

Abstract
The adenine nucleotide translocase (ANT) of the mitochondrial inner membrane exchanges ADP for ATP. Mitochondria were isolated from human vastus lateralis muscle (n=9). Carboxyatractyloside titration of O2 consumption rate (Jo) at clamped [ADP] of 21 μM gave ANT abundance of 0.97 ± 0.14 nmol ANT/mg and a flux control coefficient of 82% ± 6%. Flux control fell to 1% ± 1% at saturating (2 mM) [ADP]. The KmADP for Jo was 32.4 ± 1.8 μM. In terms of the free (−3) ADP anion this KmADP was 12.0 ± 0.7 μM. A novel luciferase-based assay for ATP production gave KmADP of 13.1 ± 1.9 in the absence of ATP competition. The free anion KmADP in this case was 2.0 ± 0.3 μM. Targeted proteomic analyses showed significant acetylation of ANT Lysine23 and that ANT1 was the most abundant isoform. Acetylation of Lysine23 correlated positively with KmADP, r = 0.74, P = 0.022. The findings underscore the central role played by ANT in the control of oxidative phosphorylation, particularly at the energy phosphate levels associated with low ATP demand. As predicted by molecular dynamic modeling, ANT Lysine23 acetylation decreased the apparent affinity of ADP for ANT binding.
Keywords: Flux control, energy metabolism, bioenergetics, acetylation
Introduction
The expansive aerobic scope of mammalian skeletal muscle operates across a narrow range of cytosolic free ADP concentration ([ADP]f) and free energy of ATP (ΔGATP) [1–6]. The matching of mitochondrial ATP synthesis to the rate of breakdown depends on a combination of feedback and feedforward (or parallel) mechanisms [4, 7]. Feedback control is important in skeletal muscle [8, 9] and depends on signals related to the products of ATP breakdown, ATP → ADP + Pi. These energy phosphates are, in turn, linked to the cellular creatine pool by the equilibrium maintained by the creatine kinase (CK) reaction, ATP + Cr ⇆ ADP + PCr [10, 11] (the protons involved in these two reactions, which tend to cancel each other, are omitted here for simplicity). Two basic feedback models that describe the control of oxidative flux are based on the signals [ADP]f (also inorganic phosphate) and ΔGATP [12]. The ADP-based model describes the hyperbolic or sigmoidal relationship of oxidative flux against cytosolic [ADP]f:
| (1) |
where v is the rate of O2 consumption (Jo) or ATP production (Jp), Vmax is the maximum rate, KmADP is the [ADP]f at ½Vmax, and nH is the Hill coefficient. Obviously, when the Hill coefficient equals 1.0, Equation 1 is the Michaelis-Menten (MM) equation. Here ADP kinetics will be called “simple MM” when nH equals 1.0 or nearly so. Sigmoidal character in the relationship emerges as nH exceeds unity and will be referred to as “higher order” control [9].
ATP free energy based control models describe the near-linear relationship between oxidative flux and ΔGATP according to non-equilibrium thermodynamics (NET) [4, 7, 13–15]:
| (2) |
in which oxidative flux, ν, rises linearly according to the product of the fall (“relaxation”) in ATP free energy and a phenomenological proportionality constant, the “conductance” of the oxidative pathway, which also includes the stoichiometry of the transduction [7]. Due to the near equilibrium maintained at CK, at constant pH these two feedback models are fundamentally related to each other [12].
Non-invasive measurement of intact skeletal muscle energy phosphates using 31P-MRS yields estimates of apparent KmADP in the range of 20–40 μM. Earlier studies suggested simple MM ADP kinetics [8], but more recently higher order control by ADP, with Hill coefficients (nH) approaching or exceeding 2.0, has been consistently observed in human [16, 17] and rodent [18] skeletal muscle. Across most of the dynamic control region, these same data sets also conform well to the NET model [4, 9, 19].
In vitro studies of isolated skeletal muscle mitochondria report KmADP values similar to that of 31P-MRS, 20–40 μM [7, 20–22]. However, unlike intact muscle, in this case simple MM kinetics provide an adequate fit to the data. When ATP is also included in the polarography incubation medium and ΔGATP is determined, a near linear flow-force relationship is observed [7, 15, 23, 24].
In marked contrast, in vitro experiments using permeabilized muscle fibers often report apparent KmADP values well into the triple digit micromolar range [25]; KmADP values as high as 1500–2000 μM have been published [26].
The adenine nucleotide translocase (ANT) carries out a 1:1 exchange of ADP for ATP across the mitochondrial inner membrane [27, 28]. Importantly, ATP4− and ADP3− are the only ionic forms that bind and undergo translocation [29]. ANT KdADP3− values from roughly 2 μM to 10 μM have been measured in both intact mitochondria and also when ANT is incorporated into liposomes possessing a phospholipid composition similar to the mitochondrial inner membrane [30–32]. This range of KdADP3− is roughly equivalent to the KmADP range observed in vivo and in isolated mitochondria when pH and Mg binding are taken into account [33], which suggests that ANT is a central locus of oxidative flux control. ANT flux control strength has not been previously evaluated in human skeletal muscle mitochondria at a constant submaximal [ADP]f and this determination was a major objective of the present study. ANT flux control and turnover were also assessed under saturating [ADP] (2 mM) conditions, when the pathway approaches its maximum rate.
Recently, Mielke et al. [34] used molecular dynamic modeling to show ADP binding to an ANT1 crystal structure with a KdADP3− of 3.7 μM, and further indicated that ADP3− binding strongly depended upon the positive charge of Lysine23 of ANT1, located near the bottom of the adenylate binding pocket in the c-form. Acetylation of this lysine, which removes the positive charge involved in the electrostatic bonding with ADP3−, increased the KdADP3− by roughly 20-fold [34]. ANT lysine acetylation and its functional consequences on mitochondrial ADP sensitivity may have particular importance in altered nutritional states and in metabolic diseases. The overall acetylation of mitochondrial proteins correlates with muscle insulin resistance, and the extent of deacetylation of Lysine23 of ANT1 after acute exercise is also related to insulin sensitivity [34].
In the current study, lean healthy (n=9) volunteers underwent percutaneous biopsies of the vastus lateralis muscle to accomplish two objectives. The first was to use mitochondria isolated from the skeletal muscle to determine ANT abundance, turnover, flux control strength, and ADP kinetics for both O2 consumption and ATP synthesis. The second objective was to analyze the relationship between ADP kinetics and the acetylation of ANT Lysine23.
Materials and Methods
Ethical Approval
Human Subjects. A total of 9 normoglycemic volunteers took part in this study, which was approved by the Institutional Review Boards of Arizona State University and the Mayo Clinic. All studies were conducted at the Clinical Studies Infusion Unit at the Mayo Clinic in Arizona. Informed, written consent was obtained from all participants.
In Vivo Methods
All of the participants were sedentary, and they did not report a change in body weight for at least 6 months before participation. Participants were instructed to not exercise for 48 hours before study and to maintain their usual diet. A medical history, physical examination, 12-lead electrocardiogram, and chemistry panel were obtained, and a 75-g oral glucose tolerance test was performed. No one was taking any medication known to affect glucose metabolism.
After completing screening studies, volunteers returned to the Clinical Studies Infusion Unit at about 7 AM after an overnight fast. Euglycemic clamps and muscle biopsies were performed as described [35]. A stable isotope ([6,6-2H] glucose) was used to trace glucose metabolism, and steady state equations were used to calculate rates of glucose appearance and disposal.
After a 10-h overnight fast, a percutaneous biopsy of the vastus lateralis muscle was obtained with a Bergstrom cannula under local anesthesia one hour before the start of insulin infusion (80 mU m−2 min−1). Biopsied muscle, about 100–150 mg, was promptly cleaned of visible fat and connective tissue and placed into a pre-weighed 10 ml beaker containing 2.0 ml of ice-cold modified Chappell-Perry buffer (Solution I; mM): 100 KCl, 40 Tris-HCl, 10 Tris-Base, 5 MgCl2, 1 EDTA, 1 ATP, pH 7.5. The wet mass of biopsied muscle was obtained by reweighing and transferred into a clean 5 ml tube containing 9.0 volumes of Solution I (10% w/v muscle tissue suspension). Mitochondrial isolation was then carried out as described below. A separate approximately 50 mg portion of the muscle biopsy was promptly frozen and stored in liquid nitrogen for mass spectrometry analysis at a later date.
