

Abstract
The COP9/signalosome (CSN) multi-protein complex regulates the activity of cullin-RING ubiquitin ligases (CRLs), including the DDB2 and CSA CRL4 ligases (CRL4DDB2 and CRL4CSA), which are involved in the repair of UV-induced DNA damages. In the present study, we demonstrated that the protein kinase ATM, a key component of the DNA damage response (DDR), phosphorylates CSN1 and CSN7a, two subunits of the CSN complex, in a UV-dependent manner. The phosphorylation of CSN1 on serine 474 was detected as early as 3 h after UV-exposure, peaked at 8 h and persisted until 48 h post-UV irradiation. Such a time course suggests a role in late DDR rather than in DNA repair. Consistently, overexpression of a phosphorylation-resistant S474A CSN1 mutant reduced UV-induced apoptosis. Thus, CSN1 appears to play a role not only in DNA repair but also in UV-induced apoptosis.
Keywords: COP9/signalosome, CRL4 E3 ubiquitin ligase, ATM, NER, apoptosis
Introduction
The COP9 signalosome (CSN) is an evolutionarily conserved complex of 8 to 9 protein subunits that regulates numerous cellular and biological processes such as embryonic development, cell cycle, DNA damage repair, checkpoint repair control, signal transduction, circadian rhythm, T-cell development and autophagy (1). CSN accomplishes these various functions by controlling cullin-RING-E3 ligases (CRLs), the most prominent class of E3-enzymes, which promote ubiquitination of a variety of regulatory proteins subsequently targeted to proteasomal degradation (2). All CRLs are composed of a constitutive cullin RING-based catalytic core and of exchangeable substrate recognition modules (3). The human genome encodes at least seven cullins (Cul1, 2, 3, 4A, 4B, 5 and 7), which share some sequence homology and a similar elongated arc-shaped structure (4,5). The substrate recognition module is specific to each cullin, and consists of a substrate recognition subunit connected to the cullin by an adaptor subunit, such as DDB1 in CRL4 ligases. Despite their considerable diversity, all CRLs are activated by conjugation of the ubiquitin-like protein Nedd8 to the cullin subunit (neddylation) (3,6). CSN inactivates cullin proteins both by hydrolyzing the cullin-Nedd8 conjugates (7) and by inhibiting CRL activity as a direct Cullin-Roc1 binding partner, thereby preventing substrate recruitment (8,9). The association of CSN with CRL is highly dynamic, and context- as well as substrate-dependent (10,11).
Previously, we discovered that two Cullin 4-Roc1-DDB1 ligases (CRL4s) are involved in the repair of ultraviolet (UV)-induced DNA lesions by the nucleotide excision repair (NER) pathway.
NER repairs helix-distorting forms of single-strand DNA damage such as UV-created cyclobutane pyrimidine dimers (CPDs) and 6-4 pyrimidone phosphoproducts (6-4PPs) or chemotherapy generated bulky adducts. NER consists of two main sub-pathways, global genomic NER (GG-NER) that repairs the whole genome on one hand and transcription-coupled NER (TC-NER) that is restricted to actively transcribed genes. Mutations in the NER pathway cause two well-known genetic diseases: the developmental disorder Cockayne syndrome is caused by mutations in TC-NER; the skin cancer predisposition syndrome Xeroderma pigmentosum is the consequence of an impairment of GG-NER. TC-NER and GG-NER are similar, and the main differences are in the mechanism of DNA lesion recognition, a step at which NER-associated CRL4 ligases are involved. DDB2 (DNA damage binding protein 2) and CSA (Cockayne Syndrome protein A) are the substrate recognition receptors of these CLR4 ligases (CRL4DDB2 and CRL4CSA). DDB2 is the smaller subunit of DDB, a heterodimeric protein implicated in the ethiology of Xeroderma pigmentosum group E (XPE), while CSA is a TC-NER component and a complementation factor for the Cockayne syndrome disease (12). CRL4DDB2 and CRL4CSA are regulated by CSN, albeit in a different manner (10). Immediately after UV-irradiation, CRL4DDB2 is released from CSN and activated by neddylation (10,13). By contrast, CRL4CSA is silenced by CSN at the beginning of the repair process and is activated at later stages, upon dissociation of CSN (10,14).
