

Abstract
Receptor tyrosine kinases (RTKs) play important roles in cell growth, motility, differentiation, and survival. These single-pass membrane proteins are grouped into subfamilies based on the similarity of their extracellular domains. They are generally thought to be activated by ligand binding, which promotes homodimerization and then autophosphorylation in trans. However, RTK interactions are more complicated, as RTKs can interact in the absence of ligand and heterodimerize within and across subfamilies. Here, we review the known cross-subfamily RTK hetero-interactions and their possible biological implications, as well as the methodologies which have been used to study them. Moreover, we demonstrate how thermodynamic models can be used to study RTKs and to explain many of the complicated biological effects which have been described in the literature. Finally, we discuss the concept of the RTK interactome: a putative, extensive network of interactions between the RTKs. This RTK interactome can produce unique signaling outputs; can amplify, inhibit, and modify signaling; and can allow for signaling back-ups. The existence of the RTK interactome could provide an explanation for the irreproducibility of experimental data from different studies and for the failure of some RTK inhibitors to produce the desired therapeutic effects. We argue that a deeper knowledge of RTK interactome thermodynamics can lead to a better understanding of fundamental RTK signaling processes in health and disease. We further argue that there is a need for quantitative, thermodynamic studies that probe the strengths of the interactions between RTKs and their ligands and between different RTKs.
Graphical Abstract
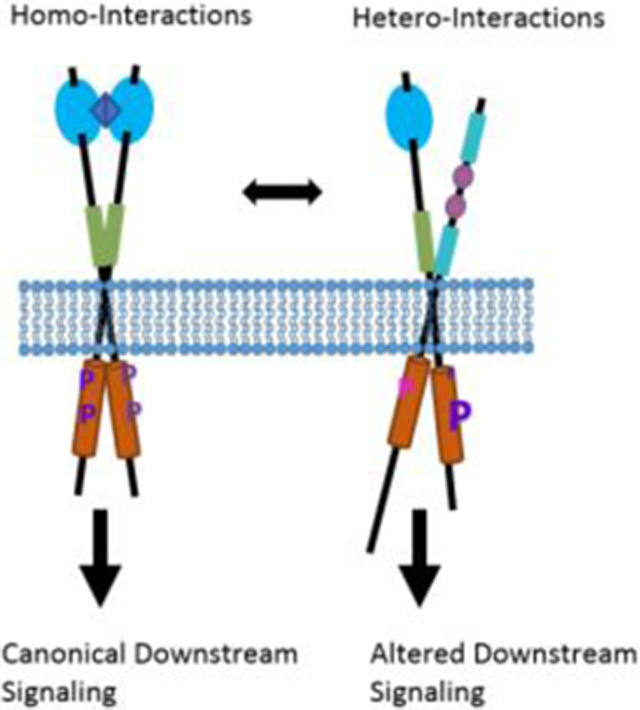
1. Introduction
Receptor tyrosine kinases (RTKs) are the second largest family of membrane receptors. There are 58 RTKs in humans, and as shown in Figure 1, they are grouped into 20 subfamilies based on the homology of their extracellular (EC) domains. Unlike G-protein coupled receptors (GPCRs), channels, and transporters, the RTKs have a single transmembrane (TM) helix which connects an N-terminal extracellular ligand-binding domain to a C-terminal intracellular (IC) kinase domain. Since a single TM helix is not efficient at transducing conformational changes across the plasma membrane, the receptors of this family rely on lateral interactions to become activated and initiate downstream signaling cascades. These signaling cascades, in turn, control many critically important biological processes, including cell growth, survival, and differentiation. Recent years have brought significant progress in our understanding of the physical interactions that regulate RTK function. RTKs are best known for forming signaling homodimers, but it is now clear that they are capable of engaging many interaction partners. While these interaction partners are from diverse classes of proteins, the focus of this review is the interactions between RTK from different subfamilies.
Figure 1.

Cartoon representation of the 58 RTKs grouped into 20 RTK subfamilies. It has long been appreciated that hetero-interactions can occur within a subfamily. This review focuses on the hetero-interactions between subfamilies. Key RTK features and domains are depicted as distinct shapes as explained in the legend. The plasma membrane is shown in blue. The N-terminal extracellular domains are shown above the membrane. RTK subfamily names are listed above the receptor, while the names of the individual RTKs in the subfamily are listed below, with common alternative names listed in parentheses. In general, the structure of all members of a given subfamily are very similar, with minor differences in the size of the full-length proteins and of the individual domains. Notable exceptions are that (i) the fifth Ig domain (third closest to the membrane) of VEGFR3 is proteolytically cleaved and held together by disulfide bonds; (ii) LTK lacks both MAM groups and the LDLa domain, and it is accordingly much shorter than the depicted ALK; and (iii) STYK1 appears to lack a signal sequences and does not seem to localize to the plasma membrane despite having a putative transmembrane domain.
*The LMTK proteins were predicted to be tyrosine kinases, but later experiments demonstrated that they only have serine/threonine kinase activity1,2. If they are not counted as RTKs, there are only be 55 total RTKs and 19 RTK subfamilies in humans.
2. Overview of the Current Paradigm of RTK Interactions
2.1. Interactions Between RTKs in the Plasma Membrane and Their Response to Ligands
The general structure of RTKs can be seen in Figure 1. Their N-terminal EC domains, which are usually several hundred amino acids long, bind activating ligands and contain characteristic arrays of structural domains.4–6 This is followed by a TM helix, a juxtamembrane (JM) segment, a kinase domain of approximately 275 amino acid residues, and in some cases a C-terminal tail that is up to 300 amino acid residues long. Contact between two kinase domains is needed to stimulate catalytic activity, which results in the cross-phosphorylation of receptor molecules and phosphorylation of cytoplasmic substrates, ultimately activating signaling cascades that control cell behavior.5,7–9
After the discovery of RTKs in the 1970s,10–12 RTK research was governed by the canonical model of activation. This model postulates that RTKs are monomeric in the absence of ligand and only form dimers upon ligand binding to the EC domain. However, it has now been shown that RTKs can form dimers even in the absence of ligand (Figure 2A).13–20 Different RTKs form unliganded dimers with different energies of interaction, but their existence appears to be largely universal. Unliganded dimers are stabilized through lateral interactions between the kinase, JM, and TM domains, while the EC domains usually inhibit dimerization.21–25
Figure 2.

(A) A simplified model of RTK dimerization and activation for a generic RTK and ligand (blue diamond). Inactive monomers dimerize to form unliganded dimers with basal activity. Ligand binding induces a conformational change and enhances phosphorylation (purple P), leading to full signaling activity. The process can be fully described by thermodynamic cycles such as those in Figure 3. (B) Cartoon depicting the law of mass action for RTKs which exist in a monomer-dimer equilibrium. The single circles represent the RTK monomers, and the overlapping circles represent the RTK dimers (D). Three different RTK concentrations are depicted. As the total RTK concentration increases from left to right, so does the fraction of receptors which are dimeric. Dimeric fraction is defined as the fraction of total RTKs (T) which exist as a dimer: .
In accordance with the law of mass action, RTK expression levels control the relative distribution of monomers and dimers (Figure 2B). As interactions are required for RTK activity, the value of the two-dimensional dissociation constants and the expression levels exert control over activity. This means that even in the absence of ligand, increased expression—which is common in many cancers26–28—can shift the equilibrium from a predominantly monomeric to a predominantly dimeric population, triggering signaling cascades.
Even though RTK activity in the absence of ligand is possible, ligands are still important for normal function. The ligands are usually polypeptides, and they are commonly referred to as “growth factors.” They usually bind to the receptors picomolar to nanomolar affinity,29–34 and they stabilize the dimers by directly interacting with two copies of the receptor and/or by causing conformational changes. Ligands have been shown to induce structural changes that arguably propagate along the entire length of the RTK, through the TM domain, and ultimately affect the kinase domain (Figure 2A). In general, the kinase domain can exist in both a catalytically active and inactive state, and ligand binding results in the kinase domain being converted from its inactive state into its active state.35,36
Studies on endothelial growth factor receptor (EGFR) and other ErbB (erythroblastic oncogene B) family members, fibroblast growth factor receptors (FGFRs), and vascular endothelial growth factor receptor 2 (VEGFR2) have indicated that ligand binding leads to a conformational switch in the TM helix, implying that the EC and TM domains are structurally coupled.13,21,37,38 A study by Sinclair et al. further demonstrated that the TM and JM domains of EGFR are coupled, as different ligands induce different TM and JM conformations.39 Moreover, data by Bell et al. suggest that the TM and IC domains are also structurally coupled, and that the TM domain dimer structure controls kinase activity.40 There is also evidence that the conformation of the kinase domain can be sensed by the EC domain, as different inhibitors binding to the EGFR kinase domain have different effects on EGF binding.41
However, others believe that ligand-induced structural changes in the EC domain are not propagated to the IC domain, because the linkers between the different regions are unstructured. For example, Springer et al. have argued that a single ligand-bound EGFR conformation can be coupled to multiple kinase domain arrangements.42 Furthermore, a single EGFR kinase domain arrangement can couple to two different EC states,43 suggesting that the EC and IC domains of EGFR can change conformations independently of each other. One possible explanation is that the different ligands differentially stabilize RTK dimers, leading to different kinetic lifetimes and signaling.44 It has also been proposed that ligand binding causes changes in the EC domain that alter the local cell membrane, and these alterations are sensed by the intracellular domain.45
The debate over the physical effects of ligand binding, briefly overviewed above, highlights the fact that many of the most fundamental questions about RTK activity are still unanswered, despite intense research since the 1970s. There is remarkable consensus, however, that lateral interactions between the RTKs are absolutely critical for RTK activation. This has led to the development of models that explain how RTK homo-interactions occur and regulate biological activity. Despite the fact that a wide range of hetero-interactions have been described in the literature, these interactions are rarely taken into account when developing mechanistic models. We argue here for the importance of updating these models to account for the numerous hetero-interactions which are known to occur and to affect RTK activity.
2.2. RTK Interactions Regulate RTK Function
In order for an RTK to become active, its kinase domain must be phosphorylated, and this occurs when the two kinases in a dimer cross-phosphorylate each other on select tyrosines. Accordingly, productive lateral interaction is required for RTK activity, and the unphosphorylated monomers are inactive. Once phosphorylated, the activity of the kinase domain is enhanced. As a result, the kinase domain can bind adaptor proteins9,46–50 and phosphorylate other molecules,51–53 and this causes activation of downstream signaling pathways. Although RTKs have a diverse range of roles, the pathways they mediate often lead to cell growth and proliferation, and abnormally high phosphorylation is linked to many cancers.54–57 In fact, several RTKs were originally identified as products of oncogenes.
Recent work has shown that unliganded dimers are often phosphorylated on selected tyrosines,21,58,59 which is known as “basal activity,” and this appears to play important roles in pathogenesis. Many cancers exhibit increased expression of RKTs in the absence or even loss of ligand.55,56,60,61 Furthermore, there are pathogenic mutations that predominantly affect the basal phosphorylation of the receptor. The G380R mutation in FGFR3, which is the genetic cause of achondroplasia, the most common form of human dwarfism, increases FGFR3 dimerization and phosphorylation in the absence of ligand.62 Intriguingly, the formation of EphA2 unliganded dimers actually inhibits oncogenic signaling.59 EphA2’s oncogenic activity is caused by the soluble kinase Akt phosphorylating serines on the EphA2 monomers. The formation of the unliganded dimers decreases serine phosphorylation, increases tyrosine phosphorylation, and decreases EphA2-controlled cell migration, which generally correlates with metastasis and invasiveness.
Although unliganded dimers are important, ligands are a vital part of RTK signaling. RTKs often require ligand to initiate downstream signaling cascades. For example, FGFR2 has been shown to be a Grb2-stabilized dimer in the absence of ligand, and it is phosphorylated to a low degree.58,63,64 Ligand binding leads to Grb2 phosphorylation, which drives the dissociation of Grb2, allowing other proteins to bind, and this triggers downstream signaling by the receptor.
3. Thermodynamics of RTK Interactions
The use of thermodynamic cycles allows for rigorous analysis of RTK interactions. These cycles account for all possible receptor-receptor and receptor-ligand interactions, and for all of the possible pathways: from monomers, which are inactive, to liganded dimers, which are signaling-competent and active. These thermodynamic cycles can be used to interpret experimental data and predict the concentrations of the different types of dimers using measured equilibrium constants and the total concentrations of receptors and ligands.
One such thermodynamic cycle is shown in Figure 3A. This is the so-called “binding in an aggregating system” model, described in the classical text by Wyman and Gill,3 and it is applicable for monomeric ligands which bind to a receptor which homodimerizes. The interactions are governed by three dimerization constants—K1, (dimerization of unliganded monomers), K2 (dimerization of a liganded monomer with an unliganded monomer), and K3 (dimerization of liganded monomers)—and three ligand binding constants—L1 (ligand binding to a monomer), L2 (ligand binding to an unliganded dimer), and L3 (ligand binding to a dimer with one ligand already bound); these constants are fully defined to the right of the cycle in Figure 3A. All paths along the cycle which share a beginning and ending state are thermodynamically equivalent, and therefore, the constants are inter-dependent on each other. For instance, K1*L2 = L1*K2 and K2*L3 = L1*K3.
Figure 3.

Thermodynamic cycles which allow for rigorous analysis of RTK interactions. The interactions are governed by the RTK dimerization constants, Ki, and the ligand binding constants, Li; these constraints are defined to the right of the cycles. The concentrations of the receptors are in molecules per unit area while the concentrations of the ligands are in molecules per unit volume. These constants are inter-dependent on each other, as paths along the cycle which share a beginning and ending state are thermodynamically equivalent. Once the dimerization and ligand binding constants are known, it is possible to predict the concentrations of monomers and dimers—and in particular, the concentration of the signaling-competent, liganded dimers—for any given receptor and ligand concentrations. (A) The “binding in an aggregating system” model,3 describing the homodimerization of a receptor (X) which binds monomeric ligand (L). The receptor can form homodimers (XX) and bind its ligand as either a monomer (LX), a dimer (LXX), or a liganded dimer (LLXX). (B) A model depicting the homodimerization of a receptor (X) which binds a dimeric ligand (L). The liganded monomer (LX) can interact with an unliganded monomer (X) to form the liganded dimer (LXX). Alternatively, two liganded monomers (LX) can interact to form the liganded dimer (LXX) while releasing a ligand (L) into solution. (C) A model describing the homo- and heterodimerization of receptors X and Y, (XX, YY, and XY) where X binds monomeric ligand (L), but Y does not bind ligand. (D) A model describing the homo- and heterodimerization of receptors X and Y, (XX, YY, and XY) where X binds dimeric ligand, but Y does not bind ligand. (E) A model describing the heterodimerization of X and Y (XY) and X and Z (XZ), where X binds dimeric ligand (L), but neither Y nor Z bind ligand, nor can they interact with each other; the three receptors also form homodimers (XX, YY, and ZZ). (F) A model describing the homo- and heterodimerization of receptors X and Y (XX, YY and XY), where both X and Y bind dimeric ligand (LX, LXX, LY, and LYY), and the heterodimer does so as well (LXY).
The equilibrium constant K1 can be determined by experiments conducted in the absence of ligand that report on the two-dimensional concentrations of monomers, [X], and dimers, [XX], in the membrane. Experiments can also be performed in the presence of ligand, and these can measure the two-dimensional concentration of the RTK in the membrane, [X]Total, and the concentration of the ligand bound to the receptors in the membrane, [L]Bound. These two measurable parameters depend on the equilibrium constants; [X]; and on the free, soluble ligand concentration, [L]:
| (1) |
| (2) |
When [X] is determined from equation (2) and substituted into (1), equation (1) provides a connection between all the measurable parameters and the three unknowns: L1, L2, and L3. Thus, measurements of [L] and [X]Total, performed at four or more different ligand concentrations, allow for L1, L2, and L3 to be determined using least square fitting procedures. The dimerization constants K2 and K3 can be then calculated from K1, L1, L2, and L3. It is important to note that the plasma membrane is best viewed as a two-dimensional structure; accordingly, the receptor concentrations are given in receptors per unit area (e.g., mol/μm2), not receptors per unit volume (e.g., mol/μm3). The unbound ligand is, however, in three dimensions, and hence some of the association constants have rather unusual units.
The utility of the thermodynamic cycles approach was first demonstrated in a comprehensive study of EGF binding to EGFR, in which both the concentration of the ligand and the concentration of the receptor were varied.65 By fitting the model to a large experimental data set, the researchers were able to determine all the thermodynamic constants for homoassociation and ligand binding. This work demonstrates that the behavior of RTKs in cells can largely be explained by a relatively simple physical-chemical model. Once the dimerization and ligand binding constants are known, it is possible to predict the concentrations of monomers and dimers, and in particular, the concentration of the signaling-competent liganded dimers, for any given receptor and ligand concentration.
While EGF is monomeric, many RTKs are activated by dimeric ligands. For instance, VEGF is a disulfide linked dimer, and one VEGF ligand binds to and activates the VEGFR2 dimer.66,67 The relevant thermodynamic cycle in the case of a dimeric ligand is shown in Figure 3B. In this case, a liganded monomer can interact with an unliganded monomer to form the fully liganded dimer. Alternatively, two liganded monomers can interact to form the liganded dimer while releasing a ligand into solution. The equilibrium constraints are the same as in the monomeric ligand case, except that K3 involves two liganded monomers releasing a bound ligand upon dimerization, and there is no L3. All the equilibrium constants can be determined in a manner similar to that of the monomeric ligand case.
Thermodynamic cycles can be also used to account for heterodimerization. Figures 3C and D correspond to the cases of monomeric and dimeric ligands, respectively. In these models, we assume that there is a second RTK (Y), which can participate in both homodimerization (YY) and heterodimerization (XY), but it does not bind any ligand. Similar cycles can be created to describe more complex interaction models, such as the formation of higher order oligomers, the binding of multiple ligands to an RTK, the binding of a ligand to multiple RTKs, the binding of a ligand to both RTKs and the heterodimer (Figure 3E), and the occurrence of multiple hetero-interactions (Figure 3F). Such thermodynamic cycles can explain how hetero-interactions can decrease the concentration of liganded homodimers, how changes in the total amount of a receptor can change the relative amounts of monomers and dimers, and how drugs which decrease the stability of homodimers can increase the concentration of heterodimers. These models could guide our understanding of the effect of hetero-interactions on biological function, as discussed in the “Using Thermodynamic Models to Understand Hetero-interactions” section.
For the thermodynamic cycles to provide accurate predictions of the concentration of the different types of dimers, the association constants describing the strengths of interaction need to be experimentally determined. There are many methods which can be used to study protein-protein interactions, but few of them work with membrane proteins. Still fewer are suitable for making detailed quantitative measurements of interaction constants. Below, we discuss methods that are used to study membrane protein interactions, and we highlight methods that can produce quantitative information.
4. Methods to Study Protein-Protein Interactions in the Membrane
4.1. Affinity Capture
Many methods for determining protein-protein interactions (and other proteins-biomolecule interactions) can be described as affinity capture. The basic idea is that a protein of interest is modified such that it can be selectively purified in a way that preserves interactions with other proteins. In a related strategy, the protein of interest marks nearby proteins, and the marked proteins are selectively captured. Once the proteins are isolated, the identity of unknown proteins can be determined. One of the most common methods for determining the identity of the purified proteins is mass spectroscopy. It is beyond the scope of this review to discuss the details of different types of mass spectroscopy (MS) experiments and analyses, but this has been extensively reviewed elsewhere.68–72 Affinity capture techniques vary from describing the interactions between two proteins with a high degree of detail to high throughput methods that generate thousands of pairs of interactions that are analyzed with statistical machine learning methods and other big data analytic techniques. In general, these methods provide evidence of interactions and provide qualitative information on how these interactions change under different condition, but they cannot provide quantitative information about the strength of interactions.
4.1.1. Coimmunoprecipitation
Coimmunoprecipitation involves isolating a protein from cell lysates.73,74 An antibody is bound to the protein of interest, and then the antibody-protein complex is precipitated with beads conjugated to antibody binding proteins (usually protein A or protein G). After washing off the rest of the cell lysate and separating the complex from the bead, the presence or absence of a specific interaction partner is determined, typically using a western blot. Of note, this is not a high throughput method, and it only provides binary or semiquantitative (e.g., the amount of the binding partner detected via western blot increased in cells treated with ligand relative to untreated cells) information.
This is one of the few methods which does not need tagged versions of the proteins and hence can be used to study proteins as they are naturally expressed by the cells (endogenous proteins), but it does require extracting the protein out of its native environment. It cannot distinguish between two proteins directly interacting and the proteins interacting as part of a larger complex. Weak interactions might not be detected, as the interactions have to be strong enough to persist through the cell lysing process and initial precipitation. This is a particular concern for membrane proteins, as interactions might be lost moving from the hydrophobic membrane to the aqueous assay conditions. Crosslinking agents can be used to stabilize the interactions, although this raises concerns about spurious interactions appearing.
It is possible to get information about the oligomer size by comparing how the precipitated complex runs on a gel in comparison to a molecular ladder of known weight (i.e., does the weight match a dimer, trimer, tetramer, etc.). However, this is not the most robust means of determining oligomer size, as the shape of the complex affects how it runs on the gel, and this process generally requires crosslinking to increase stability. Furthermore, differences in post translational glycosylation makes it difficult to know the “true” weight of a given oligomer size. Despite these limitations, coimmunoprecipitation can provide evidence of interactions, it is widely performed in many labs due to the lack of expensive equipment and use of commercially available reagents, and it has been the most commonly used technique to study RTK hetero-interactions.
4.1.2. Two-Hybrid Screening
The first two-hybrid assay to study protein-protein interaction was developed in 1989 by Fields and Song using yeast.75 Known as yeast two-hybrid (Y2H), the basic idea is that a reporter gene will only be transcribed if the protein of interest (bait) interacts with another protein (prey). This is accomplished by splitting the Gal4 transcriptional activator into its two domains and attaching the bait to the DNA-binding domain and the prey to the activating domain. Transcription of the reporter gene—for example, a gene which encodes for an essential nutrient lacking from the media—will only occur if the DNA-binding domain and the activating domain are in close proximity (importantly, direct binding is unnecessary), which can only happen if the bait and prey interact with each other. By creating large cDNA libraries encoding for prey fused to the activation domain, Y2H can be used as a high throughput method to quickly determine thousands of protein interactions. Accordingly, Y2H and closely related variants have been used to create large scale interactome maps, including a map of human RTK-phosphatase interactions.76,77 The technical details of all the different two-hybrid systems are beyond the scope of this review, and they have been reviewed elsewhere.78,79
It is important to note the limitations of Y2H. Fusing the proteins to the DNA-binding and activating domains could affect their ability to interact with other proteins. The proteins must still be able to fold and interact properly in an environment different from where the proteins are normally found, as the interactions are being studied in the nucleus of yeast cells; moreover, the normal associated proteins and post-translational modifications will be lacking. Studying membrane proteins in an aqueous environment is virtually impossible, as they are prone to misfold and aggregate. This aggregation contributes to Y2H having a high rate of false positives.
Not all of these issues can be eliminated, but advancements in the methodology have helped to reduce their effect, particularly in regard to membrane proteins. A variant known as membrane yeast two-hybrid (MYTH) solves the aqueous environment problem by allowing the interactions to be studied in the membrane.80,81 MYTH uses ubiquitin split into two stable moieties, and the bait and prey are tagged with these moieties. The tag on the prey is fused with a reporter molecule (the E. coli DNA-binding domain LexA connected to the herpes simplex virus VP16 transcriptional activation domain), and when the bait and the prey interact, the two moieties become in close contact, allowing for the formation of a pseudoubiquitin. This pseudoubiquitin is recognized by deubiquinating enzymes (DUBs), which release the reporter molecule, freeing it to enter the nucleus and activate the reporter gene. Since DUBs are only found in the cytosol, MYTH is only applicable to membrane proteins that contain a cytosolic portion which can be tagged. A variant of this method for use in mammalian cells called mammalian-membrane two-hybrid (MaMTH) has been developed using a different reporter molecule and reporter gene.82
MYTH and MaMTH allow membrane proteins to be studied in the membrane. Moreover, using mammalian cells allows for mammalian proteins to be studied in an environment where relevant adaptor proteins and post-translational modifications may be present. As with the original Y2H system, concerns related to the effect of tagging the proteins and using an overexpression system still apply. None of these methods can be used to obtain interaction strengths or other quantitative information, but they can screen thousands of potential interactions to reveal hits that can then be investigated using quantitative techniques.
4.1.3. BioID
A high-throughput method for detecting potential protein-protein interactions is known as proximity-dependent biotin identification (BioID).83 In this method, a biotin ligase is fused to the protein of interest, and then the modified protein is introduced into cells. By supplementing the culture media with biotin, proteins that are proximal to the protein of interest become biotinylated, and then can be isolated (e.g., using a streptavidin pull-down) and identified, typically using MS. The details of how to implement BioID and couple it with traditional affinity purification methods have been reviewed elsewhere,84,85 but a brief discussion of the method is given below.
BioID allows for protein interactions to be studied in live cell, and a large number of interactions can easily be identified. However, this method does not directly probe for interactions, but rather detects proteins which are nearby the target. It has been estimated that approximately 50% of detections occur within 20–30 nm of the target protein, but the exact resolution is unknown.83 Moreover, the biotin ligase used in the development of BioID (BirA*) can only react with primary amines, meaning interaction partners lacking these cannot be detected. There is some possibility of non-specific binding, and larger proteins could be more likely to be identified by MS. All this means that BioID results should not be taken as proof of protein-protein interaction, but as positive hits in a screen which warrant further investigation.
As with any method involving a fusion protein, the effects of this modification need to be kept in mind. The biotin ligase is 35 kD, which is a little larger than GFP, and whether or not this affects protein interaction or function must be assessed for each protein of interest. The biotinylation of the interacting proteins could alter the secondary modifications or the interactions of the proteins, and the addition of biotin to the media could alter biological activity. Since the fusion protein needs to be introduced into cells, typically by transient or stable transfection, BioID cannot study fully endogenous protein interactions, although the biotinylated interaction partners can be endogenous.
4.2. Proximity Ligation Assay
The proximity ligation assay (PLA) is a method for detection of specific proteins and their interactions.86,87 It works by attaching DNA strands to the proteins of interest such that when the proteins come in close contact, a ligation reaction between the two DNA strands can occur. This DNA ligation product can be PCR amplified, allowing for sensitive detection of protein interactions. By attaching the DNA probes to primary or secondary antibodies against the proteins of interest, this assay can be performed using commercially available kits for a wide variety of targets.
A modified version of PLA was developed to work in situ.88,89 The DNA probes are designed such that their linkage creates a circular DNA strand. This serves as a template for a rolling-circle amplification (RCA) reaction, allowing for a large amount of rolling circle product (RCP) to be generated. In around an hour, DNA polymerase can form an RCP almost one micron in dimeter, which is near the detection limit of conventional light microscopy. By hybridizing the RCP to fluorescently labeled oligonucleotides complementary to the detection probe, the RCP becomes labeled with hundreds of fluorophores, making for easy visualization.
PLA allows for the detection of protein-protein interaction in a relatively native environment, although the need for antibody-staining generally requires the cells to be fixed and permeabilized. The exact distance limit for detection varies based on the size of the antigen binding agent and the oligonucleotide sequence, but it has been roughly estimated that if the probes are within a few tens of nm, the interaction can be detected.88 It is important to emphasize that PLA does not directly report on protein-protein interaction, but it indicates that two proteins of interest are within close proximity. Accordingly, it provides no information on the strength, geometry, or stoichiometry of interactions; furthermore, it cannot distinguish between proteins which directly interact with each other, and those which are proximal due to mutual association with a third molecule or as part of a complex.
4.3. FRET
Förster resonance energy transfer (FRET) involves the non-radiative transfer of energy from one fluorophore (the donor) to another (the acceptor). This energy transfer decreases as a function of the distance between the two fluorophores to the sixth power, making it highly sensitive to small changes in distance.90 It is accordingly commonly used as a conformational probe, since a change from an open or extended conformation to a closed or compact conformation can easily be seen as a change from low to high FRET (given appropriate labeling). FRET can be also used to study the association of proteins, both in solution and in the plasma membrane.
Furthermore, quantitative FRET methodologies exist which take into account the concentrations of the donor and acceptor that can be used to determine homo- and hetero-interaction strengths. These methodologies work in both cell-derived and live-cell systems, and typically the proteins of interest are genetically tagged with the donor and acceptor fluorophores and introduced using stable or transient transfection. Transient transfection is advantageous in these experiments, as it allows a broad range of concentration to be sampled—receptor concentration ranges spanning two to three orders of magnitude have been reported.91–93
Quantitative Imaging FRET (QI-FRET) is one such quantitative FRET method, and it can be performed using a traditional confocal microscope.94–96 The appropriately labeled sample is imaged three times: (i) a donor scan which gives the donor fluorescence when the donor is excited, (ii) a FRET scan which gives the acceptor fluorescence when the donor is excited, and (iii) an acceptor scan which gives the acceptor fluorescence when the acceptor is excited. These three scans impose a major constraint on the fluorophores which can be used, as the donor should not be excited by the acceptor excitation source, and there must be minimal bleed through between the two emission channels. A common choice for QI-FRET is to use a member of the YFP family as the donor and mCherry as the acceptor.92,97 This fluorophore constraint is eliminated in the similar Fully Quantified Spectral Imaging FRET (FSI-FRET), which uses spectrally resolved two-photon imaging to acquire two scans—a FRET scan and an acceptor scan—and can use any two fluorophores which form a FRET pair.98 The method has both high sensitivity and a high signal-to-noise ratio, but it requires a specialized microscope.99
By measuring donor and acceptor concentrations and FRET efficiencies, association curves can be generated with the QI-FRET and the FSI-FRET methods, and these can be fit to different oligomerization models to determine the best fit model and the ΔG of interaction. In the case of membrane proteins, which are confined to the effectively two-dimensional plasma membrane, such fits require that the contribution of “proximity” or “stochastic” FRET is also taken into account.100–102 Detailed protocols to correct for this contribution are available in the literature.102
Although to date quantitative FRET has mostly been used to study homo-interactions, it is easily adapted to study hetero-interactions. In the homo-interaction case, a portion of the proteins of interest are labeled with the donor, and the rest are labeled with the acceptor. In the hetero-interaction case, all of one protein is labeled with the donor, and all of the other with the acceptor. The only specific interactions which will result in FRET are the hetero-interactions. Thus, for membrane proteins, specific hetero-interactions will result in FRET that is higher than the proximity FRET. The calculation of hetero-dimerization constants requires that all homo- and hetero-interactions are taken into account; detailed protocols can be found in the literature.97,103
FRET has also been used to gain information about the size of the oligomer, by analyzing the dependence of FRET on the donor-to acceptor ratio.102,104,105 Although two-color, quantitative-FRET techniques are good at distinguishing between monomers (i.e., no interactions), dimers, and higher order oligomers, they struggle to precisely determine the size of oligomers larger than dimers.102 In some cases, oligomer size and geometry have been determined by histogramming the pixel-level apparent FRET values for a cell.106,107 This histogram is then fit to one or more Gaussians, and the fit for hundreds of cells which exhibit single peaks can be histogrammed into a “meta-histogram.” By fitting the meta-histogram to multiple Gaussians and comparing the number of peaks and the fit parameters to the theoretical FRET values for different donor and acceptor configurations, oligomer size and geometries are determined.
Overall, FRET has provided valuable information about the interactions of several RTKs. However, FRET requires labeling, and thus it cannot be used to study endogenous proteins. The fluorescent proteins are large (~27 kDa), and they may affect RTK interactions or function. Accordingly, the effect of the label must be assessed for each RTK of interest through control assays comparing RTK function with and without labels. FRET only assess the interactions between the labeled proteins, and other interaction partners will be missed if they are unlabeled.
4.4. Statistical and Correlational Fluorescence Methods
There are several fluorescent methods which rely upon statistics and correlated movement or correlated fluorescence to provide information about the structure and interaction of molecules. These methods do not directly report on interactions per se, but rather, they indicate the apparent size of or number of fluorophores in the labeled complex. This indirect nature precludes the methods from determining if the proteins are directly interacting or part of a larger complex. Although these methods have generally been applied to homo-interactions, they can be adapted to study hetero-interactions. As with all techniques which require labeling the protein of interest, they cannot be used study endogenous proteins, and it is possible that the modification affects normal protein function or interactions. Some of these methods are briefly discussed below, and ideally a combination of two or more methods should be used in order to get a reliable estimate of the oligomer size.
4.4.1. Fluorophore Localization Imaging with Photobleaching
Fluorophore localization imaging with photobleaching (FLImP) involves using single particle tracking to determine the distance between two fluorophores.108,109 This is accomplished by using changes in the diffraction-limited image spots when one of them photobleaches and fitting the point spread function. The distances of thousands of traces are histogrammed, and the histogram is decomposed into different peaks. The peaks correspond to different distances between the fluorophores, and this provides information about the oligomer size and geometry of the labeled proteins. FLImP is able to determine lateral distances between identical fluorophores that are within about 60 nm, although fixation is required to obtain resolution below 10 nm. There are, of course, limitations on how close two distances can be and still be resolved into two separate peaks, and the number of peaks cannot always be unambiguously determined. Although FLImP can be performed on commercially available microscopes, it is computationally intense, and the analysis is rather technical and requires care to ensure that accurate conclusions are reached. It is a highly statistical method, and errors—e.g., sample drift, autofluorescence, and the crowded cell environment—must carefully be taken into account. As FLImP uses single particle tracking, relatively low concentrations are required.
4.4.2. Number and Brightness
Number and brightness (N&B) is based on the fact that although the same number of fluorophores will give the same average fluorescence intensity regardless of oligomer size, the variance in the intensity fluctuations will be different for different oligomer sizes.110 As an illustration of this concept, a dimer diffusing out of the imaging window will cause a larger fluctuation in fluorescent intensity than a monomer. There is also a two color version which uses the cross-variance of the intensity fluctuations of the two fluorescent channels, and this is well suited to study hetero-interactions.111 Experimentally, N&B works by rapidly taking a stack of images of the same region, and then computing the average florescence intensity and the variance across the stack for each pixel. This allows for the number of particles and the molecular brightness of each pixel to be determined. In theory, the brightness of a dimer is twice that of a monomer and higher order oligomers scale linearly. However, in practice, issues with fluorophore maturation, quenching, and other complications can cause the brightness of a dimer to be less than double the brightness of a monomer. Accordingly, the most accurate results will be obtained when experimental results are compared controls of known oligomer size rather than just scaling everything relative to the monomer. Care must also be taken to account for photobleaching and cell movement.