Isolation of Mitochondria
The following description of the mitochondrial isolation procedure is based on a hypothetical wet muscle mass of 100 mg, which would be suspended in 900 μl Solution I. Every step of the isolation was carried out on ice or at 273 – 277 K and all centrifugations were 10 min in duration (Eppendorf Model 5415R). The muscle tissue 10% suspension was minced with scissors. Nagarse (bacterial proteinase Type XXIV, Sigma, P-8038) was then added (0.5 mg per 100 mg muscle) and the mince was incubated 7.0 min. Digestion was terminated by adding an equal volume of Solution I (1.0 ml), and the 2.0 ml of diluted digest was gently homogenized by hand with 5 passes in a ground glass-to-glass Potter-Elvehjem homogenizer. The homogenate was transferred into two 1.5 ml tubes and centrifuged at 1000 × g. Supernatants were transferred to clean tubes and centrifuged at 14,000 × g to obtain mitochondrial pellets. The supernatants were then discarded and each mitochondrial pellet resuspended in 500 μl Solution II (mM): 100 KCl, 40 Tris-HCl, 10 Tris-Base, 5 MgCl2, 1 EDTA, 0.2 ATP, and 1.5% BSA, pH 7.5. Following centrifugation at 7000 × g, the supernatants were discarded and the mitochondrial pellets were resuspended and combined in a total of 500 μl of Solution III (identical to Solution II, but without BSA). After the final centrifugation at 4000 × g and removal of supernatant, the final mitochondrial pellet was resuspended in mannitol sucrose (M-S) buffer (mM) 220 Mannitol, 70 Sucrose, 10 Tris-HCl, 1 EGTA, pH 7.40, 1 μl M-S per mg of original wet muscle (thus, 100 μl M-S in this example). The protein concentration of the final mitochondrial suspension was determined with the Lowry assay [36].
Polarography
Mitochondrial O2 consumption (Jo) was measured (Oxygraph, Hansatech Instruments, UK) at 310 K as described previously [22]. On average about 25 μg of mitochondrial protein were incubated in 500 μl respiration buffer (referred to as “buffer” below) containing (in mM) 100 KCl, 50 MOPS, 20 glucose, 10 K·P04, 5 MgCl2, 1 EGTA, and 0.2% w/v BSA, pH 7.0. Oxidative fuel was provided to mitochondria as succinate + glutamate (S+G), each at a final concentration of 10 mM.
Conventional indices of the structural and functional integrity of mitochondria were assessed [37]. After the addition of mitochondria and S+G to 500 μl respiration medium, Jo was followed continuously (see Figure 1). After S+G addition stimulated state 2 Jo (proton leak Jo), State 3 (maximal) Jo was elicited with a bolus addition of 250 nmol ADP (500 μM final). Mitochondria returned to resting (state 4) Jo following ADP phosphorylation. The total O2 consumed during State 3 was determined to calculate the phosphorylation ratio (ADP/O) and the respiratory control ratio (RCR) was calculated (RCR = State 3Jo/State 4Jo).
Figure 1.

Representative experimental record from the O2 electrode during an assay for conventional indices of mitochondrial function, ADP kinetics using a CK energetic clamp, and flux control and abundance determinations of the adenine nucleotide translocase (ANT) by titrating out Jo with additions of carboxyatractyloside (CAT).
Kinetics of Respiratory Control
In the same experimental run, the kinetics of ADP control of respiration were assessed by incubating the mitochondria at fixed [ADP]f established with a progressive creatine kinase (CK) energetic clamp [7]. At the conclusion of the conventional indices assessment, the cap of the respiration chamber was removed and the medium allowed to re-oxygenate (see Figure 1).
While the cap was off the CK clamp was primed by adding to the medium 4.5 mM ATP (combined with the previous ADP bolus yields a final total adenylate concentration of 5.0 mM), 2.5 mM phosphocreatine (PCr), and 5.0 mM creatine (Cr). The cap was then repositioned to seal the chamber and Jo was recorded for several minutes to ensure steady state conditions. Following the measurement of steady state Jo, excess CK (40 units) was added to bring the buffer energy phosphate pool into equilibrium. At the free magnesium under these conditions, calculated to be 1.1 mM [33], and pH = 7.0, the equilibrium constant of the CK reaction, Kck, has a value of 177 [38]:
| (3) |
Rearranging gives the free ADP concentration after CK addition:
| (4) |
The Gibbs free energy of ATP hydrolysis (ΔGATP) was calculated as:
| (5) |
where R is the gas constant (8.314 J K−1 mol−1), T is 310 K, and gamma (Γ) is the mass action ratio of the ATP hydrolysis reaction:
| (6) |
Under these conditions the equilibrium constant, KATP, of the ATP hydrolysis reaction is 219,000 [38]. After measuring the steady state Jo in the presence of CK, PCr was added stepwise at ~3 minute intervals, giving the following [ADP]f (μM): 107, 75, 55, 35, and 21 (Figure 2). Steady state Jo was measured at each of these ADP levels. The corresponding ATP free energies (ΔGATP in kJ/mol) were: −53.54, −54.66, −55.71, −56.75, and −57.70, respectively.
Figure 2.

The CK clamp and CAT titration region of Figure 1 is shown in greater detail.
The [ADP]f:Jo data were analyzed according to Equation 1 using non-linear regression (Solver, Excel, Microsoft Corp, USA) to determine the parameter values KmADP and nH (Vmax was a known parameter in these analyses, obtained from the measurement of state 3 Jo). Kinetic parameters in terms of the ADP3− ion were determined by taking into account free [Mg2+] [33].
The same data were also analyzed using the Eadie-Hofstee transform of the MM equation, by plotting Jo against Jo/[ADP]f. Finally, Jo was also plotted against ΔGATP to obtain the NET flow-force relationship [7].
Flux Control Analysis and Determination of ANT Abundance
As illustrated in Figure 2, the CK clamp was followed by stepwise additions of CAT, a high affinity inhibitor (Kd ~ 1 nM) of ANT [39]. CAT titration of Jo reveals the extent to which ANT controls the entire pathway of mitochondrial O2 consumption. The endpoint of the titration reflects the abundance of the protein. The flux control coefficient of an enzyme, CJ, is defined as the fractional change in flux (ΔJ/J) that results from a fractional change in functional enzyme abundance (ΔE/E) [40, 41]:
| (7) |
The first two CAT additions were delivered in amounts predicted to each match approximately 5% of the ANT dimers present in the respiring mitochondria. CAT titrations were performed under two conditions: 1) As shown in Figure 2, in which the extramitochondrial [ADP]f was fixed by the CK clamp at 21 μM, and 2) also in the presence of saturating, 2.0 mM, ADP (not shown). The kinetic response to CAT addition was analyzed using the nonlinear model of Gellerich et al. [39], which evaluates both the flux control located at ANT (CJANT, which spans from zero to 1.0) and ANT abundance (nmol ANT/mg mitochondrial protein).
Luciferase ADP Kinetic Assay
Oxidative phosphorylation rate (Jp) was measured at 8 levels of [ADP]: 0, 1.5, 3.0, 6.0, 12.5, 25, 50, and 125 μM, in a microplate based assay (Tecan, Genios, Switzerland) using a firefly luciferase/luciferin luminescence system (Molecular Probes, USA) to monitor the accumulation of pmol quantities of ATP. Mitochondria, roughly 0.25 μg protein, were assayed at 298 K for up to 10 min in a final volume of 200 μl of respiration buffer after the addition of G+S to start the reaction. All assay progress curves were linear with time (R2 ≥ 0.99) and ATP standard curves, up to 1000 pmol (5 μM), were linear with ATP addition (R2 ≥ 0.99). Pilot experiments showed that, because assays also contained 20 μM diadenosine pentaphosphate, background ATP production from adenylate kinase was abolished [42].
Mass spectrometry
Whole muscle lysates were processed as previously described [35]. Muscle lysate proteins were resolved by one-dimensional SDS-PAGE. The band corresponding to the molecular weight of ANT1 was excised and used for analysis. HPLC-ESI-MS/MS was performed in positive ion mode on a Thermo Scientific Orbitrap Elite Velos Pro hybrid mass spectrometer fitted with an EASY-Spray Source (Thermo Scientific, San Jose, CA). NanoLC was performed using a Thermo Scientific UltiMate 3000 RSLCnano System with an EASY Spray C18 LC column (Thermo Scientific, 50cm × 75 μm inner diameter, packed with PepMap RSLC C18 material, 2 μm, cat. # ES803). All solvents were liquid chromatography mass spectrometry grade. Spectra were acquired using XCalibur, version 2.1.0 (Thermo Scientific). A “top 15” data-dependent MS/MS analysis was performed (acquisition of a full scan spectrum followed by collision-induced dissociation mass spectra of the 15 most abundant ions in the survey scan). Tandem mass spectra were extracted from Xcalibur ‘RAW’ files and charge states were assigned using the ProteoWizard 2.1.x msConvert script using the default parameters. The fragment mass spectra were then searched against the human SwissProt database using Mascot (Matrix Science, London, UK; version 2.5.0) with the default probability cut-off score. The search variables used were 10 ppm mass tolerance for precursor ion masses and 0.5 Da for product ion masses; digestion with trypsin; a maximum of two missed tryptic cleavages; variable modifications of oxidation of methionine and phosphorylation of serine, threonine, and tyrosine. Cross-correlation of Mascot search results with X! Tandem was accomplished with Scaffold (version Scaffold_4.4.0; Proteome Software, Portland, OR, USA).
