

Purpose of review
Primary immunodeficiency disorders (PIDs) are no longer defined by infections alone. First clinical sign or sequelae of PID may include autoimmunity, such as cytopenias, arthritis or enteropathy. This review addresses the latest in multidisciplinary approaches for expanding clinical phenotypes of PIDs with autoimmunity, including new presentations of known entities and novel gene defects. We also discuss diagnostic tools for identifying the distinct changes in immune cells subsets and autoantibodies, mechanistic understanding of the process, and targeted treatment and indications for hematopoietic stem-cell transplantation (HSCT).
Recent findings
In the past years, increased awareness and use of genetic screening, confirmatory functional studies and immunological biomarkers opened the door for early recognition of PIDs among patients with autoimmunity. Large cohort studies detail the clinical spectrum and treatment outcome of PIDs with autoimmunity with specific immune genes (e.g., CTLA4, LRBA, PI3Kδ, NFKB1, RAG). The benefit of early recognition is initiation of targeted therapies with precise re-balancing of the dysregulated immune pathways (e.g., biologicals) or definitive therapy (e.g., HSCT).
Summary
Clinical presentation of patients with PID and autoimmunity is highly variable and requires in-depth diagnostics and precision medicine approaches.
Keywords: autoimmunity, immune dysregulation, primary immunodeficiency, targeted therapy, tolerance
INTRODUCTION
Primary immunodeficiencies (PIDs) are no longer defined by tendency for infections alone. PID patients with noninfectious complications are increasingly recognized with features of ‘immune dysregulation’ including autoimmunity, inflammation, lymphoproliferation or malignancy (Fig. 1) [1].
FIGURE 1.
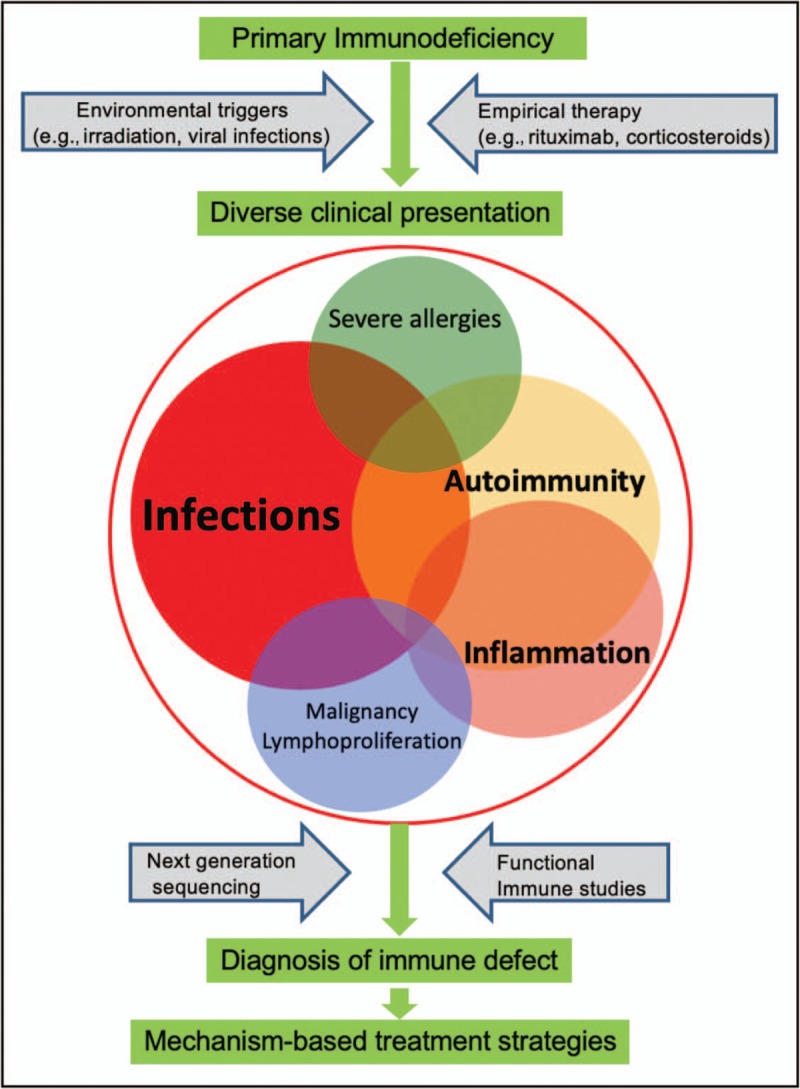
Precision medicine therapy for the diverse disease spectrum of primary immunodeficiency (PID). The variable clinical presentation of PID is influenced by environmental triggers as wells as empirical therapy. For instance, rituximab therapy eliminates B cells and thus reduces autoantibodies but also increases risk of infections, whereas viral infections can stimulate dysregulated B cells and result in autoimmunity. The use of next-generation sequencing and functional immune studies can identify the precise molecular basis of disease and enable optimal treatment which targets the molecular defect and minimizes adverse effects.
However, identifying an underlying PID in a heterogenous group of patients with a variety of autoimmune disorders can be a daunting task. Most pediatricians or specialists taking care of patients with autoimmune disorders may not consider immune evaluation in the initial workup and assume low probability. Therefore, it is not uncommon that the specific diagnosis of highly vulnerable patients with genetic immune deficiency disease is delayed.
In this review, we will focus on recent understanding of the most common clinical entities with PID where autoimmune complications dominate and highlight features that may distinguish these patients from the general population of autoimmune patients. We will also introduce a basic description of immune and genetic diagnostic tools that are essential for understanding the underlying immune defect. We will discuss the pathomechanism of autoimmunity in immune-deficient background, including both intrinsic and extrinsic factors. Lastly, we will cite current literature on the decision-making process for targeted therapy and importance of a multidisciplinary approach for autoimmune disorders.
Box 1.

no caption available
INCREASING AWARENESS FOR AUTOIMMUNITY: THE JANUS FACE OF PRIMARY IMMUNODEFICIENCY DISORDER
In the latter half of the 20th century, two rare classical endocrine monogenic autoimmune disorders were described. These two very early onset disorders are specific defects in regulatory T cells and/or elimination of autoreactive T cells, and are known as the syndromes of IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) and APECED (autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy) (Fig. 2). APECED and IPEX are examples when pediatricians partnered in a multispecialty approach with endocrinologists and gastroenterologists for the care for complex monogenic autoimmune diseases. Once the underlying immune defect of autoimmune regulator (AIRE) (causing APECED) [2,3] and forkhead box P3 (FOXP3) (causing IPEX) [4,5] were discovered in 1997 and 2000 respectively, studies were initiated for insight into the disease biology, which informed clinical immunologists for both diagnosis and management of large numbers of patients [6] and eventually enabled transplant specialists to justify the use of HSCT for IPEX cases [6,7]. Similarly, clinical and research collaboration between pediatric hematologists/oncologists and immunologist began to properly care for patients with persistent multilineal cytopenias, lymphoproliferation, and tendency for malignancy with defects in immune cell apoptosis now termed as ALPS (autoimmune lymphoproliferative syndrome). Once genetic defects, biomarkers, and pathomechanism were better understood, diagnostic approaches and targeted therapies were developed successfully [8].