4.4.3. Spatial Intensity Distribution Analysis
Spatial intensity distribution analysis (SpIDA) is a spatial method which works by fitting super Poisson distributions to intensity histograms.112,113 One or more regions of interest are analyzed from a single image, and oligomer size is determined by comparing the determined brightness to the brightness of a monomer control. As with N&B, it is possible that the brightness does not scale linearly with oligomer size, and best results are obtained when the experimental brightness is compared to controls of multiple oligomer sizes. SpIDA can be applied to both live cells and fixed samples, and thus endogenous proteins can be studied using immunofluorescent staining.
4.4.4. Pulsed Interleaved Excitation Fluorescence Cross-Correlation Spectroscopy
In general, fluorescence correlation spectroscopy (FCS) uses the temporal fluorescent intensity fluctuations through a small excitation volume coupled with correlational analysis to determine the diffusion coefficients and the concentration of a fluorophore.114–117 This is expanded in fluorescence cross-correlation spectroscopy (FCCs), which uses the auto- and cross-correlation information of two different colors.118–120 If the fluctuations are occurring simultaneously in both channels, it means that the fluorophores must be moving together as part of a complex. For more detailed information about FCS and FCCS, please see one of the many extensive reviews on the subjects.121–125 A further expansion of this methodology is pulsed interleaved excitation FCCS (PIE-FCCS), which uses rapid alternation between multiple excitation sources such that the fluorescence emission generated from one excitation pulse is complete before the next excitation pulse arrives.126,127 Accordingly, spectral crosstalk can be eliminated, as the excitation source of each detected photon is known, and hence higher resolution is obtained. PIE-FCCS does not directly report on oligomer size or stoichiometries, but rather, it allows for a determination of the relative size of co-diffusing species.
4.4.5. Raster Image Correlation Spectroscopy
Another fluorescence correlation method is raster image correlation spectroscopy (RICS).128 This method involves repeated raster scanning over a viewing window to create an image stack. The 2D spatial correlation of the fluorophore is then calculated using the image stack, and this yields diffusion coefficients and concentration. There is also a two-color, cross-correlation version which is more suitable for heterointeractions.129 This can all be done on a commercial laser scanning microscope. Care needs to be taken to account for photobleaching and cell movement. Furthermore, as is the case with PIE-FCCS, RICS does not directly report on oligomer size or stoichiometries, but comparisons can be made using the size information that can be determined from the diffusion coefficients.
4.5. Multistep Photobleaching
A single molecule technique using total internal reflection florescence (TRIF) microscopy for determining oligomer size is known as multistep photobleaching.130,131 An area containing a small number (~50–200) of fluorescent spots is photobleached by repeated imaging, and for each spot, the number of bleaching steps is counted. If each subunit is fluorescently labeled, the number of bleaching steps corresponds to the oligomer size of the complex. Using single color labeling, it is possible to obtain some information about hetero-interactions by comparing both individually-labeled cases to the dual-labeled case. It should be possible to study hetero-interactions by using two-color labeling where each component is labeled with a different color,132 although we are unaware of this having been applied to live cells.
Of note, in some cases, a smaller number of bleaching steps will be observed than the oligomer size due to a fluorophore being non-fluorescent, a subunit not being labeled, or a photobleaching event not occurring during the imaging time; all of this must be accounted for in the analysis. Multistep photobleaching does not directly report on interactions, but rather, everything which sequentially photobleaches is assumed to be part of the same complex, as it remained within a small volume during the image acquisition time. Furthermore, it struggles to distinguish discrete bleaching steps for complexes with more than five labeled subunits. As with all signal molecule techniques, it can only be used with low concentrations of fluorescent molecules.
4.6. Biomolecular Fluorescence Complementation
Biomolecular fluorescence complementation (BiFC) involves two non-fluorescent protein fragments coming together to form a fluorescent molecule.133,134 It is a type of protein-fragment complementation assay, which is where the proteins of interest are fused to fragments of a third protein, and when those fragments combine, a detectable reaction occurs. There are many possible choices of the third protein which can be spilt into fragments—e.g., ubiquitin135, β-galactosidase,136 luciferase,137 and TEV138—but we focus on BiFC due to its ability to directly visualize interactions and its applicability to living cells. (Of note, two-hybrid screening is also a type of complementation assay, but this was described earlier in the “Affinity Capture” section due to the way the results are typically analyzed.) To study homo-interactions, a portion of the protein of interest is labeled with one fluorophore fragment (A), and the rest with the other fluorophore fragment (B); to study hetero-interactions, all of one protein of interest is labeled with one fluorophore fragment (A), and all of the other protein of interest is labeled with the other fluorophore fragment (B). In both cases, when the labeled proteins of interest interact, the two fluorophore fragments combine (AB) into a fluorescent protein. Multiple interactions can be studied simultaneously, as a multicolor BiFC variant exists where a protein of interest is labeled with a fluorophore fragment (C) which can combine with two different fluorophore fragments (D and E) such that these combinations (CD and CE) gives different fluorescence,139 and split fluorophores have been developed that have a wide array of colors.140
BiFC has many strengths, including that it can be used to directly study interactions in live cells using conventional fluorescent microscopes without the need for specialized software. It can detect weak interactions, has good spatial resolution, and the range of colors available allows for simultaneous visualization of multiple interactions. However, it still has many drawbacks. It requires modified proteins, so it cannot be used to study endogenous proteins, and the modifications can affect normal protein function or interactions. Once the two fragments have combined, the fluorophore may take almost an hour to mature (i.e., after formation, it may not be fluorescent until almost an hour later); this means that BiFC cannot be used to study shortlived interactions or protein dynamics in real time. Moreover, since the two fluorophore fragments combine irreversibly, the proteins effectively become permanently linked. Accordingly, interactions involving dynamic association and dissociation will be disrupted, and hence thermodynamic calculations cannot be made. Although BiFC directly reports on interactions, it cannot determine whether two molecules are directly interacting with each other or whether the two molecules are part of a larger complex. Moreover, determining the concentration of the protein of interest is difficult, as doing so requires a secondary label.
5. Interaction Databases
There are several online databases that curate the literature and facilitate searches for biomolecular interactions. Depending on the database, these contain hundreds of thousands of protein-protein and protein-biomolecule interactions based on experiments, homology modeling, and computational predictions, as well as post translational modifications. Although the databases are not specific for RTKs or membrane proteins, they can be useful starting points for trying to understand all the possible RTK interactions. Some of the larger databases are MIntAct141 (a merger of MINT142 and IntAct,143 which also provides training to researchers to use the platform and emphasizes the adaption of standards), BioGRID144 (Biological General Repository for Interaction Datasets, which includes protein, genetic, and chemical interactions for major model organisms), IID145 (Integrated Interactions Database, which focuses on tissue specific protein interactions), DIP146 (Database of Interacting Proteins, only experimentally determined protein interactions), HPRD147 (Human Protein Reference Database, manually curated and only including human protein interactions), MIPS mammalian protein-protein interaction database148 (mammalian protein-protein interactions focusing on individually performed experiments), and Reactome149 (general biological pathways and reactions).
6. RTK Hetero-Interactions Within the Same Subfamily
Most of the work in the field has focused on RTK homodimerization or homo-oligomerization. The studies on hetero-interactions have generally investigated heterodimers between two members of the same RTK family, as it has long been appreciated that these can form due to sharing a common ligand. Heterodimerization is viewed as a means to enhance diversity in signaling by a ligand which is capable of binding two or more related receptors. Indeed, it has been shown that ligand binding to RTK homodimers and heterodimers leads to the phosphorylation of different tyrosines, and to the recruitment of different adaptor proteins which mediate different biological responses (overviewed below).
For example, the members of the ErbB family form same family heterodimers depending on their expression levels and which ligands are present, and this allows for increased signaling complexity and an enhanced ability to respond to changing stimuli.35,150,151 There are four members of the ErbB family—EGFR, ErbB2, ErbB3, and ErbB4—and they are important for cell division, survival, and migration; organ growth and development; and maintenance of adult tissue.152–157 ErbB overexpression and mutations are associated with many cancers,158–162 making the ErbBs major drug targets.163–166 The receptors and their ligands are associated with several other disorders, including decreased ErbB4 activity playing a role in schizophrenia167,168 and a link to psoriasis.169,170
Different ligands bind to different ErbBs, and this can cause different dimer pairings to form. Endothelial growth factor (EGF) binds to EGFR, while Neu differentiation factor (NDF, which is a form of neuregulin-1) binds to ErbB3 and ErbB4; in fibroblast cells, EGF promotes EGFR-ErbB2 heterodimers while NDF promotes ErbB2-ErbB3 and ErbB2-ErbB4 heterodimers.171 Different interactions cause different tryosines to become phosphorylated and different adaptor proteins to bind. For instance, EGFR homodimers bind c-Cbl, while EGFR-ErbB2 heterodimers do not.172 While EGFR homodimers and EGFR-ErbB4 heterodimers both bind Shc, only the homodimer binds Grb2.171 Intriguingly, ErbB2 has no known EGF-like ligand, and it appears to be largely dependent on heterodimerization for its activity, which may explain why ErbB2 is the preferred binding partner of the other three ErbBs.173 ErbB3 is also highly dependent on heterodimerization for its signaling, as its kinase activity is impaired, and hence it requires heterodimerization to become phosphorylated.174
This dependence on heterodimerization has been seen in numerous cell types. For instance, in hematopoietic cells, variants expressing only one ErbB could not be mitotically activated, but a variant with both ErbB2 and ErbB3 (and to a lesser extent, one with EGFR and ErbB3) had strong lamellipodia activity in response to ligand.175 A similar result has been seen in the neoplastic transformation of fibroblast cells, were ErbB2 or ErbB3 alone do not have a large effect, but in combination are transformative, and this is associated with increased ErbB3 phosphorylation.176 The presence of heterodimers can affect the time frame of signaling, as in myeloid cells, EGFR homodimers are quickly degraded, terminating the signal, but EGFR-ErbB2 and -ErbB3 heterodimers are recycled to the cell surface, prolonging in the signal.177
Another RTK family where hetero-interactions play a key role is the VEGFRs. There are three VEGFRs—VEGFR1, VEGFR2, and VEGFR3—and they are critical for angiogenesis,66,178,179 with VEGFR2 being of particular importance, while VEGFR3 is key for the development of the lymphatic system.180,181 Inhibiting the VEGFRs has been the focus of major clinical effort, because many cancers overexpress VEGFRs or VEGF ligands or have VEGFR mutations,182–186 and there are many such inhibitors now on the market.187 Furthermore, aberrant angiogenesis is part of the pathology of numerous other diseases, including macular degeneration,188,189 diabetic retinopathy,190,191 and rheumatoid arthritis.192,193
The role of VEGFR1 during development is somewhat nonintuitive, as mice which do not express VEGFR1 have abnormally organized vasculature and die in utero,194 but mice with a truncated version which completely lacks the kinase domain appear to develop normally.195 VEGFR1 is capable of binding the primary ligand of VEGFR2, VEGFA, as well as forming heterodimers with VEGFR2.196 Accordingly, it is believed that the primary role of VEGFR1 is to serve as a negative regulator of VEGFR2 by tightly controlling the amount of free VEGFA and VEGFR2 homodimers.66,197,198 This mechanism of regulation is more complicated than just sequestration, as the heterodimers do have some activity. They have been found to induce migration and PI3 and PLCγ phosphorylation in response to VEGFA, although to a different degree than VEGFR2 homodimers.199
Moreover, VEGFR1 can directly phosphorylate VEGFR2, as a kinase dead version (i.e., the kinase domain can be phosphorylated as normal, but it cannot phosphorylate another molecule) of VEGFR2 is phosphorylated by VEGFR1.200 A variant of VEGFA which can only bind VEGFR1 induces different activity than the VEGFR1 specific ligand placenta growth factor (PlGF). This VEGFA variant is unable to rescue PlGF−/− mice, and it suppresses VEGFR2 phosphorylation while PlGF enhances it; the two ligands cause different VEGFR1 tyrosines to become phosphorylated and induce different gene expression profiles. A ligand that binds specifically to the VEGFR1-VEGFR2 heterodimer (a dimer of PlGF and VEGFE, a VEGFR2 specific ligand) also has a unique effect: it causes VEGFR2 phosphorylation, but does not appear to affect heterodimer formation, and relative to VEGFA or VEGFE, it only weakly activates ERK1/2 and does not induce cell proliferation.201 Of note, VEGFR1-VEGFR2 heterodimers can form in the absence of ligand, but both VEGFA and a VEGFA-PlGF dimer—which had previously been found in the media of several human tumor lines and has a mitogenic effect on cells, but significantly less than that of VEGFA202,203—increase heterodimer formation.200
VEGFR2 is also capable of forming a heterodimer with VEGFR3. There are five tyrosines on the carboxy tail of VEGFR3 which are normally phosphorylated in homodimers, but only three of them are phosphorylated in VEGFR2-VEGFR3 heterodimers.204 In endothelial cells which naturally have both VEGFR2 and VEGFR3, VEGFR2-VEGFR3 heterodimers could not be detected by coimmunoprecipitation after addition of VEGFA, which only very weakly binds to VEGFR3, but could be detected after addition of VEGFC, which binds to both receptors205. However, using PLA, a small number of VEGFR2-VEGFR3 heterodimers were observed in the absence of ligand, with a small increase occurring after VEGFA addition, and a large increase occurring after VEGFC addition. This emphasizes the importance of using multiple techniques to study hetero-interactions, and it demonstrates how immunoprecipitation in particular is liable to miss weak interactions. The same study found that after VEGFA addition, there were approximately eight-fold more VEGFR2 homodimers than heterodimers, and after VEGFC addition, there were approximately two-fold more VEGFR3 homodimers than heterodimers. In three dimensional embryoid bodies, VEGFA, and to a lesser extent VEGFC, induce angiogenic sprouting. Heterodimers, as seen by PLA, where significantly more concentrated in these sprouts than the stalk. Since the degree of heterodimerization is dependent on the concentrations of the receptors and ligands, different distributions are likely to be seen during different stages of angiogenesis, and hence the formation of heterodimers allows for increased signal complexity and fine-tuning, and it provides a means for the same set of receptors to cause different functional outputs during different processes.
Several other families have important hetero-interactions, and we briefly discuss them here. (For more information on RTK families not described here, see their first appearance in the “Specifics of known RTK Cross-Subfamily Hetero-Interactions” section.) PDGFRα and PDGFRβ can dimerize to form an αβ heterodimer, and this interaction results in a different tyrosine being phosphorylated than in the homodimer cases206 and unique downstream effects.207 In our lab, we have shown that truncated versions of the FGFRs containing the EC and TM domains can form heterodimers in the absence of ligand, and that their stabilities are similar to the homodimer stabilities.97 MET and Ron form heterodimers and directly phosphorylate each other, amplifying and sustaining the signaling of both pathways.208,209 Many hetero-interactions are known to occur between members of the Eph family, including that EphB1 and EphB4 can cross-phosphorylate and activate EphB6;210,211 EphB2 co-clusters with and phosphorylates EphA3, and the interactions appears to modulate cell retraction and segregation signaling;212 EphB6 suppresses EphA2 phosphorylation on serine 897 and anti-apoptosis signaling;213 and EphA4 interacts with EphB2 and enhances ephrin-B2 induced phosphorylation, and this interaction is important for regulating cell mitogenic activity and may play a role in the differential effect of the different ephrin ligands.214 ROR1 and ROR2 form heterodimers, and it is believed that this interaction helps regulate Wnt-5a signaling, which is critical for the formation of synapses in hippocampal neurons.215 Tyro3 and AXL heterodimerize, and the interaction appears to amplify the signaling of both receptors.216 InsR and IGF-1R frequently heterodimerize in many tissues, and the heterodimers often form to a higher degree than expected by a simple expression level analysis, especially in cancer.217–220 The two Tie (tyrosine kinase with Ig and EGF homology domains) receptors, which are important for vasculature development and adult homeostasis, also form heterocomplexes.221,222 Tie1, which has no known activating ligand, appears to negatively regulate Tie2 by forming ligand-independent dimers with it, and these heterodimers decrease Tie2 phosphorylation and downstream signaling; different Tie2 ligands stabilize or destabilize these heterodimers to different degrees, allowing for fine control over Tie2 activity.223–226 It is clear that there are a wide range of different RTK hetero-interactions within the same subfamilies. Many, but not all, are caused by ligands which bind to both receptors, and they are important for signaling regulation, amplification, and diversification.
7. Ligands Binding to Multiple Subfamilies
As evidenced by RTK hetero-interactions from the same subfamily, heterodimers are often caused by a ligand which is capable of binding two different receptors. There are several known instances of a ligand associated with one RTK subfamily interacting with another subfamily, and we briefly overview some of these interactions here. The membrane bound ephrins typically interact with the Eph receptors, but they can also interact with several other RTKs. For instance, Ret is necessary for proper ephrin-A growth signaling in neurons, and Ret knockout mice have inhibited peroneal axon projections.227 Direct interaction between Ret and ephrin-A2 and ephrin-A5 can be seen in neurons by coimmunoprecipitation and PLA. In neurons, addition of ephrin-A5 enhances neuronal branching and synaptic density induced by the TrkB ligand brain-derived neurotrophic factor (BDNF), and RNAi silencing of TrkB diminishes this effect.228 All three Trks coimmunoprecipitate with ephrin-A5 and ephrin-A7 when a receptor and ligand are exogenously expressed in CHO cells. Moreover, in a neuronal cell line, addition of the TrkA ligand nerve growth factor (NGF) induces interaction between TrkA and ephrin-A5, and the binding appears to enhance Akt signaling. Normally, ephrin-B1 causes dissociation of embryonic cells in Xenopus embryos, but addition of FGFs inhibits this process.229 Ephrin-B1 becomes phosphorylated after FGF addition as long as kinase active FGFR1 is present, and ephrin-B1 coimmunoprecipitates with phosphorylated FGFR1 and FGFR2. Furthermore, ephrin-B1 is phosphorylated after addition of platelet-derived growth factor (PDGF), and this appears to be due to an interaction with a platelet-derived growth factor receptor (PDGFR).230
PDGFRα and PDGFRβ have also been found to interact with VEGFA. In MSCs that have the PDGFRs but not the VEGFRs, addition of VEGFA increases migration and proliferation, and inhibition or knockdown of the PDGFRs abolishes this effect; VEGFA and the PDGFRs were seen to interact via coimmunoprecipitation that was stabilized by crosslinking.231 This interaction was also seen using an isotope labeled version of VEGFA, and VEGFA was found to competitively inhibit PDGFs from binding, although it was able to activate PDGFRα to some degree.232
Interestingly, there is some evidence for the existence of hetero-ligands composed of ligands associated with two different subfamilies. PDGF-BB was found to interact with FGF2 by surface plasmon resonance (SPR), and this interaction lead to the formation a PDGF-BB(FGF2)2 trimer, as determined by both steady-state fluorescence and solid-phase immunoassay, with an estimated one-step dislocation constant in the pico- to femtomolar squared (pM2-fM2) range.233 Computational modeling indicates that a VEGF-EGF dimer could exist, and that it could bind to EGFR with normal affinity, but it would have impaired binding to a VEGFR.234 In experiments using a synthetic VEGF-EGF hetero-ligand purified from yeast, the hetero-ligand binds to EGFR with almost ten-fold higher affinity than EGF, and it binds to VEGFR2 with about the same affinity as VEGF, as determined by ELISA.235 Additionally, the hetero-ligand induces phosphorylation of both VEGFR2 and EGFR, and a version with a radio labeled cargo is successfully internalized by cells.
Such multiple subfamily binding ligands could result in cross-subfamily hetero-interactions similar to the hetero-interactions seen within a family. Moreover, given that RTKs dimerize in the absence of ligand, it is not unreasonable to expect that RTKs that do not share a common ligand could physically interact with each other. Cross-subfamily interactions are made more plausible by the fact that the kinase domains across the RTK subfamilies are closely related;36 in fact, drugs designed to inhibit RTK kinase domains often inhibit several RTKs.236,237 Accordingly, kinase-kinase interactions help stabilize homodimers in the absence of ligand,13,21 and the kinase domains of RTKs from different subfamilies are likely to also help stabilize hetero-interactions. The TM domains may also be contributing to cross-subfamily hetero-interactions. Below, we explore the literature on RTK hetero-interactions form different subfamilies, and we discuss their possible biological significance.
8. Overview of Known RTK Cross-Subfamily Hetero-Interactions
RTK cross-subfamily interactions have been reported for over a dozen different subfamilies, and these interactions involve around half of the RTKs. These interactions occur in a wide variety of circumstances, and their function is varied and often not well understood. However, it is clear that these interactions have important biological consequences, particularly in regard to cancer progression and its treatment, and hopefully future quantitative experiments will help clarify their nature and function. Table 1 at the beginning of the section provides a list of RTKs known to engage in cross-subfamily hetero-interactions. Below is a detailed overview of the contents of Table 1. Readers primarily interested in an overview of cross-subfamily hetero-interactions and a discussion of their possible effects may proceed to the “Using Thermodynamic Models to Understand Hetero-Interactions” section.
Table 1.
A List of Known Interactions Between RTKs of Different Subfamilies
| RTK Subfamily | RTK | Known Cross-Subfamily Hetero-Interactions |
|---|---|---|
| Eph | EphA2 | EGFR,253,254 ErbB2255,256 |
| EphA4 | FGFR1,257–259 FGFR2,257,259 FGFR3,257,259 FGFR4257,259 | |
| EphA7 | RYK260 | |
| EphB2 | RYK260,261 | |
| EphB3 | RYK260–262 | |
| ErbB | EGFR | AXL,263–265 EphA2,253,254 FGFRla,266 FGFR3a,267 IGF-1R,268,269 MET,270–278 PDGFRα,279,280 PDGFRβ,281–283 Ron,276,284–287 ROR1288,289, STYK1a,290 TrkAa,291 TrkBa292–295 |
| ErbB2 | AXL,263,296,297 EphA2,255,256 FGFR1a,266 FGFR3a,298 IGF-1R,299–301 MET,273,302 TrkA,303 TrkB304 | |
| ErbB3 | FGFRla,266,305 FGFR2a,306 FGFR3a,298 IGF-1R,301,307,308 MET,272,273 ROR1288,289,309 | |
| ErbB4 | AXL263 | |
| FGF | FGFR1 | EphA4,257–259 EGFRa,266 ErbB2a,266 ErbB3a,266,305 IGF-lRa,305 METa,266,305 PDGFRα266,305,310 |
| FGFR2 | EphA4,257,259 ErbB3a306 | |
| FGFR3 | EGFRa,267 EphA4,257,259 ErbB2a,298 ErbB3a,298 KIT311 | |
| FGFR4 | EphA4257,259 | |
| Ins | IGF-1R | EGFR,268,269 ErbB2,299–301 ErbB3,301,307,308 FGFRla,305 METa,312,313 Ron314,315 |
| MET | MET | AXL,263,316 EGFR,270–278 ErbB2,273,302 ErbB3,272,273 FGFR1a,266,305 IGF-1Ra,312,313 RET,273 RQR1,289,317 VEGFR2318 |
| Ron | EGFR,276,284–287 IGF-1R,314,315 PDGFRβ319 | |
| MuSK | MuSK | ROR1320 |
| PDGF | KIT | FGFR3311 |
| PDGFRα | EGFR,279,280 FGFR1266,305,310 | |
| PDGFRβ | AXL,263 EGFR,281–283 Ron,319 VEGFR2321,322 | |
| PTK7 | PTK7 | ROR2,323,324 VEGFR1,325 VEGFR2326,327 |
| RET | RET | MET,273 TrkAa,328,329 TrkBa,330 VEGFR2331,332 |
| ROR | ROR1 | EGFR,288,289 ErbB3,288,289,309 MET,289,317 MuSK320 |
| ROR2 | PTK7323,324 | |
| RYK | RYK | EphA7,260 EphB2,260,261 EphB3260–262 |
| STYK1 | STYK1 | EGFRa290 |
| TAM | AXL | EGFR,263–265 ErbB2,263,296,297 ErbB4,263 MET,263,316 PDGFRβ,263 VEGFR2a333 |
| TYRO3 | MET316 | |
| Trk | TrkA | EGFRa,291 ErbB2,303 RETa328,329 |
| TrkB | EGFRa,292–295 ErbB2,304 RETa330 | |
| VEGF | VEGFR1 | PTK7325 |
| VEGFR2 | AXLa,333 MET,318 PDGFRβ,321,322 PTK7,326,327 RET331,332 |
There is indirect evidence for this interaction, but no direct experimental demonstration that this specific interaction occurs.
8.1. Eph Receptors Interact with Multiple RTKs
We begin our overview of known interactions between RTKs of different subfamilies with the Eph receptors. The Eph receptors (erythropoietin-producing human hepatocellular carcinoma) are the largest class of RTKs, having 14 members in humans. They are split into nine EphAs and six EphBs, based on their ability to bind the Ephrin-A and Ephrin-B ligands. Ephrin ligands are membrane proteins located on adjacent cells, and the Eph-Ephrin interaction causes bidirectional signaling: Eph receptor dimerization and higher order oligomerization followed by phosphorylation causes forward signaling, while the Ephrins can dimerize and trigger reverse signaling in the adjacent cell.238–240 This complex signaling is important for many cellular processes,241 such as neuronal development and axon guidance,242,243 migration and proliferation,214,244,245 inflammation,246 and cardiovascular development.247 Additionally, aberrant Eph signaling is associated with many medical conditions including cancer,248–250 bone and joint disorders,251 and cardiovascular disease.252
8.1.1. EphB2 and EphB3 Interact with RYK
The first discovered cross-subfamily hetero-interactions involving an Eph receptor are those of EphB2 and EphB3 interacting with RYK (related to tyrosine kinase). RYK is a kinase dead receptor which binds Wnts and Frizzled334 and is important for Wnt signaling.335 It is generally involved in planar cell polarity,336,337 axon guidance,338 neuronal differentiation,339 and stem cell maintenance.340 The interactions between RYK and EphB2 and RYK and EphB3 were discovered due to the similarity between the phenotype of RYK null mice and mice deficient in both EphB2 and EphB3.260 RYK null mice have craniofacial deformities consistent with a complete cleft of the secondary platelet, shortened limbs, and most die on the day of birth. In transient transfection cell culture experiments, hetero-complexes of both RYK-EphB2 and RYK-EphB3 (mice RYK with human EphB2 and EphB3) coimmunoprecipitated from HEK 293T cells, and tyrosine phosphorylation of the kinase dead RYK was observed in both cases. (Of note, Ryk-EphA7 hetero-complexes also coimmunoprecipitated, but no RYK phosphorylation was detected.) This cross-phosphorylation appears to be unidirectional, as co-expressing RYK with a kinase dead mutant of EphB3 did not cause detectable RYK phosphorylation. Importantly, RYK, EphB2, and EphB3 (along with EphA7) all overlap spatiotemporally in the developing palatal shelves and tongue, and hence Ryk-EphB2 and Ryk-EphB3 interactions appear critical for proper murine craniofacial development.
Additionally, RYK, EphB2, and EphB3 (along with ephrin-B1 and ephrin-B2) all express in the cerebellum of mice and rat brains.262 In a transient transfection experiment involving COS-7 cells, rat RYK coimmunoprecipitated with EphB3, and mutational studies indicated that the leucine rich motifs of the extra cellular domain are critical for interaction, while the kinase domain is not. When GFP-labeled RYK was overexpressed in embryonic cortical brain slices, cell migration was inhibited, but overexpression of GFP-labeled RYK without the leucine rich domains did not have this effect. These data indicate that RYK may regulate cortical cell migration through its interactions with the Eph receptors and ligands.
Key differences have been observed between human RYK and the murine analog. In a coimmunoprecipitation study also performed in HEK 293T cells, it was found that although human RYK interacts with both EphB2 and EphB3, neither is able to phosphorylate RYK.261 Murine RYK coimmunoprecipitates with AF-6260—a cell junction-associated scaffold protein which is the target of activated Ras members341 and can associate with EphB2 and EphB3342—while human RYK does not.261 It is possible that these differences reflect a difference in the role of RYK in humans and mice, but it is also possible that the HEK 293T cells lack a co-receptor or a posttranslational modification required for the interactions.
Although the exact role of the RYK-EphB2 and RYK-EphB3 interactions remains to be determined, it has been proposed that these interactions regulate the Eph receptor signaling that becomes distorted in human craniofrontonasal syndrome.343 This is an X-linked syndrome involving mutations in the gene that encodes for ephrin-B1 that results in severe craniofacial distortions, but it affects females significantly more than males.344 The idea is that RYK normally modulates EphB2 and EphB3 activity by altering EphB2 and EphB3 homodimerization potential, affinity for ephrin ligand, and/or the confirmation of the EphB2 and EphB3 dimer in a way which alters phosphorylation. In craniofrontonasal syndrome, the interactions between ephrin-B1 and EphB2 and EphB3 are altered. In heterozygous females, the mosaic pattern due to x-inactivation interferes with cell-cell interaction (ephrin-B1 is membrane bound and interacts with EphB2 and EphB3 from an adjacent cell), while homozygous males have uniformly altered signaling which is less deleterious. The role of RYK in this process has not been experimentally validated, but it emphasizes the need to study the role of hetero-complexes in RTK signaling. Furthermore, as this proposed disease mechanism involves alterations of normal interactions, it is well suited to be studied using quantitative, thermodynamic approaches.
8.1.2. EphA4 Interacts with the FGFRs
EphA4 has been found to interact with the FGFRs. The FGFRs are important for proper musculoskeletal development,345–347 and their misfunction is associated with numerous growth and neurological disorders.238,348–351 A yeast two-hybrid screen using the juxtamembrane region of FGFR3 as bait revealed the intracellular domain of EphA4 as a potential interaction partner.257 Co-expressing EphA4 and FGFR1, −2, −3, or −4 in HEK 293 cells showed that all four hetero-interactions occur in the absence of ligand via coimmunoprecipitation. Phosphorylation studies of a mutated version of EphA4 that is kinase dead with wild type FGFR1 or vice versa showed that increasing concentrations of the wild type receptor causes increasing phosphorylation of the kinase dead receptor. This indicates that there is bidirectional cross-phosphorylation between EphA4 and the FGFRs. In neuronal cells endogenously expressing FGFR1, −2, and −3 and EphA4, interactions could only be seen via coimmunoprecipitation in the presence of ephrin-A1; moreover, addition of ephrin-A1 potentiated the FGF induced phosphorylation of the FGFR adaptor protein FRS2α and the downstream signaling molecule MAPK. Similar results have been seen in a glioblastoma line, where addition of EphA4 lacking the kinase domain inhibited FGFR1 phosphorylation and proliferation in response to FGF2.258 The same is true in neural stem/progenitor cells, where expressing EphA4 lacking the intracellular domain or FRS2α lacking phosphorylation sites decreased the mitogenic effects of FGF2 and ephrin-A1.259 A study involving immunoprecipitation coupled with MS to identify the pull-down partners found that FGFR3 activity is correlated with EphA4 (and several other Eph receptors) being phosphorylated.352
8.1.3. EphA2 Interacts with EGFR and ErbB2
Interactions with EphA2 and EGFR and ErbB2 have also been observed. Addition of EGF increases EphA2 levels in both a human head and neck carcinoma cell line and in a human epidermoid carcinoma cell line that overexpress EGFR.253 The receptors colocalize on the plasma membrane by immunofluorescence, and they coimmunoprecipitate in the absence of ligand, although addition of EGF increases the observable amount of interaction. (It is unclear if EGF increases hetero-interactions through direct binding to hetero-complexes, or if EGF induced EphA2 upregulation results in more heterocomplexes through mass action.) In the absence of EGF, EphA2 is not phosphorylated, but it is phosphorylated one hour after adding EGF. Addition of Ephrin-A1 causes EphA2, but not EGFR, to be internalized, and it inhibits EGF induced migration. Moreover, EphA2 is commonly overexpressed in colorectal cancer and correlates with poor prognosis, and increased EphA2 levels correlates with a poor response to the EGFR inhibitor cetuximab.254 This suggests that EphA2 may be able to restore the activity EGFR when it is inhibited by cetuximab or that the hetero-interaction inhibits cetuximab binding or activity.
The interaction between EphA2 and ErbB2 has been observed in the context of breast cancer. In mice models of ErbB2 positive breast cancer, the lack of EphA2 results in decreased tumorigenesis and metastasis, and the effect appears to be due to modulation of Ras/ERK signaling.255 EphA2 and ErbB2 coimmunoprecipitate in the absence of ligand, both in an exogenesis overexpression model and in primary mammary tumor cells that endogenously express both proteins. Inhibiting the ErbB2 kinase decreases EphA2 phosphorylation. In humans, high EphA2 levels in ErbB2 positive breast cancer correlates with poor patient prognosis, and in an EphA2 positive human breast cancer line, the exogenous expression of EphA2 is sufficient to confer resistance to the anti-ErbB2 drug trastuzumab.256 This effect appears to require EphA2 phosphorylation, as expressing a kinase dead version of EphA2 does not confer resistance. Moreover, the inhibition of the basal level EphA2 phosphorylation decreases proliferation and potentiates trastuzumab. Further evidence of cross-phosphorylation is that co-expression of EphA2 and ErbB2 is sufficient for EphA2 to be phosphorylated, and the phosphorylation is blocked by PP2 inhibition of Src. Accordingly, it appears that the EphA2-ErbB2 interaction has an oncogenic effect and can result in anti-ErbB2 drug resistance.
8.2. ROR1 Interacts with Multiple RTKs
RTK-like orphan receptor 1 (ROR1) is known to interact with members of several RTK families. It is part of the ROR family consisting of ROR1 and ROR2, and it is important during embryonic development for proper musculoskeletal, nervous system, and organ formation, and is involved in the Wnt signaling pathway.335,353,354 Relatively recent research has shown that both ROR receptors are associated with numerous cancers, which has made them attractive therapeutic targets.355–359 Whether or not ROR1 has intrinsic kinase activity is unclear, as mutations in the kinase domain and an apparent inability to autophosphorylate or phosphorylate substrates generally leads to ROR1 being classified as kinase dead,309,360–362 but several groups have reported some kinase activity.288,363
The interaction between ROR1 and MET was discovered by knocking down ROR1 in a large array of cancer lines.289 Only two lines exhibited greater than 50% growth inhibition, and although there was no correlation between ROR1 expression and inhibition, the inhibited lines were the only two that exhibited ROR1 phosphorylation. Both lines have MET amplification, and chemical inhibition or knockdown of MET abolishes ROR1 phosphorylation. In cell lines with high MET but negligible ROR1, exogenously expressing ROR1 results in ROR1 being phosphorylated. The transphosphorylation appears to be unidirectional, as downregulating ROR1 in cells which express both ROR1 and MET does not affect MET phosphorylation. This appears to be a direct interaction, as ROR1 coimmunoprecipitates with MET. (ROR1 also coimmunoprecipitates with EGFR and ErbB2, but neither appears to be able to transphosphorylate ROR1.)