To determine linearity and reproducibility of spectral counting for relative quantification of ANT, a single, pooled lysate of human skeletal muscle was processed as described above. The resulting solution of tryptic peptides underwent mass spectrometry analysis using 3 separate aliquots each of 1, 2, and 4 μl of purified peptide solution (total of 9 runs). Data were expressed as spectral counts normalized to the individual total sample spectra (normalized spectral counts). To obtain a measure of ANT acetylation relative to total ANT, spectra for acetylated ANT were divided by all ANT spectra (acetylated and unacetylated). We termed this the ANT acetylation index. Because of differences in the behavior of various peptide ions derived from ANT in this analysis, spectrum counts are a measure of relative and not absolute abundance. We previously showed a strong relationship between spectrum counts and protein abundance [35]. Therefore, although this acetylation index may not reflect precise stoichiometric relationships, the index is highly likely to be directly related to stoichiometry of acetylation.
Data Analysis and Statistics
Data are given as means ± SE. Kinetic parameter values were evaluated for each individual mitochondrial preparation by minimizing residual differences using the Excel Solver tool. Regression analysis was used to determine the correlation between KmADP and Lysine23 acetylation. In all statistical assessments, significance was accepted at p<.05.
Results
Human Participants
Characteristics of the participants are given in Table 1.
Table 1.
Participant characteristics. Mean ± SE, n=9 (3 females and 6 males).
| Variable | Unit | Mean | SE | n |
|---|---|---|---|---|
| Age | yr | 26.9 | 1.8 | 9 |
| Body Mass Index | kg/m2 | 21.9 | 0.7 | 9 |
| Body Fat | % | 21.5 | 1.5 | 9 |
| Total Cholesterol | mg/dL | 153 | 9 | 9 |
| HDL Cholesterol | mg/dL | 55 | 5 | 9 |
| HbA1c | % | 5.11 | 0.10 | 9 |
| Fasting Plasma Glucose | mg/dL | 83.8 | 2.2 | 9 |
| Clamp Plasma Glucose | mg/dL | 92.3 | 2.8 | 9 |
| Fasting Plasma Insulin | μU/ml | 5.06 | 1.92 | 9 |
| Clamp Plasma Insulin | μU/ml | 146 | 11 | 9 |
| Basal Glucose Disposal | mg·min−1·kg−1 | 2.46 | 0.10 | 9 |
| Clamp Glucose Disposal | mg·min−1·kg−1 | 10.3 | 1.23 | 9 |
| Clamp Endogenous Glucose Production | mg·min−1·kg−1 | 0.32 | 0.21 | 9 |
All participants had healthy [43] values for body mass index, percent body fat, total cholesterol level, fasting plasma insulin concentration, basal and insulin-stimulated rates of glucose disposal, and insulin-mediated suppression of endogenous glucose production (Table 1).
Functional Integrity of Isolated Mitochondria
Figure 1 illustrates how conventional functional parameters were measured. These data indicate the isolated mitochondria had high functional integrity and are reported in Table 2. Maximal (State 3) oxidative phosphorylation rate, Jp, was calculated as 2*(ADP/O)*(State 3 Jo).
Table 2.
Conventional parameters of mitochondrial structure and function determined at 310K. In all assays mitochondria oxidized succinate + glutamate (each at 10 mM).
| Variable | State 2 Jo | State 3 Jo | State 4 Jo | RCR | ADP/O | State 3 Jp |
|---|---|---|---|---|---|---|
| Units | nmol O2 min−1 mg−1 | nmol ATP min−1 mg−1 | ||||
| Mean | 55.3 | 371 | 36.4 | 10.9 | 2.44 | 1815 |
| SE | 7.6 | 57 | 6.4 | 0.9 | 0.06 | 277 |
| n | 9 | 9 | 9 | 9 | 9 | 9 |
Appreciable variability was evident in State 3 Jo and Jp values; the percent coefficient of variation (CV), calculated as 100*(SE/mean), was 15.4%. Biological differences undoubtedly account for some of this variability, but experimental issues might also contribute. Rates normalized to mg of isolated protein are, of course, influenced by the extent to which non-mitochondrial protein contaminates the final mitochondrial suspension [44]. When normalized to adenine nucleotide translocase (ANT) abundance, determined from the titration of Jo with CAT (see Table 3 below), state 3 Jo was 385 ± 16.6 nmol O2 min−1 nmol ANT−1, which substantially reduced the CV to 4.3%.
Table 3.
ANT abundance (nmol per mg mitochondrial protein), flux control strength, and turnover.
| Variable | ANT Content | ANT Flux Control Coefficient (CJANT) | ANT Turnover | ||
|---|---|---|---|---|---|
| at 21 μM ADP | at 2.0 mM ADP | at 21 μM ADP | at 2.0 mM ADP | ||
| Units | nmol/mg | (sec−1) | (sec−1) | ||
| Mean | 0.97 | 0.82 | 0.01 | 11.9 | 27.5 |
| SE | 0.14 | 0.06 | 0.01 | 0.5 | 1.7 |
| n | 9 | 9 | 9 | 9 | 9 |
Control of Oxidative Flux and Determination of ANT Abundance
Flux control analysis for ANT was carried out while [ADP]f was clamped at 21 μM and also at 2.0 mM (saturating) ADP. The results of these titrations are shown, respectively, in panels A and B of Figure 3.
Figure 3.

Respiration rate at 310 K is titrated out with stepwise additions of carboxyatractyloside (CAT) at a clamped [ADP]f of 21 μM (Panel A) and at saturating (2 mM) ADP (Panel B). In Panel A the regression equation of the broken line is: y = 1.00 – 1.04x, R2 = 0.994. In Panel B, the apparent breakpoint at about 0.5 total ANT was arbitrarily used to evaluate two regression equations, the first nearly flat and a second steep slope reflecting maximal ANT turnover. On the ordinates “Ji” is Jo remaining after maximum CAT inhibition and “Jmax” is Jo prior to the first CAT addition. In both panels, each point is the mean ± SE with both x- and y-error bars of 6–9 values.
Figure 3A clearly shows decreasing steady state Jo with each CAT addition when [ADP] is clamped at 21 μM. This result reflects the dominant flux control (82%, see Table 3) located at ANT under this condition. In dramatic contrast, with saturating extramitochondrial [ADP] of 2.0 mM as shown in Figure 3B, flux control at ANT is essentially zero; in fact, roughly 50% of ANT sites must be occupied before any appreciable change in Jo occurs. With [ADP] clamped at 21 μM, the endpoint of the CAT titration indicated an ANT abundance of 0.97 ± 0.14 nmol/mg protein (Note: This ANT nmol/mg mitochondrial abundance should be doubled if we assume ANT functionally operates as a dimer). Because flux control at ANT was so high when [ADP] was clamped at 21 μM, essentially identical estimates of ANT abundance were obtained from the CAT titration endpoint (“graphical analysis”) and the nonlinear regression model of Gellerich et al. [39]. From the linear portion of the CAT titration curves the ANT dimer turnover rate can be estimated. These values are reported in Table 3. Interestingly, both the ANT abundance and the ANT turnover under saturating ADP conditions are similar to rat skeletal muscle mitochondria [45].
Thermodynamic Flow-Force
The progressive fall in Jo with each PCr addition shown in Figure 2 above can also be viewed as a response to rising “ATP backpressure” (more negative ATP free energy) opposing oxidative flux [7, 15, 46]. The resulting “flow:force” relationship between Jo and ATP free energy is shown in Figure 4.
Figure 4.

Thermodynamic flow-force relationship, both absolute and relative to Vmax. The regression equations are for Panel A: y = 2130 + 34.18x, R2 = 0.998 and for Panel B: y = 5.66 + 0.091x, R2 = 0.999. All y error bars are shown; some lie within the dimensions of the symbol.
As described in Methods, the CK clamp was designed to establish 5 levels of ΔGATP spanning roughly −53.5 to −57.7 kJ/mol, which was chosen because this range lies within the near-linear region in the ΔGATP:Jp relationship observed in vivo with 31P-MRS [4, 19, 47].
Apparent KmADP for Oxidative Flux
The same CK clamp data set is used to plot the [ADP]f:Jo relationship shown in Figure 5A. Both the Eadie-Hofstee transform of the MM equation and nonlinear regression were used to evaluate kinetic parameters. The Eadie-Hofstee transform:
| (8) |
is shown in Figure 5B.
Figure 5.

Panel A shows mean values of respiration rate (Jo) at various [ADP]f of the respiration medium, established with the CK clamp. These data are then graphed as an Eadie-Hofstee plot in Panel B. The equation of the line in Panel B is: y = −34.1x + 380, R2=0.999. Each point is the mean ± SE (both x- and y-error bars) of 9 values. Panels A and B data are replotted in Panels C and D, respectively, in units relative to Vmax on the ordinate, also with x and y error bars, which often lie within the symbol.
