the complexes had similar antitumor efficacy compared with four weekly doses of VLPs alone when tested in an ID8-Defb29/Vegf-A ovarian cancer model. This type of slow-release formulation could significantly help with patient compliance should plant-based VLPs eventually be translated into the clinic.
Figure 7.

Plant virus-like particles (VLPs) for in situ anticancer vaccination. a) Nucleic acid-free VLPs are produced in plants. When delivered to tumors, the VLPs enhance neutrophil activity, which ultimately leads to tumor destruction by T lymphocytes. b, c) When used to vaccinate tumor-bearing mice, empty cowpea mosaic virus (eCPMV) particles can significantly prolong survival in a 4T1-luciferase metastatic breast cancer model (b) and an ID8-Def20/Vegf-A ovarian cancer model (c). Reproduced with permission.[164, 165] Copyright 2016, Springer Nature.
Interestingly, the antitumor immunity produced by plant VLPs is somewhat unique to the cowpea mosaic virus system. It was demonstrated that the potency induced by tobacco mosaic virus VLPs was significantly less than that of cowpea mosaic virus VLPs when used to treat B16F10 tumor-bearing mice.[167] This discrepancy could not be simply explained by the difference in viral structures, because native tobacco mosaic virus nanorods, as well as spherical nanoparticle and shorter nanorod forms, were all incapable of eliciting a strong immune response. There were no significant differences in tumor growth or survival between mice treated with any tobacco mosaic virus-derived VLP. However, when papaya mosaic virus VLPs were administered intratumorally to treat B16F10 melanoma or systemically to treat lung metastases, there was significant reduction in tumor growth and fewer metastatic nodules.[168, 169] The results indicate that some VLPs are more suitable for in situ vaccination than others, and understanding these intricate differences will be pivotal for future development of the platform.
An added advantage of VLPs is the ability to conjugate the particles with tumor-associated antigens for explicitly generating antitumor immunity. VLPs are generally small enough to enter the lymphatic system, allowing them to reach the lymph nodes for more effective antigen delivery and processing. Two vaccinations of potato virus X conjugated with a lowly immunogenic idiotypic B cell lymphoma tumor antigen was able to protect 70% of mice against intravenous challenge with BCL1 lymphoma cells.[170] In another example, subcutaneous immunizations with VLPs conjugated with an epitope of human epidermal growth factor receptor 2 induced significant amounts of antibody titer formation against the cell marker.[171] Vaccination with VLPs has also been significantly improved when used in conjunction with radiation therapy, chemotherapy, or immune checkpoint blockade therapy.[169, 172, 173] In all cases, the combination led to enhanced survival and prolonged tumor suppression compared to either treatment alone. VLP administration combined with radiation therapy has been validated in a canine preclinical trial with impressive results.[174] All canine patients treated with this combination became tumor-free, firmly supporting the clinical potential of VLPs as a therapeutic modality against cancer.
6.3. Immunogenic Cell Death
Another in situ vaccination approach relies on the concept of immunogenic cell death (ICD).[175, 176] Normally, cellular apoptosis is a silent process that goes unnoticed. However, it was recently discovered that cancer cells destroyed with certain cytostatic agents can become immunogenic and initiate antitumor immune responses. The immunogenicity is derived from the release of damage-associated molecular patterns (DAMPs) due to stress on the endoplasmic reticulum and production of reactive oxygen species. These DAMPs can be highly immunostimulatory and recruit nearby immune cells to the tumor site. APCs can then engulf the dying tumor cells, process the antigens, and present them on MHCs to activate tumor-specific T lymphocytes. An example of this strategy used mitoxantrone to induce ICD in vitro prior to treating mice with the dying tumor cells.[177] To amplify the stimulation signal, the dying cells were also conjugated with multilamellar lipid-polymer nanoparticles laden with CpG. The inclusion of this additional immune stimulus increased the survival of challenged mice from 20% to 100% in a B16F10-OVA tumor model. Similarly, 78% of mice bearing CT26 colon cancer survived when treated using the formulation combined with an anti-PD-1 checkpoint inhibitor.
There are several examples of employing biomimetic nanotechnology to induce ICD for anticancer therapy. RBC membranes have been used to coat nanomaterials to improve their circulation and to protect encapsulated cargoes.[178] In the case of certain chemotherapeutics, delivery by cell membrane-coated nanoparticles gives them a greater chance of localizing to the tumor site, where they can be released to trigger ICD. Natural killer cells have demonstrated an inherent affinity towards tumor cells and can enhance M1 macrophage polarization. To leverage these properties, natural killer cell membrane-coated nanoparticles have been used in combination with photodynamic therapy to promote ICD and significantly suppress local and distant tumor growth (Figure 8).[179] Suppression of distant tumor growth occurred through the abscopal effect and served as a sign that systemic anticancer immunity had been established. Similarly, myeloid-derived suppressor cells can naturally migrate to tumor sites. The coating of iron oxide nanoparticles with membrane from these cells led to more tumor accumulation, and this subsequently increased the effects of photothermal therapy and promoted ICD.[180]
Figure 8.
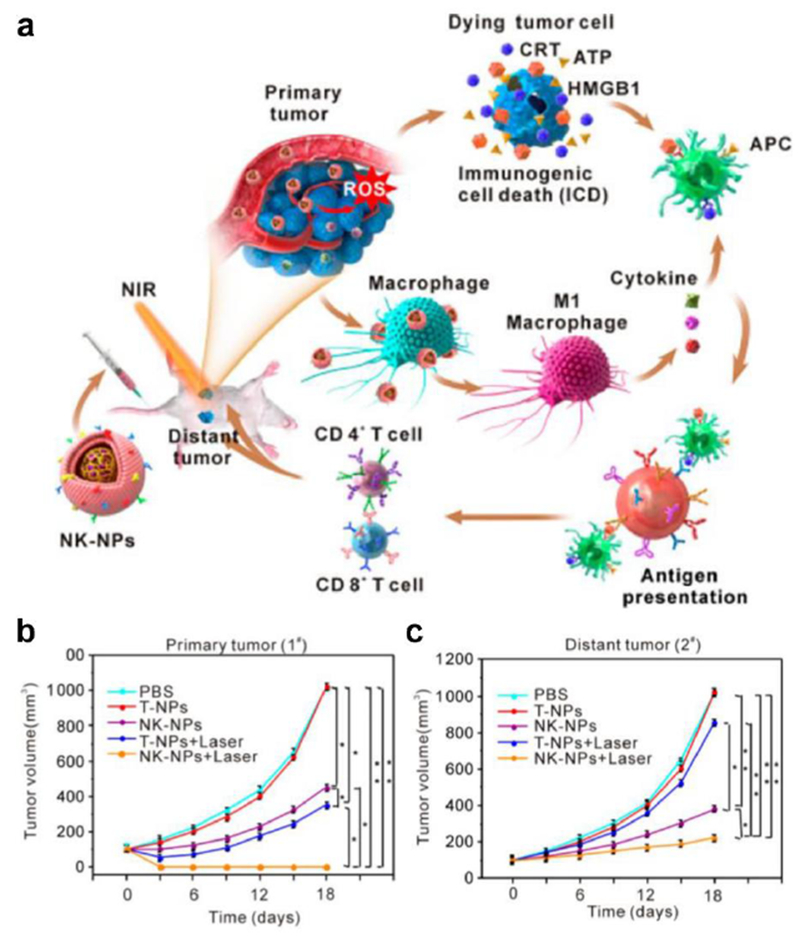
Natural killer cell membrane-coated nanoparticles (NK-NPs) for cancer therapy. a) Photosensitizer-loaded NK-NPs are administered in vivo and naturally accumulate at the tumor site. Photodynamic treatment of the tumor cells can cause immunogenic cell death and trigger the release of damage-associated molecular patterns (DAMPs) to recruit APCs for immune activation. b, c) Treatment using NK-NPs with irradiation leads to the eradication of primary tumors (b) and controls growth of distant tumors through the abscopal effect (c). Reproduced with permission.[179] Copyright 2018, American Chemistry Society.
