

Abstract
Non‐covalent molecular interactions on the basis of halogen and chalcogen bonding represent a promising, powerful catalytic activation mode. However, these “unusual” non‐covalent interactions are typically employed in the solid state and scarcely exploited in catalysis. In recent years, an increased interest in halogen and chalcogen bonding has been awaken, as they provide profound characteristics that make them an appealing alternative to the well‐explored hydrogen bonding. Being particularly relevant in the binding of “soft” substrates, the similar strength to hydrogen bonding interactions and its higher directionality allows for solution‐phase applications with halogen and chalcogen bonding as the key interaction. In this mini‐review, the special features, state‐of‐the‐art and key examples of these so‐called σ‐hole interactions in the field of organocatalysis are presented.
Keywords: Sigma-hole, Halogen-bonding, Chalcogen, Organocatalysis, Non-covalent activation
Organocatalysis: Organo‐halogen and chalcogen compounds present σ‐holes, electropositive regions, which make the X or the Ch atom adopt a Lewis acidic role and provide unique features for developing novel catalytic substrate activation modes. This Minireview provides an overview on the recent introduction and current applications of halogen and chalcogen bonding interactions in non‐covalent organocatalysis.

1. Introduction
To achieve an effective catalytic transformation, the structural design of selective catalyst structures requires the correct manipulation of the energetic and stereochemical features of intermolecular forces. Although metal‐free organocatalysis has existed for more than a century,1 only recently a tremendous interest in this area aroused, due to the many opportunities that organocatalytic systems may offer in terms of catalyst design. Thus, many organocatalytic systems rely on the cooperation of multiple non‐covalent interactions, including a large range of attractive and repulsive forces.2 Amongst others, the main interactions include ion‐pairing, ion‐dipole interactions, dipole‐dipole interactions and hydrogen bonding. Many powerful organocatalysts have been designed on the basis of the latter, as its strength and directionality opens new possibilities to access more complex molecular targets.3 However, in terms of the “hard and soft (Lewis) acids and bases” (HSAB) concept,4 catalysts derived from hydrogen bonding share one certain disadvantage. As the substrate activation relies on the interaction through the “hard” hydrogen atom,5 “soft” starting materials may be only weakly coordinated or completely disregarded. Even though a vast amount of work on hydrogen bonding catalysis has paved the way to readily understand the underlying mechanisms and allowed for the development of highly asymmetric transformations,1, 3b, 6 the strong focus on one interacting atom does not allow for much variation towards orbital sizes and polarizabilities. In light of this, halogen (X) and chalcogen (Ch) bonding have emerged as promising alternatives and have been introduced as a key interaction for the design of novel organocatalysts. While their direct origin is still under debate, the so called σ‐hole interactions are originated from an anisotropically distributed electron density along the R−X or the R−Ch bond. This results in the appearance of frequent electropositive regions, which make the X or the Ch atom adopt a Lewis acidic role. This region is predestined to form non‐covalent interactions with Lewis basic compounds and is usually referred to as the σ‐hole.7 These interactions have been observed in many different contexts, including crystal engineering,8 supramolecular9 and medicinal chemistry.8a, 10 Besides the correlation to the electrostatic potential, also charge transfer and dispersion is believed to give rise to σ‐hole interactions.11 Associated with σ*‐orbitals, the σ‐hole deepens with heavier atoms (going from the top to the bottom of the periodic table) and with a higher polarizability, which increases from right to left in the periodic table.12 Due to the existence of a second substituent, it appears that chalcogen atoms possess two σ‐holes,13 whereas one σ‐hole is available for a halogen atom (Figure 1a).8a
Figure 1.

a) σ‐Holes that are predicted to increase with heavier atoms and a higher polarizability. b) Model structures candidates explored in catalysis.14
Furthermore, electron‐withdrawing residues geminal to the X or Ch atom, respectively, increase the σ‐hole(s) and allow for the formation of strong X and Ch complexes.8a, 15 Besides their classification as soft Lewis acids, which plays an important role in substrate preference, halogen and chalcogen bonds are significantly more directional than hydrogen bonding interactions. This aspect is particularly important for a strong complexation and requires angles close to 180° for R−Ch/X⋅⋅⋅LB (Lewis base).16 This is attributed to two specific characteristics: a) the σ* interactions of the R−H bond occur via a 1s‐orbital and b) the lack of filled p‐orbitals on the valency of the latter.13c
Regarding halogen and chalcogen bonding, most of their applications concern the solid state, whereas their employment in solution has largely lagged behind. However, taking into account the similarities to hydrogen bonding, the application of halogen and chalcogen bonding in solution was a logical consequence. In general, the Xs and Chs are able to bind more strongly to the substrate and can thereby create more rigid preorganized structures, which is likely to be beneficial to the stabilization of catalytic intermediates. However, the strict geometric requirements make it extremely challenging for both, the design and the synthesis of X‐bond‐ and Ch‐bond‐based catalysts. Nevertheless, non‐covalent halogen and chalcogen bonding in solution have only recently been applied in organocatalysis. While the use of halogen8a, 9b, 17 and chalcogen13, 18 bonding has been reviewed in several articles from different perspectives, we herein wish to provide an introductory overview of halogen and chalcogen bonding and their way into non‐covalent organocatalysis. Consequently, in this minireview only organocatalytic activation modes of halogen‐bond (XB)‐donor and chalcogen‐bond (ChB)‐donor catalysis will be discussed. Thus, catalysis by elemental halides or chalcogens, as well as transient and XB‐ or ChB‐assisted catalysis will not be covered.
2. Halogen Bonding
In the last years, the research field of halogen‐bonding is gaining increasing attention in the field of organocatalysis. Generally speaking, the ability of a halogen‐bond‐donor (XB‐donor) to establish a halogen bond strongly depends on the polarizability of the XB‐donor atom (I>Br>Cl≫F), as well as on the electronic properties of the attached residue on the halogen atom.8a As a consequence, most of the reported structures in XB‐donor catalysis rely on the higher polarized iodine‐based derivatives. Moreover, in analogy to the more explored hydrogen‐bond catalysis, typical benchmark and test reactions for XB‐donors often involve the activation of neutral substrates such as carbonyls (aldehydes or ketones), imines or heteroarenes (e. g. quinolines), and more recently halide‐abstraction reactions involving an anion‐binding‐type activation. Therefore, this section is divided according to these two main activation approaches. Thus, the pioneer and the most recent examples of this type of activation in organocatalysis is presented.
2.1. Catalytic Activation of Neutral Substrates
The catalytic activation of neutral electrophiles, such as ketones, aldehydes or imines are of high synthetic significance as non‐catalytic methods often require harsh conditions, which might enable unwanted side reactions to occur, or need a stoichiometric amount of a Lewis acid. Therefore, catalytic Lewis acids are of great interest and have been explored on a broad scope. Along with the aforementioned HB‐donors, XB‐donors have recently also found application in this field and are promising candidates to overcome challenging disadvantages of their HB‐donor analogues, due to their higher directionality and usually stronger binding affinities.
2.1.1. N‐Heteroarene Reduction Reactions
Most probably, the first organocatalytic reaction employing XB‐donors was reported in 2008 by Bolm and coworkers.19 In this work, the reduction of quinolines using haloperfluoroalkanes 1 in the presence of Hantzsch ester was achieved, in which the activation of the quinoline by halogen bonding was crucial (Scheme 1).
Scheme 1.