FIGURE 2.
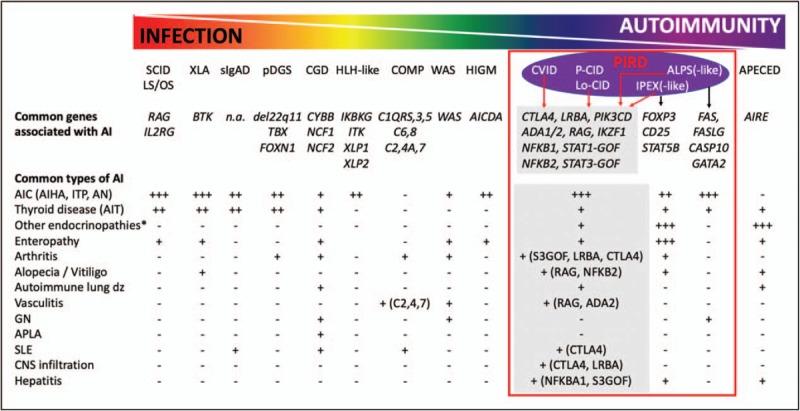
The genetic basis for infections versus autoimmunity (AI) within PIDs. An inverse gradation of infectious and autoimmune complications exists for different primary immunodeficiencies. For example, SCID patients with complete RAG deficiency or loss of IL2RG function are plagued with infections but minimal autoimmunity whereas APECED have high autoimmunity but relatively few infections. Multiple primary immune dysregulation disorders (PIRD) can be caused by the same set of genetic mutations although particular mutations favor specific diseases such as FOXP3 mutations causing IPEX-like disorder. ∗Other endocrinopathies include Addison's disease, type 1 diabetes mellitus, adrenal corticotropic hormone insufficiency, and growth hormone deficiency. COMP, complement defects; ADA1/2, adenosine deaminase 1 and 2; AIC, autoimmune cytopenia; AICDA, activation induced cytidine deaminase; AIHA, autoimmune hemolytic anemia; AIRE, autoimmune regulator; AIT, autoimmune thyroid disease; AN, autoimmune neutropenia; ALPS, autoimmune lymphoproliferative syndrome; APECED, autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy; APLA, antiphospholipid antibodies; BTK, Bruton tyrosine kinase; CASP10, caspase 10; CD25, cluster of differentiation 25 (interleukin-2 receptor α chain); CGD, chronic granulomatous disease; CNS, central nervous system; CVID, common variable immunodeficiency; CTLA4, cytotoxic T-lymphocyte antigen 4; CYBB, cytochrome B-245 β chain; FAS, FS-7 associated surface antigen; FASLG, FAS ligand; FOXN1, forkhead box N1; FOXP3, forkhead box P3; GATA2, GATA binding protein 2; GN, glomerulonephritis; HIGM, hyperimmunoglobulin M syndrome; HLH, hemophagocytic lymphohistiocytosis; IKBKG, inhibitor of nuclear factor κ B kinase subunit gamma; IKZF1, IKAROS family zinc finger 1; IL2RG, interleukin-2 receptor gamma subunit; ITP, immune thrombocytopenia; LS, leaky severe combined immunodeficiency; Lo-CID, late-onset combined immunodeficiency; LRBA, lipopolysaccharide responsive beige-like anchor protein; NCF1/2, neutrophil cytosolic factor 1 and 2; NFKB1/2, nuclear factor κ B subunits 1 and 2; OS, Omenn syndrome; P-CID, profound combined immunodeficiency; pDGS, partial DiGeorge syndrome; PIK3CD, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta; RAG, recombinase activating gene; SCID, severe combined immunodeficiency; sIgAD, secretory immunoglobulin A deficiency; SLE, systemic lupus erythematosus; STAT5B, signal transducer and activator of transcription 5B; STAT1/3-GOF (S1/3GOF), signal transducer and activator of transcription 1 and 3 gain-of-function mutations; TBX, T-box transcription factor; WAS, Wiskott Aldrich syndrome; XLP1/2, X-linked lymphoproliferative disease 1 and 2; XLA, X-linked agammaglobulinemia.
The need for a multidisciplinary approach for immune dysregulation among patients with autoimmunity continues on. Beyond IPEX, APECED, and ALPS, disorders with similar presentations (‘IPEX- or ALPS-like’) but different genetic causes have been steadily resurfacing (Fig. 2). This group of Mendelian multiorgan autoimmune diseases are now coined as primary immune regulatory disorders (PIRD) with subgroups of dominantly regulatory T-cell defects (T-regopathies) [9], and late-onset or profound combined immunodeficiency disorders (LoCID, P-CID) [10,11] where both T and B cells compartments are intrinsically autoimmune prone (Fig. 2). An excellent case-based review by Chandrakasan et al.[12▪] brings us close to the stories of immune dysregulation in PID patients with intricacies on the varying features of IPEX, ALPS, and/or LoCID-like disease. This report highlights the fact that isolated clinical autoimmune manifestations present early (even before infections) and the clinical course worsens with age [12▪].
A recent French national study, which is the largest to date and includes all types of PID and autoimmune manifestations, has been published by Fischer et al.[13▪▪]. One or more autoimmune and inflammatory manifestations were noted in 26.2% of 2183 retrospectively screened PID patients. In particular, immune manifestation of dysregulation, such as autoimmune cytopenias, enteropathy, and skin disease, occurred in 50% of PID patients with T-cell deficiency (CID) by 40 years of age and 10–30% of children below 18 years of age depending on the underlying immunodeficiency (innate, B-cell or T-cell defects). The study highlighted increased risk for autoimmune hemolytic anemia (AIHA) (830 times), inflammatory bowel disease (80 times), and arthritis (40 times) in children with PID compared with the general population. Although autoimmune manifestations occurred in patients with all types of PIDs, those with T-cell defects or common variable immunodeficiency (CVID) tended to have the highest risk for autoimmunity. Overall, this study well demonstrates that autoimmunity is a significant component of clinical presentation of all types of PIDs [13▪▪].
