

Abstract
The desire to analyse limited amounts of biological material, historic samples and rare cell populations has collectively driven the need for efficient methods for whole genome sequencing (WGS) of limited amounts of poor quality DNA. Most protocols are designed to recover double-stranded DNA (dsDNA) by ligating sequencing adaptors to dsDNA with or without subsequent polymerase chain reaction amplification of the library. While this is sufficient for many applications, limited DNA requires a method that can recover both single-stranded DNA (ssDNA) and dsDNA. Here, we present a WGS library preparation method, called ‘degraded DNA adaptor tagging’ (DDAT), adapted from a protocol designed for whole genome bisulfite sequencing. This method uses two rounds of random primer extension to recover both ssDNA and dsDNA. We show that by using DDAT we can generate WGS data from formalin-fixed paraffin-embedded (FFPE) samples using as little as 2 ng of highly degraded DNA input. Furthermore, DDAT WGS data quality was higher for all FFPE samples tested compared to data produced using a standard WGS library preparation method. Therefore, the DDAT method has potential to unlock WGS data from DNA previously considered impossible to sequence, broadening opportunities to understand the role of genetics in health and disease.
INTRODUCTION
Whole genome sequencing (WGS) has radically changed medical diagnostics and research and is a rapidly evolving technology platform (1). Illumina sequencing technologies facilitated expanding investigations from a single-region, single-gene approach to interrogating the whole genome simultaneously. While this approach is cost effective, WGS of fragmented genomic DNA is associated with sequencing and mapping artefacts, which are significantly more prevalent in formalin-fixed paraffin-embedded (FFPE) material (2,3). FFPE treatment is routinely used to preserve clinical specimens; however, it can result in extensive DNA damage (particularly DNA cross-links and deamination of cytosines) and fragmentation, leading to poor quality sequencing data, which renders many samples unusable for WGS. Consequently, large sequencing efforts such as ‘The 100,000 Genomes Project’ led by Genomics England have proposed that collection of fresh tissue should be standard of care in modern cancer diagnostics (4). Nevertheless, for retrospective studies FFPE tissues are often the only material available; therefore, there remains a need to develop new methodology that can improve sequencing quality.
There are numerous WGS library preparation methods available to researchers, and these differ in their price, preparation time and recommended input material (5). Most library preparation methods for WGS rely on attaching short double-stranded DNA (dsDNA) oligos to fragmented genomic dsDNA isolated from a fresh or FFPE sample of choice. The gold standard methods for WGS library preparation sold by major biotech companies continue to be improved over time in order to be applicable for very low amounts of input DNA, provided this material is of good quality (such as that isolated from fresh tissues or cells). One limitation of these kits is that the adaptor ligation step is inefficient (6) and will not recover single-stranded DNA (ssDNA).
In this study, we modified an existing method for DNA methylation analysis (7,8) to circumvent several inefficient steps associated with adaptor ligation-based library preparation methods. The degraded DNA adaptor tagging (DDAT) method utilizes random priming that can amplify ssDNA in addition to dsDNA that is captured by other current commercially available kits. Here, we investigate the efficiency of the DDAT method on FFPE samples of varying quality, evaluating library quality and yield, and directly comparing it to a standard preparation method that utilizes adaptor ligation.
MATERIALS AND METHODS
Sample information
FFPE blocks anonymized to the researchers were provided by collaborators at University College London Hospitals Biobank (REC approval 11/LO/1613) and Oxford University Hospitals (REC approval 10/H0604/72).
Genomic DNA extraction
DNA was extracted from FFPE colorectal cancer samples using the High Pure FFPET DNA Isolation Kit (Roche Diagnostics Ltd) according to the manufacturer’s protocol. DNA was quantified using the Qubit® 3.0 fluorometer (Life Technologies) and quality was estimated using a multiplex polymerase chain reaction (PCR)-based assay as previously described (9).