Pioneer XB‐bond‐catalyzed reduction of quinolines by Bolm et al.
In 2014, the group of Tan followed this approach and carried out some attempts towards an asymmetric reduction of quinolines by employing bidentate dihydroimidazolines of type 2 as halogen bond donors.20 Even though, no enantioselectivity was observed, the XB‐donor catalyzed the reaction in very good to excellent yields of up to 99 % with catalyst loadings of just 2 mol % (Scheme 2). Both electron‐withdrawing and electron‐donating substituents were well tolerated. Additionally, this method could be extended to more challenging pyridine and imine derivatives, which were also reduced successfully employing the same XB‐donor catalyst.
Scheme 2.
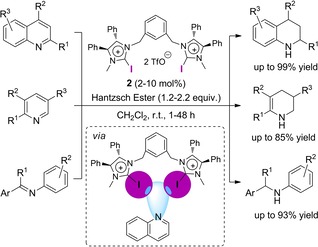
XB‐bond‐catalyzed reduction of quinolines, pyridines and imines.
2.1.2. Activation of Michael Acceptors and Carbonyls
In 2014 and 2018, Huber and coworkers investigated the catalytic activation of methyl vinyl ketone as neutral substrate in the Diels‐Alder reaction with cyclopentadiene (Scheme 3).21 In this case, a bidentate bis‐iodo‐imidazolium XB‐donor catalyst 3‐BArF (BArF=Tetrakis[3,5‐bis(trifluoromethyl)phenyl]borate) led to the quantitative formation of the product at room temperature after 3 h, while no reaction was observed in the absence of the catalyst.
Scheme 3.

Diels‐Alder reaction catalyzed by bidentate iodo‐imidazolium salts.
Another type of reported reactions comprises XB‐donor catalyzed Michael addition reactions. Inspired by elemental iodine catalyzed conjugated additions,22 the group of Huber reported a Michael addition of indole to α,β‐unsaturated ketones based on an iodo‐triazolium salt catalyst 4‐BArF, which was used as an activator providing conversions of up to 68 % after 36 h (Scheme 4, bottom left).23 This work was followed by Breugst and coworkers, which reported the Michael reaction with the XB‐donor catalyst 5 (Scheme 4, bottom middle).24 The iodobenzimidazolium salt 5 was able to catalyze the Michael reaction of trans‐crotonophenone with indole in 44 h. Although the reaction was significantly faster with elemental iodine and N‐bromo or N‐iodo succinimides, Huber and Breugst could successfully show that their imidazolium‐based halogen‐bond‐donor catalysts can also be used for the activation of Michael acceptors.
Scheme 4.

XB‐donor‐catalyzed Michael additions by Huber and Breugst et al.
Alternatively, an example for the activation of carbonyl groups was presented in 2017 by the group of Sekar using commercially available CBr4 as a XB‐donor catalyst to selectively activate benzaldehydes (Scheme 5).25 Although here one can argue if the symmetrical CBr4 can act as a XB‐donor, α,β‐unsaturated ketones could be easily synthesized upon Aldol‐ or Knoevenagel‐condensation reactions at mild and solvent‐free reaction conditions, hence tolerating sensitive functional groups. A variety of functional groups, including electron‐withdrawing and electron‐donating groups on both, the acetophenone and the benzaldehyde moieties, could be efficiently employed. Furthermore, the reaction could be performed on gram scale and the method applied for the synthesis of biologically active licochalcones.
Scheme 5.

Activation of benzaldehydes: CBr4‐catalyzed Aldol and Knoevenagel condensation reactions.
In 2017, Takemoto and co‐workers reported on the first application of XB‐bond donors for an electrophilic activation of iodonium(III) ylides for cross‐enolate coupling reactions (Scheme 6).26 With this umpolung reaction, various 1,4‐dicarbonyls were obtained by activating the iodonium ylides with the iodo‐imidazolium XB‐donor catalyst 6 and subsequently coupling them with different silyl enol ethers. Furthermore, this catalytic system proved to be able to couple in situ base‐activated 3‐substituted oxindoles with iodonium ylides, without observing the deactivation of the XB‐donor catalyst.
Scheme 6.

Cross‐enolate coupling catalyzed by monodentate XB‐donor 6.
2.1.3. Activation of Imines
The aza‐Diels‐Alder reaction between imines and substituted 1,3‐butadienes represents an important synthetic transformation and therefore has also awaken the attention as model reaction in XB‐donor catalysis, first introduced in 2014.27a In this regard, Fukuzawa and coworkers investigated the impact of steric bulk on the activity of iodotriazolium tetrafluoroborates as XB‐donor catalysts in the aza‐Diels‐Alder reaction between imines and 2‐siloxy‐1,3‐butadienes (Scheme 7),27b which could not be catalyzed by the corresponding HB‐donors or iodine. For this purpose, different aromatic substituents into 5‐iodo‐1,2,3‐triazolium‐salts were introduced by differing the residues on the azide and alkyne moieties. Eventually they found out that the derivative 7 bearing two mesityl groups was the most effective, while less sterically hindered phenyl‐ or the more hindered DIPS‐groups had a negative impact on the reaction outcome. Therefore, choosing the appropriate steric bulk around the catalytic center seems to be of utmost importance. In contrast to that and supported by DFT‐calculations, the mesityl groups are almost perpendicular towards the triazolium core. That leads to a lack of conjugation between the two units, thus providing a more localized positive charge, while also minimizing the steric repulsion of the methyl groups and the iodine‐atom. Finally, when conducting the reaction in dichloromethane at 30 °C for 14 h, the XB‐donor 7 afforded the product in a high 93 % yield.
Scheme 7.
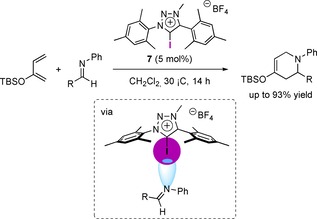
XB‐donor 7‐catalyzed aza‐Diels‐Alder reaction of imines and 2‐siloxy‐1,3‐butadienes.
Recently, the group of Kanger has notably contributed on the synthesis of chiral XB‐donor catalysts,28 as solely enantioselective XB‐donor catalysis remains challenging. Based on this, the group underwent efforts towards the development of an enantioselective dihydropyridinone synthesis by activation of an imine with chiral halo‐1,2,3‐triazolium salts, which then can further undergo an aza‐Diels‐Alder reaction with the Danishefsky's diene (Scheme 8).29 Amongst different substitution patterns and the choice of the counteranion, the triazolium triflate salt 8 showed the best catalytical performance, allowing catalyst loadings as low as 2 mol % to achieve quantitative yields after just 2 hours, however without enantiocontrol.
Scheme 8.