Clinically the most vulnerable group of young patients with PID and autoimmunity are those with reduced but not absent T-cell immunity. Unlike in severe combined immunodeficiency (SCID), these patients with LoCID and P-CID may only emerge after external triggers (viral infections or live vaccinations) have delayed diagnosis and may not be identified by newborn screening for SCID that is now implemented nationwide in the United States. Speckmann et al.[11] have published an interim analysis of an international prospective study on natural history of 51 patients with P-CID. The age of diagnosis was delayed as late as 15 years of age, especially among those with undefined genetic cause. Curiously, within the first year of clinical presentation, over 50% of patients presented with signs of immune dysregulation and autoimmunity, especially autoimmune cytopenias (21%), which was a dominant feature and continued throughout their life [11]. Importantly, severe events of immune dysregulation resulted in hospitalizations in over half of the P-CID cases. Although autoimmune cytopenias are often dominant in the first clinical presentation, patients may eventually progress to autoimmune enteropathy, interstitial lung disease with or without granulomas or develop arthritis, alopecia, and lesions in the central nervous system. Two brief reports by Goda et al.[14] and Wu et al.[15] illustrates well the autoimmune clinical presentation of young children with P-CID secondary to partial deficiency of recombinase activating gene (RAG). In the first case, the infant developed autoimmune thrombocytopenia (ITP) after varicella infection prompted the clinicians to screen for immunological biomarkers and genetic cause for presumed P-CID [14]. The second case describes a child with history of Kawasaki-like disease, arthritis, and alopecia followed by infections [15]. Both cases highlighted the importance of immune evaluation and biomarkers, to promote early genetic diagnosis in children who have early-onset autoimmune disease [14,15].
With the advance of genetic testing, we have the ability to group PIRD patients based on the underlying immune defect (Fig. 2). In recent years, several large studies have published on clinical characteristics of specific PIRD cohorts, in particular those with cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), lipopolysaccharide (LPS)-responsive and beige-like anchor protein (LRBA), nuclear factor κ-light-chain-enhancer of activated B cells 1 and 2 (NFκB1/NFκB2), activated phosphoinositide 3-kinase delta syndrome (APDS) and hypomorphic (partial) RAG and adenosine deaminase 2 (ADA2) deficiencies.
An international cohort of 133 patients with CTLA4 deficiency [16▪▪] shows unprecedented diversity of first symptoms of presentation including autoimmune cytopenia (33%), respiratory manifestations (21%), enteropathy (IBD) (17%), type 1 diabetes (T1DM) (8%), neurological symptoms (seizures, headache, and nausea; 6%), and in less than 5% of cases autoimmune thyroid disease (AIT), arthritis, alopecia, primary biliary cirrhosis and Addison disease. Only 61% of cases had history of infections (bacterial disease, herpes reactivation, and tuberculosis). Most autoimmune manifestations (cytopenia, gastrointestinal, and lung disease) were progressive and severe. Unlike other patients with CVID, there were a high fraction of cases (28%) with involvement of the central nervous system (CNS) either secondary to lymphocytic infiltration or hematological cause (e.g., bleeding, ischemia secondary to anemia, thrombosis). Beside the presence of multiple autoimmune disorders, CNS involvement may be a clinical diagnostic clue for CTLA4 deficiency. As inheritance is autosomal dominant, subsequent generations may be involved with variable penetrance of the clinical disease.
A systematic review of clinical features of 109 cases with LRBA deficiency also highlighted that the first presentation was often autoimmunity (42%) [17▪]. Multiple autoimmune diseases occurred in 77% of patients, which were dominated by AIHA and ITP, followed by T1DM, AIT and IBD. Infections (16%) and chronic diarrhea (27%) were less frequent first signs of disease manifestation of PID. The primary clinical diagnosis of these LRBA deficient patients were CVID (43%), autoimmunity (28%), ALPS-like (8%), IPEX-like PID (7%), and LoCID (4%) [17▪]. Lastly, a systematic review of 243 APDS patients also had autoimmune complications (28%); however, infections and lymphoproliferation (70%) were more dominant [18▪].
In the LoCID group, loss-of-function NFκB1 variants were published as most common monogenic cause of CVID in Europeans [19▪▪]. Common clinical features included massive lymphadenopathy (24%), unexplained splenomegaly (48%), and autoimmune disease (48%) mainly AIHA and ITP, followed by alopecia, vitiligo, and AIT were also associated with worse prognosis. Among CID patients with immune dysregulation and hypomorphic SCID-associated genes, partial RAG deficiency tends to be the most common genetic cause. Two recent reports summarize the clinical presentation and treatment outcome of autoimmune complications. Both Delmonte et al.[20▪] and Farmer et al.[21▪] report broad clinical autoimmune presentations including autoimmune cytopenias, vitiligo, alopecia, vasculitis, and neurological disease. Farmer et al.[20▪] showed that there is significant delay in molecular diagnosis (5 years or more) and often clinical decision for HSCT is made before full understanding of underlying cause. Overall, both studies emphasize that optimal bridge therapy is yet to be defined among partial RAG deficient patients [20▪,21▪].
Unique cohorts are now described with ADA2 deficiency and may also present with CVID-like disease [22]. This is the first monogenetic vasculitis syndrome with highly pleiotropic clinical features of autoinflammation, autoimmunity (ITP, arthritis), vasculopathy, vasculitis and strokes [22,23]. Some of the earliest onset PIRDs are those with STAT3 gain-of-function (GOF) or NFκB2 variants. As high as 80% of NFκB2 deficient patients develop autoimmunity (alopecia, arthritis, cytopenia) with unique features of adrenocorticotropic hormone deficiency (44%) [24]. Fabre et al.[25] have published a comprehensive review of 42 children with STAT3 GOF pathogenic variants and very early onset autoimmunity (0.5–5 years of age) including cytopenias, interstitial lung disease, and endocrine complications (diabetes, thyroiditis, and growth failure). Lastly, Chinn et al.[26] have summarized in an excellent comprehensive review of the genetic causes of specific clinical autoimmune presentations among patients with PIDs managed by multiple specialists (hemotologist, endocrinologist, dermatologist).
In summary, PIRD groups with specific gene defects share similar features (IPEX, ALPS, CVID/CID-like) with combinations of multiple autoimmune diseases. Referral to the immunologist and diagnosis of PID is often delayed as the presenting symptoms are often autoimmunity-related with no significant history of infections.