WGS library preparation—DDAT protocol
To remove damaged bases, 2 ng of good or poor quality FFPE DNA and 10 ng of very poor quality DNA were combined with 5 U of SMUG1, 1 U Fpg, 1× NEB buffer 1 and 0.1 μg/ml bovine serum albumin (NEB) in 10 μl and incubated for 1 h at 37°C. (This enzyme digestion step was excluded in the pilot experiment.) First strand synthesis was performed immediately afterwards by combining the 10 μl reaction with 1× blue buffer, 400 nM dNTPs and 4 μM oligo 1 (5′- CTACACGACGCTCTTCCGATCTNNNNNNNNN-3′) in 49 μl. Samples were heated to 95°C for 1 min and immediately cooled on ice. 50 U of Klenow (3′ → 5′ exo-; Enzymatics) fragment was added to each sample and the tubes were incubated at 4°C for 5 min before slow ramping (4°C/min) to 37°C (i.e. 8 min for the ramping step), and then held at 37°C for 90 min. After this step, samples can be stored overnight at −20°C if required. The remaining primers were digested with 20 U of exonuclease I (NEB) at 37°C for 1 h in 100 μl before purification using AMPure XP beads (Beckman). For purification, 80 μl AMPure XP beads were added directly to the samples and incubated for 10 min at room temperature. After collecting beads on a magnet, we performed 2 × 200 μl 80% ethanol washes on the magnet. Beads were dried for 6–10 min being vigilant not to allow beads to overdry and crack. DNA was eluted in 38 μl of water before adding components for second strand synthesis (1× blue buffer, 400 nM dNTPs and 0.8 μM oligo 2 [5′-CAGACGTGTGCTCTTCCGATCTNNNNNNNNN-3′]) to the PCR tube still containing the beads. Samples were heated at 98°C for 2 min and then incubated on ice before 50 U of Klenow (3′ → 5′ exo-) was added and incubated using the same conditions as for first strand synthesis. To purify the second strand synthesis reaction, an aliquot of AMPure XP beads was centrifuged and the supernatant collected. After addition of 50 μl of water to the sample, 80 μl of bead buffer was added and mixed to resuspend the beads still within the tube and the DNA was purified as described earlier. After the final drying step, beads were resuspended in 33 μl of water and incubated for 10 min to elute the DNA. The beads were collected using a magnetic rack and the 33 μl of purified DNA transferred to a new PCR tube before adding the components for the final library PCR amplification (1× KAPA HiFi buffer, 400 nM dNTPs, 1 U KAPA HiFi Hotstart Taq, PE1.0 (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) and the indexed custom reverse primer based on the Illumina TruSeq sequence (5′-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′). For the pilot experiment, the library was dual indexed using NEBNext® Multiplex Oligos for Illumina® (NEB). Samples were amplified for 10 PCR cycles before purification of library using a 1:0.8 ratio of DNA to beads and elution in 15 μl of water. The library was quantified using the Qubit® 3.0 fluorometer, 2200 TapeStation (Agilent, Santa Clara, CA, USA) and KAPA Library Quantification Kit (Roche).
WGS library preparation—standard protocol
Good quality FFPE DNA was sonicated using the Covaris M220 focused ultrasonicator to an average fragment size of 300 bp; poor and very poor quality samples did not require further fragmentation (Supplementary Figure S1). DNA was then repaired using the NEBNext® FFPE DNA Repair Mix, according the manufacturer’s protocol (New England Biolabs, Hitchin, UK). Library preparation was performed using the NEBNext® Ultra II™ DNA Library Prep Kit for Illumina® according to the manufacturer’s protocol for FFPE samples (New England Biolabs; half volumes of all reagents were used in the pilot experiment) and 10 cycles of library amplification, during which the library was indexed using custom PE1.0 (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) and indexed reverse primer (5′-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′; index sequence underlined). For the pilot experiment, the library was dual indexed using NEBNext® Multiplex Oligos for Illumina® (NEB). The library was purified and quantified using the same methods as described for DDAT. We chose the NEBNext Ultra II DNA Library Prep Kit for comparison as although many WGS library preparation reagents are now commercially available, this kit is very widely used in the community, and as such is a relevant comparison for our DDAT methodology.