DNA damages trigger the DNA damage response (DDR) pathway, a signaling network leading to a coordinated cell response that includes cell cycle arrest, apoptosis, senescence, and DNA repair. Two related protein kinases, ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related), are essential in DDR. ATM, and its regulator the MRN (Mre11-Rad50-NBS1) complex, is the general sensor of DNA double-strand breaks (DSBs). ATR and its regulator ATRIP (ATR-interacting protein) detect the single-strand DNA (ssDNA) species generated by NER, as well as ssDNA present at stalled replication forks (15,16). UV irradiation and the NER pathway also activate ATM (17,18). The second wave of signaling involves phosphorylation of common downstream targets of ATM and ATR, e.g., H2AX, Chk1 and Chk2. The Elledge laboratory has identified over 700 new ATM and ATR targets phosphorylated at SQ/TQ sites in response to DSB-inducing ionized radiation (IR) and/or UV irradiation. Three subunits of CSN, CSN1, CSN3 and CSN7a, are phosphorylated in response to IR. CSN1 is also phosphorylated in response to UV (19).
Here, we examined phosphorylation of CSN in response to UV irradiation. We detected several new ATM-dependent phosphorylation sites in CSN1 and CSN7a. We provide evidence, based on gain- and loss-of-function experiments, that CSN1 phosphorylation on S474 is not required for DNA repair, but rather appears to be involved in DDR and participates in inducing apoptosis in response to UV. Thus, in UV-irradiated cells, CSN appears to be involved both in the DNA repair process and in cell apoptosis, a downstream cell response.
Materials and methods
Plasmid construction
CSN1 phosphomutant (S474A) was generated using the QuickChange Site-Directed Mutagenesis kit (Stratagene). Full length human CSN1 wild-type and CSN1 phosphomutant (S474A) were inserted in frame into the pREV-HTF retroviral vector as previously described (20).
Cell culture and cell lines
HeLa S3 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. HeLa S3 cell lines stably expressing FLAG and HA-tagged CSN1 or its phosphomutant were generated by retroviral transduction according to a previously established protocol (10). A HeLa cell line transduced with the empty pREV vehicle vector was used as a negative control. For UV irradiation, cells were grown on 15-cm tissue culture dishes, washed with PBS, irradiated with UV at 25 J/m2 (unless noted otherwise) and incubated in fresh medium for the indicated periods of time. Where indicated, 5 µM of the ATM-specific inhibitor, 2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one (KU-55933) was added.
Antibodies
We used the following antibodies: CSA (sc-10997; Santa Cruz Biotechnology; GTX100145; GeneTex), ATM protein kinase pS1981 (200-301-400; Rockland), CSN1 (PW8285; Enzo/Affinity), β-actin (A5441; Sigma), cyclobutane pyrimidine dimers (TDM2, D194-1, MBL) and (6-4) photoproducts (6-4M2, D195-1, MBL).
Purification of protein complexes
A solubilized chromatin fraction was prepared as described previously (10).
Briefly, 1×108 cells were suspended in hypotonic Micrococcal Nuclease (MNase) buffer (20 mM Tris, pH 8.0, 5 mM NaCl, 2.5 mM CaCl2) and disrupted by freezing/thawing cycles. Nuclei were collected by centrifugation at 1,000 × g for 5 min, washed once with 1 ml of MNase buffer and resus-pended in 400 µl of MNase buffer. MNase (N3755; Sigma) was added at 10 U/ml and the samples were incubated for 10 min at room temperature. MNase reaction was terminated with 10 mM EGTA. The samples were centrifuged at 5,000 × g for 5 min and the supernatants were collected (solubilized chromatin fraction). Protein complexes were immunoprecipitated with anti-FLAG M2 agarose (Sigma) and eluted with FLAG peptide. They were either immediately analyzed, or further affinity purified with anti-HA-conjugated agarose and eluted with the HA peptide. Complexes were resolved by SDS-PAGE and stained using the Silver Quest kit (Invitrogen).