GENETIC AND IMMUNOLOGICAL DIAGNOSTIC TOOLS
A genetic revolution has occurred in recent years regarding PID diagnosis. Panels with 200–300 PID genes are now more affordable and broadly available on clinical grounds. These panels are helpful to identify the most common PIDs linked to autoimmunity and discover ‘new face of old disorders’ (such as RAG deficiency). However, the quest often needs to continue in ‘PID-panel’ negative cases with whole exome and/or genome sequencing approaches as novel genes are being discovered. For example, Fernandez et al.[27▪] recently described a homozygous IL2RB pathogenic variant from two infant siblings, which manifested as multisystem autoimmunity (enteropathy, lymphocytic interstitial pneumonitis, Coombs+) and susceptibility to cytomegalovirus (CMV) infection. There are specific clinical target groups with autoimmunity where genetic testing proved to be fruitful. Among pediatric patients with Evans syndrome, there is a high probability of finding an underlying PID based on recent studies from France [28▪▪,29]. In a prospective cohort of 203 pediatric patients with Evans syndrome, 80 patients underwent genetic testing, which revealed 32 (40%) had an underlying pathogenic mutation. The affected genes included TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS[28▪▪]. One of the largest studies on genetic defects in PID patients has been the ‘Houston project’ by the teams of Jordan Orange and James Lupski (2017). They investigated 278 families from 22 countries by whole-exome sequencing. This project highlighted that 5% of patients had two distinct PID disease genes shaping their clinical and immune phenotype (mixed phenotype). Genes linked to autoimmune diseases spanned several categories (autoimmune disease, CID, CVID, defects of innate immunity) and were identified in more than 23 cases (8%). Genetic causes included COPA, CTLA4, FOXP3, STAT1, STAT3, RAG1, CCDC40, CASP10, PLCG2, FALG, and CBL[30]. Lastly, an eloquent case report of a mother and child by Le Coz et al.[31] highlights CD40L duplication in human autoimmune diseases including autoimmune cytopenias (AICs) and AIT, and discusses the relevance of epigenetic control in disease progression, CD40L overexpression and CD40/CD40L interaction as previously seen in lupus.
In case of variants of uncertain significance, functional studies are needed to confirm the link between the clinical phenotype and genetic findings. Therefore, immunologists and other specialists need to be prepared to initiate evaluation for protein expression and function such as CTLA4 or LRBA expression, STAT1 or STAT3 phosphorylation, B or T-cell repertoire or in vitro recombination activity testing (RAG and DNA deficiencies). Unfortunately, most of these assays are only available on a research basis in collaboration with expert centers. A practical approach to genetic testing for PID has been recently summarized by the Clinical Immunology Society [32,33].
Immune phenotyping is of utmost importance in identifying or confirming the underlying PID among the heterogenous group of patients with autoimmunity. Immunoglobulin levels can identify patients with CVID and CID, and a simple test of the ratio of naïve and memory T cells (CD45RA/CD45RO) may distinguish those with LoCID or P-CID [10,11,34].
There are specific subsets of T and B cells that have been linked to PID with autoimmunity. These include the expansion of TCRαβ CD4−CD8− (double negative) T cells in ALPS, CD19hi21lo B cells in CVID with autoimmunity, abnormal count of regulatory T cells (Treg) in Tregopathies, Th17 cells in STAT1 GOF patients, and expanding follicular helper T cells (Tfh) in CTLA4 and LRBA deficiency [35▪▪]. Changes in these subsets may also predict progression of autoimmune complications or response to therapy.
Patients with PID tend to have broad selection of autoantibodies as seen in RAG deficiency with AICs [20▪]. Further, in some PIDs, particular antibodies with unique self-reactivity occur and may serve as biomarkers. Beyond antibodies to IFNα, IFNω and IL-12 in patients with partial RAG deficiency [14,36] and APECED [37], Rosenberg et al.[38] reported neutralizing anti-IFN α antibodies among patients with IPEX in two large cohorts from Seattle Children's Hospital and San Rafaelle Hospital (Milan, Italy). Anti-IFN ω antibodies were recently described in a patient with NFκB2 deficiency after herpes virus infection [39]. In addition, novel autoantibodies against lung-specific bactericidal/permeability-increasing fold-containing protein B1 (BPIFB1) and the potassium channel regulator protein (KCNFG), have been linked to onset of autoimmune pneumonitis in disorders of central T-cell tolerance (APECED, RAG deficiency, and thymoma) [39]. It is intriguing that presence of anticytokine antibodies are also proposed as markers of prognosis in APECED, as they noted negative correlation of anti-IFN α antibodies and the incidence of T1DM [40,41].
PATHOMECHANISM OF AUTOIMMUNITY IN IMMUNE-DEFICIENT BACKGROUND
Many patients with immunodeficiency harbor autoreactive T and B cells secondary to abnormal pruning of naturally occurring polyreactive clones in the bone marrow (B cells) or thymus (T cells). As these autoreactive cells fail central tolerance checkpoints secondary to intrinsic abnormalities, they spill to the periphery. If peripheral tolerance checkpoints are intact, these clones can become dormant. Additional triggers (extrinsic factors) may revive these clones (Fig. 3). In particular, autoreactive B cells have a second chance to undergo somatic hypermuation (SHM) of their B-cell receptor (BCR) during a process known as clonal redemption and lose self-reactivity [42]. If this process is impaired, autoreactive B cells remain activated and convert into plasma cells to generate autoantibodies. In particular, among patients with CVID and AIC, translocated microbiome or its components (e.g., LPS) have been proposed as triggers for self-reactive immunoglobulin heavy chain variable gene segment 4-34 (IgH-V4-34) expressing B-cell clones capable of binding both commensal bacteria and red blood cells [43▪▪]. Likewise, herpes virus infections as triggers are also well described and linked to onset of autoimmune complications [14,20▪]. In many of these patients, germinal centers are hyperplastic but likely inefficient as B cells display low SHM of BCR suggestive of abnormal clonal redemption [43▪▪]. Tfh cells are being recognized as important cellular players in immune dysregulation [44], as overproduction of Tfh cells has been associated with the generation of autoantibodies and autoimmunity. The phenomenon of Tfh expansion has been highlighted in a mouse model [45] and among patients with CVID with AIC [43▪▪], LRBA [35▪▪], CTLA4 deficiencies [46] and APDS [47]. Thauland et al.[47] have highlighted the importance of a new pathway for expansion of Tfh cells in APDS, which involved intracellular osteopontin and p85α.
FIGURE 3.
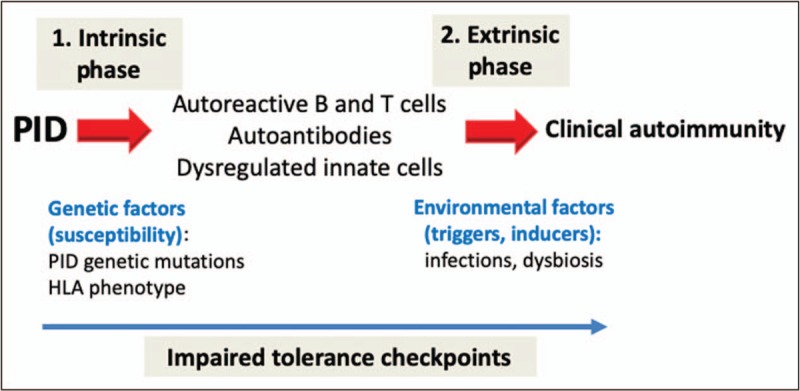
Intrinsic and extrinsic phases of autoimmunity in PID. Autoimmunity in PID arises because of impaired tolerance checkpoints resulting from both intrinsic (genetic susceptibility) and extrinsic (environmental) causes.