Bioinformatics analysis pipeline
For each sample, the paired-end sequence reads were initially quality checked with FastQC v0.11.5 to investigate base quality scores, sequence length distributions and additional features of the data. The reads were then aligned to the reference human genome hg19 (for the pilot experiment) and hg38 (for the samples in Supplementary Table S1) by the BWA-MEM algorithm used in Burrows–Wheeler Aligner v0.7.8. The resulting SAM file was processed into a BAM file using Samtools v1.3.1, and then sorted and indexed with PCR duplicates marked using Picard v2.6 and v2.12 (for the pilot experiment and samples in Table 1, respectively). The final BAM files were quality checked using BamQC v0.1 and Picard v2.12 to investigate mapping qualities, coverage uniformity, percentage of soft clipped reads (reads that require some trimming from 5′ or 3′ to remove bases that are of low quality) and other basic statistics of the processed data shown in Table 2 and Supplementary Table S2. Coverage statistics and mapped insert size histograms (reads/base) were calculated with DepthofCoverage tool in GATK v3.6 and Picard v2.12. VCF files were generated using GATK v4.0 Mutect2.
Table 1.
Sample and preparation details for WGS comparing standard versus DDAT library preparation
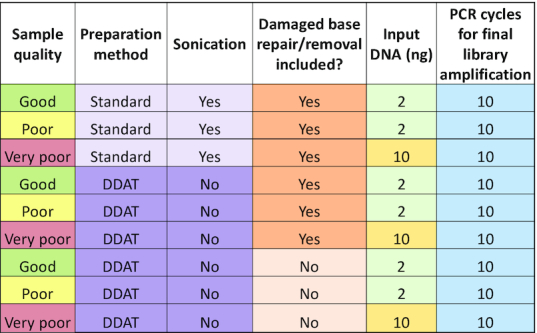
|
Table 2.
Sample alignment metrics for WGS comparing standard versus DDAT library preparation
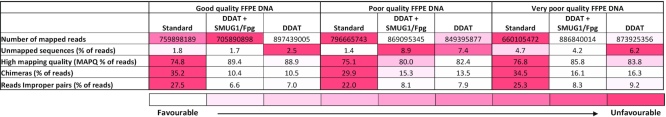
|
Statistical analysis
Significance testing was performed using Prism (v.5.04) and one-way ANOVA with Bonferroni post-hoc tests as specified in the figure legend. Where applicable, data are plotted as mean ± standard error of the mean (SEM).
RESULTS
DDAT library preparation improves sequencing quality and increases depth compared to standard methods
We first performed a pilot experiment to compare WGS data generated using the DDAT and the NEBNext Ultra II (‘standard’) library preparation methods, using a representative FFPE colorectal cancer DNA sample. We anticipated that the DDAT method would be of greatest benefit for FFPE DNA that was substantially degraded, as these samples contain more ssDNA that is inaccessible using the standard method. We therefore used poor quality FFPE DNA for our first test of the DDAT method (see Supplementary Figure S1 for assessment of FFPE DNA quality using multiplex PCR).
The DNA input for both library preparation methods was 2 ng and both used 10 PCR cycles of final library amplification. In the pilot experiment, we did not use the ‘Damaged base removal’ step in the DDAT method (Figure 1). After preparing libraries and performing quality controls (Supplementary Figure S2, Supplementary Table S1), we sequenced samples on Illumina's HiSeq X Ten, achieving close to 440 million raw reads in both cases.
Figure 1.

Work flow of the standard versus DDAT library preparation method. To generate WGS libraries from low-input, degraded DNA, the complete protocol starts with the addition of enzymes SMUG1 (single-strand-selective monofunctional uracil-DNA glycosylase) and Fpg (formamidopyrimidine [fapy]-DNA glycosylase) to the input DNA (A and B) that remove damaged bases such as deoxyuracil and 8-oxoguanine, caused by the FFPE treatment. A short denaturation step (B) is followed by the first strand synthesis; during this step, the genomic DNA, primers and Klenow fragment (3′ → 5′ exo-) are gradually heated from 4 to 37°C with a slow ramping speed of 4°C/min, which is an essential reaction condition (see ‘Discussion’ section), before incubation at 37°C for a further 1.5 h (C). The primers contain nine random nucleotides from the 3′-end, in addition to the standard Illumina adaptor sequence, and will anneal to complementary DNA sequences present in the DNA sample. After the first strand synthesis, any remaining primers or short ssDNA fragments are digested with exonuclease I and the dsDNA is purified with AMPure XP beads. Next, the dsDNA is denatured to carry out the second strand synthesis using a second adaptor primer also containing nine random nucleotides, with the same conditions as the first synthesis, followed by bead purification (C). Finally, 10 PCR cycles are carried out using standard Illumina p5 and p7 indexed primers (D). The library is purified and assessed using standard quality control methods.
