

SUMMARY
Over 80% of pancreatic ductal adenocarcinoma (PDA) patients are diagnosed with non-resectable late-stage disease that lacks effective neoadjuvant therapies. Stereotactic body radiation therapy (SBRT) has shown promise as an emerging neoadjuvant approach for treating PDA, and here, we report that its combination with local interleukin-12 (IL-12) microsphere (MS) immunotherapy results in marked tumor reduction and cures in multiple preclinical mouse models of PDA. Our findings demonstrate an increase of intratumoral interferon gamma (IFNγ) production following SBRT/IL-12 MS administration that initiates suppressor cell reprogramming and a subsequent increase in CD8 T cell activation. Furthermore, SBRT/IL-12 MS therapy results in the generation of systemic tumor immunity that is capable of eliminating established liver metastases, providing a rationale for follow-up studies in advanced metastatic disease.
In Brief
Mills et al. demonstrate a marked antitumor response following radiotherapy and IL-12 microsphere combination treatment (SBRT/IL-12 MS) of murine pancreatic cancer. The IFNγ-dependent mechanism repolarized myeloid suppressors and promoted robust T cell activation. SBRT/IL-12 MS elicited in situ tumor “vaccination” and an abscopal effect capable of eliminating hepatic metastases.
Graphical Abstract
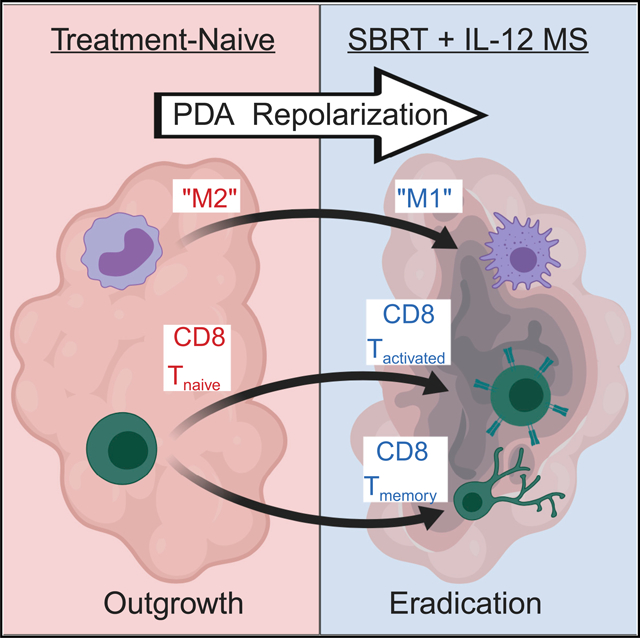
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is the twelfth most common malignancy worldwide; however, it is the seventh leading cause of cancer-associated deaths due to disease aggressiveness (Bray et al., 2018; Siegel et al., 2018). Poor survival statistics are the result of common dysfunctions in core signaling pathways, including cell growth (KRAS), DNA damage control (TP53), and cell cycle regulation (CDKN2A) (Bailey et al., 2016). These aberrations drive rapid disease progression prior to symptom onset, increasing the prevalence of locally advanced PDA at the time of primary intervention (Hidalgo et al., 2015). Currently, the only cure for PDA is surgical resection. Unfortunately, 80%–90% of newly diagnosed patients are deemed inoperable, and neoadjuvant therapy is unsuccessful in downstaging approximately 90% of unresectable lesions (Gillen et al., 2010).
