

Abstract
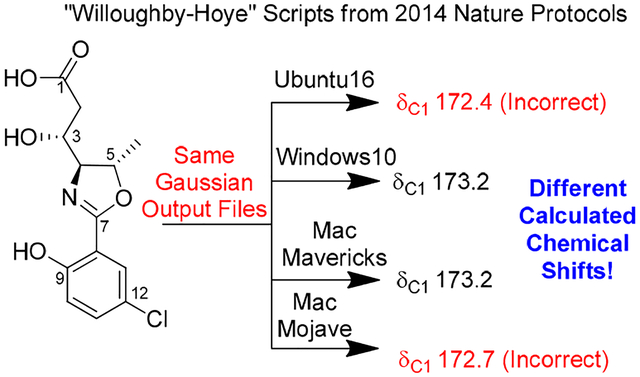
Bioactivity-guided examination of a Leptolyngbya sp. led to the isolation of leptazolines A-D (1-4), from the culture media, along with two degradation products (5-6). DFT NMR calculations established the relative configurations of 1-2 and revealed the calculated shifts depended on the operating system when using the “Willoughby-Hoye” python scripts to streamline the processing of the output files; a previously unrecognized flaw that could lead to incorrect conclusions.
Graphical Abstract
As part of our long-standing interest in cyanobacterial natural products, we recently began screening strains within our culture collection against pancreatic adenocarcinoma (PANCA).1 This screen flagged several media extracts. While metabolites from cells are well-studied, there are comparatively few reports of cyanobacterial secondary metabolites excreted in the culture medium to any appreciable degree.2 Excreted metabolites are likely to be more polar and structurally different from those isolated from the cell mass. We report here the results from the examination of strain O-2–5, a Leptolyngbya sp, i.e., the isolation and characterization of leptazolines A-D (1-4), and two hydrolysis products (5–6).3 Assignment of the relative configurations of 1 and 2 involved calculating NMR chemical shifts using a widely-cited protocol outlined by Willoughby et al.,4 which revealed a surprising operating system dependence on the calculated values due to issues with one of the python scripts.
The molecular formula of leptazoline A (1) was established as C13H14ClNO5 from its HRESIMS signal at m/z 300.0637 [M+H]+ and isotopic patterns indicative of a chlorine atom, i.e. a 3:1 ratio of m/z 300 and 302. The 1H and 13C NMR spectra (Table 1 and Table S1; MeOH-d4) revealed the presence of three aromatic proton signals consistent with a 1,2,4-trisubstituted benzene ring, i.e., δH 7.02 (d, J = 8.8 Hz), 7.47 (dd, J = 8.8, 2.7 Hz), and 7.53 (d, J = 2.7 Hz). Analysis of the NMR data suggested that a chlorine atom, an oxygen atom and
Table 1.
NMR Spectroscopic Data of Leptazoline A (1) in DMSO-d6
| no. | δC, type | δH (J) | COSY | HMBC or CIGAR |
|---|---|---|---|---|
| 1 | 173.8,C | |||
| 2 | 39.0,CH2 | 2.46, dd (15.4, 3.8) 2.37, dd (15.3, 8.6) |
1, 3, 4 | |
| 3 | 68.0,CH | 3.99, dt (8.6, 3.8) | 2, 4 | 1, 2c, 5 |
| 4 | 75.7,CH | 3.93, dd (6.3, 3.8) | 2, 3, 6, 7, 8c | |
| 5 | 77.7,CH | 4.77, p (6.3) | 4, 6 | 3, 4c, 7 |
| 6 | 20.5,CH3 | 1.38, d (6.3) | 4, 5 | |
| 7 | 163.4,C | |||
| 8 | 111.7,C | |||
| 9 | 158.2,C | |||
| 10 | 118.5,CH | 7.02, d (8.8) | 11 | 7w, 8, 9, 12, 13w |
| 11 | 133.2,CH | 7.47, dd (8.8, 2.7) | 8w, 9, 12, 13 | |
| 12 | 122.2,C | |||
| 13 | 126.6,CH | 7.53, d (2.7) | 11w | 7, 9, 10c, 11, 12 |
| NH | −170.4a | 12.28, brs |
Nitrogen chemical shift from 1H−15N HMBC
15N referenced to MeNO2 (IUPAC std). Referenced to NH3 it resonates at 210.1 ppm
CIGAR only
Weak correlations
a sp2 hybridized carbon were attached to this ring at C-12, −9 and −8, respectively. These assignments were consistent with the carbon chemical shifts reported for 5-chlorosalicylic acid, with the same substitution pattern,5 and yielded the lowest mean average error (MAE) of the calculated 13C NMR shifts given the other possible isomers (Table S2).