After filtering and mapping the reads to the human genome, we assessed the alignment metrics and found that the DDAT method gave a mean 2.5-fold increase in coverage (Figure 2A, Supplementary Table S2) and 80% of these reads had a high mapping quality (MAPQ ≥ 20) compared to 70% using the standard method (Supplementary Table S2). The DDAT-generated library also had a larger median insert size of 162 bp compared to 96 bp (Figure 2B), another indication of an improved library preparation, with the caveat that the standard preparation includes an initial fragmentation step by sonication, which may explain this difference (Figure 1).
Figure 2.

Pilot experiment comparing DDAT and standard library preparation. (A) Comparison of genomic coverage after standard (blue) and DDAT (red) library preparation and sequencing. (B) Insert size of sequencing library prepared using the standard method (blue; 96 bp peak) or DDAT (red; 162 bp peak). (C) Sequencing reads shown on the Integrative Genomics Viewer. DDAT WGS data (upper panel) show a C>A transition (A base shown in green; chr 5: 112838399; GRCh38; total reads = 19, altered reads = 9, VAF = 0.474) resulting in a stop codon in the APC gene (p.Y935*, c.2805C>A; COSMIC19031). When using the standard library preparation method (lower panel), this region is not covered by enough reads to be identified (total reads = 2, altered reads = 2, VAF = 1).
To illustrate the utility of improved library preparation when identifying putative driver mutations in human cancers, we viewed aligned reads on the Integrative Genome Viewer (10,11) and identified a putative driver mutation in the APC gene (p.Y935*, c.2805C>A, Figure 2C). This mutation would be identified in the DDAT dataset using standard variant calling pipelines (altered reads = 9, total reads = 19, variant allele frequency [VAF] = 47.4%), but would likely have been filtered out of the data produced by the standard method due to only two reads covering the base (altered reads = 2, total reads = 2, VAF = 100%). The pilot experiment showed that we could generate superior WGS data with greater clinical value from 2 ng of poor quality FFPE DNA using DDAT compared to the standard method.
The DDAT protocol improves library yield compared to standard methods and can be used for very degraded FFPE samples
To perform a more comprehensive comparison of the two WGS library preparation methods, we selected three FFPE colorectal cancer DNA samples of variable quality (Supplementary Figure S1; samples highlighted in red). FFPE treatment of tissue commonly results in damage to DNA such as cytosine deamination to uracil; therefore, removing and/or repairing damaged DNA bases is crucial to prevent false positive mutational calls in WGS data (12). Since the repair of the damaged base using commercially available kits is reliant on a complementary strand template (as is the case for the standard method), damaged bases within ssDNA cannot be repaired since there is no opposite strand. We therefore wanted to assess whether excision of the damaged base, without repair, would improve the quality of the WGS data from FFPE DNA. To do this, we modified the DDAT protocol to include an initial enzyme digestion step using commercially available SMUG1 (excises deoxyuracil and deoxyuracil derivatives) and Fpg (an N-glycosylase and an AP-lyase that removes damaged based such as 8-oxoguanine). These enzymes create an abasic site in ssDNA and dsDNA, and the AP-lyase activity of Fpg creates a nick in the DNA backbone (13–15). In the standard method, a polymerase would then repair the gap in a dsDNA fragment by adding the missing complementary base; in contrast, in the DDAT method the missing base is not added and a heat denaturation separates the DNA strands, creating shorter ssDNA fragments where a damaged base has been removed. Table 1 summarizes the experimental set-up for this series of tests. We increased the input quantity of the very poor quality sample to 10 ng as the DNA was substantially degraded (Supplementary Figure S1).