As can be seen in Figure 5B, Eadie-Hofstee analysis provided an excellent fit to mean data. These same data are shown in Figures 5C and 5D in flux units relative to Vmax and again with error bars in both dimensions. Kinetic parameters from Eadie-Hofstee analysis performed for each individual mitochondrial preparation are reported in Table 4.
Table 4.
ADP kinetic parameters from both the polarographic (Jo) and luciferase (Jp) assays.
| Polarography Assay (CK clamp) | Luciferase Assay | |||||
|---|---|---|---|---|---|---|
| Variable | State 3 Jo | E-H Vmax | KmADP | KmADP3− | KmADP | KmADP3− |
| Units | nmol O2 min−1 mg−1 | μM | μM | μM | μM | |
| Mean | 371 | 380 | 32.4 | 12.0 | 13.1 | 2.0 |
| SE | 57 | 63 | 1.8 | 0.7 | 1.9 | 0.3 |
| n | 9 | 9 | 9 | 9 | 6 | 6 |
The KmADP for respiration was 32.4 ± 1.8 μM, determined in the presence of 5 mM ATP and 1.1 mM free Mg2+. As can be seen in Figure 5 panel B, the Eadie-Hofstee plot of mean data resulted in a y-intercept value that was almost identical to State 3 Jo measured separately with saturating ADP (value from Table 2 repeated here in Table 4). Table 4 also shows that kinetic assessment based on the concentration of ADP3−, the ionic form of ADP that binds to ANT, gave an apparent KmADP3− of 12.0 ± 0.7 μM, again in the presence of 5 mM ATP, which under these conditions converts to an ATP4− concentration range of 320–340 μM.
During the course of this study a luciferase-based assay to determine ADP kinetics of oxidative phosphorylation rate was developed. Six of the nine mitochondrial preparations were assayed using this procedure at several ADP concentrations below and above the KmADP and in the absence of ATP competition for ANT binding (Figure 6). Under these conditions the KmADP was much lower, 13.1 ± 1.9 μM. Further, because this assay was also carried out with a buffer [Mg2+] of 5.0 mM, but with essentially zero ATP, the free [Mg+2] was calculated to be 3.95 mM. Thus, in the absence of ATP competition and taking Mg binding to ADP into account, the KmADP3− was 2.0 ± 0.3 μM (Table 4 and Figure 6C). Nonlinear kinetic analysis indicated a Hill coefficient value of 1.05 ± 0.02, consistent with simple MM kinetics and the excellent fit of the Eadie-Hofstee transform to mean data shown in Figure 6B.
Figure 6.

Luciferase assay to evaluate ADP and ADP3− kinetics of oxidative phosphorylation rate (Jp). Filled symbols, all n=6, show mean ± SE (error bars in both x and y dimensions). In panels A and C, open symbols and dashed lines show nonlinear fit of Equation 1 to the data. Panel A: Analyzed kinetic parameters were KmADP = 13.1 ± 1.9 μM and Hill coefficient, nH = 1.05 ± 0.02. Panel B: Eadie-Hofstee plot of the same data shown in Panel A. The equation of the line for mean data was: y = −12.6x + 1.01, R2 = 0.993. Panel C: Analyzed kinetic parameters in terms of the ADP3− ion were: KmADP3− = 2.0 ± 0.3 μM and Hill coefficient, nH = 1.05 ± 0.02. Rates are expressed in units relative to Vmax.
Mitochondrial Creatine Kinase
As illustrated in Figure 2, in the presence of buffer concentrations of 5 mM ATP, 2.5 mM PCr, and 5.0 mM Cr, but prior to the addition of 40 units of CK (80 μmol min−1 ml−1), the incubated mitochondria respired at a steady state Jo that was about four-fold higher than state 4 Jo (Table 5). After CK was added and brought the buffer PCr, Cr, ATP, and ADP into equilibrium, Jo promptly increased to a steady state value of 297 ± 49.9 nmol O2 min−1 mg−1, a delta Jo of 135 ± 39.9 nmol O2 min−1 mg−1. The PCr additions that followed caused stepwise reductions in buffer [ADP]f and Jo fell with the MM kinetics reported in Table 4. Although we do not know the buffer [ADP]f prior to the addition of CK, we can use the kinetic parameters of Table 4 and Equation 9 to infer the “effective” [ADP]f that mitochondrial CK and diffusional restrictions of the outer membrane were making available to ANT in the intermembrane space:
| (9) |
Table 5.
Mitochondrial Jo and Jp before (Pre-CK) and after (Post-CK) saturating (40 U) CK activity was added to the respiration buffer (Mean ± SE).
| Variable | Units | Pre-CK | Post-CK | Delta | n |
|---|---|---|---|---|---|
| Jo | nmol O2 min−1 mg−1 | 161.3 ± 21.8 | 296.7 ± 49.9 | 135.0 ± 39.9 | 9 |
| Jp | nmol ATP min−1 mg−1 | 755 ± 89 | 1388 ± 215 | 633 ± 132 | 9 |
| [ADP]f | μM | 26.4 ± 2.6 | 108.6 ± 0.1 | 82.2 ± 2.6 | 9 |
As reported in Table 5, prior to the addition of CK to the respiration buffer, mitochondria respired at 161 ± 21.8 nmol O2 min−1 mg−1, which is about 41% of Vmax and the Jo corresponding to a buffer [ADP]f of 26.4 μM (Table 5). Promptly after CK addition, respiration jumped to the steady state Jo of 296.7 ± 49.9 nmol O2 min−1 mg−1, about 78% of Vmax and corresponding to a buffer [ADP]f of 108.6 μM. The addition of CK activity to the buffer therefore apparently increased the effective steady state [ADP]f by over four-fold, increasing it by 82.2 ± 2.6 μM (Table 5).
Proteomic Analysis of ANT
Proteomic analysis indicated that ANT1 was the predominant isoform. Table 6 reports the analysis of a typical experiment.
Table 6.
ANT isoform abundance of a typical muscle biopsy.
| ANT Isoform | Unique exclusive peptides | Unique exclusive spectra | Total spectra |
|---|---|---|---|
| ANT1 | 52 | 79 | 546 |
| ANT2 | 3 | 3 | 9 |
| ANT3 | 3 | 5 | 5 |
We assessed acetylation of ANT1 using mass spectrometry-based proteomics techniques. Based on normalized spectral counts, relative quantification of ANT1 was linear across about a four-fold range of abundance. As expected, spectral counts were approximately equivalent when normalized to the total spectral count in the samples (Figure 7), indicating the ability of spectrum counting techniques to accurately reflect relative changes in protein and peptide abundance. The coefficient of variation across the nine separate determinations of normalized spectral counts was approximately 6%.
Figure 7.
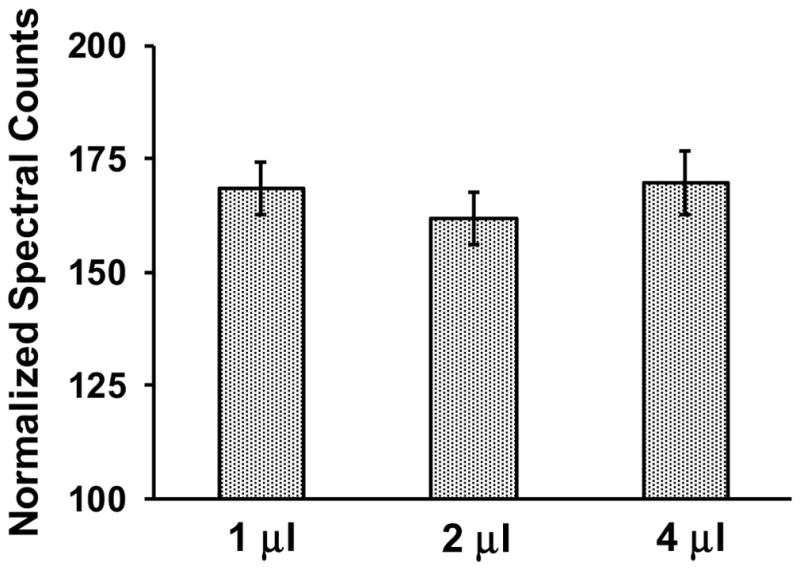
In mass spectrometry-based proteomic assessments, the relative abundance of ANT1 was independent of the sample volume analyzed across a four-fold range with a coefficient of variation of approximately 6%.
The ANT1 Lysine23 acetylation index varied across a range of roughly 1–4%. Figure 8 shows that ANT Lysine23 acetylation positively correlated with KmADP (r = 0.74, P = 0.022).
Figure 8.