A COSY correlation connected the lone methyl doublet (H-6) to H-5 (4.77 ppm), while an HMBC correlation from H-6 to C-4 (δC 75.7) as well as a COSY correlation between H-5 and H-4 (δH 3.93) extended the chain. A further network of COSY correlations from H-4 to H-3 and then to H-2 completed the proton-coupled spin-system. The large geminal coupling constant (>15 Hz) for the methylene protons H-2 indicated an n-acceptor substituent, presumably a carbonyl moiety given the molecular formula, was at C-1,6–8 which was supported by an HMBC correlation from H-3 to C-1 (δC 173.8), giving a hexanoyl fragment.
The hexanoyl fragment and the 5-chlorosalicylic acid derivative were connected by an oxygen atom between C-5 and C-7, based on their chemical shifts and an HMBC correlation from H-5. At the time, based on 13C NMR chemical shifts and the molecular formula, we assumed C-7 was an ester carbonyl carbon. An HMBC correlation existed between H-4 and C-7 as well but we opted for the C-7-O-C-5 connection based on the relatively upfield chemical shift of H-4 (3.93 ppm) compared to H-5 (4.77 ppm) that was more consistent with the presumed ester.
Thus, the main structural framework of the molecule was established but the additional heteroatoms, exchangeable protons and the remaining degree of unsaturation – a ring – could not be placed with certainty based on the available spectroscopic data. The carbon chemical shifts of C-3, C-4 and C-9 all suggest oxygenation, which given the atoms remaining, then suggested C-1 might be an amide assuming C-7 was an ester.
Compound 1 was acetylated to determine which carbons bore heteroatoms with exchangeable protons and which were within the ring system. This reaction resulted in the formation of two products 7-8, each of which showed nominal m/z values of 342 [M+H]+ indicating monoacylation. Purification of these products by HPLC and analysis of the resulting 1H NMR spectra showed a downfield shift of H-3 in 7 consistent with acetylation of a hydroxyl group, while the other product 8 had acetylation of the phenolic OH. Given C-5 and C-7 were already connected via an oxygen atom and C-3 and C-9 had acetylated, the unassigned ring must therefore cyclize via some combination of C-1, C-4 and C-7.
It was readily apparent at this point that the location and identity of the nitrogen containing functional group was the key to this elucidation. A 1H−15N HMBC experiment resulted in a single correlation from H-3 to a nitrogen atom resonating at −170.4 ppm, which clearly indicated an amide or amine was not present.9 Assuming a 3-bond correlation, these observations indicated the nitrogen atom was attached to C-4, and C-1 was a carboxylic acid. A broad singlet signal at 12.28 ppm was observed in the 1H NMR spectrum obtained in DMSO-d6 further supporting this conclusion.10 As stated earlier, the chemical shift of C-4 (75.7 ppm) suggested attachment to an oxygen atom, but it was also in reasonable agreement for a carbon atom in an oxazoline ring (Figure 1). While cyanobacteria of the genus Leptolyngbya are known to produce various bioactive metabolites, such as the palmyrolides11 and the coibamides12, it does not appear that oxazoline-containing compounds have been reported from this genus previously.
Figure 1.

Structure of 1 deduced from COSY (Bold) and 1H−13C (red) and 1H−15N HMBC (Blue) correlations.
Due to degradation of 1, the relative configuration was established through DFT calculations of the NMR chemical shifts.2 The lowest energy conformers for each diastereomer of 1 were identified using MacroModel (Maestro/Schrodinger), optimized in Gaussian 0913 and their zero-point energy calculated before the NMR shielding tensors of the 1H and the 13C nuclei were computed. The chemical shift values for each diastereomer were obtained using published intercepts and slopes in consideration of the Boltzmann distribution of the conformers.