6.4. Artificial Antigen-Presenting Cells
Vaccination strategies that directly provide tumor antigen material can ensure the generation of specific antitumor responses. However, most of these antigens are lowly immunogenic and are very similar to antigens found on healthy cells, thus it is often difficult to produce immune responses with sufficient potency to combat tumor growth. Rather than relying on the activation of APCs for training T cells with the correct specificities, recent aAPC technology has attempted to bypass endogenous antigen presentation by directly engineering nanoparticles to replicate all of the signals necessary for T cell stimulation.[181] To design aAPCs, two essential components must be included. The first is the presentation of a peptide epitope in the context of MHC, which provides antigen specificity and binds to the cognate TCR on effector T cells. The second is a costimulatory molecule, such as CD80 or CD86, that must concurrently bind to T cells for successful activation and proliferation. By mimicking the cellular functions of APCs, the goal of aAPCs is to elicit tumor-specific responses without the need for delivering antigens and adjuvants that require subsequent processing by endogenous immune machinery.
One example of nanoscale aAPCs involved the use of iron oxide nanoparticles or quantum dots as the core material.[182] The iron oxide nanoparticles were coated with a layer of dextran to help conjugate the two biological signals. Briefly, iron–dextran surfaces were functionalized with anti-biotin antibodies, and the aAPCs were then manufactured by using biotinylated peptide-MHC complexes and anti-CD28 antibodies. Quantum dot aAPCs were generated in a similar fashion by using commercially available avidin-coated nanocrystals. When exposed to antigen-specific effector cells, iron–dextran aAPCs were able to induce greater than a 15-fold expansion of T lymphocytes expressing the cognate TCR. Quantum dot aAPCs were equally capable of inducing T cell proliferation compared to noncognate controls. The aAPCs were used to activate antigen-specific T cells in culture, and the cells were able to significantly control tumor growth in a mouse model of melanoma upon infusion. Cell membrane coating nanotechnology has also been used to aid in the fabrication of aAPCs.[183] Magnetic nanoclusters of iron oxide nanoparticles were functionalized with an azide-engineered leukocyte membrane coating (Figure 9). The membrane coating helped stabilize the nanocluster structures, provided functional groups for conjugation, and increased the overall biocompatibility of the system. These engineered biomimetic aAPCs were able to successfully stimulate CTLs, visually guide adoptively transferred CTLs to tumors sites, and inhibit tumor growth.
Figure 9.

Artificial APCs (aAPCs) for direct stimulation of T cells. a) The two signals required for immune cell activation are conjugated onto magnetic nanoclusters coated with azide-engineered leukocyte membrane. Once administered, the magnetic aAPCs enable manual guidance of CTLs to the tumor. b) Co-incubation of aAPCs with CD8+ T cells induces activation and proliferation. c) Treatment with aAPC-guided adoptively transferred CTLs significantly controls tumor growth. Reproduced with permission.[183] Copyright 2017, American Chemistry Society.
The physical properties of aAPCs can have a major impact on their ability to stimulate T cells. Since the dimensions of nanoparticle-based aAPCs are vastly different compared to native APCs, manipulating their structure to more closely mimic cell surfaces can enhance activation. As an example, elongating aAPCs into an elliptical shape can decrease surface curvature and bring the two signals closer together. This close proximity increases the probability that both receptors can simultaneously bind to their respective ligands on CTLs, resulting in increased cellular proliferation.[184] Nanoparticle size also plays a similar role, as increases in diameter can tighten the gap between the two signals to promote enhanced interactions with TCRs by providing multivalent binding.[185] However, because larger nanoparticles may exhibit decreased lymphatic drainage, it has been recently demonstrated that decoupling the two signals and including them on different nanoparticles can maximize T cell stimulation while still maintaining the advantages of nanoscale aAPCs.[186] The fluidity of the components on the substrate surface may also play a major role in the efficiency of the antigen presentation process.[187] Overall, aAPC systems hold significant potential and may eventually see widespread usage as a method for directly stimulating antigen-specific CTLs in vivo to potentiate antitumor responses.
aAPCs may ultimately be employed for personalized therapeutics by utilizing cancer neoantigens, which arise from tumor-specific mutations that generate novel protein structures otherwise nonexistent in heathy tissue.[188] Neo-epitopes are highly patient-specific, and their identification requires genomic analysis of a patient’s cancerous cells as compared to their healthy cells. Personalized vaccines that utilize conventional APCs to generate neoantigen-specific CTLs have already had significant success in the clinic.[189–191] In a similar fashion, aAPCs loaded with personalized neoantigens may be used in the future to prime CTLs in vivo while bypassing the need to activate endogenous APCs.
6.5. Whole Cell Vaccinations
One hurdle in treating cancer with single-antigen immunotherapy is the extensive heterogeneity within tumors.[192] Antigenic diversity can lead to immune escape, as tumor cells that have limited expression of the antigen being targeted can continue to proliferate and drive tumor growth. In addition, tumors can lose the expression of immunogenic antigens as a mechanism of immune evasion, rendering the attack by CTLs generated from single-antigen vaccination ineffective.[193] One strategy to address this issue is by using the entirety of a cancer cell as the source of antigenic material.[194] Presenting a large spectrum of different antigens represented in a tumor allows for the development of a diverse repertoire of CTLs that are more difficult to completely escape.
Following this logic, several groups have combined the benefits of using tumor cell lysate as an antigen along with the benefits of nanoparticle delivery systems. In some cases, lysate-loaded nanoparticles with or without adjuvant have been used to enhance in vitro uptake and presentation of antigens by dendritic cells for cell-based immunotherapies.[195–197] Superior delivery of multiantigenic material has been demonstrated in human immune cells as well.[198] Dendritic cells derived from human peripheral blood mononuclear cell samples efficiently took up polymeric nanoparticles loaded with lysates from an epithelial ovarian cancer line. Further, co-cultures of particle-pulsed dendritic cells with human-derived CTLs exhibited a strong proinflammatory signature as indicated by cytokine profiling and immunophenotyping. This approach has also been applied in vivo using B16 melanoma lysate loaded into chitosan nanoparticles and modified with mannose for in vivo dendritic cell targeting.[199] To achieve this, B16 cells were lysed using a freeze-thaw method, mixed with chitosan, then added to a mannose-alginate solution to form nanoparticles through electrostatic interactions. Encapsulation of the lysate enabled superior uptake into bone marrow-derived dendritic cells in vitro and better transport to lymph nodes in vivo compared to free lysate, with the mannose moiety further enhancing this effect. The tumor lysate nanoparticles induced a significant increase in CTL formation in the draining lymph nodes and spleen, and mice treated prophylactically with the formulation had significantly reduced tumor burdens.