A follow-up study by the same lab investigated the physiological role of this ROR1-MET interaction, and concluded that it diversifies MET signaling.317 MET lacking tyrosines that serve as docking sites for adaptor proteins could phosphorylate ROR1, but a kinase dead version could not. ROR1 has eight tyrosines predicted to be phosphorylatable—three in the kinase domain and five in the proline-rich domain of the post-kinase tail—and deletion studies indicate that ROR1 phosphorylation is lost once the proline rich domain is removed, although all truncated versions except the complete removal of the intracellular domain still immunoprecipitated with MET. However, mutating all five tyrosines in the proline-rich domain to phenylalanine (ROR15F) did not completely abolish MET induced phosphorylation, and the complete loss of phosphorylation requires the three tyrosines in the kinase domain to be mutated to phenylalanines as well. It appears that Src interacts with ROR1 (as well as MET), and this interaction requires the proline-rich domain to be present and results in the three tyrosines in the kinase domain becoming phosphorylated. Intriguingly, both ROR1 and a mutant where the three tyrosines in the kinase domain are mutated to phenylalanines (ROR13F) inhibit apoptosis and increase proliferation in cells with high MET levels, but ROR15F does not. Furthermore, ROR1 induces invasiveness, but neither ROR15F nor ROR13F has this effect. The ROR1-MET interaction accordingly is able to increase the signaling capacity of MET by separately allowing for increased survival and growth or invasiveness.
ROR1 is also able to interact with EGFR.288 Addition of EGF results in interactions between ROR1 and EGFR, as seen via coimmunoprecipitation, in both lung adenocarcinoma cells endogenously expressing the proteins and COS-7 cells exogenously expressing them. The interaction requires the cysteine rich domain of the extracellular domain, as deleting it eliminates the interaction, but not the kinase or proline-rich domains. Moreover, in lung adenocarcinomas, knocking down ROR1 results in significant growth inhibition, even in cell lines resistant to anti-EGFR drugs. Exogenous expression of ROR1, but not a fully kinase dead variant, enhances growth. This seems to be due in part to an effect on ErbB3, as ROR1 knockdown decreases ErbB3 phosphorylation and EGF-induced ErbB3-EGFR interaction, as seen via coimmunoprecipitation. The ErbB3 effect does not require Src activity, ROR1 kinase activity, or the presence of its proline-rich domain. Accordingly, it appears that ROR1 can affect growth both through an interaction with EGFR and though an alteration of ErbB3 activity, as well as a separate Src-dependent mechanism.
A later study found a direct interaction between ROR1 and ErbB3 in triple-negative breast cancer (TNBC) cells.309 ROR1 is amplified in many TNBC patients, and levels correlate with poor patient prognosis. CRISPER-Cas9 was used to create ROR1 knockout versions of TNBC cells. These cells had reduced proliferation, migration, and invasiveness compared to the wild type; expressing wild type, but not a fully kinase dead variant, ROR1 restored the wild type phenotype to the knockout cells. Following neuregulin-1 stimulation in TNBC cells, ROR1 and ErbB3 coimmunoprecipitated, and ErbB3 was phosphorylated on a novel tyrosine, Tyr1307, and four tyrosines known to be phosphorylated by EGFR. Chemical inhibition of EGFR removes phosphorylation of those four sites but not Tyr1307, while ROR1 knockdown abolishes Tyr1307 phosphorylation but not that of the other four tyrosines. Knocking down EGFR does not affect the ROR1-ErbB3 interaction, but overexpression of a fully kinase dead mutant of ROR1 abolishes Tyr1307 phosphorylation. By investigating downstream signaling molecules, the authors conclude that the ROR1-ErbB3 interaction triggers a signaling cascade that modulates the Hippo-YAP pathway, and this results in tumor cell proliferation and bone metastasis.
One last known interaction partner of ROR1 is muscle-specific kinase (MuSK).320 MuSK is found in skeletal muscles and in neurons, and it is critical for formation and maintenance of neuromuscular synapses.364–367 Not surprisingly, dysregulation of the MuSK signaling pathway is associated with several neuromuscular disorders.368,369 ROR1 and MuSK coimmunoprecipitate when exogenously expressed in Cos-7 cells.320 Moreover, ROR1 is phosphorylated, but only if Dok-7 (which binds to Musk and activates it and also coimmunoprecipitates with ROR1) is also present. Of note, ROR1 coimmunoprecipitates with kinase dead MuSK but is not phosphorylated, indicating that either MuSK directly phosphorylates ROR1 or activated MuSK binds or phosphorylates a protein which does. Similar to what was seen with the ROR1-MET interaction,317 deletion of the proline rich domain does not affect coimmunoprecipitation, but the deletion does abolish ROR1 phosphorylation; however, in this case, the role of Src or other adaptor proteins has not been investigated. Although the effect of this ROR1-MuSK interaction is currently unknown, it appears to be physiologically important, as ROR1 and MuSK coimmunoprecipitate in mouse myogenic cells differentiated into myotubes that endogenously express ROR1, MuSK, and Dok-7.
8.3. PTK7 Interacts with Multiple RTKs
Another RTK with known hetero-interactions is protein tyrosine kinase 7 (PTK7), which is a kinase dead RTK which is the only member of its family. It is important for a wide range of cell-cell communication and migration processes such as tissue homeostasis, morphogenesis, planar cell polarity, and wound healing, as well as being involved in the Wnt signaling pathway.370,371 PTK7 is overexpressed or mutated in many cancers,372,373 making it a therapeutic target374,375 and potential prognostic biomarker.376,377
There is evidence that PTK7 interacts with the VEGFR family, but the exact nature of the interaction is still a source of debate. The Lee lab found that inhibiting PTK7 by using either the soluble extracellular domain as a decoy (i.e., a competitive inhibitor) or siRNA knockout results in human umbilical vein endothelial cells (HUVECS)—which endogenously express PTK7, VEGFR1, and VEGFR2—having decreased capillary-like tube formation, migration, and invasiveness in response to addition of the VEGF ligand.326 This lab later showed that in HUVECs, inhibiting PTK7 reduces VEGF induced phosphorylation of VEGFR2 and its downstream signaling molecules, but not VEGFR1.327 Moreover, PTK7 was found to coimmunoprecipitate with VEGFR2 but not VEGFR1 in both HUVECs and HEK 293 cells which were transfected with the proteins.
However, the Dana lab found the opposite results, as in several vascular endothelial cell lines, including HUVECs, PTK7 coimmunoprecipitated with VEGFR1, but not VEGFR2 or VEGFR3, and the amount of hetero-complex increased after VEGFA addition.325 Knocking down PTK7 via siRNA decreased VEGFA induced phosphorylation of VEGFR1 and its downstream molecules; however, inhibition did not affect VEGFR2 phosphorylation. It is possible that this difference in hetero-formation can be explained by differences in cell media affecting receptor expression levels or which ligands are present. The population of heterodimers can be highly dependent on expression levels (see Figure 4D for a thermodynamic explanation), and the interactions with ligand can magnify this effect. This emphasizes the importance of a thermodynamic understanding of RTK interactions, as well as the need for careful attention to conditions which can affect expression and interactions.
Figure 4.
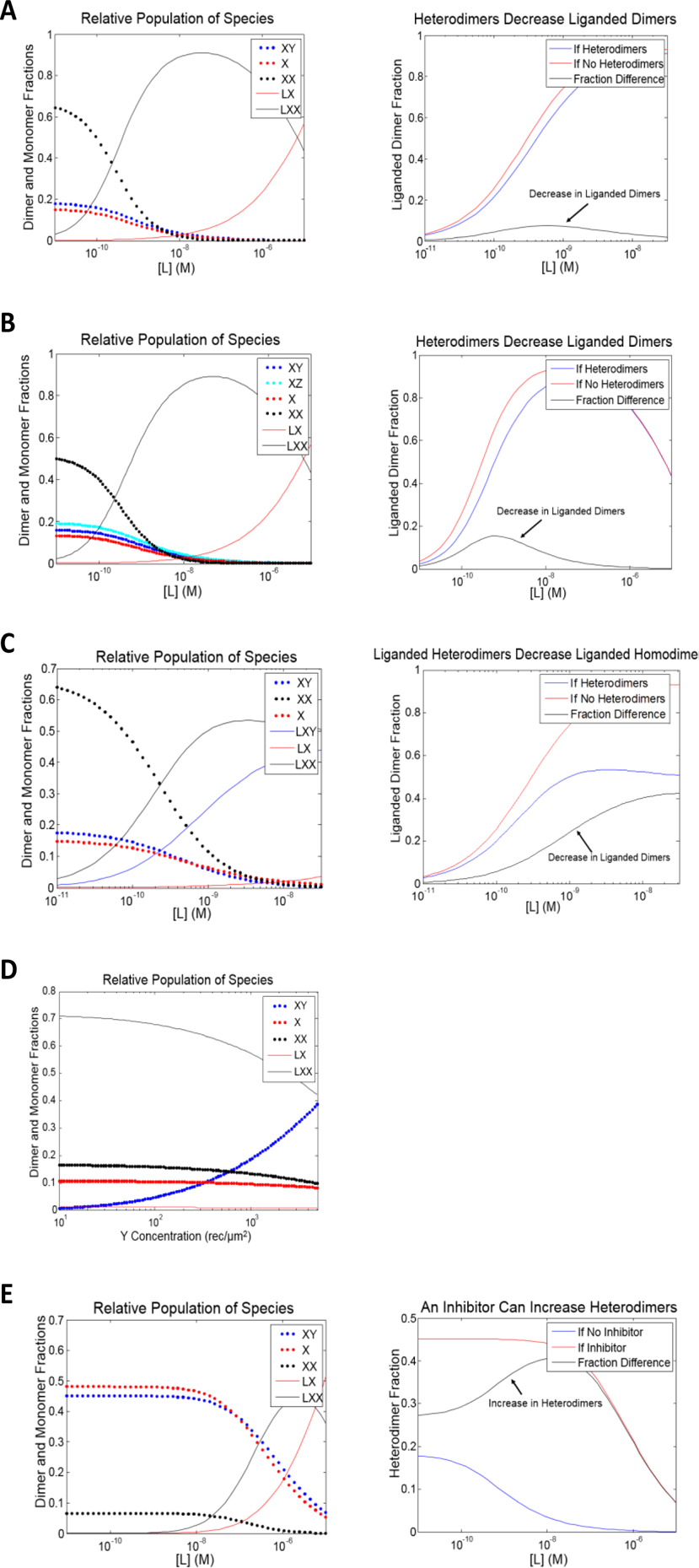
Predictions based on the thermodynamic cycles in Figure 3, produced with MATLAB. These predictions can help explain many of the complicated biological effects which have been described in the literature (see the “Overview of Known RTK Cross-Subfamily Hetero-Interactions” section). All receptors are assumed to have concentrations of 500 rec/μm2 unless otherwise stated. Values of the constants used in the predictions are estimates for VEGFR2 dimerization; VEGFA binding to VEGFR2; EGFR dimerization; and in the case of 4B, EphA2 dimerization. The K’s (receptor-receptor interactions) and L’s (ligand-receptor interactions) are association constants in units of μm2/rec and M−1, respectively. (A) A prediction based on the thermodynamic cycle in Figure 3D: KX = .029 μm2/rec, KY = .0088 μm2/rec, KXY = .0094 μm2/rec, L1 = 9.6*107 M−1, and L2 = 4.3*109 M−1. The left plot shows the different dimeric and monomeric fractions as a function of ligand concentration. The right plot compares the fraction of X receptors which exist as liganded dimers, for the case modeled in the left plot (blue), and for the case where X and Y cannot heterodimerize (red), as a function of ligand concentration. The black curve is the difference between the red and the blue curves, and it depicts the decrease in liganded dimers due to the presence of heterodimers. (B) A prediction based on the thermodynamic cycle in Figure 3E: KX = .029 μm2/rec, KY = .0088 μm2/rec, KZ = .0049 μm2/rec, KXY = .0094 μm2/rec, KXZ = .0092 μm2/rec, L1 = 9.6*107 M−1, and L2 = 4.3*109 M−1. The left plot shows the different dimeric and monomeric fractions as a function of ligand concentration. The right plot compares the fraction of X receptors which exist as liganded dimers, for the case modeled in the left plot (blue), and for the case where the heterodimers XY and XZ cannot form (red), as a function of ligand concentration. The black curve is the difference between the red and the blue curves, and it depicts the decrease in liganded dimers due to the presence of heterodimers. (C) A prediction based on the thermodynamic cycle in Figure 3F: KX = .029 μm2/rec, KY = .0088 μm2/rec, KXY = .0094 μm2/rec, LX1 = 9.6*107 M−1, and LX2 = 4.3*109 M−1, LY1 = 9.6*106 M−1, LY2 = 4.3*108 M−1, and LXY = 4.3*109 M−1. The left plot shows the different dimeric and monomeric fractions as a function of ligand concentration. The right plot compares the fraction of X receptors which exist as liganded dimers, for the case modeled in the left plot (blue), and for the case where X and Y do not form heterodimers (red), as a function of ligand concentration. The black curve is the difference between the red and the blue curves, and it depicts the decrease in liganded dimers due to the presence of heterodimers. (D) A prediction based on the thermodynamic cycle in Figure 3D, which shows the different dimeric and monomeric fractions as a function of Y concentration for a fixed X concentration: the concentration of X is 250 rec/μm2, the concentration of L is 1 nM, KX = .029 μm2/rec, KY = .0088 μm2/rec, KXY = .0094 μm2/rec, L1 = 9.6*107 M−1, and L2 = 4.3*109 M−1. The concentration of liganded homodimers decreases and the concentration of the heterodimers increases as the concentration of Y increases. (E) A prediction based on the thermodynamic cycle in Figure 3D, which models the effect of an inhibitor of homodimerization by assuming that the homodimerization constant of the RTK which binds ligand, X, and the ligand binding constants are reduced one hundred-fold: KX = .00029 μm2/rec, KY = .0088 μm2/rec, KXY = .0094 μm2/rec, L1 = 9.6*105 M−1, and L2 = 4.3*107 M−1. These decreases mimic the effect of a targeted inhibitor which decreases the ability of an RTK to form homodimers and to bind ligand. The left plot shows the different dimeric and monomeric fractions as a function of ligand concentration. The right plot shows the difference between the fraction of X receptors which is in a heterodimer in the “no inhibitor case,” depicted in blue (identical conditions to Figure 4A) and in the case when the inhibitor is present (modeled on the left), depicted in red. The black curve is the difference between the red and the blue curves, and it shows a large increase in heterodimers in the inhibitor case.
PTK7 is also known to interact with ROR2. Coimmunoprecipitation using transiently transfected MCF7323 or HEK 293T324 cells indicates that PTK7 interacts with ROR2. Furthermore, the HEK 293T study found no interaction with ROR1, and truncation experiments showed that the ROR2 interaction requires part of the PTK7 extracellular domain to be present, but none of the intracellular domain. This interaction was also seen by coimmunoprecipitation in mouse embryonic fibroblast (MEF) cells that endogenously express both proteins, and knockdown studies in developing xenopus indicate a functional interaction between PTK7 and ROR2.324 In xenopus neural crest cells, fluorescently labeled PTK7 and ROR2 colocalize, and addition of ROR2 could rescue migratory deficiencies caused by PTK7 knockout, but a kinase dead mutant of ROR2 could not.323 Although the role of the PTK7-ROR2 interaction is not well understood, it appears to have a functional role in development and is sometimes able to rescue impaired PTK7 function.
8.4. STYK1 and EGFR Crosstalk
The term “crosstalk” has generally been used in the literature to indicate the overlap of two separate signaling pathways. In this review, we use “crosstalk” to indicate evidence of a potential interaction which has not been directly verified. There is some indirect evidence that STYK1 (Serine/threonine/tyrosine kinase 1) can interact with EGFR.290 STYK1 differs somewhat from the other RTKs, as it almost completely lacks an extracellular domain, and so it does not bind ligand. It also lacks a membrane signal sequence, so it behaves as a cytosolic protein despite having a canonical single pass “transmembrane” domain.378 Overexpression of STYK1 has been found to cause tumorigenesis and metastasis in mice models.379 It is overexpressed in many human tumors, and it may be a good biomarker in lung cancer.380
In transient transfection experiments, immunofluorescent staining revealed that STYK1 expression is cytosolic, and it forms small, dot-like fluorescence and larger, aggregate-like fluorescence.290 The aggregates largely colocalize with early endosomes, while the dot-like fluorescence does to a smaller degree. Immunohistochemical staining comparing cervical and breast cancer tissue to healthy tissue shows that STYK1 levels are much higher in cancerous tissue and STYK1 is mostly aggregated, indicating that a similar behavior may occur in more native biological systems. Deleting the small extracellular domain had no effect, but deleting the transmembrane domain significantly reduced aggregation and abolished endosomal colocalization. Stimulation with EGF resulted in a high degree of colocalization with EGFR, although the colocalization decreased over time. This evidence is suggestive of an interaction between STYK1 and EGER. Since little is known about the biological role of STYK1, an interaction between STYK1 and EGFR is a promising avenue of investigation.
8.5. IGF-1R Interacts with the ErbBs
The insulin/insulin-like growth factor receptors are preformed, disulfide linked dimers (each monomer is composed of an α and β chain, and the dimer is a βα-αβ complex) which are important for regulating metabolism, proliferation, and differentiationref. Insulin receptor (InsR) binds insulin and is primarily involved in metabolic activity. Insulin-like growth factor 1 receptor (IGF-1R) binds insulin-like growth factor 1 and 2 (IGF-1 and IGF-2) and is important for cell proliferation and differentiation.381 IGF-1R is overexpressed in many cancers, and the role that the interaction between it and the ErbBs plays in cancer has garnered a lot of attention382,383. Moreover, general overlap between the signaling axes of IGF-1R and VEGFR, MET, PDGFR, and ALK has been observed in cancer.384,385 Evidence of direct interactions between the insulin receptors and ErbB receptors are one of the earliest examples of interactions between different RTK families, as chimeric studies in the early nineties indicated potential interactions.386 Since then, numerous examples of direct interactions between the two families have been described.
In a screen of nine different non-small cell lung carcinoma (NSCLC) cell lines, it was found that the cell lines have differing sensitivity to the anti-EGFR drug erlotinib, and that the anti-IFGF-1R drug AG1024 synergistically enhances erlotinib inhibition.268 A resistant derivative of one of the erlotinib sensitive cell lines was created by culturing it with the drug, and it has significantly higher IGF-1R levels than the parent line. Notably, in both the parent and resistant line, IGF-1R and EGFR coimmunoprecipitate, but more heterocomplexes are present in the resistant line, and IFGR-1R does not coimmunoprecipitate with either ErbB2 or ErbB3. Adding erlotinib increases the amount of IGF-1R-EGFR heterocomplexes in several cell lines (see Figure 4E for a possible thermodynamic explanation), as seen via coimmunoprecipitation, although heterocomplexes were not found in one of the highly sensitive lines in the presence or absence of erlotinib. This suggests that formation of an IGF-1R-EGFR heterocomplex confers resistance to erlotinib, because more resistant cell lines have more heterocomplexes and the drug induces heterocomplex formation in resistant lines. A possible mechanism of resistance is that the heterocomplex increases expression of the anti-apoptotic protein survivin: the resistant cells, but not the sensitive ones, had an increase in survivin levels upon erlotinib addition.
Furthermore, IGF-1R-EGFR heterocomplexes have been linked to resistance to the anti-EGFR and -ERBB2 drug afatinib.269 The EGFR T790M mutation is the most common mutation that causes resistance to anti-EGFR therapy in NSCLC; afatinib binds tightly to EGFR T790M, but does not improve patient survival. An afatinib resistant NSCLC cell line which has the EGFR T790M mutation, as well as the common EGFR L858R mutation, was created. In both the parent and resistant lines, IGF-1R and EGFR coimmunoprecipitate and afatinib increases IGF-1R phosphorylation in a dose dependent manner. However, the resistant line has higher basal IGF-1R phosphorylation levels than the parent line. Moreover, although linsitinib inhibition of IGF-1R does not affect the parent line, it reduces growth of the resistant line, and shRNA knockdown of IGF-1R restores afatinib sensitivity in the resistant line. This suggests that the afatinib resistant cells are dependent on IGF-1R activity, possibly due to the IGF-1R-EGFR hetero-interaction enabling EGFR activity.
Interactions between IGF-1R and ErbB2 have also been observed. In several breast cancer lines, down regulating IGF-1R decreases ErbB2 phosphorylation, and IGF-1R and ErbB2 coimmunoprecipitate and colocalize via immunofluorescence.299 In a breast cancer line which expresses both receptors but not their ligands, IGF-1R and ErbB2 only faintly coimmunoprecipitate, but addition of either IGF-1 or NDF substantially increases the amount of heterocomplexes. Complementary results were seen in a comparison between a breast cancer cell line that is sensitive to the anti-ERBB2 drug trastuzumab and the same line but with induced drug resistance.300 IGF-1R and ErbB2 coimmunoprecipitate in the resistant line, but not the parent line, and the addition of IGF-1 causes a small decrease in ErbB2 phosphorylation in the parent cells, but a significant increase in the resistant line. Interestingly, IGF-1 did not detectably affect hetero-interaction in the resistant line, but it appears to cause a small degree of hetero-interaction in the parent line. The IGF-1R inhibitor I-OMe-AG538 did not affect ErbB2 phosphorylation in the parent line, but in the resistant line, it did decrease ErbB2 phosphorylation and restore trastuzumab sensitivity. It is possible that IGF-1 induces heterocomplex formation by binding to both IGF-1R and ErbB2, but given that ErbB2 has no known activating ligands, it seems more probably that IGF-1R activation promotes the hetero-interaction. This is similar to the manner in which ErbB2 heterodimerizes with other ErbBs (see the “RTK Hetero-Interactions Within the Same Family” section for more detail).
Finally, interactions have also been seen between IGF-1R and ErbB3. In a breast cancer line that is sensitive to trastuzumab and the same line but with induced drug resistance, IGF-1R and ErbB3 coimmunoprecipitate and colocalize via fluorescence in the resistant line, but not the parent line.301 (Although trastuzumab is specific to ErbB2, ErbB3 is kinase dead and is largely dependent on hetero-interactions, especially with ErbB2, for its activity.174,176) ErbB2 and ErbB3 coimmunoprecipitated in both the parent and resistant lines, but significantly more in the resistant line, and shRNA knockdown of either IGF-1R or ErbB3 sensitized the resistant cells to trastuzumab. Loose evidence that heterotrimers form was seen in the fact that immunodepletion of IGF-1R removes ErbB2-ErbB3 interactions, and complexes corresponding by weight to a trimer were seen on a native PAGE. Additional evidence of interaction between IGF-1R and ErbB3 has been seen in other cancers. A comparison of an ovarian cancer cell line with resistance to trastuzumab to the non-resistant parent line found that the resistant line had increased proliferation but decreased EGFR and ErbB2 expression.307 Instead, IGF-1R and ErbB3 were significantly upregulated, and inhibiting either inhibits cell proliferation. Furthermore, in pancreatic cancer, IGF-1R, ErbB3, and their ligands are often overexpressed, and in cell lines, the bispecific antibody inhibitor of IGF-1R and ErbB3 istiratumab sensitizes the cells to traditional chemotherapeutics.308 All in all, there is evidence that IGF-1R interacts with EGFR, ErbB2, and ErbB3 under a wide range of conditions, and these heterocomplexes appear to confer resistance to several anti-ErbB cancer therapeutics.
8.6. PDGFRs Interact with EGFR and VEGFR2
PDGFRα and PDGFRβ have been found to interact with members of several different RTK families, including EGFR, VEGFR2, and FGFR1 (see “FGFRs Interactions with Multiple RTKs in Cancer”). These receptors are two members of the PDGF family, and they are important for the development of mesenchymal cells in numerous organs during development, as well as wound healing in adults.387 Overexpression and mutations of both receptors and the PDGF ligands are associated with numerous cancers, and they have accordingly received a lot of interest as clinical targets.388–391
For PDGFRα and EGFR, both receptors are sometimes overexpressed in glioblastomas (GBM). In patient-derived GBM sphere lines expressing both receptors, EGFR inhibition with gefitinib causes dephosphorylation of PDGFRα, and this is true even in lines with low EGFR expression.280 A later study by the same lab determined that surgically resected primary GBM tumor sphere lines have a higher degree of heterogeneity for EGFR and PDGFRα expression than the commercially available tumor lines, and this also occurs in the absence of receptor amplification.279 In these primary lines, EGF stimulation results in increased PDGFRα phosphorylation, and interaction with EGFR was seen via coimmunoprecipitation and PLA. Although the interaction could be seen in the absence of EGF, its addition increases hetero-interaction. Moreover, EGF is required for PDGFRα phosphorylation in the heterocomplex; this phosphorylation is blocked by gefitinib. This hetero-interaction appears to affect downstream signaling, as the heterogeneous receptor expression correlates with the phosphorylation of Akt and ERK and cell proliferation. Increased PDGFRα corelates with decreased efficacy of EGFR inhibition. The PDGFRα-EGFR interaction appears to enable activated EGFR to phosphorylate PDGFRα and influences the signaling pathways and drug resistance in GBM.
PDGFRβ is also known to interact with EGFR. In a fibroblast cell line which endogenously expresses both PDGFRβ and EGFR, addition of EGF or EGFR overexpression increases PDGFRβ phosphorylation, and they coimmunoprecipitate in the presence and absence of ligand.281 Furthermore, PDGFRβ phosphorylation increases with increasing EGFR activity, indicating that PDGFRβ is phosphorylated by activated EGFR—either directly or through a larger complex—or EGFR upregulates PDGFRβ or its ligand. Similar results were seen in rat aortic smooth muscle cells (VSMCs) which endogenously express both receptors.282 EGFR phosphorylation significantly increases in response to PDGF-BB, and the receptors coimmunoprecipitate in the presence and absence of ligand. Notably, the PDGFR kinase inhibitor AG1295 did not prevent PDGF-BB induced activation of EGFR, but the EGFR inhibitor AG1478 did; moreover, inhibiting Src kinases with PP2 both decreases PDGF-BB induced EGFR phosphorylation and hetero-interactions, indicating a role of the Src kinases in formation of the hetero-species. This interaction also appears to be important for ERK activation through metalloproteases, as in MEFs, PDGF-BB addition results in increased ERK and EGFR phosphorylation, but both can be blocked by chemical inhibition of metalloproteases or EGFR.283 These studies indicate that the PDGFRβ-EGFR complex is functioning as part of a larger complex, but further studies are needed to confirm and clarify these interactions.
Whereas the PDGFRα- and PDGFRβ-EGFR interactions have an activating effect, the PDGFRβ interaction with VEGFR2 has an inhibitory effect.321,322 In both mice and chicken embryo models, separately adding either VEGFA or PDGFR-BB promotes neovascularization, but adding both ligands together abolishes this effect; the same phenomenon was seen in primary human VSMCs with regard to proliferation and migration. This inhibitory effect was not seen in cell lines which only express VEGFR2 or PDGFRβ, and inhibiting VEGFR2 but not VEGFR1 eliminates it as well. Addition of VEGFA suppresses PDGF-BB induced phosphorylation of PDGFRβ in VSMCs—although phosphorylation could be recovered by titrating in more PDGF-BB—and transfecting cells that only endogenously express PDGFRβ with VEGFR2 causes the same inhibition, although only minimally if a truncated version of VEGR2 lacking the kinase domain is used. In the VSMCs, PDGFRβ and VEGFR2 coimmunoprecipitate when both VEGFA and PDGF-BB are present—but not if only one or neither are—but this was not seen with PDGFRα. The same results were seen by transfecting HEK 293 cells with both receptors and using PLA, but hetero-interactions were not observed if VEGFR2 lacking the kinase domain or kinase dead VEGFR2 were used. Comparable results were seen in human aorta-derived VSMCs, as transfection with heme oxygenase-1 (HO-1) increases VEGFR2 and VEGFA expression and decreases PDGF-BB induced migration and PDGFRβ phosphorylation. After addition of both VEGFA and PDGF-BB, the receptors coimmunoprecipitate, and knockdown of VEGFR2 or VEGFA reduces the inhibitory effect of HO-1. The exact mechanism of this inhibitor effect is unknown, but it is clear that it requires activated receptors, which is suggestive of the heterocomplex recruiting a phosphatase that deactivates one or both receptors. However, it is also possible that the heterocomplex can only form when both PDGFRβ and VEGFR2 are phosphorylated, and the heterocomplex blocks binding of key adaptor proteins or results in nonproductive phosphorylation. Further experiments are required to elucidate the details of this interaction.
Although to the best of our knowledge no direct interactions between wild type PDGFRα and VEGFR2 have been reported, a chimeric fusion between a large portion of the VEGFR2 extracellular domain and the PDGFRα transmembrane and intracellular domains was found in a surgical glioblastoma sample.392 Transfection of NIH 3T3 cells with the fusion causes a tumorigenic phenotype. The fused receptor is constitutively active in the absence of ligand, and phosphorylation of downstream molecules is similar to that of PDGF-AA activated wild type PDGFRα. Immunoprecipitation studies indicate that the fused product can form homodimers, as well as heterodimers with both VEGFR2 and PDGFRα, and the hetero-dimerizing partners are phosphorylated.
8.7. RET and VEGFR2 Interaction
An interaction between Ret and VEGFR2 has been observed. RET (rearranged during transfection) is important for neuronal and tissue development,393,394 and gain-of-function mutations are common in many cancers, leading to large efforts to develop inhibitors.395 In a ureteric bud cell line which endogenously expresses both VEGFR2 and RET, VEGFR2 phosphorylation increases in response to addition of the Ret ligand glial cell-derived neurotrophic factor (GDNF), and RET phosphorylation increases in response to VEGFA.331 A similar interaction was seen in response to ligand with respect to both proliferation and branching morphogenesis, and RET and VEGFR2 coimmunoprecipitate in the presence and absence of VEGFA. Furthermore, GDNF is necessary for VEGFA to increase vascular profusion in ischemic skeletal muscle,332 and the efficacy of the anti-cancer drug sorafenib has been linked to its dual inhibition of Ret and VEGFR2.396
8.8. Trks Interact with Multiple RTKs
Another family of RTKs with multiple known hetero-interactions is the tropomyosin-related kinases (Trks). This family of RTKs is important for the development of the primary and periphery nervous system; the survival, maintenance, and differentiation of neurons; and the transduction of sensory signals.397–399 They primarily bind neurotrophins—which are NGF, BDNF, neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/5) in mammals—but can also be activated by a wide range of other factors400. Trk overexpression and mutations are associated with a wide variety of cancers, making them an attractive therapeutic target,401,402 as well as a wide range of neurological disorders403 including Alzheimer’s,404,405 depression,406 and schizophrenia.407
TrkA was found to coimmunoprecipitate with ErbB2 in a prostate cancer cell line after addition of NGF.303 Addition of NGF also increases ErbB2 phosphorylation, and dual inhibition with the anti-Trk drug lestaurtinib (CEP-701) and the anti-ErbB2 drug pertuzumab inhibits proliferation significantly more than either drug individually. There is also evidence of crosstalk between TrkA and EGFR.291 In human monocytes, EGF increases NGF expression and phosphorylation of TrkA, and NGF increases the phosphorylation of EGFR. Moreover, chemical inhibition of either EGFR or TrkA decreases EGF and NGF induced activation of the cognate receptor. These results are indicative of bidirectional activation between TrkA and EGFR and ErbB2, although the evidence is currently tenuous.
TrkB has also been found to directly interact with ErbB2.304 Tumor samples of breast cancer brain metastasis (BBM) have much higher TrkB and ErbB2 phosphorylation than primary breast cancer samples, and immunofluorescence and cryo-electron microscopy with antibody labeling indicated colocalization of TrkB and ErbB2. Addition of BDNF increases ErbB2 phosphorylation and TrkB-ErbB2 colocalization. Of note, the brain microenvironment has high BDNF levels, and flow cytometry with antibodies showed that about half of the BBM cells have both TrkB and ErbB2. Accordingly, these interactions are likely to occur in the native biological system. In the presence and absence of both BDNF and NGF, TrkB and ErbB2 coimmunoprecipitate from BBM, and in silico modeling of the kinases of the receptors indicates that physical interaction between the two is plausible. Inhibiting TrkB with cyclotraxin and ErbB2 with lapatinib decreases the interaction between the two receptors and inhibits proliferation to a significantly greater degree than either drug individually. This suggests that the TrkB-ErbB2 interaction might increase the activity of ErbB2, and that the interaction could be oncogenic.
Although to the best of our knowledge there is no direct evidence of interaction between TrkB and EGFR, there is significant evidence of a potential interaction. In NSCLC, TrkB expression correlates with metastasis and poor patient prognosis, and in an NSCLC cell line, addition of EGF causes an increase in TrkB phosphorylation.292 Similar crosstalk was seen in a human ovarian cancer cell line, were addition of EGF increases TrkB phosphorylation, and addition of BDNF increases EGFR phosphorylation.293 Chemical inhibition of EGFR with PD153035 or TrkB with k252a inhibits both receptors’ response to EGF and BDNF, as well as ligand induced Akt phosphorylation and proliferation. There is evidence that TrkB leads to anti-EGFR drug resistance in colorectal cancer. Indeed, in a colorectal cancer cell line, addition of BDNF blocks the antiproliferation effect of the anti-EGFR antibody cetuximab and addition of k25a potentiates the effect of cetuximab.294 Under noncancerous conditions, primary cortical precursor cells harvested from mice brains have a low TrkB response to BDNF and NT3, but high phosphorylation in response to EGF. The phosphorylation was not blocked by BDNF neutralizing antibodies, indicating that EGF was not simply upregulating BDNF.295 During development, high levels of TrkB are seen in the cerebral cortex before BDNF levels reach the levels seen in adult brains, and TrkB and EGFR are co-expressed in neuronal cells in the forebrain. It is clear that the interaction between TrkB and EGFR is important for nervous system development and cancer therapy. It remains to be determined whether or not there exists a direct, physical interaction, and quantitative studies of the interaction can help explain this complicated biology.