XB‐donor 8‐catalyzed aza‐Diels‐Alder reaction between imines and the Danishefsky's diene.
Furthermore, two plausible mechanistic pathways were proposed based on HMBC and HRMS analysis (Scheme 9). Accordingly, the reaction either proceeds by a) a Mannich‐Michael pathway or by b) a pericyclic [4+2]‐aza‐Diels‐Alder mechanism to obtain the desired dihydropyridinone products.
Scheme 9.

Plausible reaction mechanism via a) a Mannich‐Michael or b) a [4+2]‐aza‐Diels‐Alder pathway.
In 2009, Bew and coworkers reported on the synthesis of aziridines using a N‐fluoropyridinium salt 9 as catalyst to activate different N‐aryl amines to subsequently react with ethyl diazoacetate (Scheme 10).30 They were able to synthesize racemic aziridines in up to 83 % yield. It can be envisioned that this reaction is catalyzed through XB‐donor interactions, even though it was initially not proposed to proceed by halogen‐bonding.3
Scheme 10.

XB‐donor 9‐catalyzed aziridine synthesis.
2.1.4. Si‐X Bond Activation
In 2015, the group of Takemoto reported a direct dehydroxylative coupling reaction of alcohols and organosilanes like allyl TMS, by activating a Si−X bond by the XB‐donor catalyst (Scheme 11).31 Assisted by the halogen‐bond donor 10, in situ formed TMS‐I groups were capable of abstracting benzylic alcohol groups to form an ion pair and undergo a subsequent nucleophilic attack by TMS‐substituted nucleophiles. Electron donating and withdrawing groups were well tolerated on the phenyl ring and beside allyl‐TMS, alkinyl‐, cyano‐ and enolate‐TMS nucleophiles were also successfully used in this transformation.
Scheme 11.

XB‐donor‐catalyzed, TMS‐I induced activation of alcohols.
2.1.5. Miscellaneous
In 2016, the group of Sugita reported on the activation of thioamides by a XB‐catalyst based on a relatively simple, neutral iodoalkyne 11 (Scheme 12).32 In the presence of the catalyst, various thioamides were reacted with 2‐aminophenol leading to differently substituted benzoxazoles in good to quantitative yields. Apart from the simple catalyst design, further advantages of this neutral catalyst are the good solubility in organic solvents and superior stability compared to often labile and less soluble cationic XB‐donor structures, which decompose under these reaction conditions.
Scheme 12.

Iodoalkyne 11‐catalyzed benzoxazole formation.
More recently, the group of Sekar further published on the oxidation of heteroarlymethanes using N‐bromosuccinimide as XB‐donor catalyst towards the selective synthesis of ketones (Scheme 13).33 Initially, benzylic pyridines, which could undergo a XB‐assisted imine‐enamine tautomerism, were investigated. Thus, upon bromination of the enamine and a subsequent substitution by DMSO, the corresponding ketones were obtained in up to 96 % yield (Scheme 13, top). In the following, the active NBS catalyst is regenerated from succinimide and HBr. This method was further expanded for the oxidation of 2‐benzylbenzothiazoles in good yields (Scheme 13, bottom). In both cases, electron donating and withdrawing groups were well tolerated. However, mechanistic studies revealed that beside the imine‐enamine tautomer mechanistic pathway, another possible pathway might occur, at least partially: the halogen‐bond‐assisted electron transfer towards an heterobenzylic radical and subsequent trapping by oxygen, which is not shown here.
Scheme 13.

NBS‐catalyzed benzothiazole formation.
2.2. XB‐Donor Anion‐Binding Catalysis
2.2.1. Reactions with Benzhydryl Halides
Amongst the benchmark reactions for XB‐donor activators, the solvolysis of benzhydryl halides with wet acetonitrile constitutes one of the most favored and studied transformation as there are usually no background and side reactions like hidden Brønsted catalysis, which can easily be ruled out. However, due to the catalyst inactivation by binding of the halogen‐atom, in most cases stochiometric amounts of the activating reagents are still required.34 Nevertheless, considering the importance of this stoichiometric methodologies for the understanding and further development of XB‐donor catalysis, these examples are initially discussed in this section. Furthermore, this reaction is a good indicator for the strength of the established halogen‐bonds, as their hydrogen‐bond donor analogues are not able to successfully promote this reaction, i. e. establish too weak bonding for the abstraction of bromine or even more challenging chlorine atoms.
Initially, Huber et al. introduced various families of iodo‐based XB‐donors for the solvolysis of benzhydryl bromides (Scheme 14).34 The best results were achieved with bis‐/tri‐dentated XB‐donors such as p‐substituted(diimidazolium) benzene 12, tris(5‐iodo‐1,2,3‐triazolium)benzene 13 or 4,4′‐azobis(halopyridinium) salts 14.
Scheme 14.

Early XB‐donor‐mediated solvolysis of benzhydryl bromides.
More recently, Huber and coworkers explored cyclic iodolium salts as catalytically active, non‐covalent Lewis acids35 and explicit multivalent XB‐donor catalysts (Scheme 15).21b While hypervalent iodine(III) species are well‐established organic compounds, for example in the oxidation of functional groups,36 they also proved to be effective as XB‐donor catalysts. Thus, the reported monodentate iodolium salt 15 was used as an activating agent in the reaction of benzhydryl chloride with wet acetonitrile to obtain the acetamide in 80 % yield, thereby showing even an enhanced activity compared to previously reported bidentate XB‐donors such as 12–14.34 Furthermore, a multivalent halogen‐bonding activation mode with the catalyst 15 could be successfully confirmed. As a consequence, substitutions in the ortho‐positions relative to the iodine led to a complete inactivation of the catalysts by blocking the electrophilic axes of the iodine‐atom, hence preventing XB‐interactions.
Scheme 15.

Stoichiometric hypervalent iodine(III) X‐donor 15 in the solvolysis of benzhydryl chloride.
Based on preliminary studies from 2012,37a in 2018, the groups of Beer and Huber jointly investigated the role of charge in triazolium based XB‐donor activators, by comparing compounds such as neutral bistriazole 16, bicationic bis‐triazolilum salt 17, and monocationic pyridinium‐triazole derivative 18 (Scheme 16).37b Their activating strength in a Friedel‐Crafts alkylation of 1,3,5‐trimethoxybenzene with benzhydryl bromide as a benchmark reaction was then evaluated. Furthermore, the influence of the counteranion of the catalyst on the binding strengths for the monocationic species of 18 and its derivatives was also determined. While neutral XB‐donors of type 16 gave no observable formation of the product, compounds of type of 17 and 18 seemed to be equally active, showing similar binding affinities to anions and giving the product after five hours in 54 % and 42 % yield, respectively. Moreover, these results could be improved to up to 69 % yield when increasing the reaction time to 12 hours. They were then able to show that charge has a very important role in the activity of XB‐donor motifs, while the location of these charges does not seem to have a significant effect for this particular model reaction. Finally, XB‐donors of type 18 offer advantages over 17 as the dicationic catalysts often show low solubility in organic solvents and the anion exchange is generally facilitated in the monocationic species.
Scheme 16.