Another strategy is to use nanovesicles generated from the disruption of cancer cells directly as delivery vehicles. Adjuvant-loaded B16F10 cancer cell nanovesicles were made by mixing CpG and OVA with the cells, followed by sonication to induce vesiculation, and this was followed by the addition of a second adjuvant MPLA onto the surface of the vesicles.[200] T cells derived from mice vaccinated with the formulation produced a robust immune response against the parent B16-OVA cells as indicated by IL-2 production. The introduction of polyethylene glycol (PEG) onto cancer cell nanovesicles loaded with adjuvant has better enabled the in vivo use of this platform.[201] Cultured B16-OVA cells were lysed by freeze-thawing and then sonicated to form nanovesicles. To enhance immunogenicity, the nanovesicles were loaded with cholesterol-conjugated CpG, then finally PEGylated using a lipid-PEG conjugate to improve transport through the lymphatic system. After two prophylactic vaccinations, mice immunized with the nanovesicles had a significant increase in OVA-specific T cells and 50% of mice challenged with B16-OVA cells had no tumor for at least 80 days. In a therapeutic setting, 63% of mice treated with the formulation along with anti-PD-1 therapy had complete remissions.
6.6. Cell Membrane-Coated Nanovaccines
Cell membrane-coated nanoparticles, with their unique properties, have recently been leveraged in the design of anticancer vaccines.[202–204] RBC-NPs are known for their reduced host immune interactions and have been used extensively as a delivery vehicle.[117, 205–207] Along these lines, RBC-NPs were used as a carrier for glycoprotein 100 (gp100), a tumor-associated antigen enriched in B16F10 melanoma.[208] The gp100 was conjugated to the polymer with a disulfide bond to encourage antigen release within the lysosomal compartment of dendric cells. The nanoparticle surface was also modified with mannose for dendritic cell targeting, and MPLA was further incorporated into the cell membrane coating as an adjuvant. In both prophylactic and therapeutic B16F10 cancer models, C57BL/6 mice administered with the nanovaccine had significantly higher rates of tumor growth inhibition.
Cancer cells have been used as a source for membrane coating in order to introduce multiantigenic material into nanovaccines. As previously mentioned, single-antigen formulations can provide focused immunity, but tumor heterogeneity and antigenic loss can lead to immune evasion and progression of tumor growth despite the successful expansion of antigen-specific T cells. Material derived directly from cancer cells can combat this by providing a large spectrum of antigens as training cues to promote the development of a diverse T cell repertoire that can detect a broader range of targets. However, the strength of immunity generated by traditional whole cell vaccines is often diluted by the presence of a large amount of housekeeping genes, nucleic acids, lipids, and other antigens.[209] Using purified cancer cell membrane as an antigen source is one way to maximize the benefits of multiantigenic vaccination while also producing a strong and more focused immune response.
The first cancer cell membrane-coated nanoparticle (CCNP) used membrane derived from a B16F10 murine melanoma cell line to coat over a polymeric nanoparticle core.[109] The CCNPs were able to retain several important cancer cell membrane proteins and could effectively mature bone marrow-derived dendritic cells when MPLA was incorporated into the cell membrane coating. This work was further developed by using a CpG-loaded double emulsion nanoparticle as the core, which was coated in a similar manner with B16F10 cancer cell membrane (Figure 10).[210] The CpG was chosen to take advantage of the fact that nanoparticles are generally taken up by dendritic cells into the endosomal compartment, where the adjuvant can then be released to interact with its receptor TLR9. This was confirmed by showing that dendritic cells incubated with CpG nanoparticles showed significantly higher levels of activation, as indicated by upregulation of proinflammatory cytokines, compared with CpG in its free form. The successful coating of CpG nanoparticles with the B16F10 membrane was shown by TEM imaging, which revealed a characteristic core–shell structure. Western blot analysis of the CCNPs showed the presence of at least two prominent B16F10 antigens, gp100 and tyrosinase-related protein 2 (TRP2), and mice vaccinated with these particles showed an increase in CTLs specific for both. In a prophylactic setting, 86% of mice immunized with the CpG-loaded CCNPs survived tumor-free for at least 150 days after B16F10 challenge. When combined with anti-PD-1 and anti-CTLA-4 checkpoint blockades in a therapeutic setting, the nanovaccine could extend survival to 32 days compared with 18 days for untreated mice. Additionally, 50% of mice receiving the combination treatment survived until the end of the study, while only 20% survived for the checkpoint blockade only group and none survived in the unvaccinated group.
Figure 10.

Cancer cell membrane-coated nanoparticles (CCNPs) for multiantigenic anticancer vaccination. a) Cancer cell membrane is coated onto polymeric nanoparticles loaded with the adjuvant CpG, and the resulting CpG-CCNP formulation can be used to stimulate multiantigenic antitumor immunity. b) Vaccination with CpG-CCNPs induces CTLs specific for the gp100 and TRP2 melanoma-associated antigens. c) Mice vaccinated with CpG-CCNPs are able to better reject B16F10 tumor challenge. Reproduced with permission.[210] Copyright 2017, Wiley-VCH.
Similar works have confirmed and expanded upon the utility of the CCNP platform for anticancer vaccination. In one example, polymeric nanoparticles were loaded with the TLR7 agonist R837 and coated with B16-OVA cell membrane as the antigen.[211] Importantly, the surface of the cell membrane was modified with mannose for improved APC delivery. In vitro, bone marrow-derived dendritic cells took up the mannose-decorated nanoparticles at twice the rate of the unmodified formulation. Intradermal injection of the mannose-functionalized nanoparticles also led to better retention in the lymph nodes, presumably due to increased interaction with APCs. In another study, CCNPs were coloaded with multiple adjuvants.[212] To make the formulation, calcium phosphate nanoparticle cores were loaded with CpG, followed by coating with B16-OVA membrane-associated antigens as well as DAMPs consisting of the heat shock protein αHSP70p inserted into the bilayer membrane. Mice vaccinated with this formulation were able to induce TRP2-specific T cells and OVA-specific T cells at a 7.2-fold higher frequency than the formulation without αHSP70p. The formulation also outperformed all other controls in reducing lung metastases after two B16F10 tumor challenges, and it caused almost complete tumor regression in a therapeutic model when combined with anti-PD-1 checkpoint blockade therapy. In a final example, a CCNP-based vaccine was designed using a core material that could serve as an intrinsic adjuvant.[213] Specifically, thermally oxidized porous silicon was coated with a spermine-modified acetalated dextran using glass capillary microfluidic nanoprecipitation to make the immunostimulatory core. Membrane vesicles from human MDA-MB-231 breast cancer cells were then co-extruded with the cores to make the final nanovaccine. Incubation of the resulting nanoformulation with human peripheral blood monocytes led to a strong immunostimulatory response, as well as an increased ability to inhibit the proliferation of MDA-MB-231 cells.