We measured total yield of each sequencing library and found that the DDAT method (including damaged DNA removal) gave higher library yields compared to the standard method for all samples (Figure 3; good: 52-fold, poor: 9.8-fold and very poor: 23-fold). Library yields using the standard method were lower than expected; we hypothesized that this is due to the highly damaged and fragmented nature of the FFPE DNA. To test this, we extracted dsDNA from a cell line (i.e. not FFPE treated) and compared the library yield from 2 ng of intact dsDNA and sonicated DNA (Figure 4A), using either the standard method or DDAT. This head-to-head comparison of the two methods showed that the DDAT method can generate libraries from high molecular weight DNA (i.e. not sonicated; 26 nM), whereas the standard method cannot. Sonicating input DNA is required to produce a library using the standard method, and using DDAT only marginally improved the yield (yield: DDAT, 34 nM; standard, 7.1 nM; Figure 4B and C). However, based on these data using non-FFPE-treated and sonicated DNA, we cannot conclude that DDAT outperforms the standard method. The strength of DDAT is in generating higher yield libraries from FFPE-treated DNA, with improved sequencing quality and increased depth compared to the standard method. The addition of the damaged base removal step caused a slight decrease in library yield of the DDAT method (good: 1.35-fold, poor: 1.8-fold and very poor: 1.3-fold). When assessing insert size, the DDAT method (with or without damaged base removal) gave higher median insert sizes for all samples compared to the standard method (Supplementary Figure S3), which indicates better library quality. In general, the increased library yields and insert size indicated that the DDAT method was capturing more of the input DNA compared to the standard method, validating the results of the pilot experiment.
Figure 3.
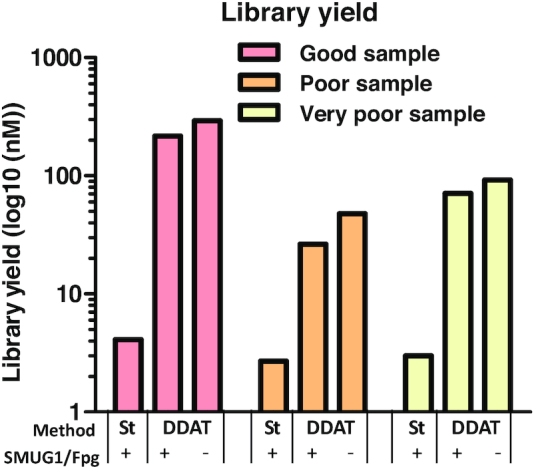
Library yield after standard versus DDAT method. The graph shows yield after standard or DDAT method from good (pink), poor (orange) and very poor (yellow) FFPE samples, with or without enzyme repair (n = 1). St = standard library preparation method.
Figure 4.

Library preparation from non-FFPE DNA. (A) TapeStation data showing size distribution of sonicated genomic DNA from KG-1 cell line used as the input for DDAT or standard library preparation. (B) Library yield after standard versus DDAT method using genomic DNA from cell line. Graph shows yield after standard or DDAT method from dsDNA (pink) or sonicated DNA (orange) (n = 1). (C) TapeStation data showing the size distribution of libraries generated using DDAT and dsDNA (left), sonicated DNA (middle) or the standard method and sonicated DNA (right).
Genome coverage for DDAT is up to 3.7-fold higher compared to the standard method
After sequencing the samples and aligning the reads to the human genome, we assessed the alignment metrics (Table 2). In general, data from the DDAT libraries were of higher quality than those from the standard libraries. The DDAT method resulted in higher mapping quality and lower proportions of chimeras and improper read pairs for all three samples. The addition of the damaged DNA removal step to the DDAT method did not have a consistent effect on the quality of the sequencing data, based on MAPQ scores. However, it is notable that in these samples the DDAT method resulted in a higher percentage of unmapped reads (see ‘Discussion’ section).