Crosstalk between the Trks and RET has also been seen. In neuroblastomas, the RET ligand GDNF increases TrkA expression.328 Furthermore, in mature sympathetic neurons, NGF increases Ret phosphorylation, and this effect is blocked by inhibiting TrkA with k25a.329 An analogues crosstalk between TrkB and Ret has also been observed. In neuroblastomas, BDNF increases RET phosphorylation and siRNA knockdown of TrkB blocks RA induced RET phosphorylation.330
8.9. FGFRs Interactions with Multiple RTKs in Cancer
A large number of hetero-interactions with the FGFRs have been identified as being involved with cancer. In general, FGFR mutations which cause gene amplification, increased activity, oncogenic fusions, increased ligand expression, and aberrant signaling activity have all been observed in cancers.57,408 This has led to an intense effort to develop FGFR based cancer treatments, but the clinical results have been mixed.409
One difficulty in targeting the FGFRs stems from the fact that even when treatment is initially effective, drug resistance often develops, and there is evidence that this resistance is often caused by the activity of other RTKs. An RTK phosphorylation assay of FGFR1-amplified lung cancer cell lines with FGFR inhibition resistance found that different cell lines have high phosphorylation of different RTKs.266 One cell line has high PDGFRα phosphorylation, and several have high phosphorylation of EGFR, ErbB2, ErbB3, and MET. Treating the cells with an FGFR inhibitor and an inhibitor for the coactivated RTK results in significantly increased apoptosis relative to either inhibitor individually, and only both drugs in combination cause suppressed phosphorylation of the FGFR1 signal mediating adaptor protein FRS2.
The mechanism of this resistance was directly probed by investigating the effects of an FGFR inhibitor on six FGFR1-amplified lung cancer cell lines, and in all cases, the FGFR inhibitor by itself only had a minimal effect.305 HCC95 cells have high ErbB3 activity, and co-inhibition of both FGFR and ErbB3 causes long-term inhibition of ERK and AKT phosphorylation. Other cell lines require FGFR and IFGF-1R or FGFR and MET inhibition to significantly inhibit ERK phosphorylation. A similar interaction between FGFR1 and PDGFRα was observed in NCI-H170 cells, as inhibiting both causes long-term suppression of ERK phosphorylation, and, intriguingly, inhibition of either FGFR1 or PDGFRα increases phosphorylation of the other. Unlike the indirect nature of the above interactions, there is evidence that the FGFR1- PDGFRα interaction is direct, as FGFR1 and PDGFRα coimmunoprecipitate from these cells. Of note, FGFR1 and PDGFRα were previously found to interact in a noncancerous, human endothelial cell line, where it was noted that PDGF-BB inhibits the effects of FGF2, and both ligands being present appears to increase hetero-interaction.310
Moreover, RTK phosphorylation crosstalk has also been seen with FGFR2 using an RTK phosphorylation assay of FGFR2-amplified cancers.306 In all cases, a significant decrease in phosphorylation of ErbB3 and FGFR3 was seen in response to FGFR2 inhibition. Notably, the authors found that FGFR2 overexpression activates PI3K in a manner which requires ErbB3 transphosphorylation, presumably by FGFR2. Under the right conditions, it appears that many RTKs can rescue impaired FGFR1 or FGFR2 function.
This ability of other RTKs to compensate for an inhibited FGFR has also been observed in the case of FGFR3. A high throughput parallel siRNA screen was performed against all known protein kinases and phosphatases in 11 FGFR-mutated cancer lines.267 Although the FGFR1 and FGFR2 cell lines were inhibited by FGFR siRNA, the FGFR3 lines did not exhibit any negative growth effects. In all the FGFR3-mutated cell lines, of all the tested siRNAs, knockdown of EGFR resulted in the greatest sensitivity to the FGFR inhibitor PD173074. The anti-EGFR drug gefitinib blocks the ERK phosphorylation restoration that is seen when PD173074 is used alone. In another study, an FGFR3-mutant cancer line was made resistant to the FGFR inhibitor BGJ398 or the general kinase inhibitor ponatinib by incubating the cells with a stepwise increasing concentration of the drug.298 Relative to the parent cells, the resistant ones have a more metastatic phenotype and increased ErbB2 and ErbB3 phosphorylation (detected by pan RTK phosphorylation assays) and expression relative to the parent cells. Additionally, the parent cells are insensitive to the ErbB inhibitors AZD8931 and lapatinib, while the resistant cells are sensitive to both. The anti-FGFR drug resistance and metastatic phenotype are reduced by inhibiting either ErbB2 or ErbB3 by shRNA and are abolished by growing the cells in the absence of the drug for two to four weeks. Although the exact mechanism remains to be determined, it is clear that the ErbBs are able to compensate for inhibited FGFR3 function in some cancers.
It has also been found that FGFRs are the cause of anti-RTK drug resistance in some cases. NSCLC often overexpress FGFRs and their ligands, and in an NSCLC cell line, FGFR2 and FGFR3 mRNA levels increased after treatment with the EGFR inhibitor gefitinib.410 A similar study found that treatment of a panel of NSCLC cell lines with gefitinib to create resistant lines results in several cell lines having increased mRNA levels of FGFR1.411 In both these cells and in the FGFR2 and FGFR3 overexpressing cells, ERK phosphorylation and anchorage-independent growth could only be reduced using an FGFR inhibitor. A different study using a chronic myeloid leukemia (CML) cell line found that the FGFR ligand FGF2 confers resistance to the general tyrosine kinase inhibitor imatinib412. This resistance is abolished with anti-FGFR inhibitors, and siRNA knockdowns indicates that it is specifically FGFR3 which is responsible for the drug resistance. In a comparison between a gastrointestinal stromal tumor cell line normally sensitive to imatinib due to mutated KIT (a member of the PDGF family) and an insensitive variant, an siRNA screen revealed knockdown of FGFR3 to have the largest inhibitory effect.311 Inhibiting either KIT or FGFR3 reduces phosphorylation of the other, and KIT and FGFR3 coimmunoprecipitate when both are ectopically expressed in HEK 293 cells. Addition of FGF2 reduces the efficacy of imatinib in the sensitive line, but not if KIT or FGFR3 is knocked down, and inhibiting FGFR3 restores imatinib sensitivity in the resistant line.
Most of the above studies do not directly investigate hetero-interactions. In some cases, coimmunoprecipitation did not show any interactions, but it is important to note that, especially with membrane proteins, weak interactions are often missed due to the washes and non-native conditions. Moreover, others studies have found hetero-interactions between RTKs in cancer cells using coimmunoprecipitation only after the cells have become fully drug resistant.272,301 This highlights the need for quantitative studies that directly probe FGFR hetero-interactions.
8.10. MET Interactions with Multiple RTKs in Cancer
Another RTK that has many hetero-interactions associated with cancer is MET (for the interaction between MET and ROR1, see the “ROR1 Interacts with Multiple RTKs” section). MET (named because its gene was discovered after a human sarcoma line was further transformed by N-methyl-N′-nitroso-guanidine) plays important roles in epithelial cell proliferation and migration, embryogenesis, and angiogenesis.413–417 It has long been known that increased levels of MET and its ligand hepatocyte growth factor (HFG) are associated with many cancers.418,419 In more recent years, there has been interest in targeting MET in cancer therapy,61,420–423 with a particularly large clinical effort to treat NSCLC using MET and HFG inhibitors.424 However, these drugs have only been moderately successful.
Both EGFR and MET are often overexpressed in NSCLC, and in cell culture, adding EGF increases MET phosphorylation, while inhibiting EGFR decreases it.270,271 Furthermore, there is evidence that MET can confer anti-EGFR drug resistance to NSCLCs. A gefitinib resistant NSCLC cell line was created by prolonged incubation with the drug, and unlike in the parent cells, both EGFR and ErbB3 are phosphorylated in this cell line in the presence of gefitinib.272 MET expression is amplified but not mutated, and although the MET inhibitor PHA-665752 has little effect on its own, in combination with gefitinib, it causes a significant reduction in cell proliferation and ErbB3 phosphorylation and an increase in apoptosis. Overexpression of MET is sufficient to confer resistance to gefitinib in the parent cells, and in the resistant but not the parent cells, MET and ErbB3 coimmunoprecipitate (see Figure 4D for how a change in expression levels can increase heterodimers). Across multiple NSCLC lines, combinations of MET, EGFR, ErbB2, ErbB3, and RET were found to be highly phosphorylated.273 PHA-665752 inhibition of MET reduces phosphorylation of all these receptors, increases apoptosis, and decreases cell proliferation, but gefitinib, lapatinib (anti-EGFR and -ERBB2), and vandetanib (anti-EGFR, -VEGFR2, and -RET) all have little effect. This indicates that MET causes the phosphorylation the other receptors, and in these cell lines, EGFR, ErbB2, ErbB3, and RET all coimmunoprecipitate with MET. In these cancer cells, the evidence indicates that MET is able to directly phosphorylate inhibited ErbBs (and RET) to rescue inhibited function.
Similar MET hetero-interactions occur in many other cancers, and the interactions often appear to cause MET activation. It has been found that EGFR and MET are often co-overexpressed in laryngeal cancer, and MET levels can serve as a predictor of patient outcome.425 In healthy human hepatocyte cell lines, increased levels of the EGFR ligand TGFα lead to increased MET phosphorylation, and in hepatoma cell lines, EGFR is highly expressed and endogenous levels of TGFα are sufficient to cause high MET phosphorylation in the absence of HGF; additionally, EGFR inhibition decreases MET phosphorylation.274 The reverse is not true, as HGF does not lead to the phosphorylation of EGFR, indicating the effect of this hetero-interaction is at least somewhat unidirectional. Notably, in the cancer cell lines, EGFR and MET coimmunoprecipitate regardless of which ligands are present, but they do not coimmunoprecipitate in normal cells. It is difficult to say whether these differences are caused by differences in receptor expression, different ligand expression, or the presence of an unknown adaptor protein, but quantitative studies of protein-protein interactions could help create a model which can explain them.
Analogous results were seen in anaplastic thyroid carcinoma cells, as inhibiting EGFR leads to a decrease in MET phosphorylation275. In canine osteosarcoma cells, MET and EGFR coimmunoprecipitate and TGFα reduces MET inhibition by crizotinib and increases MET phosphorylation.276 Furthermore, both MET and ErbB2 are expressed in many breast cancers, and increased MET and HGF levels in tumors correlate with resistance to the anti-ErbB2 drug trastuzumab.302 Breast cancer cell lines that express both receptors exhibit increased proliferation and trastuzumab resistance in response to HGF, and MET inhibition increases trastuzumab sensitivity. In a pancreatic cancer cell line, crosstalk between MET and IFGF-1R was observed, as addition of HGF and IGF-1 synergistically increases cell migration and invasiveness, and downregulating MET abolishes the effects of IGF-1 addition312. A comparable result was seen in a prostate cancer cell line, as IGF-1 leads to MET phosphorylation, although slower than that of IGF-1R phosphorylation, and IGF-1R or MET knockdown abolishes IGF-1 induced MET activation313. These results demonstrate the complicated and widespread interactions that occur between MET and other RTKs, as MET can activate and be activated by numerous RTKs, and these interactions can be important for drug resistance and disease prognosis.
In glioblastomas, EGFR amplification or mutations are seen in 40–50% of cases, and the most common mutated form of EGFR is EGFRvIII, which cannot bind ligand but is constitutively active. By sorting GBM cells by EGFRvIII expression level using fluorescence-activated cell sorting (FACS) and then using MS to determine phosphorylation sites, it was found that MET phosphorylation significantly increases with increasing EGFRvIII levels.277 Inhibiting EGFR results in decreased MET phosphorylation, and inhibiting MET reduces EGFRvIII induced drug resistance. Inducing increased EGFRvIII expression with tetracycline in an engineered GBM cell line increases MET phosphorylation, but addition of EGF decreases it and restores sensitivity to temozolomide without affecting EGFRvIII.278 In the absence of EGF, MET and EGFRvIII coimmunoprecipitate, but this interaction is lost upon EGF addition if wild type EGFR is also present. The authors propose that there is a conformational change in the EGFRvIII-EGFRwt heterodimers which prevents MET interaction. This may indicate that anti-EGFR treatment could increase MET activity, as the treatment could decrease EGFRvIII-EGFRwt heterodimers, and this in turn increases MET-EGFRvIII heterodimers (see Figure 4E for a possible thermodynamic explanation).
Intriguingly, the oncogenic effect of MET appears to be regulated by VEGFR2 in some cases.318 In an array of mouse astrocytoma cell lines with varying amounts of VEGF, increasing VEGF levels correlates with decreasing invasiveness and MET phosphorylation, despite that fact that MET levels remain constant. Inhibiting VEGFR2 but not VEGFR1 removes this effect, and the addition of HGF does not affect VEGFR2 phosphorylation. In human GBM samples, MET and VEGFR2 coimmunoprecipitate, and the interaction can also be seen by PLA. This interaction requires the kinase domain of VEGFR2, as cells transfected with a truncated version of VEGR2 lacking the intracellular domain do not exhibit an interaction with MET by coimmunoprecipitation or PLA. Neither VEGF nor HGF is required for the formation of a heterocomplex, but HGF is needed to observe MET phosphorylation within the heterocomplex. Addition of the general tyrosine phosphate inhibitor sodium orthovanadate removes the VEGF effect on MET phosphorylation, as does inhibiting the nonreceptor protein tyrosine phosphatase 1B (PTP1B). Coimmunoprecipitation studies indicate that VEGF enables recruitment of PTP1B to the MET-VEGFR2 complex, and then PTP1B dephosphorylates MET, regulating its oncogenic activity.
8.11. Ron Interacts with Multiple RTKs
The other RTK in the same family as MET is Recepteur d′Origine Nantais (Ron). It shares a high degree of structural similarity with MET, and it is also known to have multiple hetero-interactions. Ron is canonically activated by binding to its ligand, macrophage stimulating protein (MSP, also known as hepatocyte growth factor-like or HGFL), and it is important for embryonic and physiological development.417 Overexpression and mutations of Ron are associated with a wide variety of cancer, which has resulted in a growing interest in using it as a therapeutic target.426–428 Ron and Met can heterodimerize and transphosphorylate each other, triggering downstream signaling cascades.208,209
Ron has been found to interact with EGFR in a variety of systems. Using the mouse fibroblast cell line NIH 3T3, a phenotypic analysis done by overexpressing Ron and modulating the activity of endogenous EGFR indicated a functional link between Ron and EGFR, and they coimmunoprecipitate in the presence and absence of MSP and EGF284. This Ron-EGFR link has also been found in dog osteosarcoma cells276 and head and neck squamous cell carcinomas (HNSCCs).285 The majority of HNSCC tumor samples tested overexpress Ron, and increasing Ron levels correlates with increasing EGFR. In HNSCC cell lines, MSP stimulation causes increased EGFR phosphorylation. EGFR and Ron coimmunoprecipitate in the presence of MSP, but only weakly in its absence. This interaction has also been seen in human bladder cancer cell lines, where inhibiting either EGFR or Ron decreases the phosphorylation of the other, and they coimmunoprecipitate.286 As determined by immunofluorescence, serum starvation results in the majority of the plasma membrane Ron being translocated to the nucleus.287 The nuclear fraction is not phosphorylated, but it does colocalize with EGFR, and siRNA knockdown of Ron decreases nuclear EGFR expression. A ChIP-chip analysis indicated that Ron and EGFR are both associating with stress response pathways, suggesting that under cancerous conditions, the Ron-EGFR axis is acting as a transcription factor to promote survival. In addition to enhancing signaling through bidirectional cross-phosphorylation, the Ron-EGFR interactions appears to affect signaling by altering the spatial distribution of EGFR, which leads to altered gene expression.
A link between Ron and IGF-1R was first seen when a screen of childhood sarcoma lines revealed variable sensitivity to the IGF-1R inhibitor BMS-536924.314 Using an siRNA library against tyrosine kinases found that knocking down Ron results in the greatest sensitivity to BMS-536924 in resistant cell lines. This connection has also been seen in pancreatic cancer cell lines, where Ron and IGF-1R were seen to interact in the presence and absence of MSP and IGF-1 via coimmunoprecipitation and PLA.315 The Ron inhibitor BMS-777607 decreases hetero-interaction. MSP does not result in IGF-1R phosphorylation, but IGF-1 does cause Ron phosphorylation. Altogether, these results suggest that the Ron-IGF-1R interaction is able to contribute to the pathology and potentially the drug resistance of several cancers.
Additionally, Ron has also been found to interact with PDGFRβ.319 In human mesangial cells (HMCs), Ron phosphorylation increases with addition of PDGF-BB, although the phosphorylation kinetics observed by western blotting are different than that of PDGFRβ. Inhibiting PDGFRβ with imatinib reduces Ron phosphorylation, and Ron and PDGFRβ coimmunoprecipitate and colocalize on the plasma membrane via immunocytochemistry. Similar to the Ron-EGFR interaction seen in bladder cancer, Ron can localize to the nucleus, but unlike that case, it is the phosphorylated Ron which primarily does so. Moreover, the Ron-PDGFRβ interaction is at least partly cell dependent, as in both human epidermal keratinocytes and peripheral blood-derived adherent monocytes expressing both PDGFRβ and Ron, addition of PDGF-BB does not result in increased Ron phosphorylation.
8.12. AXL Interacts with Multiple RTKs
One final RTK with known cross-family hetero-interactions is AXL. AXL is a member of the TAM family (named for the three members which compose it, TYRO3, AXL, and MER), and unlike most RTKs, it is not primarily involved in development, but rather maintaining adult tissue.429 This maintenance role is important for processes such as the nervous system development,430 inhibition of the innate immune system,431–433 and phagocytosis of apoptotic cells.434,435 Unsurprisingly, disfunction of AXL is linked to many diseases, including cancer,436,437 autoimmune diseases,438–440 and infectious diseases.441,442
An interaction between AXL and EGFR appears to play a role in drug resistance in several cancers. AXL expression was identified as a strong predictor of ErbB inhibitor resistance in cancer by applying a machine learning algorithm to publicly available cancer databases.263 The interaction was investigated using TNBC cells which endogenously express both AXL and EGFR. Inhibiting EGFR with erlotinib does not inhibit cell viability, but addition of the AXL inhibitor R428 does. Both MET and AXL have increased phosphorylation upon EGF addition, although the AXL ligand Gas6 does not activate EGFR, and siRNA silencing of AXL decreases EGF, TGFα, and HGF induced phosphorylation of downstream signaling molecules. A crosslinking coimmunoprecipitation assay indicated that EGFR, ErbB2, ErbB4, MET, and PDGFRβ all interact with AXL.
This AXL-EGFR interaction has been seen in several other cancers and drug resistances. In a GBM cell line, EGF increases AXL phosphorylation on the same time scale as EGFR phosphorylation increases, and inhibiting AXL with BGB324 does not affect EGF induced phosphorylation, but inhibiting EGFR with gefitinib does.264 AXL and EGFR interact in the absence of ligand as seen by both coimmunoprecipitation and BiFC, and inhibiting AXL blocks EGF induced invasiveness. Across a range of squamous cell carcinoma cell lines, increasing AXL expression correlates with increasing resistance to the PI3Kα inhibitor BYL719.265 The resistant cells exhibit more EGFR-AXL interaction relative to the sensitive ones as seen by coimmunoprecipitation and PLA. Moreover, overexpressing AXL induces resistance in sensitive lines, but a kinase dead version of AXL does not, and inhibiting EGFR restores sensitivity regardless of AXL levels. This indicates that there exists an interaction between AXL and EGFR which confers resistance, and it requires the kinase domain of AXL to be functional. Specifically, inhibiting AXL decreases phosphorylation of the EGFR tyrosine 1173, which is important for the PLCγ/PKC signaling axis. Several studies have indicated that AXL expression can cause resistance to multiple anti-EGFR drugs in NSCLC cell lines, and inhibiting AXL restores drug sensitivity.443–445
In addition to the AXL-EGFR interaction, an AXL interaction with ErbB2 has also been implicated in cancer drug resistance and poor patient prognosis. A study investigating the effects of increasing ErbB2 phosphorylation found that the phosphorylation of AXL increases with increasing ErbB2 phosphorylation.446 In breast cancer cells that express ErbB2, lapatinib resistant cell lines have increased AXL expression compared to sensitive lines, and inhibiting or silencing AXL restores sensitivity.296 Furthermore, AXL expression correlates with poor patient outcome in ErbB2 positive breast cancer. An AXL-ErbB2 interaction has been seen in both breast cancer cells (which exogenously express AXL and endogenously express ErbB2) and tumor samples (which endogenously express both receptors) via coimmunoprecipitation and PLA.297 Intriguingly, the AXL-ErbB2 interaction increases the concentration of AXL at the cell surface, possibly due to a decrease in endosomal degradation, an increase in recycling to the cell surface, or an increase complex stability. This same effect has been observed with the EGFR-ErbB2 interaction, which causes an increase in the concentration of EGFR on the cell surface.177 ErbB2 is able to phosphorylate kinase dead AXL, and this is inhibited by lapatinib, but AXL is unable to phosphorylate ErbB2. Coincubation of inhibitors for both AXL and ErbB2 causes increased inhibition of cell invasiveness, and a mice model indicated that AXL increased metastasis and intravasation, but tumors could still grow without it.
There is also direct evidence of an interaction between AXL and MET, as in hypothalamic Gonadotropin-releasing hormone neurons, AXL and MET coimmunoprecipitate (as do TYRO3 and MET, but not MER).316 Overexpression of AXL induces MET phosphorylation, and overexpression of a kinase dead AXL variant or silencing AXL decreases MET phosphorylation and cell migration in response to HGF. Additionally, there is crosstalk between AXL and VEGFR2, although a direct interaction has not been observed.333 In several endothelial cell lines, VEGFA promotes AXL phosphorylation, and inhibiting AXL with R428 blocks VEGFA induced activation of Akt. Furthermore, silencing AXL abolishes VEGFA induced migration, permeability, and tube formation. Src family kinases (SFK) appear to be mediating the interaction by phosphorylating the AXL juxtamembrane region after being activated by activated VEGFR2.
9. Using Thermodynamic Models to Understand Hetero-Interactions
The use of thermodynamic cycles that incorporate RTK hetero-interactions can help explain many of the complicated biological effects which have been described in the literature and are discussed above. For instance, the predictions in Figure 4 can explain how the presence of heterodimers can alter the liganded homodimer concentrations. In Figure 4A, we model the case of a dimeric ligand binding to an RTK which can heterodimerize (see Figure 3D for the schematic of this cycle). In other words, we are assuming that an RTK, X, can bind a dimeric ligand, L; can form homodimers; and can form heterodimers with another RTK, Y. The second RTK, Y, can also homodimerize, but it does not bind to the ligand.
The left plot shows the distribution of the X receptors into different types of dimers and monomers as a function of ligand concentration. There are five molecular species containing X: XY heterodimers, blue dotted line; X monomers, red dotted line; XX homodimers, black dotted line; LX liganded monomers, black dotted line; and LXX liganded dimers, black solid line. Under these conditions, almost twenty percent of the receptors exist in heterodimers when the ligand concentration is low, and the heterodimers slowly disappear as the ligand concentration increases. As expected, at low ligand concentrations, there are unliganded dimers and monomers, and as the ligand concentration increases, liganded dimers become the predominate species. Note, however, that at unphysiologically high ligand concentrations, liganded monomers have a substantial presence. Importantly, in this and all other plots in Figure 4, the magnitude of the results are strongly dependent on the values of the receptor dimerization and ligand binding constants, but the general trends are the same over a wide range of values.
In Figure 4A, we focus on the prediction for the liganded homodimers, as these are widely believed to be the most important, signaling-competent species. As seen in the right panel, the presence of heterodimers decreases the concentration of liganded homodimers. All assumptions and values for these graphs are the same as for the plot on the left. The red curve is the fraction of X receptors which exist as liganded dimers, shown as a function of ligand concentration in the absence of the Y receptors, and the blue curve is the fraction of X receptors which exist as liganded dimers in the presence of Y. The black curve is the difference between the red and blue curves, representing the decrease of the liganded homodimers due to heterodimerization. Importantly, the maximum value of the percent difference is around the expected physiological concentration of ligand.447,448 Although the effect on liganded dimers appears modest in this case, it can become much larger under some conditions, as discussed below. Furthermore, even small changes in the concentration of liganded dimers can have a large effect on signaling, as the signal is amplified through downstream signaling cascades.
A factor which furthers the decrease in liganded dimers is the presence of a second heterodimerization partner, as shown in Figure 4B. All assumptions in Figure 4B are the same as in Figure 4A, except that there exists a third RTK, Z, which can homodimerize and heterodimerize with X, but does not interact with Y or the ligand (see Figure 3E for the schematic of this cycle). The plot on the left shows the distribution of molecular species as a function of ligand concentration. It is similar to the left panel of Figure 4A, except that there is a second heterodimer, XZ (the cyan dotted line). On the right is a plot showing the effect of both receptors on the concentration of liganded dimers. As seen in the prediction describing the decrease in liganded dimeric fraction (black line), the decrease is roughly twice the decrease seen when only one heterodimerizing partner is present. This effect increases when the number of heterodimerizing partners is increased, and many cell types simultaneously express several RTKs. All these potential heterodimerizing RTKs could synergize to substantially decrease the concentration of liganded homodimers.
Another factor which leads to a large decrease in liganded dimers is the ability of the ligand to bind to both RTKs and the heterodimer, as seen in Figure 4C (for known examples, see the sections “RTK Hetero-Interactions within the Same Family” and “Ligands Binding to Multiple Subfamilies”). In this situation, in addition to the molecular species seen in Figure 4A, there exist LY liganded monomers, LYY liganded dimers, and LXY liganded heterodimers. The ligand is assumed to bind to Y an order of magnitude weaker than to its cognate receptor X, and all other assumptions are the same as in Figure 4A (see Figure 3F for the schematic of this cycle). On the left panel, we show the predicted levels of the different types of dimers and monomers. This plot of the molecular species distributions as a function of ligand looks similar to that seen in Figure 4A, except that there is a liganded heterodimer (the solid blue line). In this case, the liganded heterodimer fraction increases with increasing ligand concentration until it is above thirty percent and is close to the fraction of liganded homodimers. This model can explain why many hetero-interactions appear to increase after ligand addition. Again, the right panel shows the effect of heterodimers on liganded dimers, and in this case, the effect is fairly large, with the decrease being over forty percent at certain ligand concentrations.
There are many other, subtler factors which can have a large effect on heterodimerization and therefore, signaling. One example is seen in Figure 4D, which shows how increasing the concentration of one RTK can result in an increase in heterodimers involving another RTK. The model and assumptions used here are exactly the same as in Figure 4A, except that the concentration of the ligand binding RTK, X, is fixed while the concentration of the non-ligand binding RTK, Y, is allowed to vary, and the concentration of ligand is also fixed. The prediction is that an increase in the concentration of Y leads to an increase in the percent of X receptor population within XY heterodimers (dotted blue line), and a concomitant decrease in liganded homodimers (solid block line). This prediction applies to cases where factors such as drugs, disease states, or environmental stresses cause the upregulation of the expression of one RTK, and it explains how such conditions can increase hetero-interactions.
A particularly interesting observation is that sometimes hetero-interactions increase after the addition of an RTK inhibitor.268 This can potentially be explained by the prediction in Figure 4E, which is similar the ones in Figure 4A, except that the homodimerization association constant of the ligand binding RTK, X, and the ligand binding constants are reduced one hundred-fold. These decreases mimic the effect of a targeted inhibitor which decreases the ability of the RTK, X, to form homodimers and to bind ligand. The prediction in the left panel shows that at all but unphysiologically high ligand concentrations, almost all the RTK molecules exists as either monomers or heterodimers, with roughly equal amounts of each. The right panel shows the difference between the fraction of X receptors which are in heterodimers in the “no inhibitor case,” depicted in blue (identical conditions to Figure 4A) and in the case when the inhibitor is present, depicted in red. The black line indicates the difference (red line minus blue line), showing a large increase in heterodimers in the case where the inhibitor is present.
The discussed predictions based on thermodynamic cycles can help bring insight into a wide variety of biological systems and can be used to understand the effects of changing physiological conditions. However, the predictions are most meaningful when they are based on experimentally determined thermodynamic constants, which are rarely found in the literature. This is especially true in the case of hetero-interactions, where there are only a few measurements. Accordingly, we argue that there is a strong need for quantitative measurements of hetero-interaction strengths.
10. The RTK Interactome
Above, we described many examples of RTK hetero-interactions. It is possible that there exist many more RTK hetero-interactions that have not yet been identified. Based on the current knowledge, we cannot exclude the possibility that any two RTKs, even if they belong to different subfamilies, can engage in hetero-interactions under the right conditions. Accordingly, RTK hetero-interactions may be ubiquitous, and thus each RTK may participate in an extensive network of RTK interactions, which we call the RTK interactome. Accordingly, current models of RTK activation are likely incomplete, as they are strongly biased toward homodimer-driven signaling.
Our understanding of the scope of the various RTK interactions is presently rudimentary, and we have only a partial view of the RTK interactome. No systematic investigation of all RTK-RTK interaction partners has been undertaken, and hence many interactions are probably unknown. Furthermore, the physical mechanisms underlying RTK cross-subfamily hetero-interactions is largely unknown. It is possible that interactions between the kinase domains play a significant role, as the kinase domain is highly conserved across families,36,449 and FRET studies of RTK homodimers have shown that the deletion of their IC domains leads to dimer destabilization, both in the presence and absence of ligand.13,21 Another, not mutually exclusive, possibility is that the TM domains play a key role, as it has been argued in the literature that the interactions between RTK TM domains are promiscuous, forming many weak, nonspecific interactions.450–452 Although the EC domains of different subfamilies are often rather different (see Figure 1), they could also play a role, especially if there is a common ligand that binds to them.
There are not many measurements of stabilities of RTK homodimers and heterodimers, but these data suggest that stabilities of the homodimers and the heterodimers in the absence of ligand may be comparable.97,173 In this case, the expression levels of the different RTKs will be the main factor that determines the relative abundance of homodimers and heterodimers. Different cells express different RTKs and do so to different degrees, and thus each cell type will be characterized by its own set of RTK interactions that control its fate. Once a ligand is introduced into the interaction network, it will likely stabilize all dimers to which it binds. Notably, based on the best-estimate calculations in Figure 4, the presence of ligand will likely not abolish the heterodimers, even if the heterodimers do not bind the ligand, except at high ligand concentrations. Accordingly, ligand addition alters the interactions by enriching some dimers and depleting others, and we expect that it will also cause conformational changes which enhance phosphorylation. We cannot currently predict in a comprehensive and quantitative way exactly how this will occur, due to a lack of basic knowledge about RTK and ligand expressions in tissues and about their interaction strengths. Due to the presence of numerous hetero-interactions, studies of isolated homodimers are unlikely to be able to predict the complete signal transduction properties of a receptor in cells that express multiple RTKs.
10.1. The Biological Function of the Hetero-Interactions in the RTK Interactome
It is often assumed that the ligand-bound homodimer is the dominant signaling species and the master regulator of the signaling response. In this case, one possible role of the heterodimers is to sequester the receptor, and hence inhibit the signaling that is mediated by the liganded homodimer. As shown in the predictions in Figure 4, the presence of heterodimers decreases the concentration of the liganded homodimer. The more RTK interaction partners that are expressed, the larger the inhibitory effect. For instance, in Figure 4B, we predict that at physiological ligand concentrations in the presence of only two interaction partners, a greater than 15% decrease in the RTK liganded dimer population will occur relative to the case where there are no homodimers. If more interaction partners are expressed, the inhibitory effect will be much larger.
However, the assumption that the ligand-bound homodimer is the dominant signaling species may not be always correct. There are many possible biological functions of hetero-interactions, as depicted in Figure 5. For instance, many of the papers overviewed above demonstrate that the heterodimers are also active. In some cases, the heterodimer signals in a unique way, and thus homodimer and heterodimers mediate distinct downstream signaling cascades. Such unique heterodimer signaling has been observed in several instances, including the case of the PDGFRα-PDGFRβ206,207 and VEGFR1-VEGFR2 complexes.199 Cross-family hetero-interactions resulting in unique signaling have also been seen. For example, the ROR1-ErbB3 interaction results in the phosphorylation of a unique tyrosine on ErbB3, triggering a specific signaling cascade that modulates the Hippo-YAP pathway.309 Mechanistically, this can occur via the phosphorylation of unique tyrosines in the heterocomplex or if the heterocomplex recruits other molecules with unique activities.
Figure 5.

Depiction of the possible effects of RTK heterodimerization. Center shows a generic, activated RTK homodimer in equilibrium with an RTK heterodimer (blue diamond, ligand and purple P, phosphorylation). Starting from the top left and going clockwise, unique signaling, where the heterodimer causes signaling not seen in the homodimers, possible due to unique tyrosines being phosphorylated (pink P). Amplified signaling, where the heterodimer has a stronger downstream signal than the homodimer, possible due to increased phosphorylation or decreased degradation. Modified signaling, where the heterodimer has a different probability of phosphorylation or adaptor protein binding than the homodimer, and it is possible that some tyrosines have increased phosphorylation while other tyrosines have decreased phosphorylation. Inhibited signaling, where the heterodimer has a weaker downstream signal than the homodimer, possible due to the heterodimer being inactive or recruitment of a molecule which directly dephosphorylates the RTK. Signaling back-up, where the homodimer has been inhibited, possibly by a drug (red cross), but signaling can continue due to direct phosphorylation by the heterodimerization partner. Note that the heterodimers are shown as unliganded, which may not always be the case (i.e., some heterodimers could be liganded), and that the heterodimer effects are shown as only affecting signaling of one of the RTK species, although the effects could be bidirectional (i.e., the signaling of both RTK species could be affected).
There are also examples in the literature where both homodimers and heterodimers work synergistically to activate an RTK, contributing to the same downstream effects. In this case, heterodimerization works to strengthen the response without altering or diversifying it. The RTK can be efficiently phosphorylated by the partner in the heterodimer (and in some cases, the partner is also phosphorylated), and hence the outcome is signal amplification, originating from an increase in the concentration of phosphorylated receptors. Moreover, heterocomplexes can increase signaling by decreasing receptor internalization, and hence increasing the duration of the signal, as seen in the cases of ErbB2-EGFR177 and ErbB2-AXL297 heterocomplexes.