LC-MS/MS based shotgun sequencing
SDS-PAGE-resolved protein bands were subjected to trypsin-based in-gel digestion and LC-MS/MS based shotgun sequencing using an LTQ Orbitrap mass spectrometer essentially as described previously (21–25). As was carried out previously, the resulting tandem mass spectra were data-searched using the SEQUEST algorithm allowing for variable modifications on methionine (+15.9949 Da) and serine, threonine and tyrosine (+79.9663 Da) with full trypsin specificity, 2 missed cleavages and a precursor tolerance of ±1.1 Da against the UniProt human database (www.uniprot.org). Results were filtered with a ±3 ppm mass measurement accuracy cutoff (from theoretical) and a delta-Xcorr of 0.08, and all CSN phosphopeptide MS/MS spectra were manually inspected for consistency.
Analysis of UV-induced photolesions
UV-induced photolesions were quantified on a slot-blot, using TD-2 and 6-4M2 antibodies specifically recognizing CPDs and 6-4 photoproducts respectively. Irradiated cells were lysed in MNase buffer and centrifuged to obtain nuclei pellets that were subsequently treated with MNase as described above. DNA for analysis was purified by phenol-chlorophorm and measured on a NanoDrop (ND-1000; NanoDrop Technologies, Inc.). One milligram of nucleosome-associated DNA from each sample was transferred onto nitrocellulose membranes (Hybond™-N+ nylon transfer membrane 0.45 µm RPN2020B; Amersham) using the MINIFOLD I slot-blot system (Schleicher & Schuell). Immunoblot analysis was performed as previously described (14).
Results
CSN1 is phosphorylated by ATM in response to UV irradiation
To test whether CSN subunits are regulated by phosphorylation in response to UV, HeLa S3 cells expressing a Flag tag on the CSN5 subunit (10) were UV irradiated (or not for the controls). Chromatin-associated CSN complexes were affinity purified with anti-Flag antibodies 30 min or 4 h following UV exposure. Analysis of the complexes by mass-spectrometry revealed five phosphorylation sites on the CSN1 subunit and one phosphorylation site on the CSN7α subunit (Table I). Except for the CSN1 pS468 and pT479 sites, the amount of phosphorylated peptides increased at 4 h after UV exposure but not at 30 min.
Table I.
Identification of phosphorylated subunits by mass spectrometry.
| Total peptide matches after exposure to UV (min) | ||||
|---|---|---|---|---|
| Total peptide matches after exposure to UV (min) | ||||
| Subunit | Phospho-site | 0 | 30 | 240 |
| CSN1 | pS468 | 2 | 0 | 1 |
| pS474 | 6 | 5 | 9 | |
| pT479 | 4 | 4 | 4 | |
| pS483 | 0 | 0 | 2 | |
| pS485 | 0 | 0 | 2 | |
| CSN7 | pS232 | 1 | 0 | 2 |
CSN was purified using the Flag tag on the eCSN5 subunit before and after 30 and 240 min HeLa/eCSN5 cell irradiation with UV at 25 J/m2 followed by analysis of phosphopeptides analyzed by mass spectrometry.
The sequences adjacent to the S474 and S483 phosphorylation sites on CSN1 and the S232 site on CSN7a match the phosphorylation site by ATM or/and ATR kinases (SQ sites). These sites were also identified as ATM/ATR substrates in a proteomic screen for the DDR associated ATM/ATR targets performed by the Elledge laboratory (19). To confirm that the CSN1 S474Q site was phosphorylated by ATM, we used pSQ antibodies generated against ATM pS1981-Q (19): these antibodies detected ATM phosphorylation at 3 h but not at 30 min after UV irradiation (Fig. 1a). The ATM specific inhibitor Ku55933 dramatically reduced ATM phosphorylation, confirming ATM autophosphorylation. The same antibody detected a band in CSN complexes purified from chromatin. The size of the band matched the size of pCSN1. The band was detected at 3 h but not at 30 min post-UV irradiation (Fig. 1b), in good concordance with the kinetics of phosphorylation observed using mass-spectrometry. Moreover, cell pre-treatment with Ku55933 abolished the appearance of this band, supporting the idea that phosphorylation was carried out by ATM (Fig. 1b). Taken together, our results suggest that CSN1 is phosphorylated on S474 by ATM in response to UV irradiation. To confirm this hypothesis, we mutated S474 in CSN1; the resulting S474/A mutant was not phosphorylated in response to UV (Fig. 1c). The phosphorylation of ectopic CSN1 was detected 3 h post irradiation, peaked at 8 h and persisted up to 48 h. Taken together, these data indicate that CSN1 is phosphorylated in response to UV, and that CSN1 phosphorylation is a late event.