The CCNP platform has significant potential to aid in the development of personalized vaccine formulations. Since each tumor is unique, the ideal antigen profile to vaccinate against should be defined by the mutational landscape of each individual patient. This is supported by the fact that melanoma patients receiving vaccines containing a mixture of neoantigens uniquely identified by their individual tumor genes could generate T cells that specifically targeted their cancer cells.[190, 214] Further, immunity tailored to more closely reflect the composition of individual tumors could lead to significantly more cancer cell death compared to a non-personalized vaccine.[191] Cell membrane-coated nanovaccines are straightforward to synthesize, and the membrane surface of the particles can be easily modified to introduce additional functionalities. There are also established protocols for deriving single-cell suspensions from tumor material,[215] so future clinical CCNP formulations could potentially be fabricated using membrane derived from a patient’s own tumor.
7. Conclusions
In this progress report, we have provided an overview on the current progress of biomimetic nanovaccines and their potential for personalized medicine. With their unique transport kinetics, antigen profiles, immunostimulatory properties, and targeting abilities, biomimetic nanoparticles have been leveraged for the design of more efficacious vaccine formulations. In the case of infectious disease, the rise in antibiotic resistance poses unique challenges and concerns, which is further compounded by the slow development of new drugs. As a result, vaccination has become an increasingly attractive option for disease management given its ease of use, broad applicability, and ability to generate long-term protection. Biomimetic nanovaccines can be inherently multiantigenic and immunostimulatory, as is the case with OMVs and OMV-coated nanoparticles. A newly emerging approach leverages cell membrane-coated nanoparticles for neutralizing and delivering bacterial toxins for antivirulence vaccination. By vaccinating against the tools that pathogens utilize for survival, this strategy can effectively prevent colonization while limiting the direct selective pressure that drives antibiotic resistance. As the membrane coating can be easily interchanged and toxin mixtures from different bacteria can be used to supply the antigenic material, a countless number of nanotoxoid formulations can be developed, which may ultimately enable more personalized vaccines that can be tailored to individual patient populations with specific risk profiles (Figure 11).
Figure 11.

Personalized biomimetic nanovaccines. For anticancer vaccination, antigenic material can be collected directly from a patient’s resected tumor, formulated into a biomimetic nanoparticle, and then administered back into the patient to promote tumor-specific immunity. For antibacterial vaccination, strain-specific virulence factors or membrane can be immobilized onto nanoparticle substrates, and the resulting complexes can be used to vaccinate patients with an identified risk against the associated pathogen.
While a few vaccines against cancer have been successfully translated, they have not experienced the same level of success as those targeted against infectious diseases. One major challenge is the low immunogenicity of tumors and their associated antigens. Unlike with pathogens, cancer cells originate from a patient’s own healthy tissues, making it exceedingly difficult for the immune system to properly distinguish them. To address some of the obstacles facing anticancer vaccine development, there has recently been an emphasis on the use of biomimetic nanotechnology. Some platforms, such as VLPs, inherently promote inflammatory responses and can produce tumor-targeting CTLs by leveraging a patient’s own cancer cells as an in situ source of antigens. To generate specific immunity, aAPCs have been used to directly prime effector T cells by replicating the necessary biological cues. The cell membrane-coated nanoparticle platform offers some key advantages that can help to overcome tumor diversity, as CCNPs employ cancer cell membrane as a source of diverse antigenic material for eliciting multiantigenic anticancer immunity. Looking toward clinical translation, the cancer cell membrane used to coat the CCNPs may be eventually derived from a patient’s own tumor, which would ensure that the most appropriate set of antigens are used to train the immune system (Figure 11). Ultimately, continued development along the lines of biomimetic nanovaccines will help to improve upon current technologies and may significantly change the clinical landscape for the management of both infectious disease and cancer.
Acknowledgements
This work was supported by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-18-1-0014. J.Z. was supported by a National Institutes of Health 5T32CA153915 training grant from the National Cancer Institute.
Biographies

Jiarong Zhou is a graduate researcher in the Department of NanoEngineering at the University of California San Diego. He received his B.S. in NanoEngineering at the University of California San Diego in 2017. His research involves utilizing biomimetic nanoparticles for immune modulation.

Ronnie H. Fang is an Assistant Project Scientist in the Department of NanoEngineering at the University of California San Diego. He received his Ph.D. in NanoEngineering at the University of California San Diego. His research is focused on leveraging biomimetic nanoparticles for drug delivery and immunoengineering applications.

Liangfang Zhang is a Professor in the Department of NanoEngineering, Chemical Engineering Program and Moores Cancer Center at the University of California San Diego. He received his Ph.D. in Chemical and Biomolecular Engineering at the University of Illinois at Urbana-Champaign. His research aims to create cutting-edge biomimetic nanotechnologies and exploit them for various biomedical applications with a particular focus on drug delivery, biodetoxification, and vaccination.
References
- [1].Kenyon C, Cell 2005, 120, 449. [DOI] [PubMed] [Google Scholar]
- [2].Lovly CM, Carbone DP, Nat. Rev. Clin. Oncol. 2011, 8, 68. [DOI] [PubMed] [Google Scholar]
- [3].Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL, Proc. Natl. Acad. Sci. USA 2001, 98, 10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vargo-Gogola T, Rosen JM, Nat. Rev. Cancer 2007, 7, 659. [DOI] [PubMed] [Google Scholar]