XB‐donor‐mediated Friedel‐Crafts alkylation of electron rich arenes.
2.2.2. Reactions with 1‐Chloroisochroman
After the initial stoichiometric studies with benzhydryl halides, Huber and coworkers successfully applied the multidentate, neutral XB‐donor 19 38 and the dicationic, rigid 4‐OTf 39 iodo‐derivatives as catalysts in another benchmark reaction such as the halide‐abstraction reaction of 1‐chloroisochroman with a silyl ketene acetal (Scheme 17).40 While both structures were catalytically active, 4‐OTf showed stronger halogen‐bond‐donor properties. Thus, it provided the alkylated ester in very good yields up to 91 % after 6–12 h with catalyst loadings down to 0.5 mol % (vs. 10 mol % for 19).
Scheme 17.

SN1‐type nucleophilic addition to 1‐chloroisochroman.
2.2.3. Reactions with Glycosyl Halides
In 2014, the groups of Huber and Codée reported in cooperation the activation of glycosyl halides by halogen bonding (Scheme 18).41 Unfortunately, when glucosyl halides were reacted with wet CD3CN, only the more reactive β‐anomers efficiently underwent a desired condensation reaction in the presence of the bidentate diazolium salt XB‐bond activator 3‐OTf. Instead, the formation of the un‐reactive α‐anomer needed 21 days to reach a conversion of >95 %. Interestingly, the glycosylation reaction with alcohols could be achieved in the presence of a stoichiometric amount of the activator 3‐OTf upon removal of two electron withdrawing alkoxy groups on the substrates. Thus, full conversion of L‐oleandrosyl chloride with isopropanol was observed, proving that XB‐donor‐mediated glycosylation is possible.
Scheme 18.

XB‐donor‐mediated glycosylation using 3‐OTf as activating reagent.
2.2.4. XB‐Catalyzed Cationic Polymerization Reactions
Another example for an early and simple application of XB‐donor activation was reported in 2010 for the controlled ring opening polymerization (ROP) of L‐lactide at room temperature promoted by stoichiometric ICl3.42 Based on this initial XB‐catalyzed polymerization, in 2017 Takagi and coworkers reported a controlled cationic polymerization of isobutyl vinyl ether catalyzed by 3‐OTf as the most efficient XB‐donor catalyst (Scheme 19).43 Concerning the numbers of average molar mass and suppression of side reactions, the use of 1‐(1‐chloroethoxy)‐2‐methylpropane (INI) as the initiator turned out to give the best results. The reaction proceeds via an ionic intermediate I, which is stabilized by halogen bonds from the catalyst as shown in Scheme 19.
Scheme 19.

XB‐donor‐catalyzed controlled cationic polymerization reaction.
2.3. Halogen‐π‐Bonding Catalysis
While most of the reported XB‐catalysis systems focus on the interactions between the catalyst and heteroatoms, like in the activation of ketones and aldehydes or halogen anion abstraction, Arai et al. published a new methodology implying the catalytic activation of π‐electrons in [4+2]‐cycloadditions by cationic halogen‐bond donors (Scheme 20).44 They revealed that 2‐iodoimidazolium salts, such as 20, are able to form halogen‐π‐bonds in order to activate 2‐alkenylindoles to undergo [4+2] cycloadditions. The reaction conditions where applied for the cyclization of two 2‐alkenylindoles of the same kind or differently substituted, proving the product in up to 99 % yield. The introduction of both electron withdrawing and donating substituents were well tolerated, providing the syn‐product as the major diastereomer with a d.r. up to 20 : 1. Interestingly, the hydrogen‐bond catalyst analogue of 20 only provided the product in 11 % yield, while the Brønsted acid catalyst TFA preferably promoted the formation of an uncyclized byproduct II in 54 % yield.
Scheme 20.

[4+2]‐Cycloaddition of 2‐alkenylindoles catalyzed by C–I–π‐halogen‐bond‐interactions.
3. Chalcogen Bonding
Before chalcogen bonding interactions could make a mark in the non‐covalent organocatalysis world, several examples of organochalcogen compounds as covalently bound chiral nucleophilic catalysts have been reported,13b, 45 which further benefit from intramolecular Ch bonding as a tool to rigidify chiral structures. Although at first glance it may seem counter‐intuitive, chalcogen bonds also play a role in Lewis base catalysis with the Ch atom interacting perpendicular to the R−Ch−R′ axis. The different mechanisms through which Ch‐based catalysis can act will be briefly introduced before the focus will be laid on its more recent intermolecular non‐covalent applications.
3.1. Chalcogen Bond‐Based Nucleophilic Catalysts
In the last decade, sulfur‐based chiral nucleophilic catalysts have evolved as an important new class of organocatalysts, which can be traced be back to their diverse modes of activation.45b, 46, 47 Besides the covalent attachment of the catalyst to one of the substrates, various enantioselective transformations could be realized by an additional non‐covalent chalcogen bonding interaction.45b, 46, 47 In this regard, the group of Birman established the use of tetramisole or isothioureas such as 21 for this kind of chemistry.47a, 48 This concept was realized in an early report by Romo et al. in the enantioselective synthesis of cyclic β‐lactones, in which a secondary Ch⋅⋅⋅O interaction was postulated (Scheme 21).49
Scheme 21.

Intramolecular Aldol lactonization for the enantioselective preparation of β‐lactones via a secondary Ch‐bonding stabilization.
This stabilizing nσ→σ*S–C interactions was first described in 1998 by Nagao and co‐workers and fixes the intermediate into a specific conformation (Figure 2).50 In 2013, the group of Smith further evidenced this type of interaction in a solid‐state crystallographic study.25
Figure 2.

Conformational stabilization of the isothiourea moiety.19, 23
This concept was successfully expanded to a broad range of asymmetric transformations, including organocascades,51 rearrangement,47c formal cycloaddition52 and annulation reactions.47b Very recently, a chiral isothiourea catalyst was reported to efficiently catalyze the addition of 4‐nitrophenol esters to iminium intermediates.53
3.2. Organochalcogens as Lewis Basic Organocatalysts
The first applications of chalcogens in non‐covalent organocatalysis were reported in the field of Lewis base catalysis,54 in which an interaction perpendicular to the R−Ch−R′ is believed to provide the major contribution. An important example was reported in 2013 by Yeung and co‐workers, who studied the effect of Ch‐containing Lewis bases in the chloroamidation of alkenes. The haloamidation was found to proceed efficiently when diphenyl selenide was employed as activating agent (Scheme 22a).55 Only recently, Zhao et al. published the chiral selenide 22 catalyzed intermolecular difunctionallization of unactivated olefins via a stable thiiranium ion (Scheme 22b).56
Scheme 22.