Figure 1.

CSN1 is phosphorylated by ATM at Ser-474 in response to UV irradiation. Cells were irradiated with UV at 25 J/m2 and incubated in fresh media, with or without the ATM inhibitor Ku 55933 as indicated. (a) Western blotting showing ATM auto-phosphorylation; (b) the CSN1 component of CSN is phosphorylated by ATM. CSN was purified using the Flag tag on the eCSN5 subunit, and analyzed by western blotting; eCSN5 was used as a loading control. (c) Mutation of S474 abolishes CSN1 phosphorylation. Western blot analysis of affinity purified CSN1 from cells expressing wt CSN1 or S474A CSN1 mutant as indicated. Anti-ATM-pS1981 antibodies were used on all panels.
UV-dependent CSN1 phosphorylation is important for UV-induced apoptosis
The ectopic expression of a large excess of mutant CSN1 (Fig. 2a), potentially acting as a negative transdominant mutant, did not decrease the repair of 6-4PP or CPD damages. Repair of CPD damages appeared even to occur at slightly faster rates (Fig. 2b and c). Consistently, the timing of phosphorylation, which peaked at 8 h, a time at which DNA repair was completed (Fig. 2c) and its persistance for 48 h post-irradiation (Fig. 1c), suggests an involvement in downstream events. We thus tested the hypothesis that CSN1 phosphorylation could be involved in apoptosis induced by high doses of UV. Apoptosis was monitored using an Annexin V assay. Apoptosis was inhibited in HeLa cells overexpressing the S474/A mutant CSN1 (Fig. 3a); whereas 12% of HeLa cells were scored as apoptotic 24 h post-irradiation, only 6% were apoptotic in cells expressing excess amounts of the mutant. At 48 h post-irradiation, apoptotic cells could not be detected in cells expressing the mutant, whereas 6% of the cells were scored as apoptotic in the HeLa cells. Conversely, an excess of wild-type CSN1 resulted in increased apoptosis (from 12% in HeLa cells to 17% in HeLa cells expressing wild-type CSN1). This tendency was observed at various energies of irradiation (Fig. 3b). Thus, the phosphorylation of CSN1 appears to be involved in UV-induced apoptosis.
Figure 2.

UV-dependent CSN1 phosphorylation does not impact DNA repair. (a) Ectopically expressed CSN1 wt or mutant was detected by western blotting using anti-CSN1 antibodies. (b) HeLa cell lines stably expressing CSN1wt or CSN1 S474A mutant were UV-irradiated at 30 J/m2. CPD and 6-4 photo-products were detected by slot-blot, using anti-CPD or anti-6-4PP antibodies as indicated (upper panel); ethidium bromide stain of samples (lower panel). (c) Quantification of slot blots. Relative signal was calculated by dividing signals at 3 and 8 h by the signal before repair (30 min after UV irradiation).
Figure 3.
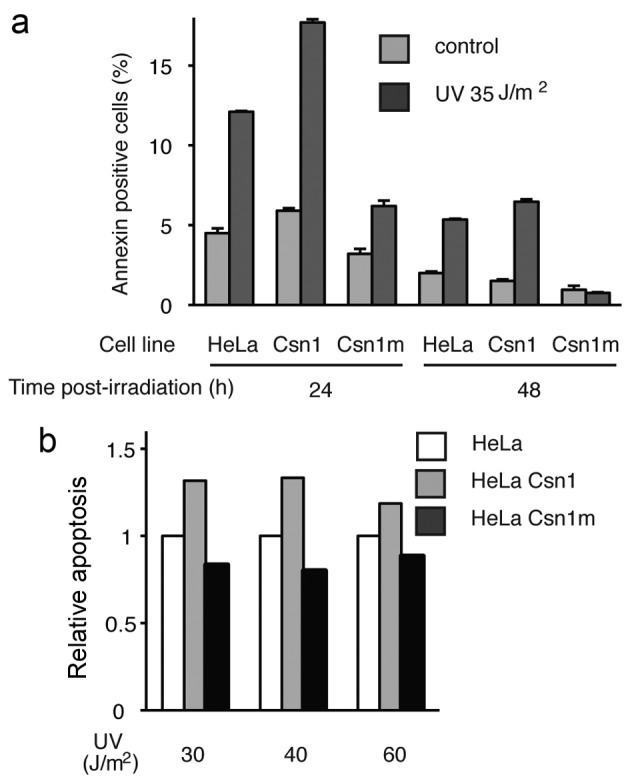
CSN1 phosphorylation by ATM impacts UV-induced apoptosis. (a and b) HeLa cell lines stably expressing CSN1wt or CSN1 S474A mutant, as well as the parental HeLa cell line, were UV-irradiated as indicated, and stained with Annexin V 24 and 48 h (a) or 24 h (b) after UV irradiation.