- [5].Nishino M, Ramaiya NH, Hatabu H, Hodi FS, Nat. Rev. Clin. Oncol. 2017, 14, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Webster RM, Nat. Rev. Drug Discov. 2014, 13, 883. [DOI] [PubMed] [Google Scholar]
- [8].Dranoff G, Nat. Rev. Cancer 2004, 4, 11. [DOI] [PubMed] [Google Scholar]
- [9].Fidler IJ, Nat. Rev. Cancer 2003, 3, 453. [DOI] [PubMed] [Google Scholar]
- [10].Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K, N. Engl. J. Med. 2000, 343, 78. [DOI] [PubMed] [Google Scholar]
- [11].Menendez JA, Lupu R, Nat. Rev. Cancer 2007, 7, 763. [DOI] [PubMed] [Google Scholar]
- [12].Ogretmen B, Hannun YA, Nat. Rev. Cancer 2004, 4, 604. [DOI] [PubMed] [Google Scholar]
- [13].Bennadi D, J. Basic Clin Pharm 2013, 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alanis AJ, Arch. Med. Res. 2005, 36, 697. [DOI] [PubMed] [Google Scholar]
- [15].Barbosa TM, Levy SB, Drug Resist. Updat. 2000, 3, 303. [DOI] [PubMed] [Google Scholar]
- [16].Levy SB, Marshall B, Nat. Med. 2004, 10, S122. [DOI] [PubMed] [Google Scholar]
- [17].Davies J, Science 1994, 264, 375. [DOI] [PubMed] [Google Scholar]
- [18].Ochman H, Lawrence JG, Groisman EA, Nature 2000, 405, 299. [DOI] [PubMed] [Google Scholar]
- [19].Meric-Bernstam F, Mills GB, Nat. Rev. Clin. Oncol. 2012, 9, 542. [DOI] [PubMed] [Google Scholar]
- [20].Ogino S, Galon J, Fuchs CS, Dranoff G, Nat. Rev. Clin. Oncol. 2011, 8, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rosenberg SA, Restifo NP, Science 2015, 348, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].de la Fuente-Nunez C, Torres MD, Mojica FJ, Lu TK, Curr. Opin. Microbiol. 2017, 37, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R, Nat. Rev. Drug Discov. 2018, 17, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hamburg MA, Collins FS, N. Engl. J. Med. 2010, 363, 301. [DOI] [PubMed] [Google Scholar]
- [25].Ginsburg GS, McCarthy JJ, Trends Biotechnol. 2001, 19, 491. [DOI] [PubMed] [Google Scholar]
- [26].Fesnak AD, June CH, Levine BL, Nat. Rev. Cancer 2016, 16, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jackson HJ, Rafiq S, Brentjens RJ, Nat. Rev. Clin. Oncol. 2016, 13, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC, Science 2018, 359, 1361. [DOI] [PubMed] [Google Scholar]
- [29].June CH, Sadelain M, N. Engl. J. Med. 2018, 379, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME, Nat. Rev. Cancer 2008, 8, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hotchkiss RS, Monneret G, Payen D, Nat. Rev. Immunol. 2013, 13, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rosenberg SA, Yang JC, Restifo NP, Nat. Med. 2004, 10, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Iwasaki A, Medzhitov R, Science 2010, 327, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Unanue ER, Annu. Rev. Immunol. 1984, 2, 395. [DOI] [PubMed] [Google Scholar]
- [35].Banchereau J, Palucka AK, Nat. Rev. Immunol. 2005, 5, 296. [DOI] [PubMed] [Google Scholar]
- [36].Reis e Sousa C, Nat. Rev. Immunol. 2006, 6, 476. [DOI] [PubMed] [Google Scholar]
- [37].Joffre OP, Segura E, Savina A, Amigorena S, Nat. Rev. Immunol. 2012, 12, 557. [DOI] [PubMed] [Google Scholar]
- [38].Gil-Torregrosa BC, Lennon-Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, van Endert P, Amigorena S, Eur. J. Immunol. 2004, 34, 398. [DOI] [PubMed] [Google Scholar]
- [39].Kumar H, Kawai T, Akira S, Biochem. J. 2009, 420, 1. [DOI] [PubMed] [Google Scholar]
- [40].Kambayashi T, Laufer TM, Nat. Rev. Immunol. 2014, 14, 719. [DOI] [PubMed] [Google Scholar]
- [41].Bousso P, Robey E, Nat. Immunol. 2003, 4, 579. [DOI] [PubMed] [Google Scholar]
- [42].Mosmann TR, Sad S, Immunol. Today 1996, 17, 138. [DOI] [PubMed] [Google Scholar]
- [43].Laidlaw BJ, Craft JE, Kaech SM, Nat. Rev. Immunol. 2016, 16, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Walport MJ, N. Engl. J. Med. 2001, 344, 1058. [DOI] [PubMed] [Google Scholar]
- [45].Batista FD, Harwood NE, Nat. Rev. Immunol. 2009, 9, 15. [DOI] [PubMed] [Google Scholar]
- [46].Mosmann TR, Coffman RL, Annu. Rev. Immunol. 1989, 7, 145. [DOI] [PubMed] [Google Scholar]
- [47].Noelle RJ, Ledbetter JA, Aruffo A, Immunol. Today 1992, 13, 431. [DOI] [PubMed] [Google Scholar]
- [48].Rajewsky K, Nature 1996, 381, 751. [DOI] [PubMed] [Google Scholar]
- [49].Grimm SK, Ackerman ME, Curr. Opin. Biotechnol. 2013, 24, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kawai T, Akira S, Nat. Immunol. 2010, 11, 373. [DOI] [PubMed] [Google Scholar]
- [51].Geddes K, Magalhaes JG, Girardin SE, Nat. Rev. Drug Discov. 2009, 8, 465. [DOI] [PubMed] [Google Scholar]
- [52].Fischer NO, Rasley A, Corzett M, Hwang MH, Hoeprich PD, Blanchette CD, J. Am. Chem. Soc. 2013, 135, 2044. [DOI] [PubMed] [Google Scholar]
- [53].Li H, Li Y, Jiao J, Hu HM, Nat. Nanotechnol. 2011, 6, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].He Q, Mitchell AR, Johnson SL, Wagner-Bartak C, Morcol T, Bell SJ, Clin. Diagn. Lab. Immunol. 2000, 7, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fernandez A, Mesa C, Marigo I, Dolcetti L, Clavell M, Oliver L, Fernandez LE, Bronte V, J. Immunol. 2011, 186, 264. [DOI] [PubMed] [Google Scholar]
- [56].Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Nat. Mater. 2011, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im EJ, Foley MH, Barouch DH, Irvine DJ, Sci. Transl. Med. 2013, 5, 204ra130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ, Nat. Mater. 2017, 16, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kuai R, Yuan W, Son S, Nam J, Xu Y, Fan Y, Schwendeman A, Moon JJ, Sci. Adv. 2018, 4, eaao1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dziubla TD, Karim A, Muzykantov VR, J. Control. Release 2005, 102, 427. [DOI] [PubMed] [Google Scholar]
- [61].Petrovsky N, Aguilar JC, Immunol. Cell Biol. 2004, 82, 488. [DOI] [PubMed] [Google Scholar]
- [62].Silva JM, Zupancic E, Vandermeulen G, Oliveira VG, Salgado A, Videira M, Gaspar M, Graca L, Preat V, Florindo HF, J. Control. Release 2015, 198, 91. [DOI] [PubMed] [Google Scholar]
- [63].Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, Moore JP, Crotty S, Langer R, Anderson DG, Chakraborty AK, Irvine DJ, Proc. Natl. Acad. Sci. USA 2016, 113, E6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Demento SL, Cui W, Criscione JM, Stern E, Tulipan J, Kaech SM, Fahmy TM, Biomaterials 2012, 33, 4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bachmann MF, Jennings GT, Nat. Rev. Immunol. 2010, 10, 787. [DOI] [PubMed] [Google Scholar]