A multitude of chemotherapy or chemoradiation regimens are incapable of downsizing advanced PDA malignancies, but an emerging strategy, stereotactic body radiation therapy (SBRT), has shown promise (Zhong et al., 2017). Unlike conventional radiation therapy (conRT) that is administered in frequent small doses over 5–6 weeks, SBRT utilizes multiple beam angles to deliver (nearly) ablative doses in oligofractions. Accordingly, a radioequivalent dose of SBRT can be delivered in 4–5 fractions (compared to 25–30 conRT fractions), affording greater tumoricidal capacity with less damage to surrounding normal tissue (Timmerman et al., 2014). By minimizing systemic lymphopenia, tumor neo-antigens unmasked during radiation-induced necrosis can be used to prime infiltrating T cells and mount a potent immunogenic response (Order, 1977). SBRT also promotes leukocyte homing by modulating the intratumoral chemoattractant milieu and altering the stromal architecture to promote extravasation (Lugade et al., 2008). Although these signals can complement direct cytotoxic effects, they also attract a variety of immunosuppressive cell types, including inflammatory monocytes (IMs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T (Treg) cells (Kalbasi et al., 2017; Walle et al., 2018; Seifert et al., 2016). Accordingly, the mixed immune-modulatory effects of SBRT make it an attractive combination for immunotherapy.
Still in its infancy, immunotherapy for treating PDA has utilized combinations of dual-checkpoint inhibitors with or without conventional chemotherapy; however, results have yet to demonstrate a notable benefit (Thind et al., 2017). Conversely, certain cytotoxic chemotherapies, such as platinum-based agents, have been shown to induce immunogenic cell death, and at high doses, they can also cause neutropenia and leukopenia (Pfirschke et al., 2016; Oun et al., 2018). Furthermore, failed T cell priming and high suppressor cell distributions in immunologically “cold” tumors, such as PDA, commonly lead to resistance to immune checkpoint blockade (Vonderheide, 2018; Jenkins et al., 2018). In contrast, the pleiotropic cytokine interleukin-12 (IL-12) is well known for its antitumor potential by means of stimulating T cell activation both directly and indirectly by increased antigen presentation and immunogenic reprogramming of both lymphoid and MDSCs, respectively (Zeh et al., 1993; Suzuki et al., 1998; Trinchieri et al., 1992; Kerkar et al., 2011). Unexpectedly, pioneering translational studies of IL-12 demonstrated underwhelming therapeutic effects, and clinical trials were short-lived due to severe toxicities resulting from bolus systemic administration (Jenks, 1996). To overcome these clinical challenges, recent studies have characterized the importance of delivering sustained intratumoral doses of the proinflammatory cytokine (Atkins et al., 1997; Colombo et al., 1996; Egilmez et al., 2000). The encapsulation of IL-12 in polylactic acid microspheres (MSs) is one such delivery method and has been shown to provide a stable, sustained-release delivery system that is superior to other modes of intratumoral administration (e.g., non-encapsulated recombinant IL-12) in murine tumor models (Egilmez et al., 2000).
In this study, we tested the hypothesis that the sustained localized delivery of adjuvant IL-12 would enhance the antitumor potency of SBRT for treating PDA. A few previous studies have shown the antitumor capacity of the combination of radiotherapy and IL-12 in heterotopic models of cancer (Sedlar et al., 2013; Deplanque et al., 2017; Wu et al., 2018); however, our report demonstrates the robust antitumor activity and cure of preclinical PDA models following local administration of SBRT/IL-12 MS. The combination immunotherapy resulted in the synergistic production of the proinflammatory cytokine interferon gamma (IFNγ), which was necessary for the immunogenic reprogramming of intratumoral suppressor cells. In addition to increases in activated CD8 T cells, the repolarization of densely populated myeloid suppressors was shown to confer functional antitumor effects and may explain the enhanced susceptibility of PDA to this therapeutic regimen. This study also demonstrated the formation of systemic immune memory with the capacity to eliminate established liver metastases following SBRT/IL-12 MS administration, providing rationale for follow-up studies in advanced metastatic disease.