In some cases, the probability of phosphorylation or the probability of an adaptor protein binding is different within the homodimers and the heterodimers. This leads to quantitative differences in the degree of activation, but it does not cause divergent signaling. In this case, heterodimerization works to modulate the strength of the response. For instance, it has been proposed that the RYK-EphB2 and RYK-EphB3 interactions modulate Eph signaling, and that this interaction becomes dysregulated in craniofrontaonasal syndrome.343 Such modulating effects exert fine control over RTK signaling, enabling small changes in receptor or ligand concentration to alter the population of homo- and heterodimers, resulting in increases or decreases in signaling as needed.
Heterodimerization can also work to inhibit signaling. As discussed above, this can occur if a receptor is inactive within the heterodimer, with heterodimerization working to sequester the receptors and prevent them for forming active ligand-bound homodimers. An example of this is the case of VEGFR1-VEGFR2 heterodimers, where VEGFR1 sequesters both the VEGFA ligand and VEGFR2 in order to tightly regulate the activity of VEGFR2.66,197,198 There are also cases where the heterodimer works to recruit a phosphatase which dephosphorylate the heterodimerized receptor. Examples of such inhibition can be seen with the MET-VEGFR2 and PDGFRβ-VEGFR2 interactions. In the MET-VEGFR2 case, the heterocomplex results in recruitment of PTP1B, which dephosphorylates MET and hence decreases MET signaling, and this effect is abolished by inhibiting VEGFR2 phosphorylation.318 Although an exact mechanism was not identified in the PDGFRβ-VEGFR2 case, it was shown that the PDGFRβ-VEGFR2 complex inhibits PDGFRβ signaling, and this only occurs in the presence of PDGF-BB and VEGFA.321,322 This is suggestive of a phosphatase being recruited, although it is possible that in the PDGFRβ-VEGFR2 heterodimers, PDGFRβ is blocked from binding adaptor proteins due to steric hindrance, thus failing to activate downstream signaling cascades.
It is further believed that heterodimerization can work to provide a signaling back-up. In this scenario, the main signaling entity is the homodimer. Yet when the homodimer is inhibited, the heterodimer assumes the signaling functions, rescuing the signaling pathway. Although this process is often thought to occur due to two different RTKs having overlapping downstream effects, it can be caused by direct phosphorylation of the inhibited RTK by the other RTK. This is mostly commonly seen in cancer, where a wide range of RTK inhibitors become ineffective or have minimal or inconsistent clinical efficacy. A drug could prevent homodimerization, block ligand binding, or suppress kinase activity, but the RTK can be still activated within a heterodimer and signal normally, since it gets phosphorylated by the partner RTK. It is possible that this signaling back-up could sometimes be beneficial, as it could allow normal signaling to occur if a mutation or deficiency reduces the ability of one RTK to homo-phosphorylate.
In general, the existence of hetero-interactions greatly increases the signaling complexity of RTKs and allows for a greater degree of regulation. Based on the current literature, it appears that the major roles of the RTK interactome are to significantly enhance the diversity of the signaling and provide signaling back-ups. It is likely that the RTK interactome mediates many additional signaling outcomes that have not yet been uncovered. Further comprehensive studies will be needed to understand the full scope of signal diversification through heterodimerization.
10.2. Implications of the RTK Interactome
Studies and mechanistic models of RTK signaling often assume that RTK homodimers are the predominant dimers in the cellular membrane, and experimental data are often interpreted under this paradigm. However, this assumption may lead to incorrect conclusions, especially in cases where other RTKs are expressed at high levels. It is possible that models of RTK activation that solely consider homodimers will never be able to correctly predict activity and cellular outcomes. Instead, we propose that the whole interactome needs to be included in order to arrive at new, comprehensive models of RTK activation with predictive power.
Furthermore, the RTK interactome concept may provide an explanation for differences in experimental data acquired in different cell lines. Usually, the literature only gives information about the expression of the receptor under investigation, and it is generally unknown what other receptors are expressed in a cell. Accordingly, even the types of possible heterodimers are unknown. Yet it is conceivable that the presence of these heterodimers greatly influences the cell signaling outcomes. It is possible that many contradictions in the literature cannot be completely resolved until we understand the entire network of RTK interactions.
The hetero-interactions also need to be considered in the design of RTK inhibitors for cancers and other diseases and disorders. Even if a drug successfully inhibits the signaling of an RTK homodimer, if a heterodimer is able to rescue its function, the drug will not have the desired effect. Therefore, the RTK expression pattern in a cell type may be a critical factor in determining the performance of the inhibitor. Since an inhibitor can affect the functions of both homodimers and heterodimers—or even influences the entire RTK interactome—it can lead to many unanticipated consequences. Without an understanding of the biological effects of hetero-interactions, it will be difficult to predict all the possible effects of an inhibitor. There are, of course, many challenges and technical hurdles in determining the strength of heterodimerization. Many RTKs, especially those in the same subfamily, are similar sizes, and hence homo- and heterodimers are indistinguishable by some techniques, particularly western blots. Fluorescence-based experiments can only follow labeled RTKs that have been introduced in the cells, and hence heterodimers may appear as monomers. Detection based on antibodies will only reveal the specific RTK being probed, and heterodimerization partners will accordingly be missed unless they are also individually probed. This is in addition to the fact that different cells express different RTKs and ligands to different degrees, and accordingly, a specific RTK hetero-interaction that does not occur under one set of conditions may occur under different conditions. Unfortunately, current biochemical and biophysical experimental techniques are not well suited to follow the entire RTK interactome in a cell. We look forward to new method development, new computational approaches, and new basic knowledge about RTK heterodimerization strengths and expressions that will move the field forward.
10.3. Beyond the RTK Interactome
There are many documented cases of interactions between RTKs and other membrane proteins, such as cell adhesion molecules, GPCRs, and other signaling receptors. For example, VEGFR2 interacts with VE-cadherin,453 and there is evidence of crosstalk between EGFR and Ecadherin.454 Numerous RTKs interact with integrins,455 including VEGFR2,456,457 PDGFRβ,458,459 and MET.460,461 A few of the RTKs are involved in Wnt signaling, for instance RYK interacts with Frizzled proteins,334 ROR2 interacts with Vangl2462 and Frizzled proteins,463 PTK7 interacts with LRP6464 and Frizzled proteins,465 and MuSK interacts with LRP4.466,467 Several RTKs interact with semaphoring receptors, including some which interact with plexins, such as MET,468,469 ErbB2,470,471 VEGFR2,472 and FGFR1;472 VEGFR2 interacts with nuerophilin-1473,474 and nuerophilin-2.475 These interactions have a wide range of important biological consequences.
Furthermore, RTKs directly interact with the plasma membrane and the cytoskeleton.45,476 There is evidence that the activity of EGFR is strongly affected by lipid composition; in particular, interactions with gangliosides477 and negatively charged lipids such as phosphatidylinositol-phosphates (PIPs)478,479 have been shown to play a key role. Numerous other RTKs have been found to interact with gangliosides, including the other ErbBs, FGFRs, TrkA, MET, PDGFRs, VEGFR2, and InsR—these interactions can be activating or inhibitory.480,481 It has further been proposed that residues in the N-terminal portion of the JM region of all RTKs interact with negatively charged lipids, in particular PIP2, and this interaction is important for proper RTK dimerization and function.482 Moreover, it has been found that the plasma membrane is sectioned into 40–300 nm compartments or corrals by the associated cortical cytosol mesh, and this can increase local receptor concentration, shifting monomer-dimer equilibria.483 The cytoskeleton also directly interacts with RTKs. For instance, EGFR binds to actin,484,485 and EphA2 exhibits directed transport due to an actin interaction.486 There is also evidence that actin plays a crucial role in the organization of InsR after ligand binding.487 These interactions could provide an additional layer of RTK signaling regulation, and they need to be considered in order to obtain a comprehensive understanding of RTK function.
It is highly likely that the RTK interactome is a part of an extensive membrane protein interactome: a network of interactions between diverse families of membrane proteins and lipids. However, little is known about the scope of these interactions, and much more work needs to be done by cell signaling researchers and membrane biophysicists before we fully understand how these interactions regulate biological function. We are looking forward to the many new discoveries in the years to come.
11. Summary and Future Perspectives
The incredible complexity of RTK hetero-interactions has only begun to be explored. These interactions can have numerous different effects, and we group these into five general categories (Figure 5). One, unique signaling outputs that increase signal diversity. Two, amplification of signaling by direct phosphorylation or by reduced internalization of the receptors, thereby increasing the signaling lifetimes. Three, modification of signaling when the probability of phosphorylation or adaptor protein binding is different in a homodimer than a heterodimer. Four, inhibition of signaling of one or both of the signaling pathways of the RTKs which compose the heterocomplex, either by sequestering on an RTK in an inactive heterocomplex or by dephosphorylating the components. Five, signaling back-up, where the signaling of an inhibited RTK is rescued by direct phosphorylation by its hetero-interaction partner. Importantly (as discussed in the “Using Thermodynamic Models to Understand Hetero-Interactions” section), the understanding of RTK hetero-interactions may help in understanding the drug resistance in cancer therapy and in designing more effective treatments.
A multitude of factors control whether and to what degree RTK hetero-interactions occur. These factors include the concentrations of the receptors, the concentrations of the ligands, and whether or not other interaction partners are present. Accordingly, different cell lines, media conditions, the degree of invasiveness of the cells, the degree of drug sensitivity of the cells, and the presence of inhibitor drugs can all affect the formation of these heterocomplexes. In turn, these interactions affect a multitude of downstream biological outcomes. Given the importance of the biological processes mediated by the 58 human RTKs, there is a great need for quantitative, thermodynamic studies that report on the strength of the interactions. As discussed above, such studies can help explain aspects of the complex RTK biology. Furthermore, close attention needs to be paid to the concentration of all RTKs, as these concentrations determine the identities of the homo- and heterodimers. Such quantitative measurements will allow for the creation of detailed thermodynamic models accounting for all relevant RTK dimers, and can ultimately predict RTK activity and the nature of the biological response.
11.1. Final Thoughts
Our understanding of RTKs has grown tremendously since they were first discovered in the 1970s, and new quantitative, physical-chemical studies of the RTK interactome will contribute to our ever-expanding knowledge of the complexity of RTK signaling. This new knowledge can empower the design of novel RTK targeted inhibitors. Ultimately, a deeper knowledge of the RTK interactome thermodynamics will lead to better understanding of fundamental biological processes in health and disease.
Acknowledgments
Supported by NIH GM068619 and NSF MCB1712740.
Biographies
Michael D. Paul received B.S. degrees in chemistry, biochemistry, and mathematics from the University of Chicago in 2014. After graduating, he joined the Johns Hopkins Program in Molecular Biophysics for his Ph.D. His current research is focused on using quantitative florescence microscopy methods and thermodynamic modeling to understand protein-protein interactions in the plasma membrane of cells.
Kalina Hristova received her B.S. and M.S. degrees in Physics from the University of Sofia, Bulgaria, and her Ph.D. degree in Mechanical Engineering and Materials Science from Duke University, USA. She did post-doctoral work at the University of California, Irvine. She is now a Professor of Materials Science and Engineering at the Institute for NanoBioTechnology at Johns Hopkins University. Dr. Hristova is the recipient of the 2007 Margaret Oakley Dayhoff award from the Biophysical Society. She was elected Fellow of the American Physical Society in 2016, and Fellow of the American Institute for Medical and Biological Engineering in 2018. The main focus of the research in her laboratory is the physical principles that underlie membrane protein folding and signal transduction across biological membranes.
Footnotes
Notes
The authors declare no competing financial interest.
References
- (1).Kawa S; Fujimoto J; Tezuka T; Nakazawa T; Yamamoto T Involvement of Brek, a Serine/Threonine Kinase Enriched in Brain, in Ngf Signalling. Genes to Cells 2004, 9, 219–232. [DOI] [PubMed] [Google Scholar]
- (2).Wang H; Brautigan DL Peptide Microarray Analysis of Substrate Specificity of the Transmembrane Ser/Thr Kinase Kpi-2 Reveals Reactivity with Cystic Fibrosis Transmembrane Conductance Regulator and Phosphorylase. Mol Cell Proteomics 2006, 5, 2124–2130. [DOI] [PubMed] [Google Scholar]
- (3).Wyman J; Gill SJ Binding and Linkage: Functional Chemistry of Biological Macromolecules; University Science Books, 1990. [Google Scholar]
- (4).Fantl WJ; Johnson DE; Williams LT Signaling by Receptor Tyrosine Kinases. Annual Review of Biochemistry 1993, 62, 453–481. [DOI] [PubMed] [Google Scholar]
- (5).Lemmon MA; Schlessinger J Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lawrence MC; Ward CW Structural Features of the Receptor Tyrosine Kinase Ectodomains. Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease 2015, 163–193. [Google Scholar]
- (7).Arteaga CL; Engelman JA Erbb Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Belov AA; Mohammadi M Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harb Perspect Biol 2013, 5, a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wagner MJ; Stacey MM; Liu BA; Pawson T Molecular Mechanisms of Sh2- and Ptb-Domain-Containing Proteins in Receptor Tyrosine Kinase Signaling. Cold Spring Harb Perspect Biol 2013, 5, a008987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Carpenter G; King L; Cohen S Epidermal Growth-Factor Stimulates Phosphorylation in Membrane Preparations Invitro. Nature 1978, 276, 409–410. [DOI] [PubMed] [Google Scholar]
- (11).Carpenter G; King L; Cohen S Rapid Enhancement of Protein-Phosphorylation in a-431 Cell-Membrane Preparations by Epidermal Growth-Factor. Journal of Biological Chemistry 1979, 254, 4884–4891. [PubMed] [Google Scholar]
- (12).Carpenter G; Cohen S Epidermal Growth-Factor. Annual Review of Biochemistry 1979, 48, 193–216. [DOI] [PubMed] [Google Scholar]
- (13).Sarabipour S; Ballmer-Hofer K; Hristova K Vegfr-2 Conformational Switch in Response to Ligand Binding. Elife 2016, 5, e13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sarabipour S; Hristova K Pathogenic Cysteine Removal Mutations in Fgfr Extracellular Domains Stabilize Receptor Dimers and Perturb the Tm Dimer Structure. J Mol Biol 2016, 428, 3903–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shen JY; Maruyama IN Nerve Growth Factor Receptor Trka Exists as a Preformed, yet Inactive, Dimer in Living Cells. Febs Letters 2011, 585, 295–299. [DOI] [PubMed] [Google Scholar]
- (16).Maruyama I; Shen JY Brain-Derived Neurotrophic Factor Receptor Trkb Exists as a Preformed Dimer in Living Cells. FASEB Journal 2012, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yu XC; Sharma KD; Takahashi T; Iwamoto R; Mekada E Ligand-Independent Dimer Formation of Epidermal Growth Factor Receptor (Egfr) Is a Step Separable from Ligand-Induced Egfr Signaling. Molecular Biology of the Cell 2002, 13, 2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tao RH; Maruyama IN All Egf(Erbb) Receptors Have Preformed Homo- and Heterodimeric Structures in Living Cells. Journal of Cell Science 2008, 121, 3207–3217. [DOI] [PubMed] [Google Scholar]
- (19).Maruyama IN Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells 2014, 3, 304–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lin CC; Melo FA; Ghosh R; Suen KM; Stagg LJ; Kirkpatrick J; Arold ST; Ahmed Z; Ladbury JE Inhibition of Basal Fgf Receptor Signaling by Dimeric Grb2. Cell 2012, 149, 1514–1524. [DOI] [PubMed] [Google Scholar]
- (21).Sarabipour S; Hristova K Mechanism of Fgf Receptor Dimerization and Activation. Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen L; Placone J; Novicky L; Hristova K The Extracellular Domain of Fibroblast Growth Factor Receptor 3 Inhibits Ligand-Independent Dimerization. Science Signaling 2010, 3, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jura N; Endres NF; Engel K; Deindl S; Das R; Lamers MH; Wemmer DE; Zhang X; Kuriyan J Mechanism for Activation of the Egf Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 2009, 137, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Thiel KW; Carpenter G Epidermal Growth Factor Receptor Juxtamembrane Region Regulates Allosteric Tyrosine Kinase Activation. Proceedings of the National Academy of Sciences 2007, 104, 19238–19243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Brewer MR; Choi SH; Alvarado D; Moravcevic K; Pozzi A; Lemmon MA; Carpenter G The Juxtamembrane Region of the Egf Receptor Functions as an Activation Domain. Molecular cell 2009, 34, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xu AM; Huang PH Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Res 2010, 70, 3857–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hamilton G; Rath B; Klameth L; Hochmair M Receptor Tyrosine Kinase Expression of Circulating Tumor Cells in Small Cell Lung Cancer. Oncoscience 2015, 2, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nagatsuma AK; Aizawa M; Kuwata T; Doi T; Ohtsu A; Fujii H; Ochiai A Expression Profiles of Her2, Egfr, Met and Fgfr2 in a Large Cohort of Patients with Gastric Adenocarcinoma. Gastric Cancer 2015, 18, 227–238. [DOI] [PubMed] [Google Scholar]
- (29).Davis S; Gale NW; Aldrich TH; Maisonpierre PC; Lhotak V; Pawson T; Goldfarb M; Yancopoulos GD Ligands for Eph-Related Receptor Tyrosine Kinases That Require Membrane Attachment or Clustering for Activity. Science 1994, 266, 816–819. [DOI] [PubMed] [Google Scholar]
- (30).Ivanisevic L; Zheng W; Woo SB; Neet KE; Saragovi HU Trka Receptor “Hot Spots” for Binding of Nt-3 as a Heterologous Ligand. J Biol Chem 2007, 282, 16754–16763. [DOI] [PubMed] [Google Scholar]
- (31).Macdonald-Obermann JL; Pike LJ Different Epidermal Growth Factor (Egf) Receptor Ligands Show Distinct Kinetics and Biased or Partial Agonism for Homodimer and Heterodimer Formation. J Biol Chem 2014, 289, 26178–26188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Klein P; Mattoon D; Lemmon MA; Schlessinger J A Structure-Based Model for Ligand Binding and Dimerization of Egf Receptors. Proceedings of the National Academy of Sciences 2004, 101, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ornitz D; Leder P Ligand Specificity and Heparin Dependence of Fibroblast Growth Factor Receptors 1 and 3. Journal of Biological Chemistry 1992, 267, 16305–16311. [PubMed] [Google Scholar]
- (34).Naylor RL; Robertson AG; Allen SJ; Sessions RB; Clarke AR; Mason GG; Burston JJ; Tyler SJ; Wilcock GK; Dawbarn D A Discrete Domain of the Human Trkb Receptor Defines the Binding Sites for Bdnf and Nt-4. Biochem Biophys Res Commun 2002, 291, 501–507. [DOI] [PubMed] [Google Scholar]
- (35).Landau M; Ben-Tal N Dynamic Equilibrium between Multiple Active and Inactive Conformations Explains Regulation and Oncogenic Mutations in Erbb Receptors. Biochim Biophys Acta 2008, 1785, 12–31. [DOI] [PubMed] [Google Scholar]
- (36).Süveges D; Jura N Structural Features of the Kinase Domain. Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease 2015, 195–223. [Google Scholar]
- (37).Scheck RA; Lowder MA; Appelbaum JS; Schepartz A Bipartite Tetracysteine Display Reveals Allosteric Control of Ligand-Specific Egfr Activation. ACS Chem Biol 2012, 7, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wilson KJ; Gilmore JL; Foley J; Lemmon MA; Riese DJ Functional Selectivity of Egf Family Peptide Growth Factors: Implications for Cancer. Pharmacology & Therapeutics 2009, 122, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sinclair JKL; Walker AS; Doerner AE; Schepartz A Mechanism of Allosteric Coupling into and through the Plasma Membrane by Egfr. Cell Chem Biol 2018, 25, 857–870 e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bell CA; Tynan JA; Hart KC; Meyer AN; Robertson SC; Donoghue DJ Rotational Coupling of the Transmembrane and Kinase Domains of the Neu Receptor Tyrosine Kinase. Molecular Biology of the Cell 2000, 11, 3589–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Macdonald-Obermann JL; Pike LJ Allosteric Regulation of Epidermal Growth Factor (Egf) Receptor Ligand Binding by Tyrosine Kinase Inhibitors. J Biol Chem 2018, 293, 13401–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lu C; Mi LZ; Grey MJ; Zhu J; Graef E; Yokoyama S; Springer TA Structural Evidence for Loose Linkage between Ligand Binding and Kinase Activation in the Epidermal Growth Factor Receptor. Mol Cell Biol 2010, 30, 5432–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lu C; Mi LZ; Schurpf T; Walz T; Springer TA Mechanisms for Kinase-Mediated Dimerization of the Epidermal Growth Factor Receptor. J Biol Chem 2012, 287, 38244–38253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Freed DM; Bessman NJ; Kiyatkin A; Salazar-Cavazos E; Byrne PO; Moore JO; Valley CC; Ferguson KM; Leahy DJ; Lidke DS et al. Egfr Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bocharov EV; Mineev KS; Pavlov KV; Akimov SA; Kuznetsov AS; Efremov RG; Arseniev AS Helix-Helix Interactions in Membrane Domains of Bitopic Proteins: Specificity and Role of Lipid Environment. Biochim Biophys Acta Biomembr 2017, 1859, 561–576. [DOI] [PubMed] [Google Scholar]
- (46).Kuriyan J; Cowburn D Modular Peptide Recognition Domains in Eukaryotic Signaling. Annual review of biophysics and biomolecular structure 1997, 26, 259–288. [DOI] [PubMed] [Google Scholar]
- (47).Schlessinger J; Lemmon MA Sh2 and Ptb Domains in Tyrosine Kinase Signaling. Sci. STKE 2003, 2003, re12. [DOI] [PubMed] [Google Scholar]
- (48).Belov AA; Mohammadi M Grb2, a Double-Edged Sword of Receptor Tyrosine Kinase Signaling. Science signaling 2012, 5, pe49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhou S; Shoelson SE; Chaudhuri M; Gish G; Pawson T; Haser WG; King F; Roberts T; Ratnofsky S; Lechleider RJ Sh2 Domains Recognize Specific Phosphopeptide Sequences. Cell 1993, 72, 767–778. [DOI] [PubMed] [Google Scholar]
- (50).Del Piccolo N; Hristova K Quantifying the Interaction between Egfr Dimers and Grb2 in Live Cells. Biophys J 2017, 113, 1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Artalejo CR; Lemmon MA; Schlessinger J; Palfrey HC Specific Role for the Ph Domain of Dynamin-1 in the Regulation of Rapid Endocytosis in Adrenal Chromaffin Cells. EMBO Journal 1997, 16, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Schlessinger J Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [DOI] [PubMed] [Google Scholar]
- (53).He L; Hristova K Physical-Chemical Principles Underlying Rtk Activation, and Their Implications for Human Disease. Biochimica et Biophysica Acta 2012, 1818, 995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Boyd AW; Bartlett PF; Lackmann M Therapeutic Targeting of Eph Receptors and Their Ligands. Nature Reviews Drug Discovery 2014, 13, 39–62. [DOI] [PubMed] [Google Scholar]
- (55).Sun C; Bernards R Feedback and Redundancy in Receptor Tyrosine Kinase Signaling: Relevance to Cancer Therapies. Trends Biochem Sci 2014, 39, 465–474. [DOI] [PubMed] [Google Scholar]
- (56).Ross JS; Slodkowska EA; Symmans WF; Pusztai L; Ravdin PM; Hortobagyi GN The Her-2 Receptor and Breast Cancer: Ten Years of Targeted Anti-Her-2 Therapy and Personalized Medicine. Oncologist 2009, 14, 320–368. [DOI] [PubMed] [Google Scholar]
- (57).Turner N; Grose R Fibroblast Growth Factor Signalling: From Development to Cancer. Nat Rev Cancer 2010, 10, 116–129. [DOI] [PubMed] [Google Scholar]
- (58).Lin CC; Melo FA; Ghosh R; Suen KM; Stagg LJ; Kirkpatrick J; Arold ST; Ahmed Z; Ladbury JE Inhibition of Basal Fgf Receptor Signaling by Dimeric Grb2. Cell 2012, 149, 1514–1524. [DOI] [PubMed] [Google Scholar]
- (59).Singh DR; Ahmed F; King C; Gupta N; Salotto M; Pasquale EB; Hristova K Epha2 Receptor Unliganded Dimers Suppress Epha2 Pro-Tumorigenic Signaling. Journal of Biological Chemistry 2015, 290, 27271–27279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Pasquale EB Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [DOI] [PubMed] [Google Scholar]
- (61).Gherardi E; Birchmeier W; Birchmeier C; Vande Woude G Targeting Met in Cancer: Rationale and Progress. Nat Rev Cancer 2012, 12, 89–103. [DOI] [PubMed] [Google Scholar]
- (62).He L; Horton WA; Hristova K The Physical Basis Behind Achondroplasia, the Most Common Form of Human Dwarfism. Journal of Biological Chemistry 2010, 285 30103–30114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ahmed Z; George R; Lin CC; Suen KM; Levitt JA; Suhling K; Ladbury JE Direct Binding of Grb2 Sh3 Domain to Fgfr2 Regulates Shp2 Function. Cellular Signalling 2010, 22, 23–33. [DOI] [PubMed] [Google Scholar]
- (64).Ahmed Z; Lin CC; Suen KM; Melo FA; Levitt JA; Suhling K; Ladbury JE Grb2 Controls Phosphorylation of Fgfr2 by Inhibiting Receptor Kinase and Shp2 Phosphatase Activity. Journal of Cell Biology 2013, 200, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Macdonald JL; Pike LJ Heterogeneity in Egf-Binding Affinities Arises from Negative Cooperativity in an Aggregating System. Proceedings of the National Academy of Sciences 2008, 105, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Olsson AK; Dimberg A; Kreuger J; Claesson-Welsh L Vegf Receptor Signalling - in Control of Vascular Function. Nat Rev Mol Cell Biol 2006, 7, 359–371. [DOI] [PubMed] [Google Scholar]
- (67).Leppanen VM; Prota AE; Jeltsch M; Anisimov A; Kalkkinen N; Strandin T; Lankinen H; Goldman A; Ballmer-Hofer K; Alitalo K Structural Determinants of Growth Factor Binding and Specificity by Vegf Receptor 2. Proc Natl Acad Sci U S A 2010, 107, 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Gingras AC; Gstaiger M; Raught B; Aebersold R Analysis of Protein Complexes Using Mass Spectrometry. Nat Rev Mol Cell Biol 2007, 8, 645–654. [DOI] [PubMed] [Google Scholar]
- (69).Gavin AC; Maeda K; Kuhner S Recent Advances in Charting Protein-Protein Interaction: Mass Spectrometry-Based Approaches. Curr Opin Biotechnol 2011, 22, 42–49. [DOI] [PubMed] [Google Scholar]
- (70).Dunham WH; Mullin M; Gingras AC Affinity-Purification Coupled to Mass Spectrometry: Basic Principles and Strategies. Proteomics 2012, 12, 1576–1590. [DOI] [PubMed] [Google Scholar]
- (71).Larance M; Lamond AI Multidimensional Proteomics for Cell Biology. Nat Rev Mol Cell Biol 2015, 16, 269–280. [DOI] [PubMed] [Google Scholar]
- (72).Aebersold R; Mann M Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- (73).Phizicky EM; Fields S Protein-Protein Interactions: Methods for Detection and Analysis. Microbiological reviews 1995, 59, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bonifacino JS; Dell’Angelica EC; Springer TA Immunoprecipitation. Current protocols in immunology 2001, 41, 8.3. 1–8.3. 28. [DOI] [PubMed] [Google Scholar]
- (75).Fields A; Song O A Novel Genetic System to Detect Protein-Protein Interactions. Nature 1989, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- (76).Parrish JR; Gulyas KD; Finley RL Jr. Yeast Two-Hybrid Contributions to Interactome Mapping. Curr Opin Biotechnol 2006, 17, 387–393. [DOI] [PubMed] [Google Scholar]
- (77).Yao Z; Darowski K; St-Denis N; Wong V; Offensperger F; Villedieu A; Amin S; Malty R; Aoki H; Guo H et al. A Global Analysis of the Receptor Tyrosine Kinase-Protein Phosphatase Interactome. Mol Cell 2017, 65, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Ito T; Chiba T; Ozawa R; Yoshida M; Hattoti M; Sakaki Y A Comprehensive Two-Hybrid Analysis to Explore the Yeast Protein Interactome. Proc Natl Acad Sci U S A 2001, 98, 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Bruckner A; Polge C; Lentze N; Auerbach D; Schlattner U Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int J Mol Sci 2009, 10, 2763–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Stagljar I; Korostensky C; Johnsson N; te Heesen S A Genetic System Based on Split-Ubiquitin for the Analysis of Interactions between Membrane Proteins in Vivo. Proceedings of the National Academy of Sciences 1998, 95, 5187–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Snider J; Kittanakom S; Damjanovic D; Curak J; Wong V; Stagljar I Detecting Interactions with Membrane Proteins Using a Membrane Two-Hybrid Assay in Yeast. Nat Protoc 2010, 5, 1281–1293. [DOI] [PubMed] [Google Scholar]
- (82).Petschnigg J; Groisman B; Kotlyar M; Taipale M; Zheng Y; Kurat CF; Sayad A; Sierra JR; Usaj MM; Snider J et al. The Mammalian-Membrane Two-Hybrid Assay (Mamth) for Probing Membrane-Protein Interactions in Human Cells. Nat Meth 2014, 11, 585–592. [DOI] [PubMed] [Google Scholar]
- (83).Roux KJ; Kim DI; Raida M; Burke B A Promiscuous Biotin Ligase Fusion Protein Identifies Proximal and Interacting Proteins in Mammalian Cells. J Cell Biol 2012, 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Hesketh GG; Youn J-Y; Samavarchi-Tehrani P; Raught B; Gingras A-C Parallel Exploration of Interaction Space by Bioid and Affinity Purification Coupled to Mass Spectrometry. Proteomics: Methods and Protocols 2017, DOI: 10.1007/978-1-4939-6747-6_1010.1007/978-1-4939-6747-6_10, 115–136. [DOI] [PubMed] [Google Scholar]
- (85).Roux KJ; Kim DI; Burke B; May DG Bioid: A Screen for Protein-Protein Interactions. Curr Protoc Protein Sci 2018, 91, 19.23.11–19.23.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Fredriksson S; Gullberg M; Jarvius J; Olsson C; Pietras K; Gústafsdóttir SM; Östman A; Landegren U Protein Detection Using Proximity-Dependent DNA Ligation Assays. Nat. Biotechnol 2002, 20, 473–477. [DOI] [PubMed] [Google Scholar]