- [66].Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF, Eur. J. Immunol. 2008, 38, 1404. [DOI] [PubMed] [Google Scholar]
- [67].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA, Nat. Biotechnol. 2007, 25, 1159. [DOI] [PubMed] [Google Scholar]
- [68].Cho NH, Cheong TC, Min JH, Wu JH, Lee SJ, Kim D, Yang JS, Kim S, Kim YK, Seong SY, Nat. Nanotechnol. 2011, 6, 675. [DOI] [PubMed] [Google Scholar]
- [69].Embgenbroich M, Burgdorf S, Front. Immunol. 2018, 9, 1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Nat. Nanotechnol. 2019, 14, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X, Porembka MR, Lea J, Frankel AE, Fu YX, Chen ZJ, Gao J, Nat. Nanotechnol. 2017, 12, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ, Nanomedicine 2018, 14, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bale SS, Kwon SJ, Shah DA, Banerjee A, Dordick JS, Kane RS, ACS Nano 2010, 4, 1493. [DOI] [PubMed] [Google Scholar]
- [74].Lin LC, Chattopadhyay S, Lin JC, Hu CJ, Adv. Healthc. Mater. 2018, 7, 1701395. [DOI] [PubMed] [Google Scholar]
- [75].Dakterzada F, Mohabati Mobarez A, Habibi Roudkenar M, Mohsenifar A, Vaccine 2016, 34, 1472. [DOI] [PubMed] [Google Scholar]
- [76].Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi J, Yethon JA, Farokhzad OC, Langer R, Starnbach MN, von Andrian UH, Science 2015, 348, aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bar-Cohen Y, Bioinspir. Biomim. 2006, 1, P1. [DOI] [PubMed] [Google Scholar]
- [78].Jiang Y, Chekuri S, Fang RH, Zhang L, Curr. Opin. Biotechnol. 2018, 58, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, Zhang Y, Ying M, Jiang Y, Kroll AV, Gao W, Zhang L, Adv. Mater. 2017, 29, 1606209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kroll AV, Fang RH, Zhang L, Bioconjug. Chem. 2017, 28, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dehaini D, Fang RH, Zhang L, Bioeng. Transl. Med. 2016, 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pierschbacher MD, Ruoslahti E, Nature 1984, 309, 30. [DOI] [PubMed] [Google Scholar]
- [83].Park J, Singha K, Son S, Kim J, Namgung R, Yun CO, Kim WJ, Cancer Gene Ther. 2012, 19, 741. [DOI] [PubMed] [Google Scholar]
- [84].Danhier F, Vroman B, Lecouturier N, Crokart N, Pourcelle V, Freichels H, Jerome C, Marchand-Brynaert J, Feron O, Preat V, J. Control. Release 2009, 140, 166. [DOI] [PubMed] [Google Scholar]
- [85].Chai Z, Hu X, Wei X, Zhan C, Lu L, Jiang K, Su B, Ruan H, Ran D, Fang RH, Zhang L, Lu W, J. Control. Release 2017, 264, 102. [DOI] [PubMed] [Google Scholar]
- [86].Wei X, Zhan C, Shen Q, Fu W, Xie C, Gao J, Peng C, Zheng P, Lu W, Angew. Chem. Int. Ed. Engl. 2015, 54, 3023. [DOI] [PubMed] [Google Scholar]
- [87].Zhan C, Li B, Hu L, Wei X, Feng L, Fu W, Lu W, Angew. Chem. Int. Ed. Engl. 2011, 50, 5482. [DOI] [PubMed] [Google Scholar]
- [88].Nirthanan S, Charpantier E, Gopalakrishnakone P, Gwee MC, Khoo HE, Cheah LS, Bertrand D, Kini RM, J. Biol. Chem. 2002, 277, 17811. [DOI] [PubMed] [Google Scholar]
- [89].Wei X, Gao J, Zhan C, Xie C, Chai Z, Ran D, Ying M, Zheng P, Lu W, J. Control. Release 2015, 218, 13. [DOI] [PubMed] [Google Scholar]
- [90].Stahl PD, Ezekowitz RA, Curr. Opin. Immunol. 1998, 10, 50. [DOI] [PubMed] [Google Scholar]
- [91].Umamaheshwari RB, Jain NK, J. Drug Target. 2003, 11, 415. [DOI] [PubMed] [Google Scholar]
- [92].Gao W, Thamphiwatana S, Angsantikul P, Zhang L, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hoshino Y, Kodama T, Okahata Y, Shea KJ, J. Am. Chem. Soc. 2008, 130, 15242. [DOI] [PubMed] [Google Scholar]
- [94].Hoshino Y, Urakami T, Kodama T, Koide H, Oku N, Okahata Y, Shea KJ, Small 2009, 5, 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hoshino Y, Koide H, Urakami T, Kanazawa H, Kodama T, Oku N, Shea KJ, J. Am. Chem. Soc. 2010, 132, 6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yoo JW, Irvine DJ, Discher DE, Mitragotri S, Nat. Rev. Drug Discov. 2011, 10, 521. [DOI] [PubMed] [Google Scholar]
- [97].Douglas T, Young M, Science 2006, 312, 873. [DOI] [PubMed] [Google Scholar]
- [98].Mastrobattista E, van der Aa MA, Hennink WE, Crommelin DJ, Nat. Rev. Drug Discov. 2006, 5, 115. [DOI] [PubMed] [Google Scholar]
- [99].Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM, Proc. Natl. Acad. Sci. USA 2002, 99, 11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kotterman MA, Schaffer DV, Nat. Rev. Genet. 2014, 15, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Waehler R, Russell SJ, Curiel DT, Nat. Rev. Genet. 2007, 8, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lichty BD, Breitbach CJ, Stojdl DF, Bell JC, Nat. Rev. Cancer 2014, 14, 559. [DOI] [PubMed] [Google Scholar]
- [103].Russell SJ, Peng KW, Bell JC, Nat. Biotechnol. 2012, 30, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC, Nat. Med. 2000, 6, 821. [DOI] [PubMed] [Google Scholar]
- [105].Zhang X, Wang C, Wang J, Hu Q, Langworthy B, Ye Y, Sun W, Lin J, Wang T, Fine J, Cheng H, Dotti G, Huang P, Gu Z, Adv. Mater. 2018, 30, 1707112. [DOI] [PubMed] [Google Scholar]
- [106].Fang RH, Kroll AV, Gao W, Zhang L, Adv. Mater. 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E, Nat. Nanotechnol. 2013, 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc. Natl. Acad. Sci. USA 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L, Nano Lett. 2014, 14, 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Copp JA, Fang RH, Luk BT, Hu CM, Gao W, Zhang K, Zhang L, Proc. Natl. Acad. Sci. USA 2014, 111, 13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wei X, Gao J, Fang RH, Luk BT, Kroll AV, Dehaini D, Zhou J, Kim HW, Gao W, Lu W, Zhang L, Biomaterials 2016, 111, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Angsantikul P, Thamphiwatana S, Zhang Q, Spiekermann K, Zhuang J, Fang RH, Gao W, Obonyo M, Zhang L, Adv. Ther. 2018, 1, 1800016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ying M, Zhuang J, Wei X, Zhang X, Zhang Y, Jiang Y, Dehaini D, Chen M, Gu S, Gao W, Lu W, Fang RH, Zhang L, Adv. Funct. Mater. 2018, 28, 1801032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wei X, Ying M, Dehaini D, Su Y, Kroll AV, Zhou J, Gao W, Fang RH, Chien S, Zhang L, ACS Nano 2018, 12, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Rao L, Bu LL, Cai B, Xu JH, Li A, Zhang WF, Sun ZJ, Guo SS, Liu W, Wang TH, Zhao XZ, Adv. Mater. 2016, 28, 3460. [DOI] [PubMed] [Google Scholar]
- [117].Hu CM, Fang RH, Zhang L, Adv. Healthcare Mater. 2012, 1, 537. [DOI] [PubMed] [Google Scholar]