RESULTS
SBRT Recruits CD8 T Cells into the Center of Human PDA Tumors and Is Superior to conRT
To assess the immune response to SBRT in human PDA, we performed immunohistochemistry on tumors resected 10–14 days following neoadjuvant SBRT (5 gray [Gy] × 5 consecutive days). All tissues analyzed were diagnosed as resectable and treatment-naive prior to SBRT-only intervention. H&E staining was used to demarcate the center and margin of each tumor (Figures S1A and S1B, unirradiated and SBRT, respectively). Immunohistochemical analysis of unirradiated (UI) tumors illustrated few CD8 T cells in the lesion center, with the greater numbers trapped in the margin (Figures 1A, 1B, and 1E). Importantly, SBRT treatment resulted in a greater infiltration of CD8 T cells into central tumor regions (Figures 1C, 1D, and 1E) as demonstrated by significantly increased center:margin cellular ratios (Figure 1F). An assessment of CD68+ myeloid cells demonstrated a uniform distribution of immunosuppressive myeloid populations across the margins and centers of tumors in both unirradiated (Figures 1G, 1H, and 1K) and SBRT-treated (Figures 1I, 1J, and 1K) groups upon center:margin quantification (Figure 1L). These clinical data suggest that SBRT results in a uniform dispersal of antitumor CD8 T cells throughout PDA tumors; however, treatment does not eliminate or alter the distribution of immunosuppressive CD68+ cells.
Figure 1. SBRT Recruits CD8 T Cells into the Center of Human PDA Tumors and Is Superior to ConRT.

(A–L) Representative (20×) CD8 (A–D) and CD68 (G–J) immunoperoxidase stains of human PDA sections that were previously untreated (n = 5 patients) or treated with SBRT only (n = 7 patients). Tumor center (UI [unirradiated], A and G; SBRT, C and I) and margins (UI, B and H; SBRT, D and J) were analyzed. The number of events per 1 mm2 high power field (HPF) as well as the center:margin distribution were calculated for CD8+ (E and F, respectively) and CD68+ (K and L, respectively) cells. Center:margin cell ratios were calculated within each sample prior to averaging; data shown as mean ± SEM, t test.
(M and N) KCKO-luc cells were orthotopically implanted on day 0 (n = 4–6) with two titanium fiducial clips for radiation therapy targeting. SBRT (6 Gy × 4 days) or conRT (2 Gy × 15 days) was delivered using the SARRP platform (or UI sham surgery with fiducial clip implantation).
(M) Bioluminescent imaging was performed using the IVIS spectrum. Values are presented as the geometric mean of maximum photon emissions (bioluminescence, BLI) within tumor regions of interest (ROIs); Holm-Sidak test, significance relative to UI/empty MS group.
(N) Corresponding survival curve of UI/SBRT/conRT mice; Grehan-Breslow-Wilcoxon test, UI/SBRT plots are representative of at least two independent experiments.
**p < 0.01. See also Figure S1.
Although SBRT is an emerging strategy to treat PDA, conRT remains the more commonly used treatment. Accordingly, we compared the efficacy of SBRT and conRT schedules preclinically in orthotopic mouse models of PDA. A derivative of the KC cell line (P48-Cre; LSL-KrasG12D) transfected with luciferase (KCKO-luc) was implanted on day 0, followed by radiotherapy beginning on day 6. Radioequivalent (isoeffective) doses of SBRT (6 Gy × 4 days) and conRT (2 Gy × 15 days) were delivered by a small animal radiation research platform (SARRP) targeted to pancreas tumors by using two fiducial titanium clips inserted on either side of the lesion during implantation. Measurement of tumor bioluminescence by using the in vivo imaging system (IVIS) confirmed greater reductions in tumor burden with SBRT scheduling relative to conRT (Figure 1M). Furthermore, the SBRT-treated group also demonstrated the greatest survival benefit (Figure 1N). These data support clinical observations that SBRT is more efficacious than conRT in reducing PDA tumor burden.