Correlation between the relative acetylation of ANT Lysine23 and the apparent KmADP determined from mitochondrial O2 consumption rates at experimentally established buffer [ADP] levels using the CK clamp. r = 0.74, P = 0.022
Discussion
Skeletal muscle cells routinely vary their rates of ATP turnover over a wide range. Optimal myocyte function requires mitochondria with high sensitivity to feedback signals so that large changes in oxidative phosphorylation rate are achieved with small changes in cytosolic [ADP] and ΔGATP [15, 48]. This steep “flow:force” near-linear relationship is shown in Figure 4, which indicates that the energetic performance of our isolated mitochondria was similar to human vastus muscle mitochondria in vivo [9]. The “static head” ΔGATP, found from the extrapolation of the ΔGATP:Jo relation to the zero flux of the x-intercept, was −62.070 ± 0.207 kJ/mol. In addition, the relative flow-force slope of 0.091 i.e., a 1.0 kJ/mol change in ΔGATP was associated with a 9% change in flux relative to maximum, is almost identical to intact muscle [9].
Three major observations about the control of metabolic flux in human skeletal muscle mitochondria were made in these studies. First, one single process, the exchange of ADP3− for ATP4− by ANT, accounted for over 80% of flux control when [ADP]f was clamped at 21 μM, a concentration associated with mild contractile activity [49]. However, at high, saturating [ADP] (2.0 mM), associated with maximum oxidative flux, control at ANT fell to essentially zero. Second, simple MM kinetics adequately described the relationship of [ADP] to oxidative flux, which is in marked contrast to the higher order kinetics observed in intact muscle using 31P-MRS [18, 49]. Third, acetylation of ANT Lysine23 reduced the sensitivity of mitochondria to the ADP3− signal.
Control of Oxidative Flux at ANT
At low [ADP] ANT accounted for 82% of the flux control of the entire oxidative pathway. Dominant control at one step is highly unusual in a long metabolic pathway [50] and emphatically identifies ANT as a major regulatory site of muscle oxidative metabolism. Such focused control implies that a decreased ANT-ADP binding affinity, due for example to lysine acetylation, would necessitate higher cytosolic [ADP] to achieve a given rate of ATP synthesis. In turn, the higher cytosolic [ADP] would be linked via the creatine kinase and adenylate kinase reactions to increased inorganic phosphate and AMP concentrations [10], which are strong positive modulators of glycolytic pathway [51] and AMP Kinase [52] activity.
It is important to emphasize that our experimental approach assesses flux control intrinsic to the mitochondrion. Conceptually similar studies of rabbit heart mitiochondria by Kholodenko et al. [53] found that ANT accounted for up to 60% of the control of oxidative flux (CJANT = 0.60), across a span of ATP turnover rates from about 20% to 50% of the aerobic capacity. In rat liver mitochondria respiring at about 50% of maximum O2 consumption rate, essentially exclusive flux control at ANT was reported by Davis and Davis Van-Thienen [54].
The evaluation of flux control strength critically depends on the dimensions of the system under investigation; clarity on this issue was a major objective of the present study. For example, much lower ANT flux control coefficients have been reported [39, 55]. In these studies mitochondrial flux (Jo) was established by adding to the incubation medium an ATP-hydrolyzing system. If this reaction has very little sensitivity to the buffer ATP/ADP ratio, which is the case for glucose + yeast hexokinase (a commonly used ATP breakdown reaction), then inhibition of ANT by CAT addition simply initiates a transient in which buffer [ADP] accumulates until it stimulates the recovery of ANT activity (and mitochondrial Jo) toward the pre-CAT rate. “Elastic recoil” was the term Davis and Davis Van-Thienen used to describe this phenomenon [54]. In contrast, the CK clamp is based on the conceptual framework of “top-down” metabolic control analysis [56]. The creatine kinase reaction acts as the “ATPase module” of the ATP-ADP loop and establishes complete concentration control. As a consequence, the incubated mitochondria, the “ATP-producing module” of the loop, account for all flux control [24]. Under these experimental conditions, any change in flux in response to the addition of a mitochondrial inhibitor unequivocally reflects intrinsic mitochondrial control of flux at the inhibited step. Our observations with human skeletal muscle mitochondria using the CK clamp, therefore, align with previous studies showing that, within the mitochondrion itself, ANT is a dominant site of flux control [53, 54] at mild to moderate rates of ATP turnover. On the other hand, ANT accounted for essentially zero flux control at the [ADP] associated with near-maximum ATP production. Thus, as ATP demand rises, mitochondrial control of oxidative flux is redistributed from ANT to other loci, such as transmembrane substrate transport, citric acid cycle enzymes, the electron transport chain (ETC), and Complex V [57].
KmADP and ANT Lysine Acetylation
The apparent KmADP determined here, 32.4 ± 1.8 μM, is similar to our previous findings in mammalian skeletal muscle mitochondria [7, 21, 22], which were also assessed in the presence of 5 mM ATP. These ADP and ATP concentrations represent all ionic forms (ΣADP and ΣATP). After adjusting for Mg2+ binding and pH [33], the corresponding KmADP3− was 12 μM, measured in the presence of roughly 320 – 340 μM ATP4−, which competes for ANT binding [27, 28]. In contrast, in the luciferase assay ATP competition is absent, and the measured KmADP3− was 2.0 μM. Thus, collectively our experimental outcomes provide a noteworthy agreement: High ANT flux control in the lower region of [ADP] was observed along with a KmADP3− fairly similar to the KdADP3− measured in purified ANT in vitro [28] and crystallized ANT in silico [34]. Moreover, acetylation of ANT Lysine23, a post-translational modification that molecular dynamic modeling has shown decreases ANT-ADP3− binding affinity by about 20-fold [34], correlated positively with apparent KmADP, even though the observed ranges of the two variables were not broad. The agreement is striking when we consider the multiple factors that influence the ANT exchange rate of ADP3− for ATP4− [48], including intermembrane space and matrix adenylate concentrations and ΔΨ [27, 28]. These factors, in turn, depend on multiple upstream processes including substrate oxidation, proton pumping, and matrix ATP synthesis [48]. Interestingly, a review by Klingenberg [28] describes data from his own and other [58] laboratories, which supports the concept that mitochondria possess two populations of ANT with KmADP that differ by an order of magnitude (below this point is further discussed in the context of computational modeling).
A few methodological caveats merit brief discussion. The acetylation index was calculated as the ratio of peptides in ANT1 containing acetylated Lysine23 divided by the sum of unacetylated plus acetylated Lysine23 containing peptides, which were measured using spectrum counting. This index is not equivalent to the percentage of ANT1 molecules that are acetylated at Lysine23, due to potentially differing behaviors of ions representing acetylated and unacetylated peptides during mass spectrometry analysis. However, we have previously shown a strong relationship between spectrum counts and protein abundance [35]. Therefore, although this acetylation index may not reflect a precise stoichiometric relationship, it is likely to be directly related to that acetylation stoichiometry. An additional caveat is that acetylation status was determined in muscle that was promptly frozen in liquid nitrogen after biopsy, while KmADP was assessed about 1.5 hr later in isolated mitochondria. Because our isolation buffers lack oxidative substrates, the mitochondria would be expected, if anything, to be less acetylated than the proteomic analysis indicated. Nevertheless, a significant relationship between acetylation index and apparent KmADP was observed in Figure 8, albeit lacking in homoscedasticity. We can say with greater confidence that the acetylation status would likely remain stable once the mitochondria were added to the incubation because respiration was fueled by succinate plus glutamate. This substrate combination delivers strong reducing power into both Complex I and Complex II, what Gnaiger [59] has termed “convergent electron flow,” and is capable of supporting both high maximum flux [60], as well as a highly reduced matrix during controlled respiration (thus depriving the SIRT3 de-acetylation reaction of its NAD+ substrate). This substrate combination also circumvents matrix Acetyl-CoA production, a major source of the acetate carbon for non-enzymatic lysine acetylation. With these important caveats in mind, the results shown in Figure 8 imply that ANT Lysine23 acetylation increases skeletal muscle KmADP.
ADP Kinetics Assessed with 31P-MRS, Isolated Mitochondria, and Permeabilized Fibers
31P-MRS assessment of intact mammalian muscle yields apparent KmADP for oxidative metabolism similar to isolated mitochondria, 20 – 40 μM [61]. But ADP kinetics in vivo are markedly sigmoidal [16–18], while our data and previous reports indicate that MM kinetics adequately describe isolated mitochondria [4, 28]. Important regulatory factors such as the cytosolic concentrations of phosphate, calcium, and oxidative substrate change with ATP turnover rate in vivo and likely account for at least some of this difference [7, 24, 62–64]. Additionally, structural changes caused by the processes of mitochondrial isolation and incubation might also be expected to alter kinetic behavior [65, 66], in particular, issues related to the intermembrane space, outer mitochondrial membrane, and interactions with the extramitochondrial environment [66]. Obviously, ADP binds to ANT in the intermembrane space, while “KmADP” refers to the extramitochondrial (cytosolic) concentration. A diffusional limitation to ADP across the outer membrane mediated by the voltage dependent anion channel (VDAC) would be particularly evident at low extramitochondrial [ADP] [67] and might therefore confer sigmoidal kinetic character to the overall apparent ADP kinetics. Gellerich and coworkers have shown that isolated rat heart mitochondria incubated in buffers supplemented with macromolecules to simulate cytosolic oncotic pressure concomitantly exhibit a contracted intermembrane space, decreased outer membrane ADP permeability, and increased apparent KmADP, a phenomenon they term dynamic compartmentation [67, 68]. Although they analyzed their data with simple MM kinetics, their graphed data, for example Figure 5 of [68], strongly suggest sigmoidal kinetics might provide a better fit.