- [118].Fang RH, Hu CM, Chen KN, Luk BT, Carpenter CW, Gao W, Li S, Zhang DE, Lu W, Zhang L, Nanoscale 2013, 5, 8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhou H, Fan Z, Lemons PK, Cheng H, Theranostics 2016, 6, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Fitzgerald JR, Foster TJ, Cox D, Nat. Rev. Microbiol. 2006, 4, 445. [DOI] [PubMed] [Google Scholar]
- [121].Nieswandt B, Watson SP, Blood 2003, 102, 449. [DOI] [PubMed] [Google Scholar]
- [122].Yeaman MR, Cell. Mol. Life Sci. 2010, 67, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Glinsky VV, Glinsky GV, Glinskii OV, Huxley VH, Turk JR, Mossine VV, Deutscher SL, Pienta KJ, Quinn TP, Cancer Res. 2003, 63, 3805. [PubMed] [Google Scholar]
- [124].Chen Z, Zhao P, Luo Z, Zheng M, Tian H, Gong P, Gao G, Pan H, Liu L, Ma A, Cui H, Ma Y, Cai L, ACS Nano 2016, 10, 10049. [DOI] [PubMed] [Google Scholar]
- [125].Cheng H, Zhu J-Y, Li S-Y, Zeng J-Y, Lei Q, Chen K-W, Zhang C, Zhang X-Z, Adv. Funct. Mater. 2016, 26, 7847. [Google Scholar]
- [126].Li SY, Xie BR, Cheng H, Li CX, Zhang MK, Qiu WX, Liu WL, Wang XS, Zhang XZ, Biomaterials 2018, 151, 1. [DOI] [PubMed] [Google Scholar]
- [127].Lewis K, Nat. Rev. Drug Discov. 2013, 12, 371. [DOI] [PubMed] [Google Scholar]
- [128].Schwechheimer C, Kuehn MJ, Nat. Rev. Microbiol. 2015, 13, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Tan K, Li R, Huang X, Liu Q, Front. Microbiol. 2018, 9, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Petousis-Harris H, Paynter J, Morgan J, Saxton P, McArdle B, Goodyear-Smith F, Black S, Lancet 2017, 390, 1603. [DOI] [PubMed] [Google Scholar]
- [131].Stevenson TC, Cywes-Bentley C, Moeller TD, Weyant KB, Putnam D, Chang YF, Jones BD, Pier GB, DeLisa MP, Proc. Natl. Acad. Sci. USA 2018, 115, E3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zhang X, Yang F, Zou J, Wu W, Jing H, Gou Q, Li H, Gu J, Zou Q, Zhang J, Vaccine 2018, 36, 1047. [DOI] [PubMed] [Google Scholar]
- [133].Wang X, Thompson CD, Weidenmaier C, Lee JC, Nat. Commun. 2018, 9, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gerritzen MJH, Martens DE, Wijffels RH, van der Pol L, Stork M, Biotechnol. Adv. 2017, 35, 565. [DOI] [PubMed] [Google Scholar]
- [135].Chen L, Valentine JL, Huang CJ, Endicott CE, Moeller TD, Rasmussen JA, Fletcher JR, Boll JM, Rosenthal JA, Dobruchowska J, Wang Z, Heiss C, Azadi P, Putnam D, Trent MS, Jones BD, DeLisa MP, Proc. Natl. Acad. Sci. USA 2016, 113, E3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Chen DJ, Osterrieder N, Metzger SM, Buckles E, Doody AM, DeLisa MP, Putnam D, Proc. Natl. Acad. Sci. USA 2010, 107, 3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Camacho AI, Irache JM, de Souza J, Sanchez-Gomez S, Gamazo C, Vaccine 2013, 31, 3288. [DOI] [PubMed] [Google Scholar]
- [138].Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, Zhang Q, Hu CM, Zhang L, Nano Lett. 2015, 15, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ, Nat. Rev. Microbiol. 2008, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Rasko DA, Sperandio V, Nat. Rev. Drug Discov. 2010, 9, 117. [DOI] [PubMed] [Google Scholar]
- [141].Mellbye B, Schuster M, MBio 2011, 2, e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Dickey SW, Cheung GYC, Otto M, Nat. Rev. Drug Discov. 2017, 16, 457. [DOI] [PubMed] [Google Scholar]
- [143].Angsantikul P, Fang RH, Zhang LF, Bioconjug. Chem. 2018, 29, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Fang RH, Luk BT, Hu CM, Zhang L, Adv. Drug Deliv. Rev. 2015, 90, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Fang RH, Jiang Y, Fang JC, Zhang L, Biomaterials 2017, 128, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hu CM, Fang RH, Copp J, Luk BT, Zhang L, Nat. Nanotechnol. 2013, 8, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hu CM, Fang RH, Luk BT, Zhang L, Nat. Nanotechnol. 2013, 8, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wang F, Fang RH, Luk BT, Hu CJ, Thamphiwatana S, Dehaini D, Angsantikul P, Kroll AV, Pang Z, Gao W, Lu W, Zhang L, Adv. Funct. Mater. 2016, 26, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chen Y, Chen M, Zhang Y, Lee JH, Escajadillo T, Gong H, Fang RH, Gao W, Nizet V, Zhang L, Adv. Healthcare Mater. 2018, 7, 1701366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Wei X, Gao J, Wang F, Ying M, Angsantikul P, Kroll AV, Zhou J, Gao W, Lu W, Fang RH, Zhang L, Adv. Mater. 2017, 29, 1701644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lapek JD Jr., Fang RH, Wei X, Li P, Wang B, Zhang L, Gonzalez DJ, ACS Nano 2017, 11, 11831. [DOI] [PubMed] [Google Scholar]
- [152].Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, Zhang S, Fang RH, Gao W, Nizet V, Zhang L, Proc. Natl. Acad. Sci. USA 2017, 114, 11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Chen W, Zhang Q, Luk BT, Fang RH, Liu Y, Gao W, Zhang L, Nanoscale 2016, 8, 10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Meacham CE, Morrison SJ, Nature 2013, 501, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Ribas A, Wolchok JD, Science 2018, 359, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Pardoll DM, Nat. Rev. Cancer 2012, 12, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Zhang X, Wang J, Chen Z, Hu Q, Wang C, Yan J, Dotti G, Huang P, Gu Z, Nano Lett. 2018, 18, 5716. [DOI] [PubMed] [Google Scholar]
- [158].Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z, Nat. Biomed. Eng. 2017, 1, 0011. [Google Scholar]
- [159].Hu Q, Sun W, Wang J, Ruan H, Zhang X, Ye Y, Shen S, Wang C, Lu W, Cheng K, Dotti G, Zeidner JF, Wang J, Gu Z, Nat. Biomed. Eng. 2018, 2, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Romero D, Nat. Rev. Clin. Oncol. 2017, 14, 140. [DOI] [PubMed] [Google Scholar]
- [161].Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, Lee SW, Gho YS, Nat. Commun. 2017, 8, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Chackerian B, Expert Rev. Vaccines 2007, 6, 381. [DOI] [PubMed] [Google Scholar]
- [163].Roldao A, Mellado MC, Castilho LR, Carrondo MJ, Alves PM, Expert Rev. Vaccines 2010, 9, 1149. [DOI] [PubMed] [Google Scholar]
- [164].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S, Nat. Nanotechnol. 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Peruzzi PP, Chiocca EA, Nat. Nanotechnol. 2016, 11, 214. [DOI] [PubMed] [Google Scholar]
- [166].Czapar AE, Tiu BDB, Veliz FA, Pokorski JK, Steinmetz NF, Adv. Sci. 2018, 5, 1700991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Murray AA, Wang C, Fiering S, Steinmetz NF, Mol. Pharm. 2018, 15, 3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Lebel ME, Langlois MP, Daudelin JF, Tarrab E, Savard P, Leclerc D, Lamarre A, J. Immunol. 2017, 198, 292. [DOI] [PubMed] [Google Scholar]