Comparison of the 1H and 13C chemical shifts (Table S3) shows that the experimental values are most closely aligned with the computed values for the (3R*,4S*,5S*)-diastereomer (1d). Based on the DP4+ probabilities14 computed from these datasets (Table S3), the predicted NMR shifts of the diastereomer 1d deviate the least from the experimental data (96.7% probability considering 1H and 13C). These calculations suggest that the (3R*,4S*,5S*)-diastereomer (1d) reflects the correct relative configuration of the compound 1.
The molecular formula of 2 was established as C16H21ClN2O7 from its HRESIMS signal at m/z 389.1110 [M+H]+. The spectral data for 2 (Table S4) had many similarities to 1 but there were a few noticeable differences, including an additional C3H7NO2 in the molecular formula. Analysis of 2D NMR data established that C-2 (73.5 ppm) was oxygenated and C-1 and C-1’ were linked through an amide bond. Evidence to support the amide bond includes the absence of the broad O-H band observed in 1 in the IR spectrum (see Figure S22), and a second nitrogen atom observed at −263.0 ppm in the 1H−15N HMBC NMR spectrum. Oxygenated carbons C-2’ and C-3’ were magnetically equivalent and attached to C-1’ based on COSY correlations from the respective protons, which formed a 2-aminopropan-1,3-diol moiety that has been found in other natural products.15, 16 These changes accounted for the difference in molecular formula.
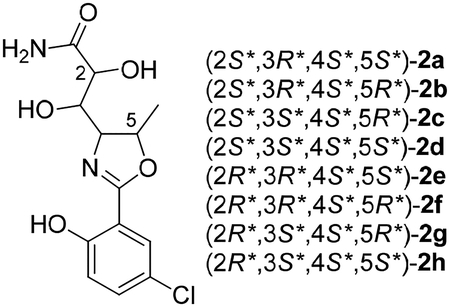
Assignment of the relative configuration of 2 relied on calculated NMR shifts. While we hypothesized that the three chiral centers shared with 1 would remain unchanged in 2, we were able to compute the 1H and 13C NMR chemical shifts of all eight diastereomers using minimal computing resources by truncating to the primary amide (2a-h) which reduced the number of conformers of each diastereomer from greater than 250 to less than 10. The calculated data for the diastereomers 2a-h and their DP4+ probabilities suggest conflicting results when 1H and 13C chemical shifts (See Table S5) are considered separately, but 2d, which corresponds to (2S*,3S*,4S*,5S*)-2, appears as the clear choice when both data sets are taken into account.
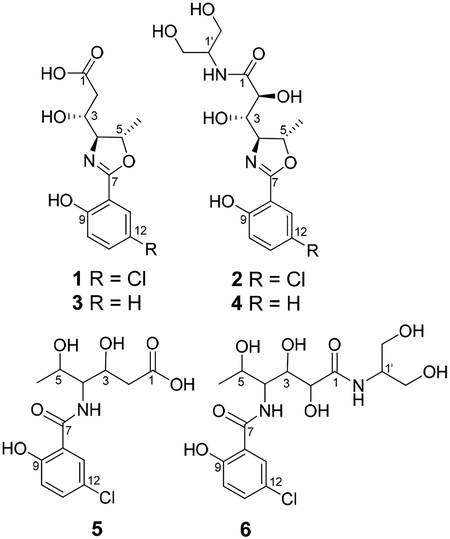
Analogs 3-6 were also isolated. Although there was insufficient material to record 13C NMR data, the planar structures and relative configurations could be determined from comparison of their HRMS and 1H NMR data with 1–2. For example, 3 and 4 had 1H NMR spectra indicative of a 1,2-disubstituted benzene ring without a chlorine atom, which was consistent with the MS data. The hydrolyzed products 5 and 6 were determined from the characteristic increase of 18 mass units in the mass spectra and examination of the 1D and 2D NMR data, and are likely the result of the inclusion of formic acid in the HPLC mobile phase.
Compounds 1 and 2 were isolated in sufficient quantities to be evaluated in our PANC-1 assay system. While 1 did not significantly inhibit the proliferation of Panc-1 cells, 2 did show inhibition of growth (GI50 10 μM). Further biological evaluation was, however, hindered due to degradation of the molecules.