- (87).Gullberg M; Gústafsdóttir SM; Schallmeiner E; Jarvius J; Bjarnegård M; Betsholtz C; Landegren U; Fredriksson S Cytokine Detection by Antibody-Based Proximity Ligation. PNAS 2004, 101, 8420–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Soderberg O; Gullberg M; Jarvius M; Ridderstrale K; Leuchowius KJ; Jarvius J; Wester K; Hydbring P; Bahram F; Larsson LG et al. Direct Observation of Individual Endogenous Protein Complexes in Situ by Proximity Ligation. Nat Methods 2006, 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- (89).Soderberg O; Leuchowius KJ; Gullberg M; Jarvius M; Weibrecht I; Larsson LG; Landegren U Characterizing Proteins and Their Interactions in Cells and Tissues Using the in Situ Proximity Ligation Assay. Methods 2008, 45, 227–232. [DOI] [PubMed] [Google Scholar]
- (90).Raicu V Efficiency of Resonance Energy Transfer in Homo-Oligomeric Complexes of Proteins. J Biol Phys 2007, 33, 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Chen LR; Novicky L; Merzlyakov M; Hristov T; Hristova K Measuring the Energetics of Membrane Protein Dimerization in Mammalian Membranes. Journal of the American Chemical Society 2010, 132, 3628–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Sarabipour S; Del Piccolo N; Hristova K Characterization of Membrane Protein Interactions in Plasma Membrane Derived Vesicles with Quantitative Imaging Forster Resonance Energy Transfer. Acc Chem Res 2015, 48, 2262–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Singh DR; Ahmed F; Sarabipour S; Hristova K Intracellular Domain Contacts Contribute to Ecadherin Constitutive Dimerization in the Plasma Membrane. J Mol Biol 2017, 429, 2231–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Li E; Placone J; Merzlyakov M; Hristova K Quantitative Measurements of Protein Interactions in a Crowded Cellular Environment. Analytical chemistry 2008, 80, 5976–5985. [DOI] [PubMed] [Google Scholar]
- (95).Del Piccolo N; Placone J; Hristova K Effect of Thanatophoric Dysplasia Type I Mutations on Fgfr3 Dimerization. Biophys J 2015, 108, 272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Sarabipour S; Hristova K Fgfr3 Unliganded Dimer Stabilization by the Juxtamembrane Domain. Journal of Molecular Biology 2015, 427, 1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Del Piccolo N; Sarabipour S; Hristova K A New Method to Study Heterodimerization of Membrane Proteins and Its Application to Fibroblast Growth Factor Receptors. J Biol Chem 2017, 292, 1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).King C; Stoneman M; Raicu V; Hristova K Fully Quantified Spectral Imaging Reveals in Vivo Membrane Protein Interactions. Integr Biol (Camb) 2016, 8, 216–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Biener G; Stoneman MR; Acbas G; Holz JD; Orlova M; Komarova L; Kuchin S; Raicu V Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation. Int J Mol Sci 2013, 15, 261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Wolber P; Hudson B An Analytic Solution to the Förster Energy Transfer Problem in Two Dimensions. Biophysical journal 1979, 28, 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).King C; Sarabipour S; Byrne P; Leahy DJ; Hristova K The Fret Signatures of Noninteracting Proteins in Membranes: Simulations and Experiments. Biophys J 2014, 106, 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).King C; Raicu V; Hristova K Understanding the Fret Signatures of Interacting Membrane Proteins. J Biol Chem 2017, 292, 5291–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Merzlyakov M; You M; Li E; Hristova K Transmembrane Helix Heterodimerization in Lipids Bilayers: Probing the Energetics Behind Autosomal Dominant Growth Disorders. Journal of Molecular Biology 2006, 358 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Veatch W; Stryer L The Dimeric Nature of the Gramicidin a Transmembrane Channel: Conductance and Fluorescence Energy Transfer Studies of Hybrid Channels. J Mol Biol 1977, 113, 89–102. [DOI] [PubMed] [Google Scholar]
- (105).Walsh SM; Mathiasen S; Christensen SM; Fay JF; King C; Provasi D; Borrero E; Rasmussen SGF; Fung JJ; Filizola M et al. Single Proteoliposome High-Content Analysis Reveals Differences in the Homo-Oligomerization of Gpcrs. Biophys J 2018, 115, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Raicu V; Stoneman MR; Fung R; Melnichuk M; Jansma DB; Pisterzi LF; Rath S; Fox M; Wells JW; Saldin DK Determination of Supramolecular Structure and Spatial Distribution of Protein Complexes in Living Cells. Nature Photonics 2009, 3, 107–113. [Google Scholar]
- (107).Singh DR; Mohammad MM; Patowary S; Stoneman MR; Oliver JA; Movileanu L; Raicu V Determination of the Quaternary Structure of a Bacterial Atp-Binding Cassette (Abc) Transporter in Living Cells. Integrative Biology 2013, 5, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Needham SR; Hirsch M; Rolfe DJ; Clarke DT; Zanetti-Domingues LC; Wareham R; Martin-Fernandez ML Measuring Egfr Separations on Cells with ~10 Nm Resolution Via Fluorophore Localization Imaging with Photobleaching. PLoS One 2013, 8, e62331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Needham SR; Roberts SK; Arkhipov A; Mysore VP; Tynan CJ; Zanetti-Domingues LC; Kim ET; Losasso V; Korovesis D; Hirsch M et al. Egfr Oligomerization Organizes Kinase-Active Dimers into Competent Signalling Platforms. Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Digman MA; Dalal R; Horwitz AF; Gratton E Mapping the Number of Molecules and Brightness in the Laser Scanning Microscope. Biophys J 2008, 94, 2320–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Digman MA; Wiseman PW; Choi C; Horwitz AR; Gratton E Stoichiometry of Molecular Complexes at Adhesions in Living Cells. Proceedings of the National Academy of Sciences 2009, 106, 2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Godin AG; Costantino S; Lorenzo LE; Swift JL; Sergeev M; Ribeiro-da-Silva A; De Koninck Y; Wiseman PW Revealing Protein Oligomerization and Densities in Situ Using Spatial Intensity Distribution Analysis. Proc Natl Acad Sci U S A 2011, 108, 7010–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Swift JL; Godin AG; Dore K; Freland L; Bouchard N; Nimmo C; Sergeev M; De Koninck Y; Wiseman PW; Beaulieu JM Quantification of Receptor Tyrosine Kinase Transactivation through Direct Dimerization and Surface Density Measurements in Single Cells. Proc Natl Acad Sci U S A 2011, 108, 7016–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Magde D; Elson E; Webb WW Thermodynamic Fluctuations in a Reacting System–Measurement by Fluorescence Correlation Spectroscopy. Physical Review Letters 1972, 29, 705–708. [Google Scholar]
- (115).Elson EL; Magde D Fluorescence Correlation Spectroscopy. I. Conceptual Basis and Theory. Biopolymers: Original Research on Biomolecules 1974, 13, 1–27. [DOI] [PubMed] [Google Scholar]
- (116).Berland KM; So P; Gratton E Two-Photon Fluorescence Correlation Spectroscopy: Method and Application to the Intracellular Environment. Biophysical Journal 1995, 68, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Kim SA; Heinze KG; Schwille P Fluorescence Correlation Spectroscopy in Living Cells. Nature methods 2007, 4, 963–973. [DOI] [PubMed] [Google Scholar]
- (118).Schwille P; Bieschke J; Oehlenschläger F Kinetic Investigations by Fluorescence Correlation Spectroscopy: The Analytical and Diagnostic Potential of Diffusion Studies. Biophysical chemistry 1997, 66, 211–228. [DOI] [PubMed] [Google Scholar]
- (119).Schwille P; Meyer-Almes F-J; Rigler R Dual-Color Fluorescence Cross-Correlation Spectroscopy for Multicomponent Diffusional Analysis in Solution. Biophysical journal 1997, 72, 1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Bacia K; Schwille P Practical Guidelines for Dual-Color Fluorescence Cross-Correlation Spectroscopy. Nature protocols 2007, 2, 2842–2856. [DOI] [PubMed] [Google Scholar]
- (121).Hess ST; Huang S; Heikal AA; Webb WW Biological and Chemical Applications of Fluorescence Correlation Spectroscopy: A Review. Biochemistry 2002, 41, 697–705. [DOI] [PubMed] [Google Scholar]
- (122).Haustein E; Schwille P Ultrasensitive Investigations of Biological Systems by Fluorescence Correlation Spectroscopy. Methods 2003, 29, 153–166. [DOI] [PubMed] [Google Scholar]
- (123).Bacia K; Kim SA; Schwille P Fluorescence Cross-Correlation Spectroscopy in Living Cells. Nature methods 2006, 3, 83–89. [DOI] [PubMed] [Google Scholar]
- (124).Digman MA; Gratton E Fluorescence Correlation Spectroscopy and Fluorescence Cross-Correlation Spectroscopy. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 2009, 1, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Ries J; Schwille P Fluorescence Correlation Spectroscopy. Bioessays 2012, 34, 361–368. [DOI] [PubMed] [Google Scholar]
- (126).Muller BK; Zaychikov E; Brauchle C; Lamb DC Pulsed Interleaved Excitation. Biophys J 2005, 89, 3508–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Huang Y; Bharill S; Karandur D; Peterson SM; Marita M; Shi X; Kaliszewski MJ; Smith AW; Isacoff EY; Kuriyan J Molecular Basis for Multimerization in the Activation of the Epidermal Growth Factor Receptor. Elife 2016, 5, e14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Brown C; Dalal R; Hebert B; Digman M; Horwitz A; Gratton E Raster Image Correlation Spectroscopy (Rics) for Measuring Fast Protein Dynamics and Concentrations with a Commercial Laser Scanning Confocal Microscope. Journal of microscopy 2008, 229, 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Digman MA; Wiseman PW; Horwitz AR; Gratton E Detecting Protein Complexes in Living Cells from Laser Scanning Confocal Image Sequences by the Cross Correlation Raster Image Spectroscopy Method. Biophys J 2009, 96, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Ulbrich MH; Isacoff EY Subunit Counting in Membrane-Bound Proteins. Nat Methods 2007, 4, 319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Chen Y; Bharill S; Isacoff EY; Chalfie M Subunit Composition of a Deg/Enac Mechanosensory Channel of Caenorhabditis Elegans. Proc Natl Acad Sci U S A 2015, 112, 11690–11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Hariri AA; Hamblin GD; Hardwick JS; Godin R; Desjardins JF; Wiseman PW; Sleiman HF; Cosa G Stoichiometry and Dispersity of DNA Nanostructures Using Photobleaching Pair-Correlation Analysis. Bioconjug Chem 2017, 28, 2340–2349. [DOI] [PubMed] [Google Scholar]
- (133).Hu C-D; Chinenov Y; Kerppola TK Visualization of Interactions among Bzip and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Molecular cell 2002, 9, 789–798. [DOI] [PubMed] [Google Scholar]
- (134).Kerppola TK Bimolecular Fluorescence Complementation (Bifc) Analysis as a Probe of Protein Interactions in Living Cells. Annu Rev Biophys 2008, 37, 465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Johnsson N; Varshavsky A Split Ubiquitin as a Sensor of Protein Interactions in Vivo. Proceedings of the National Academy of Sciences 1994, 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Rossi F; Charlton CA; Blau HM Monitoring Protein–Protein Interactions in Intact Eukaryotic Cells by B-Galactosidase Complementation. Proceedings of the National Academy of Sciences 1997, 94, 8405–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Paulmurugan R; Umezawa Y; Gambhir S Noninvasive Imaging of Protein–Protein Interactions in Living Subjects by Using Reporter Protein Complementation and Reconstitution Strategies. Proceedings of the National Academy of Sciences 2002, 99, 15608–15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Wehr MC; Laage R; Bolz U; Fischer TM; Grünewald S; Scheek S; Bach A; Nave K-A; Rossner MJ Monitoring Regulated Protein-Protein Interactions Using Split Tev. Nature methods 2006, 3, 985–993. [DOI] [PubMed] [Google Scholar]
- (139).Hu CD; Kerppola TK Simultaneous Visualization of Multiple Protein Interactions in Living Cells Using Multicolor Fluorescence Complementation Analysis. Nat Biotechnol 2003, 21, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Miller KE; Kim Y; Huh WK; Park HO Bimolecular Fluorescence Complementation (Bifc) Analysis: Advances and Recent Applications for Genome-Wide Interaction Studies. J Mol Biol 2015, 427, 2039–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Orchard S; Ammari M; Aranda B; Breuza L; Briganti L; Broackes-Carter F; Campbell NH; Chavali G; Chen C; del-Toro N et al. The Mintact Project--Intact as a Common Curation Platform for 11 Molecular Interaction Databases. Nucleic Acids Res 2014, 42, D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Chatr-aryamontri A; Ceol A; Palazzi LM; Nardelli G; Schneider MV; Castagnoli L; Cesareni G Mint: The Molecular Interaction Database. Nucleic Acids Res 2007, 35, D572–D574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Hermjakob H; Montecchi-Palazzi L; Lewington C; Mudali S; Kerrien S; Orchard S; Vingron M; Roechert B; Roepstorff P; Valencia A et al. Intact: An Open Source Molecular Interaction Database. Nucleic Acids Res 2004, 32, D452–D455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Chatr-Aryamontri A; Oughtred R; Boucher L; Rust J; Chang C; Kolas NK; O’Donnell L; Oster S; Theesfeld C; Sellam A et al. The Biogrid Interaction Database: 2017 Update. Nucleic Acids Res 2017, 45, D369–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Kotlyar M; Pastrello C; Sheahan N; Jurisica I Integrated Interactions Database: Tissue-Specific View of the Human and Model Organism Interactomes. Nucleic Acids Res 2016, 44, D536–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Salwinski L; Miller CS; Smith AJ; Pettit FK; Bowie JU; Eisenberg D The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res 2004, 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Keshava Prasad TS; Goel R; Kandasamy K; Keerthikumar S; Kumar S; Mathivanan S; Telikicherla D; Raju R; Shafreen B; Venugopal A et al. Human Protein Reference Database--2009 Update. Nucleic Acids Res 2009, 37, D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Pagel P; Kovac S; Oesterheld M; Brauner B; Dunger-Kaltenbach I; Frishman G; Montrone C; Mark P; Stumpflen V; Mewes HW et al. The Mips Mammalian Protein-Protein Interaction Database. Bioinformatics 2005, 21, 832–834. [DOI] [PubMed] [Google Scholar]
- (149).Fabregat A; Jupe S; Matthews L; Sidiropoulos K; Gillespie M; Garapati P; Haw R; Jassal B; Korninger F; May B et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res 2018, 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Olayioye MA; Neve RM; Lane HA; Hynes NE The Erbb Signaling Network: Receptor Heterodimerization in Development and Cancer. The EMBO journal 2000, 19, 3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Yarden Y; Sliwkowski MX Untangling the Erbb Signalling Network. Nature reviews Molecular cell biology 2001, 2, 127–137. [DOI] [PubMed] [Google Scholar]
- (152).Real FX; Rettig WJ; Chesa PG; Melamed MR; Old LJ; Mendelsohn J Expression of Epidermal Growth Factor Receptor in Human Cultured Cells and Tissues: Relationship to Cell Lineage and Stage of Differentiation. Cancer research 1986, 46, 4726–4731. [PubMed] [Google Scholar]
- (153).Potter C; Daele S; VIJVER M; Pauwels C; Maertens G; Boever J; Vandekerckhove D; Roels H The Expression of the Neu Oncogene Product in Breast Lesions and in Normal Fetal and Adult Human Tissues. Histopathology 1989, 15, 351–362. [DOI] [PubMed] [Google Scholar]
- (154).Sibilia M; Wagner EF Strain-Dependent Epithelial Defects in Mice Lacking the Egf Receptor. Science 1995, 269, 234–238. [DOI] [PubMed] [Google Scholar]
- (155).Miettinen PJ; Berger JE; Meneses J; Phung Y; Pedersen RA; Werb Z; Derynck R Epithelial Immaturity and Multiorgan Failure in Mice Lacking Epidermal Growth Factor Receptor. Nature 1995, 376, 337–341. [DOI] [PubMed] [Google Scholar]
- (156).Shilo B-Z -Signaling by the Drosophila Epidermal Growth Factor Receptor Pathway During Development. The EGF Receptor Family 2003, 147–156. [DOI] [PubMed] [Google Scholar]
- (157).Singh B; Carpenter G; Coffey RJ Egf Receptor Ligands: Recent Advances. F1000Res 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Menard S; Casalini P; Campiglio M; Pupa S; Agresti R; Tagliabue E Her2 Overexpression in Various Tumor Types, Focussing on Its Relationship to the Development of Invasive Breast Cancer. Annals of Oncology 2001, 12, S15–S19. [DOI] [PubMed] [Google Scholar]
- (159).Nicholson R; Gee J; Harper M Egfr and Cancer Prognosis. European journal of cancer 2001, 37, 9–15. [DOI] [PubMed] [Google Scholar]
- (160).Holbro T; Beerli RR; Maurer F; Koziczak M; Barbas CF; Hynes NE The Erbb2/Erbb3 Heterodimer Functions as an Oncogenic Unit: Erbb2 Requires Erbb3 to Drive Breast Tumor Cell Proliferation. Proceedings of the National Academy of Sciences 2003, 100, 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Hirsch F; Varella-Garcia M; Cappuzzo F Predictive Value of Egfr and Her2 Overexpression in Advanced Non-Small-Cell Lung Cancer. Oncogene 2009, 28, S32–S37. [DOI] [PubMed] [Google Scholar]
- (162).Appert-Collin A; Hubert P; Cremel G; Bennasroune A Role of Erbb Receptors in Cancer Cell Migration and Invasion. Front Pharmacol 2015, 6, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Ciardiello F; Tortora G Egfr Antagonists in Cancer Treatment. New England Journal of Medicine 2008, 358, 1160–1174. [DOI] [PubMed] [Google Scholar]
- (164).Lee CK; Brown C; Gralla RJ; Hirsh V; Thongprasert S; Tsai CM; Tan EH; Ho JC; Chu da T; Zaatar A et al. Impact of Egfr Inhibitor in Non-Small Cell Lung Cancer on Progression-Free and Overall Survival: A Meta-Analysis. J Natl Cancer Inst 2013, 105, 595–605. [DOI] [PubMed] [Google Scholar]
- (165).Swain SM; Baselga J; Kim SB; Ro J; Semiglazov V; Campone M; Ciruelos E; Ferrero JM; Schneeweiss A; Heeson S et al. Pertuzumab, Trastuzumab, and Docetaxel in Her2-Positive Metastatic Breast Cancer. N Engl J Med 2015, 372, 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Rauf F; Festa F; Park JG; Magee M; Eaton S; Rinaldi C; Betanzos CM; Gonzalez-Malerva L; LaBaer J Ibrutinib Inhibition of Erbb4 Reduces Cell Growth in a Wnt5a-Dependent Manner. Oncogene 2018, 37, 2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Corfas G; Roy K; Buxbaum JD Neuregulin 1-Erbb Signaling and the Molecular/Cellular Basis of Schizophrenia. Nat Neurosci 2004, 7, 575–580. [DOI] [PubMed] [Google Scholar]
- (168).Keri S; Szabo C; Kelemen O Uniting the Neuro Developmental and Immunological Hypotheses: Neuregulin 1 Receptor Erbb and Toll-Like Receptor Activation in First-Episode Schizophrenia. Sci Rep 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Flisiak I; Szterling-Jaworowska M; Baran A; Rogalska-Taranta M Effect of Psoriasis Activity on Epidermal Growth Factor (Egf) and the Concentration of Soluble Egf Receptor in Serum and Plaque Scales. Clin Exp Dermatol 2014, 39, 461–467. [DOI] [PubMed] [Google Scholar]
- (170).Tan F; Yang G; Wang Y; Chen H; Yu B; Li H; Guo J; Huang X; Deng Y; Yu P et al. Icotinib Inhibits Egfr Signaling and Alleviates Psoriasis-Like Symptoms in Animal Models. Biomed Pharmacother 2018, 98, 399–405. [DOI] [PubMed] [Google Scholar]
- (171).Olayioye MA; Graus-Porta D; Beerli RR; Rohrer J; Gay B; Hynes NE Erbb-1 and Erbb-2 Acquire Distinct Signaling Properties Dependent Upon Their Dimerization Partner. Molecular and Cellular Biology 1998, 18, 5042–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Muthuswamy SK; Gilman M; Brugge JS Controlled Dimerization of Erbb Receptors Provides Evidence for Differential Signaling by Homo-and Heterodimers. Molecular and cellular biology 1999, 19, 6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Graus-Porta D; Beerli RR; Daly JM; Hynes NE Erbb-2, the Preferred Heterodimerization Partner of All Erbb Receptors, Is a Mediator of Lateral Signaling. The EMBO journal 1997, 16, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Guy PM; Platko JV; Cantley LC; Cerione RA; Carraway KL Insect Cell-Expressed P180erbb3 Possesses an Impaired Tyrosine Kinase Activity. Proceedings of the National Academy of Sciences 1994, 91, 8132–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Chausovsky A; Waterman H; Elbaum M; Yarden Y; Geiger B; Bershadsky A Molecular Requirements for the Effect of Neuregulin on Cell Spreading, Motility and Colony Organization. Oncogene 2000, 19, 878–888. [DOI] [PubMed] [Google Scholar]
- (176).Alimandi M; Romano A; Curia MC; Muraro R; Fedi P; Aaronson SA; Di Fiore PP; Kraus MH Cooperative Signaling of Erbb3 and Erbb2 in Neoplastic Transformation and Human Mammary Carcinomas. Oncogene 1995, 10, 18131822. [PubMed] [Google Scholar]
- (177).Lenferink AE; Pinkas-Kramarski R; van de Poll ML; van Vugt MJ; Klapper LN; Tzahar E; Waterman H; Sela M; van Zoelen EJ; Yarden Y Differential Endocytic Routing of Homo-and Hetero-Dimeric Erbb Tyrosine Kinases Confers Signaling Superiority to Receptor Heterodimers. The EMBO journal 1998, 17, 3385–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Qutub AA; Mac Gabhann F; Karagiannis ED; Vempati P; Popel AS Multiscale Models of Angiogenesis. IEEE Eng Med Biol Mag 2009, 28, 14–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Domigan CK; Ziyad S; Iruela-Arispe ML Canonical and Noncanonical Vascular Endothelial Growth Factor Pathways: New Developments in Biology and Signal Transduction. Arterioscler Thromb Vasc Biol 2015, 35, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Kukk E; Lymboussaki A; Taira S; Kaipainen A; Jeltsch M; Joukov V; Alitalo K Vegf-C Receptor Binding and Pattern of Expression with Vegfr-3 Suggests a Role in Lymphatic Vascular Development. Development 1996, 122, 3829–3837. [DOI] [PubMed] [Google Scholar]
- (181).Aspelund A; Robciuc MR; Karaman S; Makinen T; Alitalo K Lymphatic System in Cardiovascular Medicine. Circ Res 2016, 118, 515–530. [DOI] [PubMed] [Google Scholar]
- (182).Stacker SA; Achen MG; Jussila L; Baldwin ME; Alitalo K Metastasis: Lymphangiogenesis and Cancer Metastasis. Nature Reviews Cancer 2002, 2, 573–583. [DOI] [PubMed] [Google Scholar]
- (183).Ghosh S; Sullivan CA; Zerkowski MP; Molinaro AM; Rimm DL; Camp RL; Chung GG High Levels of Vascular Endothelial Growth Factor and Its Receptors (Vegfr-1, Vegfr-2, Neuropilin-1) Are Associated with Worse Outcome in Breast Cancer. Human pathology 2008, 39, 1835–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Rodriguez-Antona C; Pallares J; Montero-Conde C; Inglada-Perez L; Castelblanco E; Landa I; Leskela S; Leandro-Garcia LJ; Lopez-Jimenez E; Leton R et al. Overexpression and Activation of Egfr and Vegfr2 in Medullary Thyroid Carcinomas Is Related to Metastasis. Endocr Relat Cancer 2010, 17, 7–16. [DOI] [PubMed] [Google Scholar]
- (185).Sitohy B; Nagy JA; Dvorak HF Anti-Vegf/Vegfr Therapy for Cancer: Reassessing the Target. Cancer Res 2012, 72, 1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Zhao D; Pan C; Sun J; Gilbert C; Drews-Elger K; Azzam DJ; Picon-Ruiz M; Kim M; Ullmer W; El-Ashry D et al. Vegf Drives Cancer-Initiating Stem Cells through Vegfr-2/Stat3 Signaling to Upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [DOI] [PubMed] [Google Scholar]
- (187).Ferrara N; Adamis AP Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat Rev Drug Discov 2016, 15, 385–403. [DOI] [PubMed] [Google Scholar]
- (188).Ohno-Matsui K; Morita I; Tombran-Tink J; Mrazek D; Onodera M; Uetama T; Hayano M; Murota S. i.; Mochizuki M Novel Mechanism for Age-Related Macular Degeneration: An Equilibrium Shift between the Angiogenesis Factors Vegf and Pedf. Journal of cellular physiology 2001, 189, 323–333. [DOI] [PubMed] [Google Scholar]
- (189).Ohr M; Kaiser PK Intravitreal Aflibercept Injection for Neovascular (Wet) Age-Related Macular Degeneration. Expert Opin Pharmacother 2012, 13, 585–591. [DOI] [PubMed] [Google Scholar]
- (190).Osaadon P; Fagan XJ; Lifshitz T; Levy J A Review of Anti-Vegf Agents for Proliferative Diabetic Retinopathy. Eye (Lond) 2014, 28, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Simo R; Sundstrom JM; Antonetti DA Ocular Anti-Vegf Therapy for Diabetic Retinopathy: The Role of Vegf in the Pathogenesis of Diabetic Retinopathy. Diabetes Care 2014, 37, 893–899. [DOI] [PubMed] [Google Scholar]
- (192).Nagashima M; Yoshino S; Ishiwata T; Asano G Role of Vascular Endothelial Growth Factor in Angiogenesis of Rheumatoid Arthritis. The Journal of rheumatology 1995, 22, 1624–1630. [PubMed] [Google Scholar]
- (193).Leblond A; Allanore Y; Avouac J Targeting Synovial Neoangiogenesis in Rheumatoid Arthritis. Autoimmun Rev 2017, 16, 594–601. [DOI] [PubMed] [Google Scholar]
- (194).Fong G-H; Rossant J; Gertsenstein M; Breitman ML Role of the Flt-1 Receptor Tyrosine Kinase in Regulating the Assembly of Vascular Endothelium. Nature 1995, 376, 66–70. [DOI] [PubMed] [Google Scholar]
- (195).Hiratsuka S; Minowa O; Kuno J; Noda T; Shibuya M Flt-1 Lacking the Tyrosine Kinase Domain Is Sufficient for Normal Development and Angiogenesis in Mice. Proceedings of the National Academy of Sciences 1998, 95, 9349–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Kendall RL; Wang G; Thomas KA Identification of a Natural Soluble Form of the Vascular Endothelial Growth Factor Receptor, Flt-1, and Its Heterodimerization with Kdr. Biochemical and biophysical research communications 1996, 226, 324–328. [DOI] [PubMed] [Google Scholar]
- (197).Rahimi N; Dayanir V; Lashkari K Receptor Chimeras Indicate That the Vascular Endothelial Growth Factor Receptor-1 (Vegfr-1) Modulates Mitogenic Activity of Vegfr-2 in Endothelial Cells. Journal of Biological Chemistry 2000, 275, 16986–16992. [DOI] [PubMed] [Google Scholar]
- (198).Zeng H; Dvorak HF; Mukhopadhyay D Vascular Permeability Factor (Vpf)/Vascular Endothelial Growth Factor (Vegf) Receptor-1 Down-Modulates Vpf/Vegf Receptor-2-Mediated Endothelial Cell Proliferation, but Not Migration, through Phosphatidylinositol 3-Kinase-Dependent Pathways. Journal of Biological Chemistry 2001, 276, 26969–26979. [DOI] [PubMed] [Google Scholar]
- (199).Huang K; Andersson C; Roomans GM; Ito N; Claesson-Welsh L Signaling Properties of Vegf Receptor-1 and-2 Homo-and Heterodimers. The international journal of biochemistry & cell biology 2001, 33, 315–324. [DOI] [PubMed] [Google Scholar]
- (200).Autiero M; Waltenberger J; Communi D; Kranz A; Moons L; Lambrechts D; Kroll J; Plaisance S; De Mol M; Bono F Role of Plgf in the Intra-and Intermolecular Cross Talk between the Vegf Receptors Flt1 and Flk1. Nature medicine 2003, 9, 936–943. [DOI] [PubMed] [Google Scholar]
- (201).Cudmore MJ; Hewett PW; Ahmad S; Wang KQ; Cai M; Al-Ani B; Fujisawa T; Ma B; Sissaoui S; Ramma W et al. The Role of Heterodimerization between Vegfr-1 and Vegfr-2 in the Regulation of Endothelial Cell Homeostasis. Nat Commun 2012, 3. [DOI] [PubMed] [Google Scholar]
- (202).DiSalvo J; Bayne ML; Conn G; Kwok PW; Trivedi PG; Soderman DD; Palisi TM; Sullivan KA; Thomas KA Purification and Characterization of a Naturally Occurring Vascular Endothelial Growth Factor Placenta Growth Factor Heterodimer. Journal of Biological Chemistry 1995, 270, 7717–7723. [DOI] [PubMed] [Google Scholar]
- (203).Cao Y; Chen H; Zhou L; Chiang M-K; Anand-Apte B; Weatherbee JA; Wang Y; Fang F; Flanagan JG; Tsang ML-S Heterodimers of Placenta Growth Factor/Vascular Endothelial Growth Factor Endothelial Activity, Tumor Cell Expression, and High Affinity Binding to Flk-1/Kdr. Journal of Biological Chemistry 1996, 271, 3154–3162. [DOI] [PubMed] [Google Scholar]
- (204).Dixelius J; Mäkinen T; Wirzenius M; Karkkainen MJ; Wernstedt C; Alitalo K; Claesson-Welsh L Ligand-Induced Vascular Endothelial Growth Factor Receptor-3 (Vegfr-3) Heterodimerization with Vegfr-2 in Primary Lymphatic Endothelial Cells Regulates Tyrosine Phosphorylation Sites. Journal of Biological Chemistry 2003, 278, 40973–40979. [DOI] [PubMed] [Google Scholar]
- (205).Nilsson I; Bahram F; Li X; Gualandi L; Koch S; Jarvius M; Soderberg O; Anisimov A; Kholova I; Pytowski B et al. Vegf Receptor 2/−3 Heterodimers Detected in Situ by Proximity Ligation on Angiogenic Sprouts. EMBO J 2010, 29, 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Rupp E; Siegbahn A; Rönnstrand L; Wernstedt C; Claesson-Welsh L; Heldin CH A Unique Autophosphorylation Site in the Platelet-Derived Growth Factor A Receptor from a Heterodimeric Receptor Complex. The FEBS Journal 1994, 225, 29–41. [DOI] [PubMed] [Google Scholar]
- (207).Wu E; Palmer N; Tian Z; Moseman AP; Galdzicki M; Wang X; Berger B; Zhang H; Kohane IS Comprehensive Dissection of Pdgf-Pdgfr Signaling Pathways in Pdgfr Genetically Defined Cells. PLoS One 2008, 3, e3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Follenzi A; Bakovic S; Gual P; Stella MC; Longati P;, C. PM Cross-Talk between the Proto-Oncogenes Met and Ron. Oncogene 2000, 19, 3041–3049. [DOI] [PubMed] [Google Scholar]
- (209).Benvenuti S; Lazzari L; Arnesano A; Li Chiavi G; Gentile A; Comoglio PM Ron Kinase Transphosphorylation Sustains Met Oncogene Addiction. Cancer Res 2011, 71, 1945–1955. [DOI] [PubMed] [Google Scholar]
- (210).Freywald A; Sharfe N; Roifman CM The Kinase-Null Ephb6 Receptor Undergoes Transphosphorylation in a Complex with Ephb1. J Biol Chem 2002, 277, 3823–3828. [DOI] [PubMed] [Google Scholar]
- (211).Truitt L; Freywald T; DeCoteau J; Sharfe N; Freywald A The Ephb6 Receptor Cooperates with C-Cbl to Regulate the Behavior of Breast Cancer Cells. Cancer Res 2010, 70, 1141–1153. [DOI] [PubMed] [Google Scholar]
- (212).Janes PW; Griesshaber B; Atapattu L; Nievergall E; Hii LL; Mensinga A; Chheang C; Day BW; Boyd AW; Bastiaens PI et al. Eph Receptor Function Is Modulated by Heterooligomerization of a and B Type Eph Receptors. J Cell Biol 2011, 195, 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Akada M; Harada K; Negishi M; Katoh H Ephb6 Promotes Anoikis by Modulating Epha2 Signaling. Cell Signal 2014, 26, 2879–2884. [DOI] [PubMed] [Google Scholar]
- (214).Jurek A; Genander M; Kundu P; Catchpole T; He X; Straat K; Sabelstrom H; Xu NJ; Pettersson S; Henkemeyer M et al. Eph Receptor Interclass Cooperation Is Required for the Regulation of Cell Proliferation. Exp Cell Res 2016, 348, 10–22. [DOI] [PubMed] [Google Scholar]
- (215).Paganoni S; Bernstein J; Ferreira A Ror1-Ror2 Complexes Modulate Synapse Formation in Hippocampal Neurons. Neuroscience 2010, 165, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Brown JE; Krodel M; Pazos M; Lai C; Prieto AL Cross-Phosphorylation, Signaling and Proliferative Functions of the Tyro3 and Axl Receptors in Rat2 Cells. PLoS One 2012, 7, e36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Soos M; Siddle K Immunological Relationships between Receptors for Insulin and Insulin-Like Growth Factor I. Evidence for Structural Heterogeneity of Insulin-Like Growth Factor I Receptors Involving Hybrids with Insulin Receptors. Biochemical Journal 1989, 263, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Bailyes EM; Hayward AC; Siddle K Insulin Receptor/Igf-I Receptor Hybrids Are Widely Distributed in Mammalian Tissues: Quantification of Individual Receptor Species by Selective Immunoprecipitation and Immunoblotting. Biochemical Journal 1997, 327, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Belfiore A; Pandini G; Vella V; Squatrito S; Vigneri R Insulin/Igf-I Hybrid Receptors Play a Major Role in Igf-I Signaling in Thyroid Cancer. Biochimie 1999, 81, 403–407. [DOI] [PubMed] [Google Scholar]
- (220).Belfiore A; Frasca F; Pandini G; Sciacca L; Vigneri R Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr Rev 2009, 30, 586–623. [DOI] [PubMed] [Google Scholar]