SBRT and IL-12 MS Combination Greatly Reduces PDA Tumor Burden and Increases Survival
Recent clinical investigations of neoadjuvant SBRT in PDA have demonstrated moderately effective downstaging, whereas the immunologically diverse infiltrate following SBRT suggests an avenue for synergy with immunotherapy. Therefore, we developed a preclinical PDA model to test the combination of SBRT with the pleiotropic proinflammatory cytokine IL-12 encapsulated in polymer MSs. Orthotopic KCKO-luc tumors were treated with a clinically relevant schedule of SBRT (6 Gy × 4 days) delivered locally by SARRP. MSs (IL-12 or empty) were intratumorally (i.t.) injected 24 h post-SBRT (detailed under STAR Methods and illustrated in Figure 2A) (Mathiowitz et al., 2000). Using AF594 fluorescently labeled MS, we demonstrated that this injection strategy results in intratumoral sequestration of MSs, whereas intraperitoneal (i.p.) injection (used to simulate MS “leakage”) led to peritoneal myeloid engulfment and subsequent trafficking into the bloodstream (Figures S2A and S2B). Furthermore, free AF594 MSs were not found in the plasma following i.t. injection, demonstrating the absence of MS spillover during the procedure (Figures S2C and S2D). We conclude that i.t. administration of MS results in local retention of the therapy.
Figure 2. SBRT and IL-12 MS Combination Greatly Reduces PDA Tumor Burden and Increases Survival.

Tumor cells were implanted on day 0 with two metal fiducial clips for SBRT targeting. SBRT was delivered (6 Gy × 4 days, or sham surgery with fiducial clip implantation) beginning on day 6 (day 3 for KPC GEMM), followed by i.t. microsphere injection of empty MS control or IL-12 MS on day 10 (day 7 for KPC GEMM).
(A) Schematic outlining orthotopic PDA mouse model and treatment scheduling. Green arrow points to tumor; white arrows point to fiducial clips.
(B and C) SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors (n = 4–5) were tracked over time using IVIS bioluminescent imaging to measure tumor growth (B), as well as survival analysis (C); representative of 2–3 independent experiments. (D and E) IVIS growth (D) and survival (E) measurements were repeated on SBRT/IL-12 MS-treated Pan02-luc orthotopic tumors (n = 5); representative of 2 independent experiments. (F) Survival analysis of the SBRT/IL-12 MS-treated KPC GEMM. KPC mice (n = 4–8) were manually palpated for pancreatic lesions beginning at 5 weeks of age, and all treatments were initiated when mice reached approximately 6 to 8 weeks of age; mice were dichotomized into treatment groups based on initial tumor weights (day 0 = clip implantation). “LTS” designates the long-term survivor further described in the supplement. Representative of 4–6 pooled independent experiments.
For each IVIS imaging analysis, values are presented as the geometric mean of maximum photon emissions within ROIs; Holm-Sidak tests. For survival analyses, Grehan-Breslow-Wilcoxon tests were performed. All significance relative to UI/empty MS group. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figures S2 and S3.
In KCKO-luc tumors treated with either SBRT or IL-12 MS alone, we observed moderate reductions in tumor burden. Remarkably, the combination of SBRT + IL-12 MS eradicated tumors by day 20 post-implantation, and lesions remained undetectable by IVIS bioluminescent imaging until measurements were terminated at day 60 (Figures 2B and S3A). Histological analyses of day 11 tumors corroborated these antitumor effects, depicting regions of marked cell death and overwhelming immune infiltration in the SBRT/IL-12 MS group (Figures S3B–S3E). Treatment with SBRT alone increased overall survival, with 20% demonstrating long-term survival greater than 120 days; however, SBRT/IL-12 MS treatment resulted in a significant benefit, with 100% of mice achieving long-term survival (Figure 2C). To generalize our findings across other PDA models, we repeated these studies using Pan02, a chemically induced and radioresistant cell line. Like KCKO-luc tumor-bearing mice, IL-12 MS delivery alone resulted in only minor reductions in tumor burden. Interestingly, combination therapy maintained synergistic antitumor effects even in the absence of strong monotherapy responses, as represented by both significantly decreased tumor burden and increased survival (Figures 2D and 2E, respectively). Furthermore, 10% of SBRT/IL-12 MS-treated Pan02-luc mice resulted in long-term survival.