The permeabilized muscle fiber technique maintains mitochondria more closely to their native cellular structural configuration [69], including proposed linkages among VDAC, mitochondrial creatine kinase (miCK), and ANT [66]. Much higher, sometimes triple-digit micromolar, estimates of KmADP [70] are reported as a simple MM parameter, although proteolytic treatment to remove putative linkages between VDAC and the cytoskeleton reduces KmADP into the double-digit micromolar range, as does the addition of 20 mM creatine [66, 71, 72]. However, diffusional issues related not to mitochondria but rather to the experimental conditions may be confounding the interpretations of this experimental approach [73]. As a technical note it is worth pointing out that permeabilized fiber preparations maintain very high bound CK activity [74], which is largely accounted for by a myofibrillar CK activity that is about 24 times higher than the CK in the mitochondrial intermembrane space [75, 76]. Thus, ADP added to an assay containing millimolar concentrations of both PCr and Cr [26, 77] will be rapidly phosphorylated to ATP at myofibrillar CK, as the reaction advances toward equilibrium (i.e., the thermodynamic basis of the CK clamp used in the present study). Accordingly, previous studies reporting KmADP in excess of 1500 μM [26] in the presence of “resting levels” of 12 mM Cr and 24 mM PCr must be viewed with caution.
We explored the potential regulatory role of the creatine pool and mitochondrial creatine kinase by adding ATP, PCr, and Cr to isolated mitochondria. In this condition, a steady state mitochondrial Jo ~ 4-fold higher than State 4 reflected the delivery of ADP to ANT via CK activity in the IMS. However, the subsequent addition of excess CK to the buffer promptly increased Jo by another 2-fold (illustrated in Figures 1 and 2). From the CK clamp determined [ADP]:Jo kinetic assessment we could back-calculate (Equation 9) that the respiration rate prior to CK addition corresponded to a buffer [ADP] of 26.4 ± 2.6 μM, which was 82.2 ± 2.6 μM away from the equilibrium [ADP] that ensued after excess CK was added. In the terms of ATP free energy, mitochondria not provided extramitochondrial CK failed to achieve energy phosphate equilibrium by about 5 kJ/mol, equivalent to almost half of the entire ΔGATP span of respiratory control. These findings in mitochondria isolated from human skeletal muscle incubated in respiration buffer lacking oncotic pressure are consistent with the previous work of Gellerich and coworkers [67, 68, 78], which showed that the diffusional characteristics of the outer membrane significantly influence the extent to which mitochondrial CK is catalytically competent to maintain equilibrium between the creatine and adenylate pools in the intermembrane space. An additional interpretation of these findings is that mitochondria depend on cytosolic CK to maintain what appears to be the near-equilibrium condition of the energy phosphate network as assessed with 31P-MRS [11].
In any case, the resting condition of skeletal muscle in vivo readily exposes gaps in our understanding of the kinetics of respiratory control. Assuming a reasonable dynamic range of resting VO2 equal to 4 ml O2 min−1 kg−1 [1, 2] and maximum VO2 equal to 250 ml O2 min−1 kg−1 [79], a KmADP of 30 μM with a Hill coefficient as high as 2.0 still predicts a resting cytosolic [ADP]f of less than 4 μM, well below the ~14 μM that 31P-MRS measurements suggest [4]. Computational models of mitochondrial metabolism offer insight into the mechanisms underlying this complex control [27, 80], but valid models must, of course, include parameter values which reflect our best estimates of binding affinity. Unfortunately, in what may be considered the best ANT kinetic model currently available [27, 81], the KdADP3− parameter (~50 μM) was obtained from a study [32] in which the authors suggested that unusual phospholipid composition of the liposomes may have inflated the value by roughly 10-fold. When this error is rectified, and a thermokinetic rate expression [82] of ANT, which includes acetylated and unacetylated forms, is incorporated into a modified computational model published by Wu et al. [80], the simulation output agrees with the data of the present study with outstanding fidelity (unpublished observations). Going forward greater care to express ADP kinetics in terms of the ADP3− ion and to consider whether binding competition from ATP is present might help achieve better clarity and consensus. Finally, VDAC permeability modulated by dimeric tubulin binding is a promising additional factor to consider, perhaps especially in the resting condition [71, 72].
Summary
In summary, the adenine nucleotide translocase plays a central and critical role in the control of ATP production rate by mitochondria isolated from human skeletal muscle. It was found that ANT dominates oxidative flux control particularly at the energy phosphate levels associated with low ATP demand. Acetylation of ANT at Lys23 was shown here to predict rising KmADP, thus decreasing the sensitivity of skeletal muscle mitochondria to the ADP respiratory signal.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants R01 DK047936 (L.J.M.) and DK066483 (L.J.M.)
Abbreviations
- ANT
adenine nucleotide translocase
- CAT
carboxyatractyloside
- CK
creatine kinase
- Cr
creatine
- Jo
rate of oxygen consumption
- Jp
rate of ATP production
- MM
Michaelis-Menten
- nH
Hill coefficient
- NET
nonequilibrium thermodynamics
- PCr
phosphocreatine
Footnotes
Author contributions
This work was performed at the Arizona Mayo Clinic in Scottsdale, AZ, USA. Author contributions: W.T.W. conceived and designed the work; acquired, analyzed, and interpreted data; drafted and revised the manuscript; D.M.-G. acquired and analyzed data; revised the manuscript; J.H. analyzed data; revised the manuscript; E.A.W. acquired and analyzed data; revised the manuscript; J.F. acquired and analyzed data; revised the manuscript; E.A.DF. acquired and analyzed data; revised the manuscript; P.R.L. acquired, analyzed, and interpreted data; revised the manuscript; C.M. conceived and designed the work; L.J.M. conceived and designed the work; analyzed and interpreted the data; drafted and revised the manuscript.
All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
The authors have no conflicts or competing interests of any type.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andres R, Cader G, Zierler KL. The quantitatively minor role of carbohydrate in oxidative metabolism by skeletal muscle in intact man in the basal state; measurements of oxygen and glucose uptake and carbon dioxide and lactate production in the forearm. J Clin Invest. 1956;35:671–682. doi: 10.1172/JCI103324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49:677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 3.Kemp GJ, Brindle KM. What do magnetic resonance-based measurements of Pi-->ATP flux tell us about skeletal muscle metabolism? Diabetes. 2012;61:1927–1934. doi: 10.2337/db11-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kemp GJ, Ahmad RE, Nicolay K, Prompers JJ. Quantification of skeletal muscle mitochondrial function by 31P magnetic resonance spectroscopy techniques: a quantitative review. Acta Physiol (Oxf) 2015;213:107–144. doi: 10.1111/apha.12307. [DOI] [PubMed] [Google Scholar]
- 5.Blomstrand E, Radegran G, Saltin B. Maximum rate of oxygen uptake by human skeletal muscle in relation to maximal activities of enzymes in the Krebs cycle. J Physiol. 1997;501(Pt 2):455–460. doi: 10.1111/j.1469-7793.1997.455bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richardson RS, Saltin B. Human muscle blood flow and metabolism studied in the isolated quadriceps muscles. Med Sci Sports Exerc. 1998;30:28–33. doi: 10.1097/00005768-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chance B, Leigh J, Clark B, Marris J, Kent J, Nioka S, Smith D. Control of oxidative metabolism and oxygen delivery in human skeletal muscle: a steady-state analysis of the work/energy cost transfer function. Proc Natl Acad Sci. 1985;82:8384–8388. doi: 10.1073/pnas.82.24.8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeneson JA, Schmitz JP, van den Broek NM, van Riel NA, Hilbers PA, Nicolay K, Prompers JJ. Magnitude and control of mitochondrial sensitivity to ADP. Am J Physiol Endocrinol Metab. 2009;297:E774–784. doi: 10.1152/ajpendo.00370.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connett R. Analysis of metabolic control: new insights using a scaled creatine kinase model. Am J Physiol. 1988;254:R949–R959. doi: 10.1152/ajpregu.1988.254.6.R949. [DOI] [PubMed] [Google Scholar]