- [169].Lebel M-È, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A, Nano Lett. 2016, 16, 1826. [DOI] [PubMed] [Google Scholar]
- [170].Jobsri J, Allen A, Rajagopal D, Shipton M, Kanyuka K, Lomonossoff GP, Ottensmeier C, Diebold SS, Stevenson FK, Savelyeva N, PLoS One 2015, 10, e0118096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Shukla S, Myers JT, Woods SE, Gong X, Czapar AE, Commandeur U, Huang AY, Levine AD, Steinmetz NF, Biomaterials 2017, 121, 15. [DOI] [PubMed] [Google Scholar]
- [172].Patel R, Czapar AE, Fiering S, Oleinick NL, Steinmetz NF, ACS Omega 2018, 3, 3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Lee KL, Murray AA, Le DHT, Sheen MR, Shukla S, Commandeur U, Fiering S, Steinmetz NF, Nano Lett. 2017, 17, 4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Hoopes PJ, Wagner RJ, Duval K, Kang K, Gladstone DJ, Moodie KL, Crary-Burney M, Ariaspulido H, Veliz FA, Steinmetz NF, Fiering SN, Mol. Pharm. 2018, 15, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G, Nat. Rev. Immunol. 2017, 17, 97. [DOI] [PubMed] [Google Scholar]
- [176].Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Nat. Rev. Cancer 2012, 12, 860. [DOI] [PubMed] [Google Scholar]
- [177].Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ, Moon JJ, Nano Lett. 2017, 17, 7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Song Q, Yin Y, Shang L, Wu T, Zhang D, Kong M, Zhao Y, He Y, Tan S, Guo Y, Zhang Z, Nano Lett. 2017, 17, 6366. [DOI] [PubMed] [Google Scholar]
- [179].Deng G, Sun Z, Li S, Peng X, Li W, Zhou L, Ma Y, Gong P, Cai L, ACS Nano 2018, 12, 12096. [DOI] [PubMed] [Google Scholar]
- [180].Yu G-T, Rao L, Wu H, Yang L-L, Bu L-L, Deng W-W, Wu L, Nan X, Zhang W-F, Zhao X-Z, Liu W, Sun Z-J, Adv. Funct. Mater. 2018, 28, 1801389. [Google Scholar]
- [181].Eggermont LJ, Paulis LE, Tel J, Figdor CG, Trends Biotechnol. 2014, 32, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Perica K, De Leon Medero A, Durai M, Chiu YL, Bieler JG, Sibener L, Niemoller M, Assenmacher M, Richter A, Edidin M, Oelke M, Schneck J, Nanomedicine 2014, 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J, Xi X, Gao X, Ma G, Xie HY, ACS Nano 2017, 11, 10724. [DOI] [PubMed] [Google Scholar]
- [184].Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ, Small 2015, 11, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Hickey JW, Vicente FP, Howard GP, Mao HQ, Schneck JP, Nano Lett. 2017, 17, 7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kosmides AK, Necochea K, Hickey JW, Schneck JP, Nano Lett. 2018, 18, 1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lin JC, Chien CY, Lin CL, Yao BY, Chen YI, Liu YH, Fang ZS, Chen JY, Chen WY, Lee NN, Chen HW, Hu CJ, Nat. Commun. 2019, 10, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Schumacher TN, Schreiber RD, Science 2015, 348, 69. [DOI] [PubMed] [Google Scholar]
- [189].Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP, Science 2015, 348, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, Nature 2017, 547, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, Tureci O, Nature 2017, 547, 222. [DOI] [PubMed] [Google Scholar]
- [192].Dagogo-Jack I, Shaw AT, Nat. Rev. Clin. Oncol. 2018, 15, 81. [DOI] [PubMed] [Google Scholar]
- [193].van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ, Nat. Rev. Cancer 2016, 16, 219. [DOI] [PubMed] [Google Scholar]
- [194].Keenan BP, Jaffee EM, Semin. Oncol. 2012, 39, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Liu SY, Wei W, Yue H, Ni DZ, Yue ZG, Wang S, Fu Q, Wang YQ, Ma GH, Su ZG, Biomaterials 2013, 34, 8291. [DOI] [PubMed] [Google Scholar]
- [196].Prasad S, Cody V, Saucier-Sawyer JK, Saltzman WM, Sasaki CT, Edelson RL, Birchall MA, Hanlon DJ, Nanomedicine 2011, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ, Mol. Pharm. 2007, 4, 47. [DOI] [PubMed] [Google Scholar]
- [198].Hanlon DJ, Aldo PB, Devine L, Alvero AB, Engberg AK, Edelson R, Mor G, Am. J. Reprod. Immunol. 2011, 65, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Shi GN, Zhang CN, Xu R, Niu JF, Song HJ, Zhang XY, Wang WW, Wang YM, Li C, Wei XQ, Kong DL, Biomaterials 2017, 113, 191. [DOI] [PubMed] [Google Scholar]
- [200].Cheung AS, Koshy ST, Stafford AG, Bastings MM, Mooney DJ, Small 2016, 12, 2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Ochyl LJ, Bazzill JD, Park C, Xu Y, Kuai R, Moon JJ, Biomaterials 2018, 182, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Fang RH, Zhang L, Annu. Rev. Chem. Biomol. Eng. 2016, 7, 305. [DOI] [PubMed] [Google Scholar]
- [203].Fang RH, Kroll AV, Zhang L, Small 2015, 11, 5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Kroll AV, Jiang Y, Zhou JR, Holay M, Fang RH, Zhang LF, Adv. Biosys. 2019, 3, 1800219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Zhuang J, Ying M, Spiekermann K, Holay M, Zhang Y, Chen F, Gong H, Lee JH, Gao W, Fang RH, Zhang L, Adv. Mater. 2018, 30, 1804693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Fang RH, Hu CMJ, Zhang LF, Expert Opin. Biol. Ther. 2012, 12, 385. [DOI] [PubMed] [Google Scholar]
- [207].Luk BT, Fang RH, Hu CM, Copp JA, Thamphiwatana S, Dehaini D, Gao W, Zhang K, Li S, Zhang L, Theranostics 2016, 6, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y, Kong M, Qi Y, Tan S, Zhang Z, ACS Nano 2015, 9, 6918. [DOI] [PubMed] [Google Scholar]
- [209].Lokhov PG, Balashova EE, J. Cancer 2010, 1, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, Zhang L, Adv. Mater. 2017, 29, 1703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Yang R, Xu J, Xu L, Sun X, Chen Q, Zhao Y, Peng R, Liu Z, ACS Nano 2018, 12, 5121. [DOI] [PubMed] [Google Scholar]
- [212].Kang T, Huang Y, Zhu Q, Cheng H, Pei Y, Feng J, Xu M, Jiang G, Song Q, Jiang T, Chen H, Gao X, Chen J, Biomaterials 2018, 164, 80. [DOI] [PubMed] [Google Scholar]
- [213].Fontana F, Shahbazi MA, Liu D, Zhang H, Makila E, Salonen J, Hirvonen JT, Santos HA, Adv. Mater. 2017, 29, 1603239. [DOI] [PubMed] [Google Scholar]
- [214].DeWeerdt S, Nature 2017, 552, S76. [DOI] [PubMed] [Google Scholar]
- [215].Leelatian N, Doxie DB, Greenplate AR, Mobley BC, Lehman JM, Sinnaeve J, Kauffmann RM, Werkhaven JA, Mistry AM, Weaver KD, Thompson RC, Massion PP, Hooks MA, Kelley MC, Chambless LB, Ihrie RA, Irish JM, Cytometry B Clin. Cytom. 2017, 92, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]