AUTOIMMUNITY INCREASES MORTALITY IN PRIMARY IMMUNODEFICIENCY DISORDER: HOW TO OPTIMIZED TREATMENT
Cunningham-Rundles et al.[48] have established high mortality among CVID patients with noninfectious complications. Recently, Farmer et al.[49] at Massachusetts General Hospital studied 142 patients with genetically undefined CVID and concluded that both presence of AIHA and/or ITP increase risk of mortality. Similarly, Fisher et al.[13▪▪] in the French national study discussed above concluded that any types of PID with autoimmunity have increased mortality and complications post-HSCT. Even carriers can be at risk. Schwab et al.[16▪▪] described that both affected CTLA4 patients and their carrier family members (‘unaffected’ or not seeking medical attention) had decreased survival compared with the general population.
It has been described that many PID patients are seen and receive immune modulatory treatment for immune dysregulation before full evaluation or discovery of the underlying immune defect [20▪]. As these treatments result in an altered immune status, it is often unclear to the specialists if immunodeficiency is induced by immune modulation or preexisted before therapy. This enigma can only be resolved if a pathogenic genetic defect causing an aberrant immune response is identified and proven by functional studies (as discussed in the genetic and diagnostic tool section). These variants may result in low (loss of function) or excessive response (gain of function) of a specific immune signaling pathway. The identification of pathogenic genetic defects can facilitate the use of targeted agents specific for the defect or group of defects that share similar pathophysiologic mechanisms.
Immune modulation is a major therapeutic challenge in the PID population and reflects the importance of precision medicine where narrow-spectrum immune modulation delicately balances infectious susceptibility. Most of biologicals are developed and received approval of the Federal Drug Administration for patients with cancer or rheumatologic conditions where safety profile is established (Table 1). In contrast, these biologicals are used off label with unclear safety profile in the PID population. There are excellent recent reviews in the literature discussing targeted therapies for immune dysregulation in PIDs [1,50–54]. Recent cohort studies highlight the use anti-IL-6 receptor and Janus kinase inhibitor biologics in STAT3GOF patients [25], CTLA4 Ig in CTLA4 deficiency [16▪▪], and a small molecule inhibitor, leniolisib, in activated protein I3 kinase delta syndrome (APDS or PI3Kδ) deficiency [55▪]. Monitoring Tfh cells is proposed as a cellular marker to assess control for immune dysregulation [35▪▪]. In patients with deficiency of adenosine deaminase 2 deficiency, TNF α blockade is first-line therapy for vasculopathy and autoinflammation [22,23].
Table 1.
Current biologicals inlcuding those most commonly used in autoimmune complications of PID
| Target | Agent | Structure | Approved indication by FDA | Used in PID patients off label or in clinical trials |
| Cellular lymphocytes | ||||
| CD52 | Alemtuzumab | anti-CD52 mAb | MS | pt with HLH |
| B cells | ||||
| CD20 | Rituximab | chimeric mouse/human anti-CD20 mAb | RA, polyJIA | ITP, AIHA, GLILD (pts with CVID/CID, APDS/PASLI) |
| Ofatumumab | human anti-CD20 mAb | adult refractory CLL | ||
| Obinutuzumab | humanized anti-CD20mAb | adult de novo CLL | ||
| CD22 | Epratuzumab | humanized anti-CD22mAb | B cell malignancies | |
| BAFF | Belimumab | human anti-BAFF mAb | SLE | |
| CD38 | Daratumumab | human anti-CD38 mAb | MM | AIHA (pts with WAS s/p HSCT) |
| Proteosome inhibitor | Bortezomib | pyrazine and boronic acid derivative | MM, MCL | |
| Carfilzomib | epoxomicinderivate | MM | ||
| Ixazomib | second generation boron containing peptide | MM, MCL | ||
| T cells | ||||
| m-TOR | Sirolimus (rapamycin) | S6K/m-TOR inhibitor | LAM, T/OR | ITP, AIHA (pts with CTLA4/LRBA def, ALPS, APDS) |
| m-TOR | Everolimus | S6K/m-TOR inhibitor | BrCA, TS, T/OR | CTLA4/LRBA def |
| IMD | Myocphenolic acid | reversible inhibitor of IMD | kidney, heart, liver transplant | ITP, AIHA (pts with CTLA4/LRBA def, ALPS, APDS) |
| CD28 | Abatacept | human CTLA4-IgG fusion protein with extracellular domain of CTLA4 and IgG1 Fc | RA, polyJIA | pts with CTLA4, LRBA def |
| p110δ | Leniolisib (CDZ173) | small-molecule inhibitor of p110δ | n.a. | pts with APDS/PASLI |
| Complement | ||||
| Complement C5 | Eculizumab | recombinant humanized IgG2 anti-C5 mAb | generalized MG, PNH | |
| Cytokines and receptors | ||||
| TNFα | Etanercept | soluable TNFα receptor IgG Fc fusion protein | RA | AInD (pts with TRAPS) |
| Infliximab | human mouse chimeric anti-TNFα mAb | RA, UC | CVID with GLILD | |
| Adalimumab | Fully human anti-TNFα mAb | RA, UC | ||
| Golimumab | Fully human anti-TNFα mAb | RA, UC | ||
| Certolizumab pegol | Humanized pegylated Fab’ fragment | RA, CD | ||
| IL-1β pathway | Anakinra | Recombinant IL-1R antagonist | RA, CAPS | CGD and AInD (pts with CAPS, FMF, TRAPS, HIDS, DIRA) |
| Rilonacept | fusion of IL-1R and IL-1R accessory protein | CAPS | AInD (CAPS such as FCAS, MWS, less effective in NOMID) | |
| Canakinumab | mAb to IL-1β | JIA, CAPS | ||
| IL-6R | Tocilizumab | Humanized IL-6R antagonist | RA, JIA | pt with STAT3 GOF |
| IL-12/IL-23 | Ustekinumab | Fully human anti-IL-12/IL-23 mAb (anti-p40) | PsA | CGD, LAD-1 |
| IL-17 | Secukinumab | Fully human anti-IL-17 mAb | PsO | |
| IFNγ | Emapalumab (NI-0501) | Fully human anti-IFNγ mAb | n.a. | AInD with HLH and NLRC4 mutation |
| JAK1/2 | Ruxolitinib | small molecule JAK (1-2) inhibitor | RA, PV | STAT1 and STAT3 GOF |
| JAK1/2 | Baricitinib | small molecule JAK (1-2) inhibitor | RA | STAT1 GOF and CANDLE syndrome |
| JAK1/3 | Tofacitinib | small molecule JAK (1-3) inhibitor | RA, PsA, UC | STAT3-GOF STAT1-GOF CANDLE syndrome |
| JAK3 | Decernotinib | small molecule JAK3 inhibitor | n.a. | RA |
| PD-1 | Nivolumab | human anti-PD-1 mAb | M | |
| PD-1 | Pembrolizumab | human anti-PD-1 mAb | UC, M, CA | |
| CXCR4 | Plerixafor | small molecule inhibitor (bicylam) | T for MM or NHL | WHIM syndrome |
| CXCR4 | Mavorixafor (X4P-001) | n.a. | n.a. | WHIM syndrome |
AInD, autoinflammatory disorder; BAFF, B cell acivating factor; BrCA, breast cancer; CA, cancer; CD, Crohns disease, CGD, chronic granulomatous disease; CTLA, cytotoxic T lymphocyte antigen; FDA, Food and Drug Administration; GLILD, granulomatous lymphocytic interstitial lung disease; IMD, inosine monophosphate dehydrogenase; JAK, janus kinase; JIA, juvenile idiopathic arthritis; IL-6R, interleukin 6 receptor; LAD-1, leukocyte adhesion defect 1; LAM, lymphangioleiomyomatosis; LRBA, lipopolysaccharide-responsive and beige-like anchor protein deficiency; m-TOR, mechanistic target of rapamycin; MCL, mantle cell lymphoma; MG, myasthenia gravis; MM, myeloma multiplex; M, melanoma; MS, mulitple sclerosis; PNH, paroxysmal nocturnal hemoglobinuria; PsA, psoriatic arthritis; Pso, psoriasis; S6K, Serine/threonine protein kinase; Pts, patients; PV, polycythemia vera; T/OR, transplant and organ rejection; TS, tuberous sclerosis; UC, ulcerative colitis.