We expanded our investigation to the P48-Cre; LSL-KrasG12D; Tp53L/L (KPC) genetically engineered mouse model (GEMM) to test this combination therapy in a more clinically relevant representation of locally advanced disease. KPC mice were enrolled in treatment at 6–8 weeks of age after the development of prominent lesions. All mice used in the study had similar tumor volume measurements at the time of fiducial marker placement (Figure S3F). SBRT/IL-12 MS, but neither treatment alone, significantly increased overall survival, demonstrating nearly triple the survival of untreated controls (Figure 2F). Histological analysis of the day 11 SBRT/IL-12 MS-treated tumor (4 days post-IL-12 MS) revealed regions of pronounced cell death and immune infiltration relative to the untreated control (Figures S3G and S3H, respectively). Of note, this model results in the malignant transformation of all P48-expressing pancreatic ductal cells, eliminating the potential for cure. Even so, one SBRT/IL-12 MS-treated mouse was afforded marked survival benefit and, upon autopsy, was found to have cleared much of the pretreatment tumor mass and displayed splenomegaly, suggesting the development of a robust antitumor immune response (Figure S3I). Taken together, these findings demonstrate a generalizable antitumor capacity of SBRT/IL-12 MS therapy that elicits survival benefits in multiple preclinical PDA models.
SBRT/IL-12 MS Therapeutic Efficacy Is Dependent upon IFNγ Function
Many of the proinflammatory biological effects of IL-12 are mediated by IFNγ. We next assayed the amount of intratumoral IFNγ following IL-12 MS treatment in our KCKO-luc orthotopic model. Luminex cytokine analysis of tumors following SBRT/IL-12 MS administration revealed significant inductions of IL-12 and IFNγ proteins only in the combination treatment group, for up to 24 (day 11) and 48 h (day 12) following MS delivery, respectively. Interestingly, the highest levels of IFNγ production were observed within the first 24 h (day 11) post-treatment. Concurrent analysis of CXCL10 levels corroborated IFNγ findings (Figure 3A). Furthermore, flow cytometric analysis of day 11 tumors confirmed a significant increase in the percent of IFNγ-positive CD45+ immune cells (Figure 3B) and CD4 T cells (Figure 3C) in the SBRT/IL-12 MS treatment group.
Figure 3. SBRT/IL-12 MS Therapeutic Efficacy Is Dependent upon IFNγ Function.

(A) SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors (n = 3) were harvested on days 11/12/13 and homogenized prior to Luminex cytokine multiplex assay analysis. Data values (pg/mL) were normalized to total protein content and are presented in pg/mg protein; Holm-Sidak test, representative of at least 2 independent experiments. (B and C) SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors (n = 5) were harvested on day 11 and digested into single cell suspensions for flow cytometric analysis. Fluorescence minus one (FMO) controls were used to identify IFNγ+ cells from CD45+ (B) and CD45+CD4+ (C) immune cell populations. Values are presented as percent IFNγ+ of total CD45+ cells identified (right panels); Holm-Sidak test, representative of at least 2 independent experiments.
(D and E) SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors (n = 5) implanted in Ifng−/− mice were measured over time using IVIS bioluminescent imaging to track tumor growth (D), as well as survival analysis (E). For IVIS imaging analysis, values are presented as the geometric mean of maximum photon emissions within ROIs; Holm-Sidak test. For survival analyses, a Grehan-Breslow-Wilcoxon test was performed. Representative of one experiment. Significance relative to UI/empty MS group.
*p < 0.05, **p < 0.01, ****p < 0.0001.
To determine if the SBRT/IL-12 MS therapeutic effect was dependent upon IFNγ signaling, we implanted KCKO-luc orthotopic tumors in IFNγ null, Ifngtm1Ts (Ifng−/−) mice. As expected, IVIS growth and overall survival measurements demonstrated a general increase in baseline tumor growth and accelerated mortality upon loss of the cytokine. IFNγ deletion also resulted in the complete abatement of the SBRT/IL-12 MS therapeutic response (Figures 3D and 3E; compare to Figures 2B and 2C). Altogether, these data demonstrate the therapeutic dependence of SBRT/IL-12 MS treatment on robust intratumoral production of IFNγ.