- (221).Partanen J; Armstrong E; Mäkelä T; Korhonen J; Sandberg M; Renkonen R; Knuutila S; Huebner K; Alitalo K A Novel Endothelial Cell Surface Receptor Tyrosine Kinase with Extracellular Epidermal Growth Factor Homology Domains. Molecular and cellular biology 1992, 12, 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Augustin HG; Koh GY; Thurston G; Alitalo K Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin-Tie System. Nat Rev Mol Cell Biol 2009, 10, 165–177. [DOI] [PubMed] [Google Scholar]
- (223).Yuan HT; Venkatesha S; Chan B; Deutsch U; Mammoto T; Sukhatme VP; Woolf AS; Karumanchi SA Activation of the Orphan Endothelial Receptor Tie1 Modifies Tie2-Mediated Intracellular Signaling and Cell Survival. FASEB J 2007, 21, 3171–3183. [DOI] [PubMed] [Google Scholar]
- (224).Marron MB; Singh H; Tahir TA; Kavumkal J; Kim HZ; Koh GY; Brindle NP Regulated Proteolytic Processing of Tie1 Modulates Ligand Responsiveness of the Receptor-Tyrosine Kinase Tie2. J Biol Chem 2007, 282, 30509–30517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Hansen TM; Singh H; Tahir TA; Brindle NP Effects of Angiopoietins-1 and −2 on the Receptor Tyrosine Kinase Tie2 Are Differentially Regulated at the Endothelial Cell Surface. Cell Signal 2010, 22, 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Seegar TC; Eller B; Tzvetkova-Robev D; Kolev MV; Henderson SC; Nikolov DB; Barton WA Tie1-Tie2 Interactions Mediate Functional Differences between Angiopoietin Ligands. Mol Cell 2010, 37, 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Bonanomi D; Chivatakarn O; Bai G; Abdesselem H; Lettieri K; Marquardt T; Pierchala BA; Pfaff SL Ret Is a Multifunctional Coreceptor That Integrates Diffusible- and Contact-Axon Guidance Signals. Cell 2012, 148, 568–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Marler KJ; Becker-Barroso E; Martinez A; Llovera M; Wentzel C; Poopalasundaram S; Hindges R; Soriano E; Comella J; Drescher U A Trkb/Ephrina Interaction Controls Retinal Axon Branching and Synaptogenesis. J Neurosci 2008, 28, 12700–12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Chong LD; Park EK; Latimer E; Friesel R; Daar IO Fibroblast Growth Factor Receptor-Mediated Rescue of X-Ephrin B1-Induced Cell Dissociation in Xenopusembryos. Molecular and cellular biology 2000, 20, 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Brückner K; Pasquale EB; Klein R Tyrosine Phosphorylation of Transmembrane Ligands for Eph Receptors. Science 1997, 275, 1640–1643. [DOI] [PubMed] [Google Scholar]
- (231).Ball SG; Shuttleworth CA; Kielty CM Vascular Endothelial Growth Factor Can Signal through Platelet-Derived Growth Factor Receptors. J Cell Biol 2007, 177, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Pennock S; Kazlauskas A Vascular Endothelial Growth Factor a Competitively Inhibits Platelet-Derived Growth Factor (Pdgf)-Dependent Activation of Pdgf Receptor and Subsequent Signaling Events and Cellular Responses. Mol Cell Biol 2012, 32, 1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Russo K; Ragone R; Facchiano AM; Capogrossi MC; Facchiano A Platelet-Derived Growth Factor-Bb and Basic Fibroblast Growth Factor Directly Interact in Vitro with High Affinity. J Biol Chem 2002, 277, 1284–1291. [DOI] [PubMed] [Google Scholar]
- (234).Lin MH; Chang CA; Fischer WB Estimating Binding Free Energy of a Putative Growth Factors Egf-Vegf Complex - a Computational Bioanalytical Study. J Biomol Struct Dyn 2016, 34, 1717–1724. [DOI] [PubMed] [Google Scholar]
- (235).Li JJ; Lan KL; Chang SF; Chen YF; Tsai WC; Chiang PH; Lin MH; Fischer WB; Shih YS; Yen SH et al. Development and Characterization of the Recombinant Human Vegf-Egf Dual-Targeting Fusion Protein as a Drug Delivery System. Bioconjug Chem 2015, 26, 2481–2496. [DOI] [PubMed] [Google Scholar]
- (236).Fabian MA; Biggs WH 3rd; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M et al. A Small Molecule-Kinase Interaction Map for Clinical Kinase Inhibitors. Nat Biotechnol 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- (237).Bantscheff M; Eberhard D; Abraham Y; Bastuck S; Boesche M; Hobson S; Mathieson T; Perrin J; Raida M; Rau C et al. Quantitative Chemical Proteomics Reveals Mechanisms of Action of Clinical Abl Kinase Inhibitors. Nat Biotechnol 2007, 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- (238).Turner CA; Eren-Kocak E; Inui EG; Watson SJ; Akil H Dysregulated Fibroblast Growth Factor (Fgf) Signaling in Neurological and Psychiatric Disorders. Semin Cell Dev Biol 2016, 53, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Gale NW; Holland SJ; Valenzuela DM; Flenniken A; Pan L; Ryan TE; Henkemeyer M; Strebhardt K; Hirai H; Wilkinson DG Eph Receptors and Ligands Comprise Two Major Specificity Subclasses and Are Reciprocally Compartmentalized During Embryogenesis. Neuron 1996, 17, 9–19. [DOI] [PubMed] [Google Scholar]
- (240).Pasquale EB Eph Receptor Signalling Casts a Wide Net on Cell Behaviour. Nat Rev Mol Cell Biol 2005, 6, 462–475. [DOI] [PubMed] [Google Scholar]
- (241).Kania A; Klein R Mechanisms of Ephrin-Eph Signalling in Development, Physiology and Disease. Nat Rev Mol Cell Biol 2016, 17, 240–256. [DOI] [PubMed] [Google Scholar]
- (242).Wilkinson DG Multiple Roles of Eph Receptors and Ephrins in Neural Development. Nature Reviews Neuroscience 2001, 2, 155–164. [DOI] [PubMed] [Google Scholar]
- (243).Xu NJ; Henkemeyer M Ephrin Reverse Signaling in Axon Guidance and Synaptogenesis. Semin Cell Dev Biol 2012, 23, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Conover JC; Doetsch F; Garcia-Verdugo J-M; Gale NW; Yancopoulos GD; Alvarez-Buylla A Disruption of Eph/Ephrin Signaling Affects Migration and Proliferation in the Adult Subventricular Zone. Nature neuroscience 2000, 3, 1091–1097. [DOI] [PubMed] [Google Scholar]
- (245).Holmberg J; Genander M; Halford MM; Anneren C; Sondell M; Chumley MJ; Silvany RE; Henkemeyer M; Frisen J Ephb Receptors Coordinate Migration and Proliferation in the Intestinal Stem Cell Niche. Cell 2006, 125, 1151–1163. [DOI] [PubMed] [Google Scholar]
- (246).Coulthard MG; Morgan M; Woodruff TM; Arumugam TV; Taylor SM; Carpenter TC; Lackmann M; Boyd AW Eph/Ephrin Signaling in Injury and Inflammation. Am J Pathol 2012, 181, 1493–1503. [DOI] [PubMed] [Google Scholar]
- (247).Adams RH; Wilkinson GA; Weiss C; Diella F; Gale NW; Deutsch U; Risau W; Klein R Roles of Ephrinb Ligands and Ephb Receptors in Cardiovascular Development: Demarcation of Arterial/Venous Domains, Vascular Morphogenesis, and Sprouting Angiogenesis. Genes & development 1999, 13, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Pasquale EB Eph Receptors and Ephrins in Cancer: Bidirectional Signalling and Beyond. Nat Rev Cancer 2010, 10, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Barquilla A; Pasquale EB Eph Receptors and Ephrins: Therapeutic Opportunities. Annu Rev Pharmacol Toxicol 2015, 55, 465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Kou CJ; Kandpal RP Differential Expression Patterns of Eph Receptors and Ephrin Ligands in Human Cancers. Biomed Res Int 2018, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Matsuo K; Otaki N Bone Cell Interactions through Eph/Ephrin: Bone Modeling, Remodeling and Associated Diseases. Cell Adh Migr 2012, 6, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Funk SD; Orr AW Ephs and Ephrins Resurface in Inflammation, Immunity, and Atherosclerosis. Pharmacol Res 2013, 67, 42–52. [DOI] [PubMed] [Google Scholar]
- (253).Larsen AB; Pedersen MW; Stockhausen MT; Grandal MV; van Deurs B; Poulsen HS Activation of the Egfr Gene Target Epha2 Inhibits Epidermal Growth Factor-Induced Cancer Cell Motility. Mol Cancer Res 2007, 5, 283–293. [DOI] [PubMed] [Google Scholar]
- (254).De Robertis M; Loiacono L; Fusilli C; Poeta ML; Mazza T; Sanchez M; Marchionni L; Signori E; Lamorte G; Vescovi AL et al. Dysregulation of Egfr Pathway in Epha2 Cell Subpopulation Significantly Associates with Poor Prognosis in Colorectal Cancer. Clin Cancer Res 2017, 23, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Brantley-Sieders DM; Zhuang G; Hicks D; Fang WB; Hwang Y; Cates JM; Coffman K; Jackson D; Bruckheimer E; Muraoka-Cook RS et al. The Receptor Tyrosine Kinase Epha2 Promotes Mammary Adenocarcinoma Tumorigenesis and Metastatic Progression in Mice by Amplifying Erbb2 Signaling. J Clin Invest 2008, 118, 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Zhuang G; Brantley-Sieders DM; Vaught D; Yu J; Xie L; Wells S; Jackson D; Muraoka-Cook R; Arteaga C; Chen J Elevation of Receptor Tyrosine Kinase Epha2 Mediates Resistance to Trastuzumab Therapy. Cancer Res 2010, 70, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Yokote H; Fujita K; Jing X; Sawada T; Liang S; Yao L; Yan X; Zhang Y; Schlessinger J; Sakaguchi K Trans-Activation of Epha4 and Fgf Receptors Mediated by Direct Interactions between Their Cytoplasmic Domains. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 18866–18871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Fukai J; Yokote H; Yamanaka R; Arao T; Nishio K; Itakura T Epha4 Promotes Cell Proliferation and Migration through a Novel Epha4-Fgfr1 Signaling Pathway in the Human Glioma U251 Cell Line. Mol Cancer Ther 2008, 7, 2768–2778. [DOI] [PubMed] [Google Scholar]
- (259).Sawada T; Jing X; Zhang Y; Shimada E; Yokote H; Miyajima M; Sakaguchi K Ternary Complex Formation of Epha4, Fgfr and Frs2alpha Plays an Important Role in the Proliferation of Embryonic Neural Stem/Progenitor Cells. Genes Cells 2010, 15, 297–311. [DOI] [PubMed] [Google Scholar]
- (260).Halford MM; Armes J; CBuchert M; Meskenaite V; Grail D; Hibbs ML; Wilks AF; Farlie PG; Newgreen DF; Hovens CM et al. Ryk-Deficient Mice Exhibit Craniofacial Defects Associated with Perturbed Eph Receptor Crosstalk. Nature Genet 2000, 25, 414–418. [DOI] [PubMed] [Google Scholar]
- (261).Trivier E; Ganesan TS Ryk, a Catalytically Inactive Receptor Tyrosine Kinase, Associates with Ephb2 and Ephb3 but Does Not Interact with Af-6. J Biol Chem 2002, 277, 23037–23043. [DOI] [PubMed] [Google Scholar]
- (262).Kamitori K; Tanaka M; Okuno-Hirasawa T; Kohsaka S Receptor Related to Tyrosine Kinase Ryk Regulates Cell Migration During Cortical Development. Biochem Biophys Res Commun 2005, 330, 446–453. [DOI] [PubMed] [Google Scholar]
- (263).Meyer AS; Miller MA; Gertler FB; Lauffenburger DA The Receptor Axl Diversifies Egfr Signaling and Limits the Response to Egfr-Targeted Inhibitors in Triple-Negative Breast Cancer Cells. Sci. Signal 2013, 6, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Vouri M; Croucher DR; Kennedy SP; An Q; Pilkington GJ; Hafizi S Axl-Egfr Receptor Tyrosine Kinase Hetero-Interaction Provides Egfr with Access to Pro-Invasive Signalling in Cancer Cells. Oncogenesis 2016, 5, e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Elkabets M; Pazarentzos E; Juric D; Sheng Q; Pelossof RA; Brook S; Benzaken AO; Rodon J; Morse N; Yan JJ et al. Axl Mediates Resistance to Pi3kalpha Inhibition by Activating the Egfr/Pkc/Mtor Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Kotani H; Ebi H; Kitai H; Nanjo S; Kita K; Huynh TG; Ooi A; Faber AC; Mino-Kenudson M; Yano S Co-Active Receptor Tyrosine Kinases Mitigate the Effect of Fgfr Inhibitors in Fgfr1-Amplified Lung Cancers with Low Fgfr1 Protein Expression. Oncogene 2016, 35, 3587–3597. [DOI] [PubMed] [Google Scholar]
- (267).Herrera-Abreu MT; Pearson A; Campbell J; Shnyder SD; Knowles MA; Ashworth A; Turner NC Parallel Rna Interference Screens Identify Egfr Activation as an Escape Mechanism in Fgfr3-Mutant Cancer. Cancer Discov 2013, 3, 1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Morgillo F; Woo JK; Kim ES; Hong WK; Lee HY Heterodimerization of Insulin-Like Growth Factor Receptor/Epidermal Growth Factor Receptor and Induction of Survivin Expression Counteract the Antitumor Action of Erlotinib. Cancer Res 2006, 66, 10100–10111. [DOI] [PubMed] [Google Scholar]
- (269).Lee Y; Wang Y; James M; Jeong JH; You M Inhibition of Igf1r Signaling Abrogates Resistance to Afatinib (Bibw2992) in Egfr T790m Mutant Lung Cancer Cells. Mol Carcinog 2016, 55, 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Xu L; Nilsson MB; Saintigny P; Cascone T; Herynk MH; Du Z; Nikolinakos PG; Yang Y; Prudkin L; Liu D et al. Epidermal Growth Factor Receptor Regulates Met Levels and Invasiveness through Hypoxia-Inducible Factor-1alpha in Non-Small Cell Lung Cancer Cells. Oncogene 2010, 29, 2616–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Salgia R; Puri N Synergism of Egfr and C-Met Pathways, Cross-Talk and Inhibition, in Non-Small Cell Lung Cancer. 2008, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Engelman JA; Zejnullahu K; Mitsudomi T; Song Y; Hyland C; Park JO; Lindeman M; Gale CM; Z. X; C. J et al. Met Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating Erbb3 Signaling. Science 2007, 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- (273).Tanizaki J; Okamoto I; Sakai K; Nakagawa K Differential Roles of Trans-Phosphorylated Egfr, Her2, Her3, and Ret as Heterodimerisation Partners of Met in Lung Cancer with Met Amplification. Br J Cancer 2011, 105, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Jo M; Stolz DB; Esplen JE; Dorko K; Michalopoulos GK; Strom SC Cross-Talk between Epidermal Growth Factor Receptor and C-Met Signal Pathways in Transformed Cells. Journal of Biological Chemistry 2000, 275, 8806–8811. [DOI] [PubMed] [Google Scholar]
- (275).Bergstrom JD; Westermark B; Heldin NE Epidermal Growth Factor Receptor Signaling Activates Met in Human Anaplastic Thyroid Carcinoma Cells. Exp Cell Res 2000, 259, 293–299. [DOI] [PubMed] [Google Scholar]
- (276).McCleese JK; Bear MD; Kulp SK; Mazcko C; Khanna C; London CA Met Interacts with Egfr and Ron in Canine Osteosarcoma. Vet Comp Oncol 2013, 11, 124–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Huang PH; Mukasa A; Bonavia R; Flynn RA; Brewer ZE; Cavenee WK; Furnari FB; White FM Quantitative Analysis of Egfrviii Cellular Signaling Networks Reveals a Combinatorial Therapeautic Strategy for Glioblastoma. Proc Natl Acad Sci U S A 2007, 104, 12867–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Li L; Puliyappadamba VT; Chakraborty S; Rehman A; Vemireddy V; Saha D; Souza RF; Hatanpaa KJ; Koduru P; Burma S et al. Egfr Wild Type Antagonizes Egfrviii-Mediated Activation of Met in Glioblastoma. Oncogene 2015, 34, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Chakravarty D; Pedraza AM; Cotari J; Liu AH; Punko D; Kokroo A; Huse JT; Altan-Bonnet G; Brennan CW Egfr and Pdgfra Co-Expression and Heterodimerization in Glioblastoma Tumor Sphere Lines. Sci Rep 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Szerlip NJ; Pedraza A; Chakravarty D; Azim M; McGuire J; Fang Y; Ozawa T; Holland EC; Huse JT; Jhanwar S et al. Intratumoral Heterogeneity of Receptor Tyrosine Kinases Egfr and Pdgfra Amplification in Glioblastoma Defines Subpopulations with Distinct Growth Factor Response. Proc Natl Acad Sci U S A 2012, 109, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Habib AA; Högnason T; Ren J; Stefánsson K; Ratan RR The Epidermal Growth Factor Receptor Associates with and Recruits Phosphatidylinositol 3Kinase to the Platelet-Derived Growth Factor B Receptor. Journal of Biological Chemistry 1998, 273, 6885–6891. [DOI] [PubMed] [Google Scholar]
- (282).Saito Y; Haendeler J; Hojo Y; Yamamoto K; Berk BC Receptor Heterodimerization: Essential Mechanism for Platelet-Derived Growth Factor-Induced Epidermal Growth Factor Receptor Transactivation. Molecular and Cellular Biology 2001, 21, 6387–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Mendelson K; Swendeman S; Saftig P; Blobel CP Stimulation of Platelet-Derived Growth Factor Receptor Beta (Pdgfrbeta) Activates Adam17 and Promotes Metalloproteinase-Dependent Cross-Talk between the Pdgfrbeta and Epidermal Growth Factor Receptor (Egfr) Signaling Pathways. J Biol Chem 2010, 285, 25024–25032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Peace BE; Hill KJ; Degen SJF; Waltz SE Cross-Talk between the Receptor Tyrosine Kinases Ron and Epidermal Growth Factor Receptor. Experimental Cell Research 2003, 289, 317–325. [DOI] [PubMed] [Google Scholar]
- (285).Keller J; Nimnual AS; Shroyer KR; Joy C; Ischenko I; Chandler CS; Dong LM; Hayman MJ; Chan EL Ron Tyrosine Kinase Receptor Synergises with Egfr to Confer Adverse Features in Head and Neck Squamous Cell Carcinoma. Br J Cancer 2013, 109, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Hsu PY; Liu HS; Cheng HL; Tzai TS; Guo HR; Ho CL; Chow NH Collaboration of Ron and Epidermal Growth Factor Receptor in Human Bladder Carcinogenesis. J Urol 2006, 176, 2262–2267. [DOI] [PubMed] [Google Scholar]
- (287).Liu HS; Hsu PY; Lai MD; Chang HY; Ho CL; Cheng HL; Chen HT; Lin YJ; Wu TJ; Tzai TS et al. An Unusual Function of Ron Receptor Tyrosine Kinase as a Transcriptional Regulator in Cooperation with Egfr in Human Cancer Cells. Carcinogenesis 2010, 31, 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Yamaguchi T; Yanagisawa K; Sugiyama R; Hosono Y; Shimada Y; Arima C; Kato S; Tomida S; Suzuki M; Osada H et al. Nkx2–1/Titf1/Ttf-1-Induced Ror1 Is Required to Sustain Egfr Survival Signaling in Lung Adenocarcinoma. Cancer Cell 2012, 21, 348–361. [DOI] [PubMed] [Google Scholar]
- (289).Gentile A; Lazzari L; Benvenuti S; Trusolino L; Comoglio PM Ror1 Is a Pseudokinase That Is Crucial for Met-Driven Tumorigenesis. Cancer Res 2011, 71, 3132–3141. [DOI] [PubMed] [Google Scholar]
- (290).Ding X; Jiang QB; Li R; Chen S; Zhang S Nok/Styk1 Has a Strong Tendency Towards Forming Aggregates and Colocalises with Epidermal Growth Factor Receptor in Endosomes. Biochem Biophys Res Commun 2012, 421, 468–473. [DOI] [PubMed] [Google Scholar]
- (291).El Zein N; D’Hondt S; Sariban E Crosstalks between the Receptors Tyrosine Kinase Egfr and Trka and the Gpcr, Fpr, in Human Monocytes Are Essential for Receptors-Mediated Cell Activation. Cell Signal 2010, 22, 1437–1447. [DOI] [PubMed] [Google Scholar]
- (292).Gotz R; Sendtner M Cooperation of Tyrosine Kinase Receptor Trkb and Epidermal Growth Factor Receptor Signaling Enhances Migration and Dispersal of Lung Tumor Cells. PLoS One 2014, 9, e100944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Qiu L; Zhou C; Sun Y; Di W; Scheffler E; Healey S; Kouttab N; Chu W; Wan Y Crosstalk between Egfr and Trkb Enhances Ovarian Cancer Cell Migration and Proliferation. International journal of oncology 2006, 29, 1003–1011. [PubMed] [Google Scholar]
- (294).de Farias CB; Heinen TE; dos Santos RP; Abujamra AL; Schwartsmann G; Roesler R Bdnf/Trkb Signaling Protects Ht-29 Human Colon Cancer Cells from Egfr Inhibition. Biochem Biophys Res Commun 2012, 425, 328–332. [DOI] [PubMed] [Google Scholar]
- (295).Puehringer D; Orel N; Luningschror P; Subramanian N; Herrmann T; Chao MV; Sendtner M Egf Transactivation of Trk Receptors Regulates the Migration of Newborn Cortical Neurons. Nat Neurosci 2013, 16, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Liu L; Greger J; Shi H; Liu Y; Greshock J; Annan R; Halsey W; Sathe GM; Martin AM; Gilmer TM Novel Mechanism of Lapatinib Resistance in Her2-Positive Breast Tumor Cells: Activation of Axl. Cancer Res 2009, 69, 6871–6878. [DOI] [PubMed] [Google Scholar]
- (297).Goyette MA; Duhamel S; Aubert L; Pelletier A; Savage P; Thibault MP; Johnson RM; Carmeliet P; Basik M; Gaboury L et al. The Receptor Tyrosine Kinase Axl Is Required at Multiple Steps of the Metastatic Cascade During Her2-Positive Breast Cancer Progression. Cell Rep 2018, 23, 1476–1490. [DOI] [PubMed] [Google Scholar]
- (298).Wang J; Mikse O; Liao RG; Li Y; Tan L; Janne PA; Gray NS; Wong KK; Hammerman PS Ligand-Associated Erbb2/3 Activation Confers Acquired Resistance to Fgfr Inhibition in Fgfr3-Dependent Cancer Cells. Oncogene 2015, 34, 2167–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Balañá ME; Labriola L; Salatino M; Movsichoff F; Peters G; Charreau EH; Elizalde PV Activation of Erbb-2 Via a Hierarchical Interaction between Erbb-2 and Type I Insulin-Like Growth Factor Receptor in Mammary Tumor Cells. Oncogene 2001, 20, 34–47. [DOI] [PubMed] [Google Scholar]
- (300).Nahta R; Yuan LX; Zhang B; Kobayashi R; Esteva FJ Insulin-Like Growth Factor-I Receptor/Human Epidermal Growth Factor Receptor 2 Heterodimerization Contributes to Trastuzumab Resistance of Breast Cancer Cells. Cancer Res 2005, 65, 11118–11128. [DOI] [PubMed] [Google Scholar]
- (301).Huang X; Gao L; Wang S; McManaman JL; Thor AD; Yang X; Esteva FJ; Liu B Heterotrimerization of the Growth Factor Receptors Erbb2, Erbb3, and Insulin-Like Growth Factor-I Receptor in Breast Cancer Cells Resistant to Herceptin. Cancer Res 2010, 70, 1204–1214. [DOI] [PubMed] [Google Scholar]
- (302).Shattuck DL; Miller JK; Carraway KL 3rd; Sweeney C Met Receptor Contributes to Trastuzumab Resistance of Her2-Overexpressing Breast Cancer Cells. Cancer Res 2008, 68, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (303).Festuccia C; Gravina GL; Muzi P; Millimaggi D; Dolo V; Vicentini C; Ficorella C; Ricevuto E; Bologna M Her2 Crosstalks with Trka in a Subset of Prostate Cancer Cells: Rationale for a Guided Dual Treatment. Prostate 2009, 69, 337–345. [DOI] [PubMed] [Google Scholar]
- (304).Choy C; Ansari KI; Neman J; Hsu S; Duenas MJ; Li H; Vaidehi N; Jandial R Cooperation of Neurotrophin Receptor Trkb and Her2 in Breast Cancer Cells Facilitates Brain Metastases. Breast Cancer Res 2017, 19, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Adachi Y; Watanabe K; Kita K; Kitai H; Kotani H; Sato Y; Inase N; Yano S; Ebi H Resistance Mediated by Alternative Receptor Tyrosine Kinases in Fgfr1-Amplified Lung Cancer. Carcinogenesis 2017, 38, 1063–1072. [DOI] [PubMed] [Google Scholar]
- (306).Pearson A; Smyth E; Babina IS; Herrera-Abreu MT; Tarazona N; Peckitt C; Kilgour E; Smith NR; Geh C; Rooney C et al. High-Level Clonal Fgfr Amplification and Response to Fgfr Inhibition in a Translational Clinical Trial. Cancer Discov 2016, 6, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Jia Y; Zhang Y; Qiao C; Liu G; Zhao Q; Zhou T; Chen G; Li Y; Feng J; Li Y et al. Igf-1r and Erbb3/Her3 Contribute to Enhanced Proliferation and Carcinogenesis in Trastuzumab-Resistant Ovarian Cancer Model. Biochem Biophys Res Commun 2013, 436, 740–745. [DOI] [PubMed] [Google Scholar]
- (308).Camblin AJ; Pace EA; Adams S; Curley MD; Rimkunas V; Nie L; Tan G; Bloom T; Iadevaia S; Baum J et al. Dual Inhibition of Igf-1r and Erbb3 Enhances the Activity of Gemcitabine and Nab-Paclitaxel in Preclinical Models of Pancreatic Cancer. Clin Cancer Res 2018, 24, 2873–2885. [DOI] [PubMed] [Google Scholar]
- (309).Li C; Wang S; Xing Z; Lin A; Liang K; Song J; Hu Q; Yao J; Chen Z; Park PK et al. A Ror1-Her3-Lncrna Signalling Axis Modulates the Hippo-Yap Pathway to Regulate Bone Metastasis. Nat Cell Biol 2017, 19, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Faraone D; Aguzzi MS; Ragone G; Russo K; Capogrossi MC; Facchiano A Heterodimerization of Fgf-Receptor 1 and Pdgf-Receptor-Alpha: A Novel Mechanism Underlying the Inhibitory Effect of Pdgf-Bb on Fgf-2 in Human Cells. Blood 2006, 107, 1896–1902. [DOI] [PubMed] [Google Scholar]
- (311).Javidi-Sharifi N; Traer E; Martinez J; Gupta A; Taguchi T; Dunlap J; Heinrich MC; Corless CL; Rubin BP; Druker BJ et al. Crosstalk between Kit and Fgfr3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res 2015, 75, 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Bauer TW; Somcio RJ; Fan F; Liu W; Johnson M; Lesslie DP; Evans DB; Gallick GE; Ellis LM Regulatory Role of C-Met in Insulin-Like Growth Factor-I Receptor-Mediated Migration and Invasion of Human Pancreatic Carcinoma Cells. Mol Cancer Ther 2006, 5, 1676–1682. [DOI] [PubMed] [Google Scholar]
- (313).Varkaris A; Gaur S; Parikh NU; Song JH; Dayyani F; Jin JK; Logothetis CJ; Gallick GE Ligand-Independent Activation of Met through Igf-1/Igf-1r Signaling. Int J Cancer 2013, 133, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Potratz JC; Saunders DN; Wai DH; Ng TL; McKinney SE; Carboni JM; Gottardis MM; Triche TJ; Jurgens H; Pollak MN et al. Synthetic Lethality Screens Reveal Rps6 and Mst1r as Modifiers of Insulin-Like Growth Factor-1 Receptor Inhibitor Activity in Childhood Sarcomas. Cancer Res 2010, 70, 8770–8781. [DOI] [PubMed] [Google Scholar]
- (315).Jaquish DV; Yu PT; Shields DJ; French RP; Maruyama KP; Niessen S; Hoover H; D AC; Cravatt B; Lowy AM Igf1-R Signals through the Ron Receptor to Mediate Pancreatic Cancer Cell Migration. Carcinogenesis 2011, 32, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Salian-Mehta S; Xu M; Wierman ME Axl and Met Crosstalk to Promote Gonadotropin Releasing Hormone (Gnrh) Neuronal Cell Migration and Survival. Mol Cell Endocrinol 2013, 374, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Gentile A; Lazzari L; Benvenuti S; Trusolino L; Comoglio PM The Ror1 Pseudokinase Diversifies Signaling Outputs in Met-Addicted Cancer Cells. Int J Cancer 2014, 135, 2305–2316. [DOI] [PubMed] [Google Scholar]
- (318).Lu KV; Chang JP; Parachoniak CA; Pandika MM; Aghi MK; Meyronet D; Isachenko N; Fouse SD; Phillips JJ; Cheresh DA et al. Vegf Inhibits Tumor Cell Invasion and Mesenchymal Transition through a Met/Vegfr2 Complex. Cancer Cell 2012, 22, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Kobayashi T; Furukawa Y; Kikuchi J; Ito C; Miyata Y; Muto S; Tanaka A; Kusano E Transactivation of Ron Receptor Tyrosine Kinase by Interaction with Pdgf Receptor Beta During Steady-State Growth of Human Mesangial Cells. Kidney Int 2009, 75, 1173–1183. [DOI] [PubMed] [Google Scholar]
- (320).Karvonen H; Summala K; Niininen W; Barker HR; Ungureanu D Interaction between Ror1 and Musk Activation Complex in Myogenic Cells. FEBS Lett 2018, 592, 434–445. [DOI] [PubMed] [Google Scholar]
- (321).Greenberg JI; Shields DJ; Barillas SG; Acevedo LM; Murphy E; Huang J; Scheppke L; Stockmann C; Johnson RS; Angle N et al. A Role for Vegf as a Negative Regulator of Pericyte Function and Vessel Maturation. Nature 2008, 456, 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Cheng C; Haasdijk RA; Tempel D; Wijnand K; Chrifi I; Blonden LA; van de Kamp EH; de Boer M; Bürgisser PE; Noorderloos A Pdgf-Induced Migration of Vascular Smooth Muscle Cells Is Inhibited by Heme Oxygenase-1 Via Vegfr2 Upregulation and Subsequent Assembly of Inactive Vegfr2/Pdgfrß Heterodimers. Arteriosclerosis, thrombosis, and vascular biology 2012, 32, 1289–1298. [DOI] [PubMed] [Google Scholar]
- (323).Podleschny M; Grund A; Berger H; Rollwitz E; Borchers A A Ptk7/Ror2 Co-Receptor Complex Affects Xenopus Neural Crest Migration. PLoS One 2015, 10, e0145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Martinez S; Scerbo P; Giordano M; Daulat AM; Lhoumeau AC; Thome V; Kodjabachian L; Borg JP The Ptk7 and Ror2 Protein Receptors Interact in the Vertebrate Wnt/Planar Cell Polarity (Pcp) Pathway. J Biol Chem 2015, 290, 30562–30572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Lee HK; Chauhan SK; Kay E; Dana R Flt-1 Regulates Vascular Endothelial Cell Migration Via a Protein Tyrosine Kinase-7-Dependent Pathway. Blood 2011, 117, 5762–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Shin WS; Maeng YS; Jung JW; Min JK; Kwon YG; Lee ST Soluble Ptk7 Inhibits Tube Formation, Migration, and Invasion of Endothelial Cells and Angiogenesis. Biochem Biophys Res Commun 2008, 371, 793–798. [DOI] [PubMed] [Google Scholar]
- (327).Shin WS; Na HW; Lee ST Biphasic Effect of Ptk7 on Kdr Activity in Endothelial Cells and Angiogenesis. Biochim Biophys Acta 2015, 1853, 2251–2260. [DOI] [PubMed] [Google Scholar]
- (328).Peterson S; Bogenmann E The Ret and Trka Pathways Collaborate to Regulate Neuroblastoma Differentiation. Oncogene 2004, 23, 213–225. [DOI] [PubMed] [Google Scholar]
- (329).Tsui-Pierchala BA; Milbrandt J; Johnson EM Jr Ngf Utilizes C-Ret Via a Novel Gfl-Independent, Inter-Rtk Signaling Mechanism to Maintain the Trophic Status of Mature Sympathetic Neurons. Neuron 2002, 33, 261–273. [DOI] [PubMed] [Google Scholar]
- (330).Esposito CL; D’Alessio A; de Franciscis V; Cerchia L A Cross-Talk between Trkb and Ret Tyrosine Kinases Receptors Mediates Neuroblastoma Cells Differentiation. PLoS One 2008, 3, e1643. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (331).Tufro A; Teichman J; Banu N; Villegas G Crosstalk between Vegf-a/Vegfr2 and Gdnf/Ret Signaling Pathways. Biochem Biophys Res Commun 2007, 358, 410–416. [DOI] [PubMed] [Google Scholar]
- (332).Shvartsman D; Storrie-White H; Lee K; Kearney C; Brudno Y; Ho N; Cezar C; McCann C; Anderson E; Koullias J et al. Sustained Delivery of Vegf Maintains Innervation and Promotes Reperfusion in Ischemic Skeletal Muscles Via Ngf/Gdnf Signaling. Mol Ther 2014, 22, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Ruan GX; Kazlauskas A Axl Is Essential for Vegf-a-Dependent Activation of Pi3k/Akt. EMBO J 2012, 31, 1692–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Lu W; Yamamoto V; Ortega B; Baltimore D Mammalian Ryk Is a Wnt Coreceptor Required for Stimulation of Neurite Outgrowth. Cell 2004, 119, 97–108. [DOI] [PubMed] [Google Scholar]
- (335).Green J; Nusse R; van Amerongen R The Role of Ryk and Ror Receptor Tyrosine Kinases in Wnt Signal Transduction. Cold Spring Harb Perspect Biol 2014, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Macheda ML; Sun WW; Kugathasan K; Hogan BM; Bower NI; Halford MM; Zhang YF; Jacques BE; Lieschke GJ; Dabdoub A et al. The Wnt Receptor Ryk Plays a Role in Mammalian Planar Cell Polarity Signaling. J Biol Chem 2012, 287, 29312–29323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Yang Y; Mlodzik M Wnt-Frizzled/Planar Cell Polarity Signaling: Cellular Orientation by Facing the Wind (Wnt). Annu Rev Cell Dev Biol 2015, 31, 623–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Bovolenta P; Rodriguez J; Esteve P Frizzled/Ryk Mediated Signalling in Axon Guidance. Development 2006, 133, 4399–4408. [DOI] [PubMed] [Google Scholar]
- (339).Lyu J; Yamamoto V; Lu W Cleavage of the Wnt Receptor Ryk Regulates Neuronal Differentiation During Cortical Neurogenesis. Dev Cell 2008, 15, 773–780. [DOI] [PubMed] [Google Scholar]
- (340).Famili F; Perez LG; Naber BA; Noordermeer JN; Fradkin LG; Staal FJ The Non-Canonical Wnt Receptor Ryk Regulates Hematopoietic Stem Cell Repopulation in Part by Controlling Proliferation and Apoptosis. Cell Death Dis 2016, 7, e2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Kuriyama M; Harada N; Kuroda S; Yamamoto T; Nakafuku M; Iwamatsu A; Yamamoto D; Prasad R; Croce C; Canaani E et al. Identification of Af-6 and Canoe as Putative Targets for Ras. J Biol Chem 1996, 271, 607–610. [DOI] [PubMed] [Google Scholar]
- (342).Hock B; Bohme B; Karn T; Yamamoto T; Kaibuchi K; Holtrich U; Holland S; Pawson T; Rubsamen-Waigmann H; Strebhardt K Pdz-Domain-Mediated Interaction of the Eph-Related Receptor Tyrosine Kinase Ephb3 and the Ras-Binding Protein Af6 Depends on the Kinase Acitivity of the Receptor. Proc Natl Acad Sci USA 1998, 95, 9779–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Truitt L; Freywald A Dancing with the Dead: Eph Receptors and Their Kinase-Null Partners. Biochem Cell Biol 2011, 89, 115–129. [DOI] [PubMed] [Google Scholar]
- (344).Wieacker P; Wieland I Clinical and Genetic Aspects of Craniofrontonasal Syndrome: Towards Resolving a Genetic Paradox. Mol Genet Metab 2005, 86, 110–116. [DOI] [PubMed] [Google Scholar]
- (345).Eswarakumar VP; Lax I; Schlessinger J Cellular Signaling by Fibroblast Growth Factor Receptors. Cytokine Growth Factor Rev 2005, 16, 139–149. [DOI] [PubMed] [Google Scholar]
- (346).Su N; Jin M; Chen L Role of Fgf/Fgfr Signaling in Skeletal Development and Homeostasis: Learning from Mouse Models. Bone Res 2014, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Ornitz DM; Itoh N The Fibroblast Growth Factor Signaling Pathway. Wiley Interdiscip Rev Dev Biol 2015, 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Muenke M; Schell U Fibroblast-Growth-Factor Receptor Mutations in Human Skeletal Disorders. Trends in Genetics 1995, 11, 308–313. [DOI] [PubMed] [Google Scholar]