Discussion
Here, we demonstrated that CSN1 is phosphorylated on S474 by ATM in response to UV irradiation. CSN1 binds to CRL4DDB2 and CRL4CSA ligases, and a tempting hypothesis was that CSN1 phosphorylation by ATM regulates the interaction between CSN1 and these ligases. However, our experiments (data not shown) indicated that this was not the case, as mutating the CSN phosphorylation site did not affect the co-immunoprecipitation of the CSN complex with CRL4DDB2 or CRL4CSA ligases. This result was consistent with the timing of phosphorylation, which occurred later than the dissociation of the CSN1/CLR4 complex. This result is also consistent with the X-ray structure of CSN, recently solved by the Thomä laboratory (28). This structure demonstrates that Ser 474 is located at the surface of the CSN C-terminal helical bundle, separated by structured linkers from the CRL4 interacting PCI ring structure. Three other COP9/CSN subunits, CSN3, CSN7a and CSN8, were phosphorylated in response to UV irradiation and these phosphosites are also located in the same C-terminal helical bundle according to CSN X-ray structure. Moreover, CSN1 itself is phosphorylated on multiple serines (19,27). Thus, eliminating only one of these phosphorylation events may not be sufficient to impact CSN/CLR4 interaction, and to achieve maximal inhibition of apoptosis.
The observation that ATM-dependent CSN1 phosphorylation is a late event correlates with a role in apoptosis rather then in DNA lesion repair. Conversely, CSN3 is phosphorylated at earlier times (from 0.5–8 h) after γ-irradiation and this phosphorylation event delayed double strands break repair in irradiated fibroblasts (27). We did not observe any effect of phosphorylation on DNA repair for S474 phosphorylation, but rather our data showed that CSN1 phosphorylation was important for cell progression toward programmed cell death (apoptosis) in response to high dose UV irradiation, as ectopic expression of the S474/A mutant reduced apoptosis. Two recent high throughput studies of cell cycle-dependent phosphorylation identified specific phosphorylation of CSN1 and CSN3 by ATM/ATR during the S phase of the cell cycle (29,30). The activation of DDR phosphorylation network mediated by ATM and ATR during the S phase of the cell cycle serves as a checkpoint and promotes the elimination of cells with unrepaired damages via apoptosis. Here, we demonstrated a role for CSN1 S474 phosphorylation in UV-induced apoptosis, as ectopic expression of the non-phosphorylated mutant reduced cell apoptosis. A similar case has been described for Ku70 in chronic lymphocytic leukemia (CLL). Upon genotoxic stress, phosphorylation of Ku70 is increased in an ATM-dependent manner in CLL cells, and mutation of this site reduced CLL cell apoptosis. By enhancing double-strand break (DSB) repair, and interfering with apoptosis, phospho-Ku70 likely contributes to the CLL drug resistant phenotype and to cancer progression. Similarly, phospho-CSN could also play a specific role in different steps of tumorigenesis. CSN has been implicated in the development of many cancers, partially via the proteasome-mediated degradation of oncogenes or tumor suppressors p53 (31), Mdm2 (32), p27 (33), Id1 and Id3 (34).