- 11.Kushmerick MJ. Energy balance in muscle activity: simulations of ATPase coupled to oxidative phosphorylation and to creatine kinase. Comp Biochem Physiol B Biochem Mol Biol. 1998;120:109–123. doi: 10.1016/s0305-0491(98)00026-1. [DOI] [PubMed] [Google Scholar]
- 12.Kemp GJ. Interactions of mitochondrial ATP synthesis and the creatine kinase equilibrium in skeletal muscle. J Theor Biol. 1994;170:239–246. doi: 10.1006/jtbi.1994.1184. [DOI] [PubMed] [Google Scholar]
- 13.Rottenberg H. The thermodynamic description of enzyme-catalyzed reactions. The linear relation between the reaction rate and the affinity. Biophys J. 1973;13:503–511. doi: 10.1016/S0006-3495(73)86004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer RA. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am J Physiol. 1988;254:C548–553. doi: 10.1152/ajpcell.1988.254.4.C548. [DOI] [PubMed] [Google Scholar]
- 15.Willis WT, Jackman MR, Messer JI, Kuzmiak-Glancy S, Glancy B. A Simple Hydraulic Analog Model of Oxidative Phosphorylation. Med Sci Sports Exerc. 2016;48:990–1000. doi: 10.1249/MSS.0000000000000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeneson JA, Wiseman RW, Westerhoff HV, Kushmerick MJ. The signal transduction function for oxidative phosphorylation is at least second order in ADP. J Biol Chem. 1996;271:27995–27998. doi: 10.1074/jbc.271.45.27995. [DOI] [PubMed] [Google Scholar]
- 17.Layec G, Haseler LJ, Hoff J, Hart CR, Liu X, Le Fur Y, Jeong EK, Richardson RS. Short-term training alters the control of mitochondrial respiration rate before maximal oxidative ATP synthesis. Acta Physiol (Oxf) 2013;208:376–386. doi: 10.1111/apha.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cieslar JH, Dobson GP. Free [ADP] and aerobic muscle work follow at least second order kinetics in rat gastrocnemius in vivo. J Biol Chem. 2000;275:6129–6134. doi: 10.1074/jbc.275.9.6129. [DOI] [PubMed] [Google Scholar]
- 19.Jeneson JA, Westerhoff HV, Brown TR, Van Echteld CJ, Berger R. Quasi-linear relationship between Gibbs free energy of ATP hydrolysis and power output in human forearm muscle. Am J Physiol. 1995;268:C1474–1484. doi: 10.1152/ajpcell.1995.268.6.C1474. [DOI] [PubMed] [Google Scholar]
- 20.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 21.Kuzmiak-Glancy S, Willis WT. Skeletal muscle fuel selection occurs at the mitochondrial level. J Exp Biol. 2014;217:1993–2003. doi: 10.1242/jeb.098863. [DOI] [PubMed] [Google Scholar]
- 22.Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, Brophy C, Meyer C, Hojlund K, Yi Z, Mandarino LJ. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes. 2010;59:2444–2452. doi: 10.2337/db10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glancy B, Barstow T, Willis WT. Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. Am J Physiol Cell Physiol. 2008;294:C79–87. doi: 10.1152/ajpcell.00138.2007. [DOI] [PubMed] [Google Scholar]
- 24.Messer JI, Jackman MR, Willis WT. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am J Physiol Cell Physiol. 2004;286:C565–572. doi: 10.1152/ajpcell.00146.2003. [DOI] [PubMed] [Google Scholar]
- 25.Guzun R, Saks V. Application of the principles of systems biology and Wiener’s cybernetics for analysis of regulation of energy fluxes in muscle cells in vivo. Int J Mol Sci. 2010;11:982–1019. doi: 10.3390/ijms11030982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry CG, Kane DA, Herbst EA, Mukai K, Lark DS, Wright DC, Heigenhauser GJ, Neufer PD, Spriet LL, Holloway GP. Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J Physiol. 2012;590:5475–5486. doi: 10.1113/jphysiol.2012.234682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metelkin E, Goryanin I, Demin O. Mathematical modeling of mitochondrial adenine nucleotide translocase. Biophys J. 2006;90:423–432. doi: 10.1529/biophysj.105.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 29.Pfaff E, Heldt HW, Klingenberg M. Adenine nucleotide translocation of mitochondria. Kinetics of the adenine nucleotide exchange. Eur J Biochem. 1969;10:484–493. doi: 10.1111/j.1432-1033.1969.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 30.Klingenberg M, Pfaff E. Metabolic control in mitochondria by adenine nucleotide translocation. Biochem Soc Symp. 1968;27:105–122. [PubMed] [Google Scholar]
- 31.Weidemann MJ, Erdelt H, Klingenberg M. Adenine nucleotide translocation of mitochondria. Identification of carrier sites. Eur J Biochem. 1970;16:313–335. doi: 10.1111/j.1432-1033.1970.tb01086.x. [DOI] [PubMed] [Google Scholar]
- 32.Kramer R, Klingenberg M. Electrophoretic control of reconstituted adenine nucleotide translocation. Biochemistry. 1982;21:1082–1089. doi: 10.1021/bi00534a040. [DOI] [PubMed] [Google Scholar]
- 33.Chinopoulos C, Vajda S, Csanady L, Mandi M, Mathe K, Adam-Vizi V. A novel kinetic assay of mitochondrial ATP-ADP exchange rate mediated by the ANT. Biophys J. 2009;96:2490–2504. doi: 10.1016/j.bpj.2008.12.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mielke C, Lefort N, McLean CG, Cordova JM, Langlais PR, Bordner AJ, Te JA, Ozkan SB, Willis WT, Mandarino LJ. Adenine nucleotide translocase is acetylated in vivo in human muscle: Modeling predicts a decreased ADP affinity and altered control of oxidative phosphorylation. Biochemistry. 2014;53:3817–3829. doi: 10.1021/bi401651e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Hojlund K, Yi Z, Mandarino LJ. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59:33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowry O, Rosebrough J, Farr A, Randall R. Protein measurement with the Folin Phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 37.Estabrook R. Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Meth Enzymol. 1967;10:41–47. [Google Scholar]
- 38.Teague WE, Jr, Golding EM, Dobson GP. Adjustment of K′ for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength. J Exp Biol. 1996;199(Pt 2):509–512. doi: 10.1242/jeb.199.2.509. [DOI] [PubMed] [Google Scholar]
- 39.Gellerich FN, Kunz WS, Bohnensack R. Estimation of flux control coefficients from inhibitor titrations by non-linear regression. FEBS Lett. 1990;274:167–170. doi: 10.1016/0014-5793(90)81355-r. [DOI] [PubMed] [Google Scholar]
- 40.Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 41.Heinrich R, Rapoport TA. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur J Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 42.Lundin A. Optimization of the firefly luciferase reaction for analytical purposes. Adv Biochem Eng Biotechnol. 2014;145:31–62. doi: 10.1007/978-3-662-43619-6_2. [DOI] [PubMed] [Google Scholar]
- 43.McLean CS, Mielke C, Cordova JM, Langlais PR, Bowen B, Miranda D, Coletta DK, Mandarino LJ. Gene and MicroRNA Expression Responses to Exercise; Relationship with Insulin Sensitivity. PLoS One. 2015;10:e0127089. doi: 10.1371/journal.pone.0127089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kras KA, Willis WT, Barker N, Czyzyk T, Langlais PR, Katsanos CS. Subsarcolemmal mitochondria isolated with the proteolytic enzyme nagarse exhibit greater protein specific activities and functional coupling. Biochem Biophys Rep. 2016;6:101–107. doi: 10.1016/j.bbrep.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis W, Dallman P. Impaired control of respiration in iron-deficient muscle mitochondria. Am J Physiol. 1989;257:C1080–C1085. doi: 10.1152/ajpcell.1989.257.6.C1080. [DOI] [PubMed] [Google Scholar]
- 46.Davis E, Davis-Van Thienen W. Force-flow and back-pressure relationships in mitochondrial energy transduction: an examination of extended state 3-state 4 transitions. Arch Biochem Biophys. 1989;275:449–458. doi: 10.1016/0003-9861(89)90391-3. [DOI] [PubMed] [Google Scholar]
- 47.Meyer R. Linear dependence of muscle phosphocreatine kinetics on total creatine content. Am J Physiol. 1989;257:C1149–C1157. doi: 10.1152/ajpcell.1989.257.6.C1149. [DOI] [PubMed] [Google Scholar]
- 48.Nicholls DG, Ferguson SJ. Bioenergetics 4. 4. Academic Press, Elsevier; Amsterdam: 2013. [Google Scholar]
- 49.Vanderthommen M, Duteil S, Wary C, Raynaud JS, Leroy-Willig A, Crielaard JM, Carlier PG. A comparison of voluntary and electrically induced contractions by interleaved 1H- and 31P-NMRS in humans. J Appl Physiol (1985) 2003;94:1012–1024. doi: 10.1152/japplphysiol.00887.2001. [DOI] [PubMed] [Google Scholar]