While preparing a manuscript, to our surprise, attempts by team members to replicate these results produced different calculated NMR chemical shifts despite using the same Gaussian files and same procedure outlined by Willoughby et al. For example, all attempts concluded 1d was the correct diastereomer but these conclusions were based on chemical shifts that appeared to depend on the computer system on which Step 15 of that protocol was performed (Table 2).
Table 2.
Variability in Calculated Carbon Chemical Shifts of 1b
| no. | LINUX (Ubuntu16) | Windows (ver. 10) | Mac (Mavericks) | Mac (Mojave) |
|---|---|---|---|---|
| 1 | 172.4 | 173.2 | 173.2 | 172.7 |
| 2 | 36.0 | 37.7 | 37.7 | 39.3 |
| 3 | 68.3 | 68.4 | 68.4 | 69.0 |
| 4 | 70.6 | 70.5 | 70.5 | 71.2 |
| 5 | 79.0 | 78.4 | 78.4 | 79.0 |
| 6 | 15.5 | 13.3 | 13.3 | 13.3 |
| 7 | 162.4 | 162.5 | 162.5 | 161.8 |
| 8 | 110.4 | 109.8 | 109.8 | 110.3 |
| 9 | 155.0 | 156.5 | 156.5 | 155.5 |
| 10 | 116.2 | 115.5 | 115.5 | 116.0 |
| 11 | 131.6 | 131.6 | 131.6 | 131.7 |
| 12 | 127.1 | 126.6 | 126.6 | 127.0 |
| 13 | 126.7 | 125.6 | 125.6 | 126.3 |
Published in 2014, this Nature Protocols manuscript provides detailed instructions aimed at enabling those with minimal theoretical knowledge of the subject area to calculate GIAO NMR chemical shifts and includes python scripts to streamline the process. It has been cited over 130 times in the last five years. Detailed investigations traced the source of the discrepancies to these python scripts which summarize the conformers, free energies, Boltzmann distributions and isotopic tensors from the various Gaussian output files and then use this information to calculate Boltzmann averaged chemical shifts (Step 15). Specifically the script “nmr-data_compilation” extracts the free energy information of each conformer and the isotopic tensors for each atom from two files, e.g. 1b-opt_freq-conf-16.out and 1b-opt_NMR-conf-16.out, respectively, and performs the necessary calculations
In theory, the chemical shift of each atom from each isomer should be properly weighted according to the free energy of the conformer, but this was not consistently the case. In the end, the inconsistency was traced to differences in the default file sorting algorithm in python across platforms, as shown in Figure 2, and that, as written, the script “nmr-data_compilation” assumes the frequency and NMR files are sorted in the same order. The Boltzmann contribution calculated from the first frequency file in the directory is simply applied to the chemical shifts calculated in the first NMR file. For example, in Windows 10 the script works as designed: two groups of output files, *opt_freq-*-ID.out containing free energies and *nmr-*-ID.out containing chemical shifts, are sorted automatically by the system such that the conformers are paired correctly (Figure 2B).
Figure 2.

Inconsistent Sorting of Files
However, in LINUX there is no default sorting of file names as this depends on the local settings, and the scripts as designed do not check that frequency and NMR files are properly matched. As a result, the free energies could be paired with chemical shifts from different conformers (Figure 2A) which leads to incorrectly calculated chemical shifts that could surely lead to a wrong final conclusion.
To overcome this issue, we amended the script to include a line in the read_gaussian_outputfiles subroutine that forces sorting before paring, and a longer file-matching check function that alerts the user when there is a potential file matching issue (see Supporting Information).17 We have tested the revised script across platforms (i.e. MacOS, LINUX, and Windows) running different versions of python and it yields consistent results.