In agreement with our pilot experiment, the samples prepared using the DDAT method had higher genomic coverage than those prepared using the standard method (Figure 5, for DDAT + SMUG1/Fpg; good: 2.45-fold, poor: 2.54-fold, very poor: 3.77-fold; Supplementary Figure S4). For the good and poor quality samples, adding the damaged base removal step in the DDAT method decreased the coverage achieved in the aligned reads (Figure 5, pink versus blue lines; Supplementary Figure S4); however, for the very poor quality sample, the coverage remained the same (Figure 5, narrow pink and blue solid lines; Supplementary Figure S4).
Figure 5.
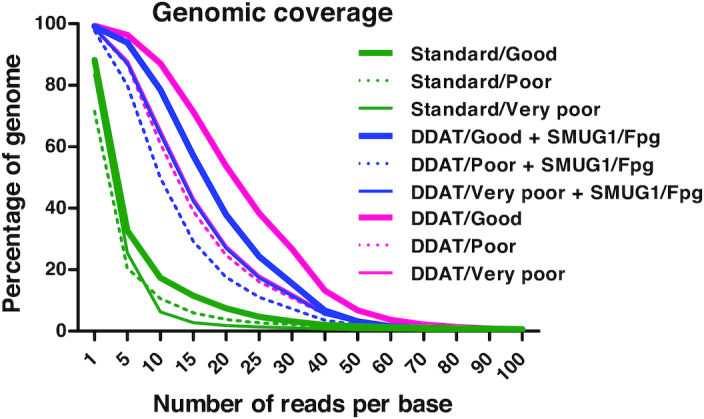
Genomic coverage after standard versus DDAT method. The figure shows genomic coverage after preparing libraries using the standard method (green), DDAT + enzyme (blue lines) or DDAT (pink lines). The coverage from libraries generated from good (thick solid lines), poor (dotted lines) and very poor (thin solid lines) FFPE samples is shown.
Glycosylase excision of damaged DNA bases reduces FFPE-induced sequencing artefacts in DDAT
To quantify whether removing the damaged DNA bases using enzymes SMUG1 and Fpg decreased the number of sequencing artefacts in the DDAT method, we calculated the ratio of C>T/A>G transitions within each dataset (Figure 6A) (16). This showed that when the damaged DNA bases were removed, the ratio decreased; therefore, including the enzyme digestion step significantly decreases the presence of C>T transitions for all FFPE samples (Figure 6B). This is comparable to the standard library preparation method that includes a DNA damage repair step. This demonstrates the importance of including SMUG1/Fpg digestion prior to the DDAT protocol to avoid FFPE-induced sequencing artefacts. Coverage uniformity was identical with or without enzyme digestion for good and very poor FFPE samples and very similar for poor sample (Figure 6C, as shown by the coefficient of determination; good sample with repair r2 = 0.87 versus without r2 = 0.87; poor sample with repair r2 = 0.75 versus without r2 = 0.72; very poor sample with repair r2 = 0.88 versus without r2 = 0.88), demonstrating that the coverage is not affected when artefacts are removed.
Figure 6.

C>T/A>G mutation ratio and coverage uniformity after standard or DDAT library preparation method. (A) The ratio of C>T/A>G mutations after DDAT (pink), DDAT + enzyme (orange) and standard (yellow) library preparation, for good (left), poor (centre) and very poor (right) FFPE DNA. (B) The average C>T/A>G ratio after combining data from all samples for DDAT (pink), DDAT + enzyme (orange) and standard (yellow). DDAT libraries prepared without enzyme digestion have a significantly higher C>T/A>G ratio indicating greater FFPE-induced artefacts in these libraries (mean ± SEM, n = 3). Statistical analysis (one-way ANOVA): F(2,8) = 12, P < 0.05. Bonferroni post-hoc test: *P < 0.05 compared with standard and DDAT + enzyme group. (C) The coverage uniformity of reads after standard library preparation (green), DDAT + enzyme (blue) and DDAT (pink). The coverage from libraries generated from good (dark shade), poor (medium shade) and very poor (light shade) FFPE samples is shown. Enz, enzyme.