- 50.Fell D. Understanding the control of metabolism. Portland Press Ltd; London: 1996. [Google Scholar]
- 51.Connett RJ. In vivo control of phosphofructokinase: system models suggest new experimental protocols. Am J Physiol. 1989;257:R878–888. doi: 10.1152/ajpregu.1989.257.4.R878. [DOI] [PubMed] [Google Scholar]
- 52.Hardie DG, Ashford ML. AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda) 2014;29:99–107. doi: 10.1152/physiol.00050.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kholodenko B, Zilinskiene V, Borutaite V, Ivanoviene L, Toleikis A, Praskevicius A. The role of adenine nucleotide translocators in regulation of oxidative phosphorylation in heart mitochondria. FEBS Lett. 1987;223:247–250. doi: 10.1016/0014-5793(87)80298-3. [DOI] [PubMed] [Google Scholar]
- 54.Davis E, Davis Van-Thienen W. Rate control of phosphorylation-coupled respiration by rat liver mitochondria. Arch Biochem Biophys. 1984;233:573–581. doi: 10.1016/0003-9861(84)90481-8. [DOI] [PubMed] [Google Scholar]
- 55.Wanders RJ, Groen AK, Van Roermund CW, Tager JM. Factors determining the relative contribution of the adenine-nucleotide translocator and the ADP-regenerating system to the control of oxidative phosphorylation in isolated rat-liver mitochondria. Eur J Biochem. 1984;142:417–424. doi: 10.1111/j.1432-1033.1984.tb08303.x. [DOI] [PubMed] [Google Scholar]
- 56.Brand M. Top down metabolic control analysis. J Theor Biol. 1996;182:351–360. doi: 10.1006/jtbi.1996.0174. [DOI] [PubMed] [Google Scholar]
- 57.Groen AK, Wanders RJ, Westerhoff HV, van der Meer R, Tager JM. Quantification of the contribution of various steps to the control of mitochondrial respiration. J Biol Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- 58.Chan SH, Barbour RL. Adenine nucleotide transport in hepatoma mitochondria. Characterization of factors influencing the kinetics of ADP and ATP uptake. Biochim Biophys Acta. 1983;723:104–113. doi: 10.1016/0005-2728(83)90014-2. [DOI] [PubMed] [Google Scholar]
- 59.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 60.Rasmussen UF, Vielwerth SE, Rasmussen HN. Skeletal muscle bioenergetics: a comparative study of mitochondria isolated from pigeon pectoralis, rat soleus, rat biceps brachii, pig biceps femoris and human quadriceps. Comp Biochem Physiol A Mol Integr Physiol. 2004;137:435–446. doi: 10.1016/j.cbpb.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 61.Kemp GJ, Meyerspeer M, Moser E. Absolute quantification of phosphorus metabolite concentrations in human muscle in vivo by 31P MRS: a quantitative review. NMR Biomed. 2007;20:555–565. doi: 10.1002/nbm.1192. [DOI] [PubMed] [Google Scholar]
- 62.Moreno-Sanchez R, Hogue B, Hansford R. Influence of NAD-linked dehydrogenase activity on flux through oxidative phosphorylation. Biochem J. 1990;268:421–428. doi: 10.1042/bj2680421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koretsky AP, Balaban RS. Changes in pyridine nucleotide levels alter oxygen consumption and extra-mitochondrial phosphates in isolated mitochondria: a 31P-NMR and NAD(P)H fluorescence study. Biochim Biophys Acta. 1987;893:398–408. doi: 10.1016/0005-2728(87)90092-2. [DOI] [PubMed] [Google Scholar]
- 64.Schmitz JP, Jeneson JA, van Oorschot JW, Prompers JJ, Nicolay K, Hilbers PA, van Riel NA. Prediction of muscle energy states at low metabolic rates requires feedback control of mitochondrial respiratory chain activity by inorganic phosphate. PLoS One. 2012;7:e34118. doi: 10.1371/journal.pone.0034118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, Romestaing C, Hepple RT. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One. 2011;6:e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guzun R, Gonzalez-Granillo M, Karu-Varikmaa M, Grichine A, Usson Y, Kaambre T, Guerrero-Roesch K, Kuznetsov A, Schlattner U, Saks V. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim Biophys Acta. 2012;1818:1545–1554. doi: 10.1016/j.bbamem.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 67.Gellerich FN, Laterveer FD, Zierz S, Nicolay K. The quantitation of ADP diffusion gradients across the outer membrane of heart mitochondria in the presence of macromolecules. Biochim Biophys Acta. 2002;1554:48–56. doi: 10.1016/s0005-2728(02)00212-8. [DOI] [PubMed] [Google Scholar]
- 68.Gellerich FN, Laterveer FD, Korzeniewski B, Zierz S, Nicolay K. Dextran strongly increases the Michaelis constants of oxidative phosphorylation and of mitochondrial creatine kinase in heart mitochondria. Eur J Biochem. 1998;254:172–180. doi: 10.1046/j.1432-1327.1998.2540172.x. [DOI] [PubMed] [Google Scholar]
- 69.Saks VA, Kuznetsov AV, Khuchua ZA, Vasilyeva EV, Belikova JO, Kesvatera T, Tiivel T. Control of cellular respiration in vivo by mitochondrial outer membrane and by creatine kinase. A new speculative hypothesis: possible involvement of mitochondrial-cytoskeleton interactions. J Mol Cell Cardiol. 1995;27:625–645. doi: 10.1016/s0022-2828(08)80056-9. [DOI] [PubMed] [Google Scholar]
- 70.Ludzki A, Paglialunga S, Smith BK, Herbst EA, Allison MK, Heigenhauser GJ, Neufer PD, Holloway GP. Rapid Repression of ADP Transport by Palmitoyl-CoA Is Attenuated by Exercise Training in Humans: A Potential Mechanism to Decrease Oxidative Stress and Improve Skeletal Muscle Insulin Signaling. Diabetes. 2015;64:2769–2779. doi: 10.2337/db14-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varikmaa M, Bagur R, Kaambre T, Grichine A, Timohhina N, Tepp K, Shevchuk I, Chekulayev V, Metsis M, Boucher F, Saks V, Kuznetsov AV, Guzun R. Role of mitochondria-cytoskeleton interactions in respiration regulation and mitochondrial organization in striated muscles. Biochim Biophys Acta. 2014;1837:232–245. doi: 10.1016/j.bbabio.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 72.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A. 2008;105:18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kongas O, Wagner MJ, ter Veld F, Nicolay K, van Beek JH, Krab K. The mitochondrial outer membrane is not a major diffusion barrier for ADP in mouse heart skinned fibre bundles. Pflugers Arch. 2004;447:840–844. doi: 10.1007/s00424-003-1214-9. [DOI] [PubMed] [Google Scholar]
- 74.Saks VA, Belikova YO, Kuznetsov AV. In vivo regulation of mitochondrial respiration in cardiomyocytes: specific restrictions for intracellular diffusion of ADP. Biochim Biophys Acta. 1991;1074:302–311. doi: 10.1016/0304-4165(91)90168-g. [DOI] [PubMed] [Google Scholar]
- 75.Kunz WS, Kuznetsov AV, Schulze W, Eichhorn K, Schild L, Striggow F, Bohnensack R, Neuhof S, Grasshoff H, Neumann HW, Gellerich FN. Functional characterization of mitochondrial oxidative phosphorylation in saponin-skinned human muscle fibers. Biochim Biophys Acta. 1993;1144:46–53. doi: 10.1016/0005-2728(93)90029-f. [DOI] [PubMed] [Google Scholar]
- 76.Bigard AX, Boehm E, Veksler V, Mateo P, Anflous K, Ventura-Clapier R. Muscle unloading induces slow to fast transitions in myofibrillar but not mitochondrial properties. Relevance to skeletal muscle abnormalities in heart failure. J Mol Cell Cardiol. 1998;30:2391–2401. doi: 10.1006/jmcc.1998.0798. [DOI] [PubMed] [Google Scholar]
- 77.Walsh B, Tonkonogi M, Soderlund K, Hultman E, Saks V, Sahlin K. The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. J Physiol. 2001;537:971–978. doi: 10.1111/j.1469-7793.2001.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gellerich FN, Schlame M, Bohnensack R, Kunz W. Dynamic compartmentation of adenine nucleotides in the mitochondrial intermembrane space of rat-heart mitochondria. Biochim Biophys Acta. 1987;890:117–126. doi: 10.1016/0005-2728(87)90012-0. [DOI] [PubMed] [Google Scholar]
- 79.Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol. 1985;366:233–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu F, Yang F, Vinnakota KC, Beard DA. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J Biol Chem. 2007;282:24525–24537. doi: 10.1074/jbc.M701024200. [DOI] [PubMed] [Google Scholar]
- 81.Metelkin E, Demin O, Kovacs Z, Chinopoulos C. Modeling of ATP-ADP steady-state exchange rate mediated by the adenine nucleotide translocase in isolated mitochondria. FEBS J. 2009;276:6942–6955. doi: 10.1111/j.1742-4658.2009.07394.x. [DOI] [PubMed] [Google Scholar]
- 82.Jin Q, Bethke CM. Kinetics of electron transfer through the respiratory chain. Biophys J. 2002;83:1797–1808. doi: 10.1016/S0006-3495(02)73945-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.