This simple glitch18 in the original script calls into question the conclusions of a significant number of papers on a wide range of topics in a way that cannot be easily resolved from published information since the operating system is rarely mentioned. In the first half of 2019 alone the protocol was referenced/used during the elucidation of several natural products,19,20,21–26 to characterize reaction products,21, 23, 27 and to understand biosynthetic pathways.28 Authors who used these scripts should certainly double-check their results and any relevant conclusions using the modified scripts in the SI. Ultimately, this example serves as a reminder of the principal caveat emptor, and that users should validate non-commercial software on their system prior to use on new applications.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge the advanced computing resources provided by the University of Hawaii Information Technology Service Cyberinfrastructure. We thank Dr. Charles O’Kelly (Cyanotech, HI) for the taxonomic analysis. This work was supported by NIH grant 5R01AG039468 to PW. Funds for the upgrades of the NMR instrumentation were provided by the CRIF program of the National Science Foundation (CH E9974921), the Elsa Pardee Foundation, and the University of Hawaii at Manoa. The purchase of the Agilent TOF LC-MS was funded by grant W911NF-04-1-0344 from the Department of Defense, and the purchase of the Agilent QTOF LC-MS was funded by MRI grant 1532310 from the National Science Foundation. We thank T. Hoye (University of Minnesota) and P. Willoughby (Ripon College) for the helpful discussions about the python scripts.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
General experimental details, NMR spectra for 1-6, NMR calculation for 2, picture of the producing organism (pdf)
Raw NMR data for all new compounds (zip)
Revised nmr-data_compilation python scripts and instructions (zip)
REFERENCES
- (1).Goess R; Friess H, A Look At The Progress Of Treating Pancreatic Cancer Over The Past 20 Years. Expert. Rev. Anticancer. Ther 2018, 18, 295–304. [DOI] [PubMed] [Google Scholar]
- (2).Neupane RP Isolation and Elucidation of Structures of Biologically Active Secondary Metabolites from Various Organisms, Including Cyanobacteria, Sponges and Areca Catechu. 2017.
- (3).Compounds 1–6 were never detected by LC-MS in the cell mass, which was also inactive in the original screen against PANC1.
- (4).Willoughby PH; Jansma MJ; Hoye TR, A Guide To Small-molecule Structure Assignment Through Computation Of (1H and 13C) NMR Chemical Shifts. Nat. Protoc 2014, 9, 643–660. [DOI] [PubMed] [Google Scholar]
- (5).Karuppagounder SS; Pinto JT; Xu H; Chen H-L; Beal MF; Gibson GE, Dietary Supplementation With Resveratrol Reduces Plaque Pathology In A Transgenic Model Of Alzheimer’s Disease. Neurochem. Int 2009, 54, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cahill R; Cookson RC; Crabb TA, Geminal coupling constants in methylene groups—II: J in CH2 groups α to heteroatoms. Tetrahedron 1969, 25, 4681–4709. [Google Scholar]
- (7).Pople JA; Bothner-By AA, Nuclear Spin Coupling Between Geminal Hydrogen Atoms. The Journal of Chemical Physics 1965, 42, 1339–1349. [Google Scholar]
- (8).Reich HJ 5-HMR-4 Geminal Proton-Proton Couplings (2JH-H). https://www.chem.wisc.edu/areas/reich/nmr/05-hmr-04-2j.htm (accessed 09/24/2019),
- (9).Typical 15N NMR chemical shifts for amides are between −210 ppm and −300 ppm or for an amine typical shifts are between −280 ppm and −380 ppm.
- (10).Initial data was collected in CD3OD (See Table S1), hence this signal was not detected until later.
- (11).Pereira AR; Cao Z; Engene N; Soria-Mercado IE; Murray TF; Gerwick WH, Palmyrolide A, An Unusually Stabilized Neuroactive Macrolide From Palmyra Atoll Cyanobacteria. Org. Lett 2010, 12, 4490–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Medina RA; Goeger DE; Hills P; Mooberry SL; Huang N; Romero LI; Ortega-Barría E; Gerwick WH; McPhail KL, Coibamide AA Potent Antiproliferative Cyclic Depsipeptide From The Panamanian Marine Cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc 2008, 130, 6324–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ, Gaussian 09, Revision B.01. In Wallingford CT, 2016. [Google Scholar]
- (14).Grimblat N; Zanardi MM; Sarotti AM, Beyond DP4: An Improved Probability For The Stereochemical Assignment Of Isomeric Compounds Using Quantum Chemical Calculations Of NMR Shifts. J. Org. Chem 2015, 80, 12526–12534. [DOI] [PubMed] [Google Scholar]
- (15).Lopez JAV; Petitbois JG; Vairappan CS; Umezawa T; Matsuda F; Okino T, Columbamides D and E: Chlorinated Fatty Acid Amides from the Marine Cyanobacterium Moorea bouillonii Collected in Malaysia. Org. Lett 2017, 19, 4231–4234. [DOI] [PubMed] [Google Scholar]
- (16).Zhang X; He H; Ma R; Ji Z; Wei Q; Dai H; Zhang L; Song F, Madurastatin B3, a rare aziridine derivative from actinomycete Nocardiopsis sp. LS150010 with potent anti-tuberculosis activity. J. Ind. Microbiol. Biotechnol 2017, 44, 589–594. [DOI] [PubMed] [Google Scholar]
- (17).The corresponding author of ref. 3 has written to us the following: “My coauthors and I are pleased to see that this flaw in our original script has been uncovered. We appreciate that the researchers here shared this information with us prior to publication and fully endorse their publishing the revised script here. Once this publication has appeared, we plan to update and correct the original Protocol (ref. 3) to further minimize the chance of compromise of future use of the method by other investigators. -TRH”
- (18).Glitch, a term of art in computer science, describes the unexpected result of a minor malfunction.