In summary, the DDAT library preparation method increases the library yield and quality of WGS data when compared to a standard method. Therefore, application of DDAT to sequencing of degraded FFPE samples is expected to recover a larger fraction of the starting DNA material than standard methods. This increases library yield, allowing for fewer PCR cycles prior to sequencing and therefore fewer PCR duplicates in the sequencing data and a 2- to 3-fold increase in genomic coverage. In addition, since the library yield is higher, a lower amount of input DNA can be used, saving precious clinical material. DDAT does not require DNA shearing or sonication as FFPE treatment in itself causes DNA fragmentation, and only a short heat step is required to denature the dsDNA rendering it accessible for random primer amplification. By using DDAT, samples considered not amplifiable with standard methods can be used to generate sequencing libraries of improved quality, and furthermore the per-sample cost of DDAT is lower than that of commercially available kits. In other words, for the same sequencing throughput, 3- to 4-fold more usable reads are produced. The quality of the DDAT sequencing data is dependent on inclusion of an enzyme digestion step to remove FFPE-induced damaged DNA bases, minimizing FFPE-associated sequencing artefacts. Finally, the quality of the sequencing is significantly improved; therefore, more robust biologically relevant information can be extracted.
DISCUSSION
We have established a new methodology for generating WGS libraries using DDAT, which gives superior library yield and quality of WGS data from FFPE DNA compared to a standard commercially available kit. The improved efficiency is due to the two random priming and extension steps that enable ssDNA and dsDNA capture. As a result, the input DNA does not require an additional DNA fragmentation step (e.g. by sonication) before using DDAT, which further maintains the integrity of the DNA. This is particularly important when the input DNA is extracted from FFPE-treated tissue that is often already highly fragmented and single stranded.
During optimization of the protocol, we discovered that the ramp rate used to reach the 37°C incubation step during the first and second strand syntheses was crucial for efficient library preparation, with a faster ramping rate (132°C/min versus 4°C/min) reducing the overall library yield (Supplementary Figure S5). The reason for this effect is unclear; however, we hypothesize that the ramping rate affects the kinetics of random primer/DNA/Klenow binding, meaning that complexes are formed more efficiently if the temperature is gradually increased.
To detect the level of DNA degradation in our FFPE DNA, we used multiplex PCR of the GAPDH gene (Supplementary Figure S1), as this has been shown to give a good prediction of the quality of data from array comparative genomic hybridization for detecting CNVs (9), but further in-depth assessment including a greater range of degraded FFPE samples is needed to establish how well multiplex PCR predicts the quality of WGS data (17).
We have shown that removing damaged DNA bases in the DDAT method is sufficient to rescue the WGS data from FFPE-induced sequencing artefacts. Removal is the only option as the damaged bases in ssDNA cannot be repaired because there is no complementary strand to use as a template. Removal rather than repair does not seem to negatively impact the resulting WGS data as the yield and quality of data from the DDAT preparation with damaged base removal are generally improved compared to the standard method; furthermore, this type of damaged based removal has been shown to be effective for low DNA input targeted sequencing (16).
We considered whether the DDAT method would have potential problems, similar to those recently identified when using the PBAT method for whole genome bisulfite sequencing (7), namely, that the random priming increases chimeric reads (https://sequencing.qcfail.com/articles/pbat-libraries-may-generate-chimaeric-read-pairs/). However, based on the alignment statistics this does not appear to be the case when using DDAT as in fact we observe a lower proportion of chimeric reads for our DDAT prepared libraries than for our standard libraries (Table 2).
Alternative methods exist that can utilize ssDNA as well as dsDNA for WGS, for example, a method for generating WGS libraries from ancient DNA (18), and for targeted sequencing from clinical samples (16). However, both these methods rely on ligation of a single-stranded adaptor to ssDNA, which is inefficient compared to the random priming used in DDAT and therefore will give inferior library yield and sequencing data from low quantities of input DNA.
In summary, we have developed DDAT as an alternative WGS library preparation method that is particularly suited to highly degraded DNA samples containing ssDNA (e.g. archival FFPE samples). DDAT increases the yield and quality of FFPE WGS data and we anticipate that this method can be applied to generate high-quality WGS data from low input quantities, particularly from good quality starting material, improving our ability to obtain relevant data from samples previously deemed unsuitable for WGS.