Organoselenides as Lewis basic organocatalysts.
3.3. Applications of Organochalcogens as Lewis Acids in Non‐Covalent Organocatalytic Reactions
As a consequence of the importance of Ch bonding in intramolecular conformation control in covalent catalysis and their role in Lewis base activation, the application of Ch bond‐based organocatalysts in intermolecular substrate activation started developing in 2017, when the group of Matile introduced Ch bond‐based catalysts that solely operate through Ch bonding interactions.57 In a proof‐of‐principle study, they examined dithienothiophene (DTT) moieties, previously successfully applied by the same group in anion‐transport processes,58 for the transfer hydrogenation of quinolines and imines with Hantzsch ester (Scheme 23).57 In the search of a suitable test reaction, DFT calculations served as a benchmark, as they indicated that nitrogen centers seem to better fit into the binding pocket than i. e. carbonyl groups.19 Several structures of catalyst candidates were tested, of which 30 mol % DDT 23 provided the desired product 2‐phenyl tetrahydroquinoline within 12 days at room temperature in 96 % yield. In accordance with computed anion binding energies for DDT 23, the addition of TBACl to the reaction mixture completely inhibited the catalyst's activity. Based on these evidential results for operational Ch bonding interactions, DDT diimides or DDIs were developed to increase the catalytic action. Thereby, a remarkable acceleration of the Hantzsch ester‐mediated transfer hydrogenation was found with kcat/kuncat=1290 for DDI 24 (for comparison: kcat/kuncat=490 for DDT 23). This resulted in a similar yield after only 7 days of reaction time and with a 10 mol % of catalyst loading.
Scheme 23.

ChB‐donor‐catalyzed transfer hydrogenation reported by Matile et al.
Based on this pioneering work, in 2017 Huber and co‐workers published the first use of selenium‐derived Ch bond donors.59 In analogy to their previously published halogen bond donors,39 cationic bis(benzimidazolium) organochalcogens 25 were found as efficient activators in the Ritter‐type solvolysis of benzhydryl bromide (Scheme 24). As mentioned before, this reaction is considered as halide abstraction (“anion binding”) benchmark reaction,34a with Lewis acids based on selenium providing a up to 30 times rate acceleration. Even though stoichiometric amounts of the Lewis acid were necessary, chalcogen bonding could be identified as the actual mode of activation by several comparative experiments. Whereas the brominated analogue provided the amide product in 35 %, Ch bond donor 25 yielded the product in 64 % after a prolonged 140 h of reaction time.
Scheme 24.

Carbon‐halogen bond activation by Se‐based Ch‐bonding reported by Huber et al.
Shortly after, the groups of Huber60 and Matile61 independently introduced other organoselenides in Ch‐catalytic applications. On the one hand, Matile and coworkers studied benzodiselenazoles (BDS) 26 and 27 as new chalcogen bonding scaffolds in the same transfer hydrogenation test reaction of quinolines as described above (Scheme 25a).61 This newly developed catalysts exhibit maximized directionality as the bite angle of 45° delivers the perfect environment for the electrophilic interplay of two selenium atoms (Scheme 25b). Going from 26 to 27, the Lewis acidity of the catalyst structure was increased by a simple oxidation of the sulfur substituents. The use of the more electron‐poor BDS 27 was translated into a more than 105 orders of magnitude higher rate enhancement with only 1.0 mol % catalyst loading compared to BDS 26, which showed no activity at all. Moreover, a cationic version of 27 was recently introduced as an active catalyst in the Ritter‐type solvolysis of benzhydryl bromide, giving up to 60 % yield after 800 h (Scheme 25).62
Scheme 25.
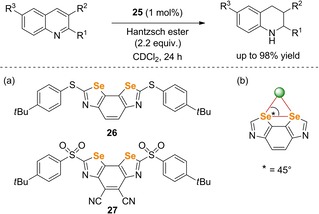
(a) ChB‐donor‐based BDS catalysts 26–27 developed by Matile et al. and (b) schematic illustration of chloride (green) bound to the BDS core showing an ideal bite angle of 45°.
On the other hand, Huber and co‐workers showed the catalytic potential of analogues of bis(benzimidazolium) 3 in a second example (Scheme 26).60 For this purpose, the reaction of 1‐chloroisochroman with a silyl ketene acetal was established as a benchmark case for chalcogen bonding. This reaction was firstly reported in 2008 with chiral hydrogen bond donors by the group of Jacobsen40 and further used to develop the first example of halogen bonding catalysis.39 It was found that the bis(benzimidazolium)‐derived catalysts of type 25 were able to provide up to 92 % yield of the product. Moreover, a bidentate mode of coordination could be confirmed with the anti‐isomer of the Ch bond donors 25 being less active than the syn‐isomers. Their outstanding catalytic activity was further underlined by a direct comparison to the brominated version, which generated the desired product in only 40 % yield.
Scheme 26.

ChB‐donor‐catalyzed reaction of 1‐chloroisochroman with a silyl ketene acetal. TBS=tert‐butyldimethylsilyl.
In 2018, the group of Matile introduced after an extensive study conceptually simple perfluorinated Ch‐bond donors in chloride‐binding catalysis.14 In this regard, they explored the halide abstraction ability of 28 and 29 in the transformation of both 1‐chloroisochroman to the corresponding esters and the Reissert‐type substitution of isoquinoline using Troc‐Cl (Troc=2,2,2‐trichloroethoxy carbonyl) as acylating agent (Scheme 27). In the latter, the tellurium‐based catalyst 28 accelerated the reaction by a factor of 52 compared to the uncatalyzed reaction. After 4–6 h, the product was formed in 48 % yield, whereas the analogue selenium compound 29 was remarkably less active giving only 6 % of the product after 55 h. Similar reactivities were obtained in the chloride abstraction reaction of 1‐chloroisochroman. To evaluate the relative strength of the chalcogen bond donor, this seminal work provides a profound evidence that σ‐hole interactions account for strong binding. In addition, a comparative assessment with homologous halogen‐ and also pnictogen‐bonding catalysts was conducted.
Scheme 27.

Chloride abstraction of 1‐chloroisochroman and Troc‐protected 1‐chloroisoquinoline by ChB‐catalysts 28 and 29.
Only recently, the group of Wang introduced Ch‐bonding catalysts with an exceptional ability to activate carbonyl groups leading to N‐heterocycles via three chalcogen‐bonding activation steps.63 In this regard, catalyst 30 was able to promote the cyclization of indoles with β‐ketoaldehydes to give the final seven‐membered N‐heterocycle in 78 % yield (Scheme 28). Thorough control experiments and mechanistic studies identified the Se⋅⋅⋅O interaction as crucial in the cyclization process with no observable product formation in absence of a Ch‐bonding catalyst.
Scheme 28.