In the era of genetic revolution, genetically defined PID patients with immune dysregulation are increasingly considered for definitive therapy with the ultimate goal to correct the underlying defect, primarily by HSCT. Hematopoietic stem cells (HSCs) may originate from a healthy donor (allogeneic) or the patient itself when genetic defect is repaired by viral transduction of the correct gene (autologous gene therapy). Furthermore, genetic defect in HSC may also be corrected via guide RNA that interacts with the cell's own DNA repair machinery [Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR associated protein 9 (CRISPR-Cas9) gene editing]. Conditioning regimens need to consider the inflammatory state of the patient as the risk of graft failure is high in these circumstances. Targeted therapy for immune dysfunction serves as a bridge until the definitive treatment with HSCT can be pursued safely. Indication to pursue HSCT in a child with autoimmune complications and underlying PID has not been fully delineated. Only a few large cohort studies and case series on HSCT for autoimmune complications have been published among patients with partial RAG and ADA2 deficiencies [20▪,22,23,56,57]. A recent report of allogeneic HSCT for chronic arthritis in children is intriguing. In a retrospective study by Silva et al.[58], allogeneic HSCT was performed in 11 patients with systemic and five with polyarticular rheumatoid factor negative juvenile idiopathic arthritis after patients failed multiple medications considered to be the standard of treatment or failed autologous HSCT, or developed life-threatening complications such as macrophage activation syndrome. Eleven of 14 surviving patients were reported to be in drug-free remission at the last follow-up. Common complications were severe infections (viral reactivation) and even autoimmunity during and after the transplant. Notably, in these reports, there was no search for underlying PID on immunological or genetic grounds. The authors argued that identifying the predictive markers of medical treatment failure in these patients would allow for early selection of candidates for HSCT, fewer HSCT-related complications and less disability.
Preventing development of autoimmunity in PID is a far-reaching goal, as it is unclear, which PID patients will progress into clinical autoimmune disease even among those with a genetic defect linked to immune dysregulation. Monitoring prognostic biomarkers of development to autoimmunity could improve early interventions and slow or reverse disease progression. These may be cellular (e.g., Tfh/Treg ratio, autoimmune prone B-cell subsets) or serum markers (e.g., antibodies targeting self-antigens including cytokines).
CONCLUSION
The face of PID is rapidly changing with increased awareness, availability of genetic testing, and in-depth immune phenotyping. A multidisciplinary approach is needed to expedite prompt diagnosis. Once multiple autoimmune complications are present, care of these patients is likely shared by multiple specialists (e.g., in hematology, rheumatology, neurology). At that point, reevaluation of the patient's immune system is of high importance. The primary pediatrician needs to advocate for prompt immune evaluation and genetic testing in this multidisciplinary setting to optimize the diagnostic and treatment approach for these vulnerable patients.
Treatment of PID patients with autoimmunity is most successful when fine-tuned immune modulation is achieved using biologicals specifically targeting the imbalanced immune pathway. Definitive therapy with HSCT or gene therapy should be considered for treatment of refractory cases.
Acknowledgements
We thank Dr. Joseph Dasso for summarizing key references, contributing to creation of the figures and editing this manuscript and Dr. Maria Chitty-Lopez for editing Table 1.
Financial support and sponsorship
J.E.W. is the Robert A. Good Chair in Immunology at the University of South Florida. She has received funding from NIH-NIAID (subcontract R01 AI100887-05). This work was also supported by the USF Foundation and Jeffrey Modell Foundation.
Conflicts of interest
J.E.W. has been an advisory board member, speaker bureau participant and site-principal investigator for a trial funded by Takeda (former Shire) outside of the submitted work.