DATA AVAILABILITY
Whole genome sequencing data that support the findings of this study have been deposited in Sequence Read Archive, accession number PRJNA531154.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Manuel Rodriguez-Justo (UCLH) and Simon Leedham (Oxford) for provision of samples.
Author Contributions: GF conceived the project; EAS and AB performed experiment; MW, KC and GF analysed bioinformatic data; GF and TAG supervised the project; all authors contributed to writing of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Medical Research Council [MR/M01892X/1 to G.F.]; Queen Mary University Proof of Concept Fund; Cancer Research UK [A19771 to A.-M.B and T.A.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shendure J., Balasubramanian S., Church G.M., Gilbert W., Rogers J., Schloss J.A., Waterston R.H. DNA sequencing at 40: past, present and future. Nature. 2017; 550:345–353. [DOI] [PubMed] [Google Scholar]
- 2. Wong S.Q., Li J., Tan A.Y., Vedururu R., Pang J.-M., Do H., Ellul J., Doig K., Bell A., MacArthur G.A. et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genomics. 2014; 7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Do H., Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 2015; 61:64–71. [DOI] [PubMed] [Google Scholar]
- 4. Turnbull C., Scott R.H., Thomas E., Jones L., Murugaesu N., Pretty F.B., Halai D., Baple E., Craig C., Hamblin A. et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ. 2018; 361:k1687. [DOI] [PubMed] [Google Scholar]
- 5. Head S.R., Komori H.K., LaMere S.A., Whisenant T., Van Nieuwerburgh F., Salomon D.R., Ordoukhanian P. Library construction for next-generation sequencing: overviews and challenges. BioTechniques. 2014; 56:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aigrain L., Gu Y., Quail M.A. Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays—a systematic comparison of DNA library preparation kits for Illumina sequencing. BMC Genomics. 2016; 17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miura F., Enomoto Y., Dairiki R., Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012; 40:e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smallwood S.a., Lee H.J., Angermueller C., Krueger F., Saadeh H., Peat J., Andrews S.R., Stegle O., Reik W., Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods. 2014; 11:817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Beers E.H., Joosse S.A., Ligtenberg M.J., Fles R., Hogervorst F.B.L., Verhoef S., Nederlof P.M. A multiplex PCR predictor for aCGH success of FFPE samples. Br. J. Cancer. 2006; 94:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson J., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Do H., Dobrovic A. Dramatic reduction of sequence artefacts from DNA isolated from formalin-fixed cancer biopsies by treatment with uracil-DNA glycosylase. Oncotarget. 2012; 3:546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alexeeva M., Moen M.N., Grøsvik K., Tesfahun A.N., Xu X.M., Muruzábal-Lecumberri I., Olsen K.M., Rasmussen A., Ruoff P., Kirpekar F. et al. Excision of uracil from DNA by hSMUG1 includes strand incision and processing. Nucleic Acids Res. 2019; 47:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haushalter K.A., Stukenberg P.T., Kirschner M.W., Verdine G.L. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 1999; 9:174–185. [DOI] [PubMed] [Google Scholar]
- 15. Howell W.M., Grundberg I., Faryna M., Landegren U., Nilsson M. Glycosylases and AP-cleaving enzymes as a general tool for probe-directed cleavage of ssDNA targets. Nucleic Acids Res. 2010; 38:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. So A.P., Vilborg A., Bouhlal Y., Koehler R.T., Grimes S.M., Pouliot Y., Mendoza D., Ziegle J., Stein J., Goodsaid F. et al. A robust targeted sequencing approach for low input and variable quality DNA from clinical samples. Genomic Med. 2018; 3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martelotto L.G., Baslan T., Kendall J., Geyer F.C., Burke K.A., Spraggon L., Piscuoglio S., Chadalavada K., Nanjangud G., Ng C.K.Y. et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat. Med. 2017; 23:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gansauge M.-T., Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013; 8:737–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing data that support the findings of this study have been deposited in Sequence Read Archive, accession number PRJNA531154.