ChB‐donor‐catalyzed assembly of small molecules: Synthesis of fused N‐heterocycles via Michael/aldol/cyclization tandem reaction.
4. Conclusion and Outlook
It was in the 90’s when the regions of positive electrostatic potential opposite to covalent C−X bonds involving halogen atoms were first reported. This observation and the interaction with an electronegative site in the so‐called halogen‐bonding (XB) interaction were further extended to compounds presenting C−Z bonds involving atoms of other groups such as chalcogens. However, only after the introduction of the term σ‐hole in 2007, the real potential of this field was recognized. As a consequence, the interest of σ‐hole interactions and their application in several research areas has experienced an exponential increase in the past few years.
Although halogen and chalcogen bonding has been already widely utilized in several areas such as crystallography, material sciences, supramolecular or medicinal chemistry, in the field of catalysis the use of X‐ and Ch‐bonding activation is still underrepresented. In this regard, and inspired by the more studied and well‐known homogeneous hydrogen bonding catalysis, the to date few reported catalytic applications have recently revealed that halogen and chalcogen bonding represent a powerful alternative. Additionally, considering the hardly exploited unique features of halogen and chalcogen bonding, it can be envisioned that in the near future fascinating new reactivities and catalytic applications will be developed.
However, some important issues still remain unsolved. In this regard, the development of enantioselective transformations based on purely XB‐ and ChB‐catalysis represent one of the most difficult current tasks. Although several attempts with chiral catalysts have been made, the strong directionality of the σ‐hole – substrate interaction (∼180°) bring important challenges for the catalyst design. Thus, besides the presented outstanding catalytic applications, deeper studies towards a better understanding of the σ‐hole interactions involved and further fundamental aspects in this type of catalysis are still needed for achieving future key advances in this research field.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Julia Bamberger, born in 1990, studied chemistry at the University of Regensburg and obtained her M.Sc. degree in 2015. In summer 2019 she obtained her Ph.D. degree from the University of Münster under the supervision of Prof. García Mancheño. Her research interests are focused on enantioselective applications and mechanistic studies in the field of non‐covalent anion‐binding organocatalysis.

Biographical Information
Florian Ostler obtained his M.Sc. degree in chemistry from the University of Regensburg in 2017, after which he started his Ph.D. in the group of Prof. García Mancheño. In 2018 he moved with her group to the University of Münster to further pursue his PhD. Since 2019 he is funded by the IRTG‐2027 WWU‐Toronto. He is working in the fields of hydrogen‐ and halogen‐bonding catalysis.

Biographical Information
Olga García Mancheño received her Ph.D. in 2005 from the Universidad Autónoma de Madrid under the supervision of Prof. J.C. Carretero. She also realized short research stays with Prof. M.T. Reetz (MPI für Kohlenforschung) and Prof. K.A. Jørgensen (University of Aarhus). After her postdoc in the group of Prof. C. Bolm at the RWTH‐Aachen (2005–2008), she started her independent career at the University of Münster. In 2013, she was appointed as joint Chemistry Professor at the University of Regensburg and TUM‐Campus Straubing. Since 2017 she is Professor for Organic Chemistry at the University of Münster.