J.E.W. has received grants as principal investigator on investigator-initiated translational studies and sponsored clinical trials of patients with WHIM syndrome funded by X4 Pharmaceuticals.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Walter JE, Farmer JR, Foldvari Z, et al. Mechanism-based strategies for the management of autoimmunity and immune dysregulation in primary immunodeficiencies. J Allergy Clin Immunol Pract 2016; 4:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECED gene. Nat Genet 1997; 17:393–398. [DOI] [PubMed] [Google Scholar]
- 3.Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 1997; 17:399–403. [DOI] [PubMed] [Google Scholar]
- 4.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20–21. [DOI] [PubMed] [Google Scholar]
- 5.Chatila TA, Blaeser F, Ho N, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 2000; 106:R75–81. PMC387260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baud O, Goulet O, Canioni D, et al. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med 2001; 344:1758–1762. [DOI] [PubMed] [Google Scholar]
- 7.Barzaghi F, Amaya Hernandez LC, Neven B, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol 2018; 141:1036–1049. e1035. PMC6050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bride K, Teachey D. Autoimmune lymphoproliferative syndrome: more than a fascinating disease. F1000Res 2017; 6:1928.PMC5668920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cepika AM, Sato Y, Liu JM, et al. Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 2018; 142:1679–1695. [DOI] [PubMed] [Google Scholar]
- 10.Bertinchamp R, Gerard L, Boutboul D, et al. Exclusion of patients with a severe T-cell defect improves the definition of common variable immunodeficiency. J Allergy Clin Immunol Pract 2016; 4:1147–1157. [DOI] [PubMed] [Google Scholar]
- 11.Speckmann C, Doerken S, Aiuti A, et al. A prospective study on the natural history of patients with profound combined immunodeficiency: an interim analysis. J Allergy Clin Immunol 2017; 139:1302–1310. e1304. PMC6311415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12▪.Chandrakasan S, Chandra S, Davila Saldana BJ, et al. Primary immune regulatory disorders for the pediatric hematologist and oncologist: a case-based review. Pediatr Blood Cancer 2019; 66:e27619. [DOI] [PubMed] [Google Scholar]; This case-based review focuses on autoimmune, lymphoproliferative, and inflammatory characteristics of monogenic immune disorders. It reviews such cases to give practical guidance as to their clinical features, immune workup, and treatment.
- 13▪▪.Fischer A, Provot J, Jais JP, et al. members of the CFPIDsg. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol 2017; 140:1388–1393. e1388. [DOI] [PubMed] [Google Scholar]; This is a large French national study on autoimmune and inflammatory manifestations, and clinical outcome in various groups of patients with primary immunodeficiencies. They conclude that 26% of patients had at least one autoimmune or inflammatory manifestation that negatively impacts treatment outcome.
- 14.Goda V, Malik A, Kalmar T, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract 2018; 6:1769–1771. e1762. PMC6072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu KY, Purswani P, Ujhazi B, et al. Arthritis in two patients with partial recombination activating gene deficiency. Front Pediatr 2019; 7:235.PMC6625222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16▪▪.Schwab C, Gabrysch A, Olbrich P, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 2018; 142:1932–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a large study of 133 patients with CTLA-4 insufficiency. Clinical features are highly variable with high prevalence of hypogammglobulinemia, lymphoproliferation, autoimmune cytopenia as wells as respriatory, gastrointestinal and neurological autoimmune complications. The incidence of malignancy is also elevated. Disease features typically occur in late childhood but can also present late in life.
- 17▪.Habibi S, Zaki-Dizaji M, Rafiemanesh H, et al. Clinical, immunologic, and molecular spectrum of patients with LPS-responsive beige-like anchor protein deficiency: a systematic review. J Allergy Clin Immunol Pract 2019; 7:2379–2386.e5. [DOI] [PubMed] [Google Scholar]; This large study of 109 patients examined clinical phenotype of LRBA deficiency. The major presentations are autoimmunity, chronic diarrhea, hypogammaglobulinemia, and recurrent infections. Although it is a monogenic immune dysregulation syndrome, in this cohort, the genotype-phenotype correlation is poor. HSCT is discussed.
- 18▪.Jamee M, Moniri S, Zaki-Dizaji M, et al. Clinical, immunological, and genetic features in patients with activated PI3Kdelta syndrome (APDS): a systematic review. Clin Rev Allergy Immunol 2019; doi: 10.1007/s12016-019-08738-9. [DOI] [PubMed] [Google Scholar]; This large study of 243 patients with APDS highlights the most common disease manifestations including recurrent respiratory infections, lymphoproliferation, autoimmunity, and enteropathy. The main immune phenotype is hyper-IgM syndrome, and B and CD4+ T-cell lymphopenia. HSCT is discussed as a promising treatment for severe or recalcitrant cases.
- 19▪▪.Tuijnenburg P, Lango Allen H, Burns SO, et al. Loss-of-function nuclear factor kappaB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 2018; 142:1285–1296. PMC6148345. [DOI] [PMC free article] [PubMed] [Google Scholar]; This large study of a European CVID cohort identified NFKB1 deficiency by whole-exome sequencing. The study claims that NFKB1 is the most common monogenic cause of CVID. Noninfectious complications were frequent including lymphadenopathy splenomegaly and autoimmunity, and correlated with worse prognosis. B cells were deficient even in carriers.
- 20▪.Farmer JR, Foldvari Z, Ujhazi B, et al. Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allergy Clin Immunol Pract 2019; 7:1970–1985.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]; This large international highly annotated cohort with RAG deficiency focuses on 63 patients with at least one autoimmune or hyperinflammatory complication. Autoimmunity frequently occurred after infections. Challenges with molecular diagnosis, treatment outcome, and indication for HSCT are discussed. AIC was the main autoimmune complication and often failed treatment, especially multileage AIC; thus, patients with RAG deficiency and AIC should be evalutated for HSCT.
- 21▪.Delmonte OM, Schuetz C, Notarangelo LD. RAG deficiency: two genes, many diseases. J Clin Immunol 2018; 38:646–655. PMC6643099. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study summarizes 429 cases of RAG deficiency from the literature. This includes 134 cases with autoimmune manifestations. The study highlights that RAG deficiency presents with diverse clinical features and immune phenotypes. Diagnosis and treatment can be challenging, especially for variants with autoimmune and/or inflammatory complications. HSCT is the preferred treatment for very severe cases, however, graft failure is common.
- 22.Hashem H, Kumar AR, Muller I, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood 2017; 130:2682–2688. PMC5731089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol 2018; 38:569–578. PMC6061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klemann C, Camacho-Ordonez N, Yang L, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol 2019; 10:297.PMC6435015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fabre A, Marchal S, Barlogis V, et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 2019; 7:1958.e9–1969.e9. [DOI] [PubMed] [Google Scholar]
- 26.Chinn IK, Orange JS. Immunodeficiency Disorders Pediatr Rev 2019; 40:229–242. [DOI] [PubMed] [Google Scholar]
- 27▪.Fernandez IZ, Baxter RM, Garcia-Perez JE, et al. A novel human IL2RB mutation results in T and NK cell-driven immune dysregulation. J Exp Med 2019; 216:1255–1267. PMC6547857. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports a novel homozygous mutation in the human interleukin-2 receptor chain (IL2RB) in two infant siblings, which manifested in autoimmune, inflammatory and infectious (CMV) complications. The defect impaired IL-2RB expression and signaling, and resulted in decreased regulatory T cells. Unlike IL2RB knockout mice, which lack NK cells, the affected infants had increased NK cells but the cells did not fully differentiate.
- 28▪▪.Hadjadj J, Aladjidi N, Fernandes H, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood 2019; 134:9–21. [DOI] [PubMed] [Google Scholar]; This is a very informative study on the importance of genetic testing in pediatric Evans syndrome (pES). Over half of the patients (65%) had mutations in genes linked to primary immunodeficiencies. The authors concluded that genetic testing should be done in pES as the results could direct treatment decisions.