- (19).Bracegirdle J; Robertson LP; Hume PA; Page MJ; Sharrock AV; Ackerley DF; Carroll AR; Keyzers RA, Lamellarin Sulfates from the Pacific Tunicate Didemnum ternerratum. J. Nat. Prod 2019, 82, 2000–2008. [DOI] [PubMed] [Google Scholar]
- (20).Xu H-C; Hu K; Sun H-D; Puno P-T, Four 14(13 → 12)-Abeolanostane Triterpenoids with 6/6/5/6-Fused Ring System from the Roots of Kadsura coccinea. Nat. Prod. Bioprospect 2019, 9, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhu JS; Li CJ; Tsui KY; Kraemer N; Son J-H; Haddadin MJ; Tantillo DJ; Kurth MJ, Accessing Multiple Classes of 2H-Indazoles: Mechanistic Implications for the Cadogan and Davis-Beirut Reactions. J. Am. Chem. Soc 2019, 141, 6247–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Guillen PO; Jaramillo KB; Jennings L; Genta-Jouve G; de la Cruz M; Cautain B; Reyes F; Rodriguez J; Thomas OP, Halogenated Tyrosine Derivatives from the Tropical Eastern Pacific Zoantharians Antipathozoanthus hickmani and Parazoanthus darwini. J. Nat. Prod 2019, 82, 1354–1360. [DOI] [PubMed] [Google Scholar]
- (23).Spaltenstein P; Cummins EJ; Yokuda K-M; Kowalczyk T; Clark TB; O’Neil GW, Chemoselective Carbonyl Allylations with Alkoxyallylsiletanes. J. Org. Chem 2019, 84, 4421–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zou Y; Wang X; Sims J; Wang B; Pandey P; Welsh CL; Stone RP; Avery MA; Doerksen RJ; Ferreira D; Anklin C; Valeriote FA; Kelly M; Hamann MT, Computationally Assisted Discovery and Assignment of a Highly Strained and PANC-1 Selective Alkaloid from Alaska’s Deep Ocean. J. Am. Chem. Soc 2019, 141, 4338–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Alvarenga ES; Santos JO; Moraes FC; Carneiro VMT, Quantum mechanical approach for structure elucidation of novel halogenated sesquiterpene lactones. J. Mol. Struct 2019, 1180, 41–47. [Google Scholar]
- (26).Neupane RP; Parrish SM; Bhandari Neupane J; Yoshida WY; Yip MLR; Turkson J; Harper MK; Head JD; Williams PG, Cytotoxic Sesquiterpenoid Quinones and Quinols, and an 11-Membered Heterocycle, Kauamide, from the Hawaiian Marine Sponge Dactylospongia elegans. Mar. Drugs 2019, 17, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kasza P; Trybula ME; Baradziej K; Kepczynski M; Szafranski PW; Cegla MT, Fluorescent triazolyl spirooxazolidines: Synthesis and NMR stereochemical studies. J. Mol. Struct 2019, 1183, 157–167. [Google Scholar]
- (28).Elkin M; Scruse AC; Turlik A; Newhouse TR, Computational and Synthetic Investigation of Cationic Rearrangement in the Putative Biosynthesis of Justicane Triterpenoids. Angew. Chem., Int. Ed 2019, 58, 1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.