Acknowledgements
The European Research Council (ERC‐CG 724695) and the Deutsche Forschungsgemeinschaft (DFG) within both the IRTG2027 and the SFB858 are gratefully acknowledged for generous support.
J. Bamberger, F. Ostler, O. G. Mancheño, ChemCatChem 2019, 11, 5198.
References
- 1. Schreiner P. R., Chem. Soc. Rev. 2003, 32, 289–296. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Knowles R. R., Jacobsen E. N., Proc. Mont. Acad. Sci. 2010, 107, 20678–20685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. MacMillan D. W. C., Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Beckendorf S., Asmus S., García Mancheño O., ChemCatChem 2012, 4, 926–936; [Google Scholar]
- 3b. Doyle A. G., Jacobsen E. N., Chem. Rev. 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]
- 4. Pearson R. G., J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- 5. Mayr H., Breugst M., Ofial A. R., Angew. Chem. Int. Ed. 2011, 50, 6470–6505. [DOI] [PubMed] [Google Scholar]
- 6. Mattson A. E., Science 2017, 358, 720–720. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Politzer P., Murray J. S., Clark T., Phys. Chem. Chem. Phys. 2013, 15, 11178–11189; [DOI] [PubMed] [Google Scholar]
- 7b. Politzer P., Murray J. S., Clark T., Resnati G., Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [DOI] [PubMed] [Google Scholar]
- 8.See for example:
- 8a. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478–2601; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Kremer A., Fermi A., Biot N., Wouters J., Bonifazi D., Chem. Eur. J. 2016, 22, 5665–5675; [DOI] [PubMed] [Google Scholar]
- 8c. Bai M., Thomas S. P., Kottokkaran R., Nayak S. K., Ramamurthy P. C., Guru Row T. N., Cryst. Growth Des. 2014, 14, 459–466; [Google Scholar]
- 8d. Werz D. B., Gleiter R., Rominger F., J. Am. Chem. Soc. 2002, 124, 10638–10639; [DOI] [PubMed] [Google Scholar]
- 8e. Li H., Eddaoudi M., O′Keeffe M., Yaghi M., Nature 1999, 402, 276–279. [Google Scholar]
- 9.See for example:
- 9a. Gleiter R., Werz D. B., Rausch B. J., Chem. Eur. J. 2003, 9, 2676–2683; [DOI] [PubMed] [Google Scholar]
- 9b. Cavallo G., Metrangolo P., Pilati T., Resnati G., Sansotera M., Terraneo G., Chem. Soc. Rev. 2010, 39, 3772–3783; [DOI] [PubMed] [Google Scholar]
- 9c. Robinson S. W., Mustoe C. L., White N. G., Brown A., Thompson A. L., Kennepohl P., Beer P. D., J. Am. Chem. Soc. 2015, 137, 499–507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Felix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122–3133. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Reid R. C., Yau M.-K., Singh R., Lim J., Fairlie D. P., J. Am. Chem. Soc. 2014, 136, 11914–11917; [DOI] [PubMed] [Google Scholar]
- 10b. Beno B. R., Yeung K.-S., Bartberger M. D., Pennington L. D., Meanwell N. A., J. Med. Chem. 2015, 58, 4383–4438. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Huber S. M., Jimenez-Izal E., Ugalde J. M., Infante I., Chem. Commun. 2012, 48, 7708–7710; [DOI] [PubMed] [Google Scholar]
- 11b. Thirman J., Engelage E., Huber S. M., Head-Gordon M., Phys. Chem. Chem. Phys. 2018, 20, 905–915; [DOI] [PubMed] [Google Scholar]
- 11c. Angarov V., Kozuch S., New J. Chem. 2018, 42, 1413–1422. [Google Scholar]
- 12.
- 12a. Bauza A., Frontera A., Mooibroek T. J., ChemPhysChem 2015, 16, 2496–2517; [DOI] [PubMed] [Google Scholar]
- 12b. Lim J. Y. C., Beer P. D., Chem. 2018, 4, 731–783. [Google Scholar]
- 13.
- 13a. Breugst M., von der Heiden D., Schmauck J., Synthesis 2017, 49, 3224–3236; [Google Scholar]
- 13b. Mahmudov K. T., Kopylovich M. N., Guedes da Silva M. F. C., Pombeiro A. J. L., Dalton Trans. 2017, 46, 10121–10138; [DOI] [PubMed] [Google Scholar]
- 13c. Vogel L., Wonner P., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [DOI] [PubMed] [Google Scholar]
- 14. Benz S., Poblador-Bahamonde A. I., Low-Ders N., Matile S., Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Iwaoka M., Tomoda S., J. Am. Chem. Soc. 1996, 118, 8077–8084; [Google Scholar]
- 15b. Iwaoka M., Isozumi N., Molecules 2012, 17, 7266–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.See for example:
- 16a. Hassel O., Roemming C., Quart. Rev. 1962, 16, 1–18; [Google Scholar]
- 16b. Weiss R., Schlierf C., Schloter K., J. Am. Chem. Soc. 1976, 98, 4668–4669; [Google Scholar]
- 16c. Row T. N. G., Parthasarathy R., J. Am. Chem. Soc. 1981, 103, 477–479; [Google Scholar]
- 16d. Metrangolo P., Meyer F., Pilati T., Resnati G., Terraneo G., Angew. Chem. Int. Ed. 2008, 47, 6114–6127; [DOI] [PubMed] [Google Scholar]
- 16e. Bulfield D., Huber S. M., Chem. Eur. J. 2016, 22, 14434–14450. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Beale T. M., Chudzinski M. G., Sarwar M. G., Taylor M. S., Chem. Soc. Rev. 2013, 42, 1667–1680; [DOI] [PubMed] [Google Scholar]
- 17b. Tepper R., Schubert U. S., Angew. Chem. Int. Ed. 2018, 57, 6004–6016; [DOI] [PubMed] [Google Scholar]
- 17c. Nagorny P., Sun Z., Beilstein J. Org. Chem. 2016, 12, 2834–2848; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Politzer P., Murray J. S., Clark T., Phys. Chem. Chem. Phys. 2010, 12, 7748–7757; [DOI] [PubMed] [Google Scholar]
- 17e. Guha S., Kazi I., Nandy A., Sekar G., Eur. J. Org. Chem. 2017, 2017, 5497–5518. [Google Scholar]
- 18. Zhao Y., Cotelle Y., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 4270–4277. [DOI] [PubMed] [Google Scholar]
- 19. Bruckmann A., Pena M. A., Bolm C., Synlett 2008, 900–902. [Google Scholar]
- 20. He W., Ge Y.-C., Tan C.-H., Org. Lett. 2014, 16, 3244–3247. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Jungbauer S. H., Walter S. M., Schindler S., Rout L., Kniep F., Huber S. M., Chem. Commun. 2014, 50, 6281–6284. [DOI] [PubMed] [Google Scholar]
- 21b. Heinen F., Engelage E., Dreger A., Weiss R., Huber S. M., Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [DOI] [PubMed] [Google Scholar]
- 22. Banik B. K., Fernandez M., Alvarez C., Tetrahedron Lett. 2005, 46, 2479–2482. [Google Scholar]
- 23. Gliese J.-P., Jungbauer S. H., Huber S. M., Chem. Commun. 2017, 53, 12052–12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. von der Heiden D., Detmar E., Kuchta R., Breugst M., Synlett 2018, 1307–1313. [Google Scholar]
- 25. Kazi I., Guha S., Sekar G., Org. Lett. 2017, 19, 1244–1247. [DOI] [PubMed] [Google Scholar]
- 26. Saito M., Kobayashi Y., Tsuzuki S., Takemoto Y., Angew. Chem. Int. Ed. 2017, 56, 7653–7657. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Takeda Y., Hisakuni D., Lin C.-H., Minakata S., Org. Lett. 2015, 172, 318–321; [DOI] [PubMed] [Google Scholar]
- 27b. Haraguchi R., Hoshino S., Sakai M., Tanazawa S.-G., Morita Y., Komatsu T., Fukuzawa S.-I., Chem. Commun. 2018, 54, 10320–10323. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Kaasik M., Kaabel S., Kriis K., Järving I., Aav R., Rissanen K., Kanger T., Chem. Eur. J. 2017, 23, 7337–7344; [DOI] [PubMed] [Google Scholar]
- 28b. Kaasik M., Kaabel S., Kriis K., Järving I., Kanger T., Synthesis 2019, 2128–2135. [DOI] [PubMed] [Google Scholar]