- 29.Besnard C, Levy E, Aladjidi N, et al. Pediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol 2018; 188:52–57. [DOI] [PubMed] [Google Scholar]
- 30.Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol 2017; 139:232–245. PMC5222743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Coz C, Trofa M, Syrett CM, et al. CD40LG duplication-associated autoimmune disease is silenced by nonrandom X-chromosome inactivation. J Allergy Clin Immunol 2018; 141:2308–2311. e2307. PMC5994181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heimall JR, Hagin D, Hajjar J, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol 2018; 38:320–329. [DOI] [PubMed] [Google Scholar]
- 33.Heimall J. Now is the time to use molecular gene testing for the diagnosis of primary immune deficiencies. J Allergy Clin Immunol Pract 2019; 7:833–838. [DOI] [PubMed] [Google Scholar]
- 34.von Spee-Mayer C, Koemm V, Wehr C, et al. Evaluating laboratory criteria for combined immunodeficiency in adult patients diagnosed with common variable immunodeficiency. Clin Immunol 2019; 203:59–62. [DOI] [PubMed] [Google Scholar]
- 35▪▪.Alroqi FJ, Charbonnier LM, Baris S, et al. Exaggerated follicular helper T-cell responses in patients with LRBA deficiency caused by failure of CTLA4-mediated regulation. J Allergy Clin Immunol 2018; 141:1050–1059. e1010. PMC5743769. [DOI] [PMC free article] [PubMed] [Google Scholar]; This mechanistic study highlights that CTLA4-Ig is an effective treatment for LRBA and CTLA4 deficiencies by decreasing numbers of circulating T-follicular helper (cTfh) cells and autoantibodies. Monitoring cTfh cell counts is advantageous for determining response to CTLA-4-Ig treatment in LRBA deficient patients.
- 36.Walter JE, Rosen LB, Csomos K, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest 2015; 125:4135–4148. PMC4639965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burbelo PD, Browne SK, Sampaio EP, et al. Anticytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood 2010; 116:4848–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg JM, Maccari ME, Barzaghi F, et al. Neutralizing anti-cytokine autoantibodies against interferon-alpha in immunodysregulation polyendocrinopathy enteropathy X-linked. Front Immunol 2018; 9:544.PMC5885158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parsons K, Cipriano SD, Rosen LB, et al. Severe facial herpes vegetans and viremia in NFKB2-deficient common variable immunodeficiency. Front Pediatr 2019; 7:61.PMC6433840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer S, Woodward M, Hertel C, et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 2016; 166:582–595. PMC4967814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiler FG, Peterson P, Costa-Carvalho BT, et al. The heterogeneity of autoimmune polyendocrine syndrome type 1: clinical features, new mutations and cytokine autoantibodies in a Brazilian cohort from tertiary care centers. Clin Immunol 2018; 197:231–238. [DOI] [PubMed] [Google Scholar]
- 42.Reed JH, Jackson J, Christ D, Goodnow CC. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J Exp Med 2016; 213:1255–1265. PMC4925023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43▪▪.Romberg N, Le Coz C, Glauzy S, et al. Patients with common variable immunodeficiency with autoimmune cytopenias exhibit hyperplastic yet inefficient germinal center responses. J Allergy Clin Immunol 2019; 143:258–265. PMC6400323. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article illuminates a possible disease mechanism of autoimmunity in CVID patients with autoimmune cytopenias. These patients have abnormal germinal center maturation involving imbalanced follicular T-helper and T-regulatory cell numbers, endotoxemia because of impaired mucosal barrier function and decreased rate of somatic hypermutation which augments autoantibodies to erythrocytes.
- 44.Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014; 41:1026–1039. PMC4309019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Preite S, Huang B, Cannons JL, et al. PI3K orchestrates T follicular helper cell differentiation in a context dependent manner: implications for autoimmunity. Front Immunol 2018; 9:3079.PMC6330320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chao G, Li X, Ji Y, et al. CTLA-4 regulates T follicular regulatory cell differentiation and participates in intestinal damage caused by spontaneous autoimmunity. Biochem Biophys Res Commun 2018; 505:865–871. [DOI] [PubMed] [Google Scholar]
- 47.Thauland TJ, Pellerin L, Ohgami RS, et al. Case study: mechanism for increased follicular helper T cell development in activated PI3K delta syndrome. Front Immunol 2019; 10:753.PMC6473200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119:1650–1657. 3286343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farmer JR, Ong MS, Barmettler S, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol 2017; 8:1740.PMC5767273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Her M, Kavanaugh A. Alterations in immune function with biologic therapies for autoimmune disease. J Allergy Clin Immunol 2016; 137:19–27. [DOI] [PubMed] [Google Scholar]
- 51.Notarangelo LD, Fleisher TA. Targeted strategies directed at the molecular defect: toward precision medicine for select primary immunodeficiency disorders. J Allergy Clin Immunol 2017; 139:715–723. PMC5369241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marciano BE, Holland SM. Primary immunodeficiency diseases: current and emerging therapeutics. Front Immunol 2017; 8:937.PMC5552668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leiding JW, Ballow M. Precision medicine in the treatment of primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol 2018; 18:159–166. [DOI] [PubMed] [Google Scholar]
- 54.Amaya-Uribe L, Rojas M, Azizi G, et al. Primary immunodeficiency and autoimmunity: a comprehensive review. J Autoimmun 2019; 99:52–72. [DOI] [PubMed] [Google Scholar]
- 55▪.Rao VK, Webster S, Dalm V, et al. Effective ‘activated PI3Kdelta syndrome’-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood 2017; 130:2307–2316. PMC5701526. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights that leniolisib trial improved immune dysregulation and decreased lymphoproliferation without significant adverse effects in six patients with activated PI3Kδ syndrome. The study demonstrates the effectiveness of therapy that targets the biochemical cause of a rare PID.
- 56.Westermann-Clark E, Grossi A, Fioredda F, et al. RAG deficiency with ALPS features successfully treated with TCRalphabeta/CD19 cell depleted haploidentical stem cell transplant. Clin Immunol 2018; 187:102–103. PMC5941932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barzaghi F, Minniti F, Mauro M, et al. ALPS-like phenotype caused by ADA2 deficiency rescued by allogeneic hematopoietic stem cell transplantation. Front Immunol 2018; 9:2767.PMC6339927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MF Silva S, Ladomenou F, Carpenter B, et al. Allogeneic hematopoietic stem cell transplantation for severe, refractory juvenile idiopathic arthritis. Blood Adv 2018; 2:777–786. PMC5894259. [DOI] [PMC free article] [PubMed] [Google Scholar]