- (349).Vajo Z; Francomano CA; Wilkin DJ The Molecular and Genetic Basis of Fibroblast Growth Factor Receptor 3 Disorders: The Achondroplasia Family of Skeletal Dysplasias, Muenke Craniosynostosis, and Crouzon Syndrome with Acanthosis Nigricans. Endocrine reviews 2000, 21, 23–39. [DOI] [PubMed] [Google Scholar]
- (350).Foldynova-Trantirkova S; Wilcox WR; Krejci P Sixteen Years and Counting: The Current Understanding of Fibroblast Growth Factor Receptor 3 (Fgfr3) Signaling in Skeletal Dysplasias. Hum Mutat 2012, 33, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Krejci P The Paradox of Fgfr3 Signaling in Skeletal Dysplasia: Why Chondrocytes Growth Arrest While Other Cells over Proliferate. Mutat Res Rev Mutat Res 2014, 759, 40–48. [DOI] [PubMed] [Google Scholar]
- (352).Balek L; Nemec P; Konik P; Kunova Bosakova M; Varecha M; Gudernova I; Medalova J; Krakow D; Krejci P Proteomic Analyses of Signalling Complexes Associated with Receptor Tyrosine Kinase Identify Novel Members of Fibroblast Growth Factor Receptor 3 Interactome. Cell Signal 2018, 42, 144–154. [DOI] [PubMed] [Google Scholar]
- (353).Minami Y; Oishi I; Endo M; Nishita M Ror-Family Receptor Tyrosine Kinases in Noncanonical Wnt Signaling: Their Implications in Developmental Morphogenesis and Human Diseases. Dev Dyn 2010, 239, 1–15. [DOI] [PubMed] [Google Scholar]
- (354).Stricker S; Rauschenberger V; Schambony A Ror-Family Receptor Tyrosine Kinases. Curr Top Dev Biol 2017, 123, 105–142. [DOI] [PubMed] [Google Scholar]
- (355).Rebagay G; Yan S; Liu C; Cheung NK Ror1 and Ror2 in Human Malignancies: Potentials for Targeted Therapy. Front Oncol 2012, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Karachaliou N; Gimenez-Capitan A; Drozdowskyj A; Viteri S; Moran T; Carcereny E; Massuti B; Vergnenegre A; de Marinis F; Molina MA et al. Ror1 as a Novel Therapeutic Target for Egfr-Mutant Non-Small-Cell Lung Cancer Patients with the Egfr T790m Mutation. Transl Lung Cancer Res 2014, 3, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Shabani M; Naseri J; Shokri F Receptor Tyrosine Kinase-Like Orphan Receptor 1: A Novel Target for Cancer Immunotherapy. Expert Opin Ther Targets 2015, 19, 941–955. [DOI] [PubMed] [Google Scholar]
- (358).Potratz J; Tillmanns A; Berning P; Korsching E; Schaefer C; Lechtape B; Schleithoff C; Unland R; Schafer KL; Muller-Tidow C et al. Receptor Tyrosine Kinase Gene Expression Profiles of Ewing Sarcomas Reveal Ror1 as a Potential Therapeutic Target in Metastatic Disease. Mol Oncol 2016, 10, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).Karvonen H; Niininen W; Murumagi A; Ungureanu D Targeting Ror1 Identifies New Treatment Strategies in Hematological Cancers. Biochem Soc Trans 2017, 45, 457–464. [DOI] [PubMed] [Google Scholar]
- (360).Bicocca VT; Chang BH; Masouleh BK; Muschen M; Loriaux MM; Druker BJ; Tyner JW Crosstalk between Ror1 and the Pre-B Cell Receptor Promotes Survival of T(1;19) Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Mendrola JM; Shi F; Park JH; Lemmon MA Receptor Tyrosine Kinases with Intracellular Pseudokinase Domains. Biochem Soc Trans 2013, 41, 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Murphy JM; Zhang Q; Young SN; Reese ML; Bailey FP; Eyers PA; Ungureanu D; Hammaren H; Silvennoinen O; Varghese LN A Robust Methodology to Subclassify Pseudokinases Based on Their Nucleotide-Binding Properties. Biochemical Journal 2014, 457, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Oishi I; Takeuchi S; Hashimoto R; Nagabukuro A; Ueda T; Liu ZJ; Hatta T; Akira S; Matsuda Y; Yamamura H Spatio-Temporally Regulated Expression of Receptor Tyrosine Kinases, Mror1, Mror2, During Mouse Development: Implications in Development and Function of the Nervous System. Genes to Cells 1999, 4, 41–56. [DOI] [PubMed] [Google Scholar]
- (364).Jennings C; Dyer SM; Burden SJ Muscle-Specific Trk-Related Receptor with a Kringle Domain Defines a Distinct Class of Receptor Tyrosine Kinases. Proceedings of the National Academy of Sciences 1993, 90, 2895–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).DeChiara TM; Bowen DC; Valenzuela DM; Simmons MV; Poueymirou WT; Thomas S; Kinetz E; Compton DL; Rojas E; Park JS The Receptor Tyrosine Kinase Musk Is Required for Neuromuscular Junction Formation in Vivo. Cell 1996, 85, 501–512. [DOI] [PubMed] [Google Scholar]
- (366).Hubbard SR; Gnanasambandan K Structure and Activation of Musk, a Receptor Tyrosine Kinase Central to Neuromuscular Junction Formation. Biochim Biophys Acta 2013, 1834, 2166–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Burden SJ; Hubbard SR; Zhang W; Yumoto N The Musk Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 359–372. [Google Scholar]
- (368).Beeson D; Higuchi O; Palace J; Cossins J; Spearman H; Maxwell S; Newsom-Davis J; Burke G; Fawcett P; Motomura M Dok-7 Mutations Underlie a Neuromuscular Junction Synaptopathy. Science 2006, 313, 1975–1978. [DOI] [PubMed] [Google Scholar]
- (369).Niks E; Van Leeuwen Y; Leite M; Dekker F; Wintzen A; Wirtz P; Vincent A; van Tol M; Jol-van der Zijde C; Verschuuren J Clinical Fluctuations in Musk Myasthenia Gravis Are Related to Antigen-Specific Igg4 Instead of Igg1. Journal of neuroimmunology 2008, 195, 151–156. [DOI] [PubMed] [Google Scholar]
- (370).Peradziryi H; Tolwinski NS; Borchers A The Many Roles of Ptk7: A Versatile Regulator of Cell–Cell Communication. Archives of Biochemistry and Biophysics 2012, 524, 71–76. [DOI] [PubMed] [Google Scholar]
- (371).Berger H; Wodarz A; Borchers A Ptk7 Faces the Wnt in Development and Disease. Front Cell Dev Biol 2017, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Dunn NR; Tolwinski NS Ptk7 and Mcc, Unfancied Components in Non-Canonical Wnt Signaling and Cancer. Cancers (Basel) 2016, 8, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Shin WS; Hong Y; Lee HW; Lee ST Catalytically Defective Receptor Protein Tyrosine Kinase Ptk7 Enhances Invasice Henotype by Inducing Mmp-9 through Activation of Ap-1 and Nf-Kb in Esophageal Squamous Cell Carcinoma Cells. Oncotarget 2016, 7, 73242–73256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Messerli SM; Hoffman MM; Gnimpieba EZ; Bhardwaj RD Therapeutic Targeting of Ptk7 Is Cytotoxic in Atypical Teratoid Rhabdoid Tumors. Mol Cancer Res 2017, 15, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Katoh M Antibody-Drug Conjugate Targeting Protein Tyrosine Kinase 7, a Receptor Tyrosine Kinase-Like Molecule Involved in Wnt and Vascular Endothelial Growth Factor Signaling: Effects on Cancer Stem Cells, Tumor Microenvironment and Whole-Body Homeostasis. Ann Transl Med 2017, 5, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Ataseven B; Gunesch A; Eiermann W; Kates RE; Hogel B; Knyazev P; Ullrich A; Harbeck N Ptk7 as a Potential Prognostic and Predictive Marker of Response to Adjuvant Chemotherapy in Breast Cancer Patients, and Resistance to Anthracycline Drugs. Onco Targets Ther 2014, 7, 1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Lhoumeau AC; Martinez S; Boher JM; Monges G; Castellano R; Goubard A; Doremus M; Poizat F; Lelong B; de Chaisemartin C et al. Overexpression of the Promigratory and Prometastatic Ptk7 Receptor Is Associated with an Adverse Clinical Outcome in Colorectal Cancer. PLoS One 2015, 10, e0123768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Liu L The Nok Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 843–859. [Google Scholar]
- (379).Liu L; Yu X-Z; Li T-S; Song L-X; Chen P-L; Suo T-L; Li Y-H; Wang S-D; Chen Y; Ren Y-M A Novel Protein Tyrosine Kinase Nok That Shares Homology with Platelet-Derived Growth Factor/Fibroblast Growth Factor Receptors Induces Tumorigenesis and Metastasis in Nude Mice. Cancer research 2004, 64, 3491–3499. [DOI] [PubMed] [Google Scholar]
- (380).Chen P; Li W-M; Lu Q; Wang J; Yan X-L; Zhang Z-P; Li X-F Clinicopathologic Features and Prognostic Implications of Nok/Styk1 Protein Expression in Non-Small Cell Lung Cancer. BMC cancer 2014, 14, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Sarfstein R; Werner H The Insr/Igf1r Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 297–320. [Google Scholar]
- (382).Jin Q; Esteva FJ Cross-Talk between the Erbb/Her Family and the Type I Insulin-Like Growth Factor Receptor Signaling Pathway in Breast Cancer. J Mammary Gland Biol Neoplasia 2008, 13, 485–498. [DOI] [PubMed] [Google Scholar]
- (383).Oliveira S; Schiffelers R; Storm G; Henegouwen P; Roovers R Crosstalk between Epidermal Growth Factor Receptor-and Insulin-Like Growth Factor-1 Receptor Signaling: Implications for Cancer Therapy. Current cancer drug targets 2009, 9, 748–760. [DOI] [PubMed] [Google Scholar]
- (384).Liu C; Zhang Z; Tang H; Jiang Z; You L; Liao Y Crosstalk between Igf-1r and Other Tumor Promoting Pathways. Current pharmaceutical design 2014, 20, 2912–2921. [DOI] [PubMed] [Google Scholar]
- (385).Simpson A; Petnga W; Macaulay VM; Weyer-Czernilofsky U; Bogenrieder T Insulin-Like Growth Factor (Igf) Pathway Targeting in Cancer: Role of the Igf Axis and Opportunities for Future Combination Studies. Target Oncol 2017, 12, 571–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Lammers R; Van Obberghen E; Ballotti R; Schlessinger J; Ullrich A Transphosphorylation as a Possible Mechanism for Insulin and Epidermal Growth Factor Receptor Activation. Journal of Biological Chemistry 1990, 265, 16886–16890. [PubMed] [Google Scholar]
- (387).Andrae J; Gallini R; Betsholtz C Role of Platelet-Derived Growth Factors in Physiology and Medicine. Genes Dev 2008, 22, 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Heldin C-H Targeting the Pdgf Signaling Pathway in Tumor Treatment. Cell Communication and Signaling 2013, 11, 97–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Noskovicova N; Petrek M; Eickelberg O; Heinzelmann K Platelet-Derived Growth Factor Signaling in the Lung. From Lung Development and Disease to Clinical Studies. Am J Respir Cell Mol Biol 2015, 52, 263–284. [DOI] [PubMed] [Google Scholar]
- (390).Wang Y; Appiah-Kubi K; Wu M; Yao X; Qian H; Wu Y; Chen Y The Platelet-Derived Growth Factors (Pdgfs) and Their Receptors (Pdgfrs) Are Major Players in Oncogenesis, Drug Resistance, and Attractive Oncologic Targets in Cancer. Growth Factors 2016, 34, 64–71. [DOI] [PubMed] [Google Scholar]
- (391).Appiah-Kubi K; Wang Y; Qian H; Wu M; Yao X; Wu Y; Chen Y Platelet-Derived Growth Factor Receptor/Platelet-Derived Growth Factor (Pdgfr/Pdgf) System Is a Prognostic and Treatment Response Biomarker with Multifarious Therapeutic Targets in Cancers. Tumour Biol 2016, 37, 10053–10066. [DOI] [PubMed] [Google Scholar]
- (392).Ozawa T; Brennan CW; Wang L; Squatrito M; Sasayama T; Nakada M; Huse JT; Pedraza A; Utsuki S; Yasui Y et al. Pdgfra Gene Rearrangements Are Frequent Genetic Events in Pdgfra-Amplified Glioblastomas. Genes Dev 2010, 24, 2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Ibanez CF Structure and Physiology of the Ret Receptor Tyrosine Kinase. Cold Spring Harb Perspect Biol 2013, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Melillo RM; Santoro M The Ret Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 559–591. [Google Scholar]
- (395).Mologni L; Gambacorti-Passerini C; Goekjian P; Scapozza L Ret Kinase Inhibitors: A Review of Recent Patents (2012–2015). Expert Opin Ther Pat 2017, 27, 91–99. [DOI] [PubMed] [Google Scholar]
- (396).Mao WF; Shao MH; Gao PT; Ma J; Li HJ; Li GL; Han BH; Yuan CG The Important Roles of Ret, Vegfr2 and the Raf/Mek/Erk Pathway in Cancer Treatment with Sorafenib. Acta Pharmacol Sin 2012, 33, 1311–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Patapoutian A; Reichardt LF Trk Receptors: Mediators of Neurotrophin Action. Current opinion in neurobiology 2001, 11, 272–280. [DOI] [PubMed] [Google Scholar]
- (398).Huang EJ; Reichardt LF Trk Receptors: Roles in Neuronal Signal Transduction. Annual review of biochemistry 2003, 72, 609–642. [DOI] [PubMed] [Google Scholar]
- (399).Hondermarck H; Demont Y; Bradshaw RA The Trk Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 777–820. [Google Scholar]
- (400).Schecterson LC; Bothwell M Neurotrophin Receptors: Old Friends with New Partners. Dev Neurobiol 2010, 70, 332–338. [DOI] [PubMed] [Google Scholar]
- (401).Chopin V; Lagadec C; Toillon RA; Le Bourhis X Neurotrophin Signaling in Cancer Stem Cells. Cell Mol Life Sci 2016, 73, 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Khotskaya YB; Holla VR; Farago AF; Mills Shaw KR; Meric-Bernstam F; Hong DS Targeting Trk Family Proteins in Cancer. Pharmacol Ther 2017, 173, 58–66. [DOI] [PubMed] [Google Scholar]
- (403).Castrén E Neurotrophins and Psychiatric Disorders. Neurotrophic Factors 2014, 461–479. [DOI] [PubMed] [Google Scholar]
- (404).Budni J; Bellettini-Santos T; Mina F; Garcez ML; Zugno AI The Involvement of Bdnf, Ngf and Gdnf in Aging and Alzheimer’s Disease. Aging Dis 2015, 6, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Canu N; Amadoro G; Triaca V; Latina V; Sposato V; Corsetti V; Severini C; Ciotti MT; Calissano P The Intersection of Ngf/Trka Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int J Mol Sci 2017, 18, 1319–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Castren E; Kojima M Brain-Derived Neurotrophic Factor in Mood Disorders and Antidepressant Treatments. Neurobiol Dis 2017, 97, 119–126. [DOI] [PubMed] [Google Scholar]
- (407).Reinhart V; Bove SE; Volfson D; Lewis DA; Kleiman RJ; Lanz TA Evaluation of Trkb and Bdnf Transcripts in Prefrontal Cortex, Hippocampus, and Striatum from Subjects with Schizophrenia, Bipolar Disorder, and Major Depressive Disorder. Neurobiol Dis 2015, 77, 220–227. [DOI] [PubMed] [Google Scholar]
- (408).Babina IS; Turner NC Advances and Challenges in Targeting Fgfr Signalling in Cancer. Nat Rev Cancer 2017, 17, 318–332. [DOI] [PubMed] [Google Scholar]
- (409).Dieci MV; Arnedos M; Andre F; Soria JC Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov 2013, 3, 264–279. [DOI] [PubMed] [Google Scholar]
- (410).Ware KE; Marshall ME; Heasley LR; Marek L; Hinz TK; Hercule P; Helfrich BA; Doebele RC; Heasley LE Rapidly Acquired Resistance to Egfr Tyrosine Kinase Inhibitors in Nsclc Cell Lines through De-Repression of Fgfr2 and Fgfr3 Expression. PLoS One 2010, 5, e14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Ware KE; Hinz TK; Kleczko E; Singleton KR; Marek LA; Helfrich BA; Cummings CT; Graham DK; Astling D; Tan AC et al. A Mechanism of Resistance to Gefitinib Mediated by Cellular Reprogramming and the Acquisition of an Fgf2-Fgfr1 Autocrine Growth Loop. Oncogenesis 2013, 2, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Traer E; Javidi-Sharifi N; Agarwal A; Dunlap J; English I; Martinez J; Tyner JW; Wong M; Druker BJ Ponatinib Overcomes Fgf2-Mediated Resistance in Cml Patients without Kinase Domain Mutations. Blood 2014, 123, 1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Johnson M; Koukoulis G; Matsumoto K; Nakamura T; Iyer A Hepatocyte Growth Factor Induces Proliferation and Morphogenesis in Nonparenchymal Epithelial Liver Cells. Hepatology 1993, 17, 1052–1061. [PubMed] [Google Scholar]
- (414).Uehara Y; Minowa O; Mori C; Shiota K; Kuno J; Noda T; Kitamura N Placental Defect and Embryonic Lethality in Mice Lacking Hepatocyte Growth Factor/Scatter Factor. Nature 1995, 373, 702–705. [DOI] [PubMed] [Google Scholar]
- (415).Birchmeier C; Gherardi E Developmental Roles of Hgf/Sf and Its Receptor, the C-Met Tyrosine Kinase. Trends in cell biology 1998, 8, 404–410. [DOI] [PubMed] [Google Scholar]
- (416).Gentile A; Trusolino L; Comoglio PM The Met Tyrosine Kinase Receptor in Development and Cancer. Cancer Metastasis Rev 2008, 27, 85–94. [DOI] [PubMed] [Google Scholar]
- (417).Gao C; Woude GF The Met Receptor Family. Receptor Tyrosine Kinases: Family and Subfamilies 2015, 321–358. [Google Scholar]
- (418).Tsao MS; Zhu H; Giad A; Viallet J; Nakamura T; Park M Hepatocyte Growth Factor/Scatter Factor Is an Autocrine Factor for Human Normal Bronchial Epithelial and Lung Carcinoma Cells. Cell Growth Differ 1993, 4, 571–579. [PubMed] [Google Scholar]
- (419).Rong S; Segal S; Anver M; Resau JH; Vande Woude GF Invasiveness and Metastasis of Nih 3t3 Cells Induced by Met-Hepatocyte Growth Factor/Scatter Factor Autocrine Stimulation. Proc Natl Acad Sci U S A 1994, 91, 4731–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Blumenschein GR Jr.; Mills GB; Gonzalez-Angulo AM Targeting the Hepatocyte Growth Factor-Cmet Axis in Cancer Therapy. J Clin Oncol 2012, 30, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Matsumoto K; Umitsu M; De Silva DM; Roy A; Bottaro DP Hepatocyte Growth Factor/Met in Cancer Progression and Biomarker Discovery. Cancer Sci 2017, 108, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Zhang Y; Xia M; Jin K; Wang S; Wei H; Fan C; Wu Y; Li X; Li X; Li G et al. Function of the C-Met Receptor Tyrosine Kinase in Carcinogenesis and Associated Therapeutic Opportunities. Mol Cancer 2018, 17, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Comoglio PM; Trusolino L; Boccaccio C Known and Novel Roles of the Met Oncogene in Cancer: A Coherent Approach to Targeted Therapy. Nat Rev Cancer 2018, 18, 341–358. [DOI] [PubMed] [Google Scholar]
- (424).Chuang JC; Neal JW The Persistent Promise of Combining Hgf/Met and Egfr Inhibition in Non-Small Cell Lung Cancer. Cancer 2017, 123, 2798–2801. [DOI] [PubMed] [Google Scholar]
- (425).Jiang M; Zhang H; Xiao H; Zhang Z; Que D; Luo J; Li J; Mao B; Chen Y; Lan M et al. High Expression of C-Met and Egfr Is Associated with Poor Survival of Patients with Glottic Laryngeal Squamous Cell Carcinoma. Oncol Lett 2018, 15, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Wagh PK; Peace BE; Waltz SE Met-Related Receptor Tyrosine Kinase Ron in Tumor Growth and Metastasis. Advances in Cancer Research 2008, 100, 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Yao HP; Zhou YQ; Zhang R; Wang MH Msp-Ron Signalling in Cancer: Pathogenesis and Therapeutic Potential. Nat Rev Cancer 2013, 13, 466–481. [DOI] [PubMed] [Google Scholar]
- (428).Faham N; Welm AL Ron Signaling Is a Key Mediator of Tumor Progression in Many Human Cancers. Cold Spring Harb Symp Quant Biol 2016, 81, 177–188. [DOI] [PubMed] [Google Scholar]
- (429).Lemke G Biology of the Tam Receptors. Cold Spring Harb Perspect Biol 2013, 5, a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Shafit-Zagardo B; Gruber RC; DuBois JC The Role of Tam Family Receptors and Ligands in the Nervous System: From Development to Pathobiology. Pharmacol Ther 2018, 188, 97–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Rothlin CV; Ghosh S; Zuniga EI; Oldstone MB; Lemke G Tam Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [DOI] [PubMed] [Google Scholar]
- (432).Lemke G; Rothlin CV Immunobiology of the Tam Receptors. Nat Rev Immunol 2008, 8, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Rothlin CV; Carrera-Silva EA; Bosurgi L; Ghosh S Tam Receptor Signaling in Immune Homeostasis. Annu Rev Immunol 2015, 33, 355–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Bosurgi L; Bernink JH; Delgado Cuevas V; Gagliani N; Joannas L; Schmid ET; Booth CJ; Ghosh S; Rothlin CV Paradoxical Role of the Proto-Oncogene Axl and Mer Receptor Tyrosine Kinases in Colon Cancer. Proc Natl Acad Sci U S A 2013, 110, 13091–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Zagorska A; Traves PG; Lew ED; Dransfield I; Lemke G Diversification of Tam Receptor Tyrosine Kinase Function. Nat Immunol 2014, 15, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Graham DK; DeRyckere D; Davies KD; Earp HS The Tam Family: Phosphatidylserine Sensing Receptor Tyrosine Kinases Gone Awry in Cancer. Nat Rev Cancer 2014, 14, 769–785. [DOI] [PubMed] [Google Scholar]
- (437).Paccez JD; Vogelsang M; Parker MI; Zerbini LF The Receptor Tyrosine Kinase Axl in Cancer: Biological Functions and Therapeutic Implications. Int J Cancer 2014, 134, 1024–1033. [DOI] [PubMed] [Google Scholar]
- (438).Lu Q; Lemke G Homeostatic Regulation of the Immune System by Receptor Tyrosine Kinases of the Tyro 3 Family. Science 2001, 293, 306–311. [DOI] [PubMed] [Google Scholar]
- (439).Hoehn HJ; Kress Y; Sohn A; Brosnan CF; Bourdon S; Shafit-Zagardo B Axl−/− Mice Have Delayed Recovery and Prolonged Axonal Damage Following Cuprizone Toxicity. Brain Res 2008, 1240, 1–11. [DOI] [PubMed] [Google Scholar]
- (440).Weinger JG; Omari KM; Marsden K; Raine CS; Shafit-Zagardo B Up-Regulation of Soluble Axl and Mer Receptor Tyrosine Kinases Negatively Correlates with Gas6 in Established Multiple Sclerosis Lesions. Am J Pathol 2009, 175, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Morizono K; Xie Y; Olafsen T; Lee B; Dasgupta A; Wu AM; Chen IS The Soluble Serum Protein Gas6 Bridges Virion Envelope Phosphatidylserine to the Tam Receptor Tyrosine Kinase Axl to Mediate Viral Entry. Cell Host Microbe 2011, 9, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Bhattacharyya S; Zagorska A; Lew ED; Shrestha B; Rothlin CV; Naughton J; Diamond MS; Lemke G; Young JA Enveloped Viruses Disable Innate Immune Responses in Dendritic Cells by Direct Activation of Tam Receptors. Cell Host Microbe 2013, 14, 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Zhang Z; Lee JC; Lin L; Olivas V; Au V; LaFramboise T; Abdel-Rahman M; Wang X; Levine AD; Rho JK et al. Activation of the Axl Kinase Causes Resistance to Egfr-Targeted Therapy in Lung Cancer. Nat Genet 2012, 44, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (444).Byers LA; Diao L; Wang J; Saintigny P; Girard L; Peyton M; Shen L; Fan Y; Giri U; Tumula PK et al. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to Egfr and Pi3k Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming Egfr Inhibitor Resistance. Clin Cancer Res 2013, 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Brand TM; Iida M; Stein AP; Corrigan KL; Braverman CM; Luthar N; Toulany M; Gill PS; Salgia R; Kimple RJ et al. Axl Mediates Resistance to Cetuximab Therapy. Cancer Res 2014, 74, 5152–5164. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (446).Bose R; Molina H; Patterson AS; Bitok JK; Periaswamy B; Bader JS; Pandey A; Cole PA Phosphoproteomic Analysis of Her2/Neu Signaling and Inhibition. Proceedings of the National Academy of Sciences 2006, 103, 9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Kut C; Mac Gabhann F; Popel AS Where Is Vegf in the Body? A Meta-Analysis of Vegf Distribution in Cancer. Br J Cancer 2007, 97, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).SHEARDOWN H; CHENG Y-L Tear Egf Concentration Following Corneal Epithelial Wound Creation. Journal of ocular pharmacology and therapeutics 1996, 12, 239–243. [DOI] [PubMed] [Google Scholar]
- (449).Yarden Y; Ullrich A Growth Factor Receptor Tyrosine Kinases. Annual review of biochemistry 1988, 57, 443–478. [DOI] [PubMed] [Google Scholar]
- (450).Lemmon MA; Engelman DM Specificity and Promiscuity in Membrane Helix Interactions. Quarterly Review of Biophysics 1994, 27, 157–218. [DOI] [PubMed] [Google Scholar]
- (451).Li E; Hristova K Role of Receptor Tyrosine Kinase Transmembrane Domains in Cell Signaling and Human Pathologies. Biochemistry 2006, 45, 6241–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Li E; Hristova K Receptor Tyrosine Kinase Transmembrane Domains: Function, Dimer Structure, and Dimerization Energetics. Cell Adhesion and Migration 2010, 4, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Coon BG; Baeyens N; Han J; Budatha M; Ross TD; Fang JS; Yun S; Thomas JL; Schwartz MA Intramembrane Binding of Ve-Cadherin to Vegfr2 and Vegfr3 Assembles the Endothelial Mechanosensory Complex. J Cell Biol 2015, 208, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Rubsam M; Mertz AF; Kubo A; Marg S; Jungst C; Goranci-Buzhala G; Schauss AC; Horsley V; Dufresne ER; Moser M et al. E-Cadherin Integrates Mechanotransduction and Egfr Signaling to Control Junctional Tissue Polarization and Tight Junction Positioning. Nat Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Somanath PR; Ciocea A; Byzova TV Integrin and Growth Factor Receptor Alliance in Angiogenesis. Cell Biochem Biophys 2009, 53, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Mahabeleshwar GH; Feng W; Reddy K; Plow EF; Byzova TV Mechanisms of Integrin-Vascular Endothelial Growth Factor Receptor Cross-Activation in Angiogenesis. Circ Res 2007, 101, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Somanath PR; Malinin NL; Byzova TV Cooperation between Integrin Alphavbeta3 and Vegfr2 in Angiogenesis. Angiogenesis 2009, 12, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Schneller M; Vuori K; Ruoslahti E Avß3 Integrin Associates with Activated Insulin and Pdgfß Receptors and Potentiates the Biological Activity of Pdgf. The EMBO journal 1997, 16, 5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Woodard AS; García-Cardeña G; Leong M; Madri JA; Sessa WC; Languino LR The Synergistic Activity of Alphavbeta3 Integrin and Pdgf Receptor Increases Cell Migration. Journal of cell science 1998, 111, 469–478. [DOI] [PubMed] [Google Scholar]
- (460).Trusolino L; Serini G; Cecchini G; Besati C; Ambesi-Impiombato FS; Marchisio PC; De Filippi R Growth Factor–Dependent Activation of Avß3 Integrin in Normal Epithelial Cells: Implications for Tumor Invasion. The Journal of cell biology 1998, 142, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (461).Rahman S; Patel Y; Murray J; Patel KV; Sumathipala R; Sobel M; Wijelath ES Novel Hepatocyte Growth Factor (Hgf) Binding Domains on Fibronectin and Vitronectin Coordinate a Distinct and Amplified Met-Integrin Induced Signalling Pathway in Endothelial Cells. BMC cell biology 2005, 6, 8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Gao B; Song H; Bishop K; Elliot G; Garrett L; English MA; Andre P; Robinson J; Sood R; Minami Y et al. Wnt Signaling Gradients Establish Planar Cell Polarity by Inducing Vangl2 Phosphorylation through Ror2. Dev Cell 2011, 20, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Grumolato L; Liu G; Mong P; Mudbhary R; Biswas R; Arroyave R; Vijayakumar S; Economides AN; Aaronson SA Canonical and Noncanonical Wnts Use a Common Mechanism to Activate Completely Unrelated Coreceptors. Genes Dev 2010, 24, 2517–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Bin-Nun N; Lichtig H; Malyarova A; Levy M; Elias S; Frank D Ptk7 Modulates Wnt Signaling Activity Via Lrp6. Development 2014, 141, 410–421. [DOI] [PubMed] [Google Scholar]
- (465).Peradziryi H; Kaplan NA; Podleschny M; Liu X; Wehner P; Borchers A; Tolwinski NS Ptk7/Otk Interacts with Wnts and Inhibits Canonical Wnt Signalling. EMBO J 2011, 30, 3729–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Kim N; Stiegler AL; Cameron TO; Hallock PT; Gomez AM; Huang JH; Hubbard SR; Dustin ML; Burden SJ Lrp4 Is a Receptor for Agrin and Forms a Complex with Musk. Cell 2008, 135, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Zhang W; Coldefy AS; Hubbard SR; Burden SJ Agrin Binds to the N-Terminal Region of Lrp4 Protein and Stimulates Association between Lrp4 and the First Immunoglobulin-Like Domain in Muscle-Specific Kinase (Musk). J Biol Chem 2011, 286, 40624–40630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Giordano S; Corso S; Conrotto P; Artigiani S; Gilestro G; Barberis D; Tamagnone L; Comoglio PM The Semaphorin 4d Receptor Controls Invasive Growth by Coupling with Met. Nature Cell Biology 2002, 4, 720–724. [DOI] [PubMed] [Google Scholar]
- (469).Conrotto P; Corso S; Gamberini S; Comoglio PM; Giordano S Interplay between Scatter Factor Receptors and B Plexins Controls Invasive Growth. Oncogene 2004, 23, 5131–5137. [DOI] [PubMed] [Google Scholar]
- (470).Swiercz JM; Kuner R; Offermanns S Plexin-B1/Rhogef-Mediated Rhoa Activation Involves the Receptor Tyrosine Kinase Erbb-2. J Cell Biol 2004, 165, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Swiercz JM; Worzfeld T; Offermanns S Erbb-2 and Met Reciprocally Regulate Cellular Signaling Via Plexin-B1. J Biol Chem 2008, 283, 1893–1901. [DOI] [PubMed] [Google Scholar]
- (472).Kigel B; Rabinowicz N; Varshavsky A; Kessler O; Neufeld G Plexin-A4 Promotes Tumor Progression and Tumor Angiogenesis by Enhancement of Vegf and Bfgf Signaling. Blood 2011, 118, 4285–4296. [DOI] [PubMed] [Google Scholar]
- (473).Whitaker GB; Limberg BJ; Rosenbaum JS Vascular Endothelial Growth Factor Receptor-2 and Neuropilin-1 Form a Receptor Complex That Is Responsible for the Differential Signaling Potency of Vegf(165) and Vegf(121). J Biol Chem 2001, 276, 25520–25531. [DOI] [PubMed] [Google Scholar]
- (474).King C; Wirth D; Workman S; Hristova K Interactions between Nrp1 and Vegfr2 Molecules in the Plasma Membrane. Biochim Biophys Acta 2018, 1860, 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Favier B; Alam A; Barron P; Bonnin J; Laboudie P; Fons P; Mandron M; Herault JP; Neufeld G; Savi P et al. Neuropilin-2 Interacts with Vegfr-2 and Vegfr-3 and Promotes Human Endothelial Cell Survival and Migration. Blood 2006, 108, 1243–1250. [DOI] [PubMed] [Google Scholar]
- (476).Bocharov EV; Sharonov GV; Bocharova OV; Pavlov KV Conformational Transitions and Interactions Underlying the Function of Membrane Embedded Receptor Protein Kinases. Biochim Biophys Acta Biomembr 2017, 1859, 1417–1429. [DOI] [PubMed] [Google Scholar]
- (477).Coskun U; Grzybek M; Drechsel D; Simons K Regulation of Human Egf Receptor by Lipids. Proc Natl Acad Sci U S A 2011, 108, 9044–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).McLaughlin S; Smith SO; Hayman MJ; Murray D An Electrostatic Engine Model for Autoinhibition and Activation of the Epidermal Growth Factor Receptor (Egfr/Erbb) Family. J Gen Physiol 2005, 126, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Michailidis IE; Rusinova R; Georgakopoulos A; Chen Y; Iyengar R; Robakis NK; Logothetis DE; Baki L Phosphatidylinositol-4,5-Bisphosphate Regulates Epidermal Growth Factor Receptor Activation. Pflugers Arch 2011, 461, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (480).Lopez PH; Schnaar RL Gangliosides in Cell Recognition and Membrane Protein Regulation. Curr Opin Struct Biol 2009, 19, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Julien S; Bobowski M; Steenackers A; Le Bourhis X; Delannoy P How Do Gangliosides Regulate Rtks Signaling? Cells 2013, 2, 751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Hedger G; Sansom MS; Koldso H The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci Rep 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Suzuki KG New Insights into the Organization of Plasma Membrane and Its Role in Signal Transduction. Int Rev Cell Mol Biol 2015, 317, 67–96. [DOI] [PubMed] [Google Scholar]
- (484).Den Hartigh JC; v. B en Henegouwen P; Verkleij AJ; Boonstra J The Egf Receptor Is an Actin-Binding Protein. The Journal of cell biology 1992, 119, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Lidke DS; Lidke KA; Rieger B; Jovin TM; Arndt-Jovin DJ Reaching out for Signals: Filopodia Sense Egf and Respond by Directed Retrograde Transport of Activated Receptors. J Cell Biol 2005, 170, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Salaita K; Nair PM; Petit RS; Neve RM; Das D; Gray JW; Groves JT Restriction of Receptor Movement Alters Cellular Response: Physical Force Sensing by Epha2. science 2010, 327, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Winter PW; Van Orden AK; Roess DA; Barisas BG Actin-Dependent Clustering of Insulin Receptors in Membrane Microdomains. Biochim Biophys Acta 2012, 1818, 467–473. [DOI] [PubMed] [Google Scholar]