- 29. Kaasik M., Metsala A., Kaabel S., Kriis K., Järving I., Kanger T., J. Org. Chem. 2019, 84, 4294–4303. [DOI] [PubMed] [Google Scholar]
- 30. Bew S. P., Ashford P.-A., Fairhurst S. A., Hughes D. L., Legentil L., Liddle J., Pesce P., Nigudkar S., Wilson M. A., Org. Lett. 2009, 11, 4552–4555. [DOI] [PubMed] [Google Scholar]
- 31. Saito M., Tsuji N., Kobayashi Y., Takemoto Y., Org. Lett. 2015, 17, 3000–3003. [DOI] [PubMed] [Google Scholar]
- 32. Matsuzawa A., Takeuchi S., Sugita K., Chem. Asian J. 2016, 11, 2863–2866. [DOI] [PubMed] [Google Scholar]
- 33. Guha S., Sekar G., Chem. Eur. J. 2018, 24, 14171–14182. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Walter S. M., Kniep F., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2011, 50, 7187–7191; [DOI] [PubMed] [Google Scholar]
- 34b. Kniep F., Rout L., Walter S. M., Bensch H. K. V., Jungbauer S. H., Herdtweck E., Huber S. M., Chem. Commun. 2012, 48, 9299–9301; [DOI] [PubMed] [Google Scholar]
- 34c. Kniep F., Walter S. M., Herdtweck E., Huber S. M., Chem. Eur. J. 2012, 18, 1306–1310. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Pinto de Magalhães H., Togni A., Lüthi H. P., J. Org. Chem. 2017, 82, 11799–11805; [DOI] [PubMed] [Google Scholar]
- 35b. Labattut A., Tremblay P.-L., Moutounet O., Legault C. Y., J. Org. Chem. 2017, 82, 11891–11896. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Richardson R. D., Wirth T., Angew. Chem. Int. Ed. 2006, 45, 4402–4404; [DOI] [PubMed] [Google Scholar]
- 36b. Singh F. V., Wirth T., Chem. Asian J., 2014, 9, 950–971. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Walter S. M., Kniep F., Rout L., Schmidtchen F. P., Herdtweck E., Huber S. M., J. Am. Chem. Soc. 2012, 134, 8507–8512; [DOI] [PubMed] [Google Scholar]
- 37b. Dreger A., Engelage E., Mallick B., Beer P. D., Huber S. M., Chem. Commun. 2018, 54, 4013–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kniep F., Jungbauer S. H., Zhang Q., Walter S. M., Schindler S., Schnapperelle I., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2013, 52, 7028–7032. [DOI] [PubMed] [Google Scholar]
- 39. Jungbauer S. H., Huber S. M., J. Am. Chem. Soc. 2015, 137, 12110–12120. [DOI] [PubMed] [Google Scholar]
- 40. Reisman S. E., Doyle A. G., Jacobsen E. N., J. Am. Chem. Soc. 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Castelli R., Schindler S., Walter S. M., Kniep F., Overkleeft H. S., van der Marel G. A., Huber S. M., Codée J. D. C., Chem. Asian J. 2014, 9, 2095–2098. [DOI] [PubMed] [Google Scholar]
- 42. Coulembier O., Meyer F., Dubois P., Polym. Chem. 2010, 1, 434–437. [Google Scholar]
- 43. Takagi K., Yamauchi K., Murakata H., Chem. Eur. J. 2017, 23, 9495–9500. [DOI] [PubMed] [Google Scholar]
- 44. Kuwano S., Suzuki T., Yamanaka M., Tsutsumi R., Arai T., Angew. Chem. Int. Ed. 2019, 58, 1–6. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. Abbasov M. E., Hudson B. M., Tantillo D. J., Romo D., J. Am. Chem. Soc. 2014, 136, 4492–4495; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45b. Robinson E. R. T., Fallan C., Simal C., Slawin A. M. Z., Smith A. D., Chem. Sci. 2013, 4, 2193–2200; [Google Scholar]
- 45c. Fukata Y., Asano K., Matsubara S., J. Am. Chem. Soc. 2015, 137, 5320–5323. [DOI] [PubMed] [Google Scholar]
- 46. Merad J., Pons J.-M., Chuzel O., Bressy C., Eur. J. Org. Chem. 2016, 5589–5610. [Google Scholar]
- 47.
- 47a. Birman V. B., Li X., Org. Lett. 2006, 8, 1351–1354; [DOI] [PubMed] [Google Scholar]
- 47b. Joannesse C., Johnston C. P., Morrill L. C., Woods P. A., Kieffer M., Nigst T. A., Mayr H., Lebl T., Philp D., Bragg R. A., Smith A. D., Chem. Eur. J. 2012, 18, 2398–2408; [DOI] [PubMed] [Google Scholar]
- 47c. Liu G., Shirley M. E., Van K. N., McFarlin R. L., Romo D., Nat. Chem. 2013, 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.
- 48a. Birman V. B., Li X., Org. Lett. 2008, 10, 1115–1118; [DOI] [PubMed] [Google Scholar]
- 48b. Yang X., Birman V. B., Adv. Synth. Catal. 2009, 351, 2301–2304; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48c. Yang X., Birman V. B., Chem. Eur. J. 2011, 17, 11296–11304. [DOI] [PubMed] [Google Scholar]
- 49. Leverett C. A., Purohit V. C., Romo D., Angew. Chem. Int. Ed. 2010, 49, 9479–9483. [DOI] [PubMed] [Google Scholar]
- 50. Nagao Y., Hirata T., Goto S., Sano S., Kakehi A., Iizuka K., Shiro M., J. Am. Chem. Soc. 1998, 120, 3104–3110. [Google Scholar]
- 51.
- 51a. Belmessieri D., Morrill L. C., Simal C., Slawin A. M. Z., Smith A. D., J. Am. Chem. Soc. 2011, 133, 2714–2720; [DOI] [PubMed] [Google Scholar]
- 51b. Liu G., Shirley M. E., Van K. N., McFarlin R. L., Romo D., Nat. Chem. 2013, 5, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith S. R., Douglas J., Prevet H., Shapland P., Slawin A. M. Z., Smith A. D. , J. Org. Chem. 2014, 79, 1626–1639. [DOI] [PubMed] [Google Scholar]
- 53. Arokianathar J. N., Frost A. B., Slawin A. M. Z., Stead D., Smith A. D., ACS Catal. 2018, 8, 1153–1160. [Google Scholar]
- 54.
- 54a. Vedejs E., Denmark S. E., Lewis Base Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2016; [Google Scholar]
- 54b. Lenardao E. J., Santi C., Sancineto L., New Frontiers in Organoselenium Compounds, Springer International Publishing: Cham, 2018. See also, e. g.: [Google Scholar]
- 54c. Denmark S. E., Beutner G. L., Angew. Chem. Int. Ed. 2008, 47, 1560–1638; [DOI] [PubMed] [Google Scholar]
- 54d. Denmark S. E., Burk M. T., Proc. Mont. Acad. Sci. 2010, 107, 20655–20660; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54e. Zhou L., Tan C. K., Jiang X., Chen F., Yeung Y.-Y., J. Am. Chem. Soc. 2010, 132, 15474–15476; [DOI] [PubMed] [Google Scholar]
- 54f. Denmark S. E., Chi H. M., J. Am. Chem. Soc. 2014, 136, 8915–8918; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54g. Jana S., Verma A., Rathore V., Kumar S., Synlett 2019, 30, 1667–1672. [Google Scholar]
- 55. Tay D. W., Tsoi I. T., Er J. C., Leung G. Y. C., Yeung Y.-Y., Org. Lett. 2013, 15, 1310–1313. [DOI] [PubMed] [Google Scholar]
- 56. Liu X., Liang Y., Ji J., Luo J., Zhao X., J. Am. Chem. Soc. 2018, 140, 4782–4786. [DOI] [PubMed] [Google Scholar]
- 57. Benz S., Lopez-Andarias J., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 812–815. [DOI] [PubMed] [Google Scholar]
- 58. Benz S., Macchione M., Verolet Q., Mareda J., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 9093–9096. [DOI] [PubMed] [Google Scholar]
- 59. Wonner P., Vogel L., Dueser M., Gomes L., Kniep F., Mallick B., Werz D. B., Huber S. M., Angew. Chem. Int. Ed. 2017, 56, 12009–12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wonner P., Vogel L., Kniep F., Huber S. M., Chem. Eur. J. 2017, 23, 16972–16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benz S., Mareda J., Besnard C., Sakai N., Matile S., Chem. Sci. 2017, 8, 8164–8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Benz S., Besnard C., Matile S., Helv. Chim. Acta 2018, 101, e1800075. [Google Scholar]
- 63. Wang W., Zhu H., Liu S., Zhao Z., Zhang L., Hao J., Wang Y., J. Am. Chem. Soc. 2019, 141, 9175–9179. [DOI] [PubMed] [Google Scholar]