References
- 1.Wei N, Serino G, Deng XW. The COP9 signalosome: More than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 3.Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66:1924–1938. doi: 10.1007/s00018-009-8712-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 5.Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 6.Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wee S, Geyer RK, Toda T, Wolf DA. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting auto-catalytic adapter instability. Nat Cell Biol. 2005;7:387–391. doi: 10.1038/ncb1241. [DOI] [PubMed] [Google Scholar]
- 8.Emberley ED, Mosadeghi R, Deshaies RJ. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J Biol Chem. 2012;287:29679–29689. doi: 10.1074/jbc.M112.352484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thomä NH. Crystal structure of the human COP9 signalosome. Nature. 2014;512:161–165. doi: 10.1038/nature13566. [DOI] [PubMed] [Google Scholar]
- 10.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 11.Hannss R, Dubiel W. COP9 signalosome function in the DDR. FEBS Lett. 2011;585:2845–2852. doi: 10.1016/j.febslet.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 12.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 13.Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 15.Shiloh Y. ATM: Expanding roles as a chief guardian of genome stability. Exp Cell Res. 2014;329:154–161. doi: 10.1016/j.yexcr.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–138. doi: 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakasugi M, Sasaki T, Matsumoto M, Nagaoka M, Inoue K, Inobe M, Horibata K, Tanaka K, Matsunaga T. Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. J Biol Chem. 2014;289:28730–28737. doi: 10.1074/jbc.M114.589747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 20.Ouararhni K, Hadj-Slimane R, Ait-Si-Ali S, Robin P, Mietton F, Harel-Bellan A, Dimitrov S, Hamiche A. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006;20:3324–3336. doi: 10.1101/gad.396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang A, Petrov KO, Hyun ER, Liu Z, Gerber SA, Myers LC. The Tlo proteins are stoichiometric components of Candida albicans mediator anchored via the Med3 subunit. Eukaryot Cell. 2012;11:874–884. doi: 10.1128/EC.00095-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hood EA, Kettenbach AN, Gerber SA, Compton DA. Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol Biol Cell. 2012;23:2264–2274. doi: 10.1091/mbc.E11-12-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Kang S, Ray S, Jackson J, Zaitsev AD, Gerber SA, Cuny GD, Glicksman MA. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry. 2011;50:9399–9408. doi: 10.1021/bi201173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sano H, Peck GR, Kettenbach AN, Gerber SA, Lienhard GE. Insulin-stimulated GLUT4 protein translocation in adipocytes requires the Rab10 guanine nucleotide exchange factor Dennd4C. J Biol Chem. 2011;286:16541–16545. doi: 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010;24:1236–1241. doi: 10.1101/gad.1920310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balaganur V, Pathak NN, Lingaraju MC, More AS, Latief N, Kumari RR, Kumar D, Tandan SK. Effect of S-methylisothiourea, an inducible nitric oxide synthase inhibitor, in joint pain and pathology in surgically induced model of osteoarthritis. Connect Tissue Res. 2014;55:367–377. doi: 10.3109/03008207.2014.953629. [DOI] [PubMed] [Google Scholar]
- 27.Füzesi-Levi MG, Ben-Nissan G, Bianchi E, Zhou H, Deery MJ, Lilley KS, Levin Y, Sharon M. Dynamic regulation of the COP9 signalosome in response to DNA damage. Mol Cell Biol. 2014;34:1066–1076. doi: 10.1128/MCB.01598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meir M, Galanty Y, Kashani L, Blank M, Khosravi R, Fernández-Ávila MJ, Cruz-García A, Star A, Shochot L, Thomas Y, et al. The COP9 signalosome is vital for timely repair of DNA double-strand breaks. Nucleic Acids Res. 2015;43:4517–4530. doi: 10.1093/nar/gkv270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 30.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, Dubiel W. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–1639. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Menon S, Lykke-Andersen K, Tsuge T, Di Xiao, Wang X, Rodriguez-Suarez RJ, Zhang H, Wei N. The COP9 signalosome inhibits p27(kip1) degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Curr Biol. 2002;12:667–672. doi: 10.1016/S0960-9822(02)00791-1. [DOI] [PubMed] [Google Scholar]
- 34.Berse M, Bounpheng M, Huang X, Christy B, Pollmann C, Dubiel W. Ubiquitin-dependent degradation of Id1 and Id3 is mediated by the COP9 signalosome. J Mol Biol. 2004;343:361–370. doi: 10.1016/j.jmb.2004.08.043. [DOI] [PubMed] [Google Scholar]