PDA Myeloid Populations Are Reprogrammed by SBRT/IL-12 MS Treatment
Radiotherapy can bolster intratumoral immunosuppressive myeloid populations in the days following treatment (Walle et al., 2018; Connolly et al., 2016). To assess SBRT/IL-12 MS effects on myeloid suppressor recruitment, we performed flow cytometry on day 11 KCKO-luc tumors. Analyses revealed SBRT-dependent increases in CD11b+Ly6C+Ly6G− IMs, CD11b+ Ly6CmodLy6G−F480+ TAMs, and CD11b+ Ly6CmodLy6G+ tumor-associated neutrophils (TANs). These responses were also generally unaffected by IL-12 MS treatment alone or SBRT/IL-12 MS (Figures 4A, 4B, left panels, and S4A). Interrogation of the day 14 time point revealed similar treatment effects on IMs; however, interestingly, SBRT-dependent increases in TAMs were found to be abrogated by the addition of IL-12 MS (Figure 4B, right panels).
Figure 4. PDA Myeloid Populations Are Reprogrammed by SBRT/IL-12 MS Treatment.

SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors were harvested on days 11 and 14 and digested into single cell suspensions for flow cytometry (n = 4–5) and RNA-seq analyses. (A) Representative flow cytometry gating strategy used to identify IM and TAM populations.
(B) IM and TAM population densities were assessed using flow cytometry and are presented as a percentage of total CD45+ cells identified; Holm-Sidak tests, representative of 2 independent experiments.
(C) FMO controls (blue) were used to gate MHCII+ (left panel: wild-type [WT] host; right panel: Ifng−/− host) IMs. Dotplot values represent mean fluorescence intensity (MFI), and percentages of positive cells are represented in the upper right corner of each plot; Holm-Sidak tests, representative of at least two independent experiments.
(D and E) IM (D, n = 3) and TAM (E, n = 2–3) populations were flow sorted prior to RNA-seq analyses. SBRT/IL-12 MS DEGs (versus UI/empty MS controls) were compared to monocyte, classical M1, and alternative M2 macrophage genesets from the Broad Institute MSigDB (MSigDB: GSE5099), and gene matches are presented in volcano plots. Blue, downregulated; red, upregulated. Representative of one experiment. (F and G) Schematic (F) of IM/TAM transplant experiment. KCKO-luc tumors (n = 4–5) from each SBRT/IL-12 MS treatment group were harvested and IMs and TAMs were flow sorted. IMs/TAMs from each group were pooled with fresh KCKO-luc cells and orthotopically implanted into naive mice (n = 4–5). No further treatment was administered. (G) Transplanted tumors were measured over time by using IVIS bioluminescent imaging to track tumor growth. The geometric means of maximum photon emissions within ROIs were normalized to KCKO-luc-only control tumors and presented as fold change relative to day 3 tumor size; Holm-Sidak test, all significance relative to UI/empty MS group, except UI/empty MS, which is relative to KCKO-luc-only control. Representative of one experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4 and Tables S1 and S2.
Having observed a robust increase in intratumoral IFNγ protein levels upon SBRT/IL-12 MS treatment, we proceeded to assess effects of this proinflammatory cytokine on myeloid polarization. Day 11 flow cytometric analysis demonstrated significantly increased percentages of MHCII+ IMs upon IL-12 MS and SBRT/IL-12 MS treatments, indicative of reprogramming (Figure 4C, left panels). Repeating analyses in Ifng−/− mice, we confirmed increases in de novo MHCII expression to be IFNγ-dependent (Figure 4C, right panels). TAMs that arise from circulating IMs are almost exclusively MHCII+, and reductions in MHCII expression have been shown to promote tumor progression (Zhu et al., 2017; Wang et al., 2011). SBRT was found to reduce the percentage of MHCII+ TAMs in KCKO-luc tumors, whereas the addition of IL-12 MS rescued the MHCII+ phenotype (Figure S4B).
For added confirmation of myeloid reprogramming, we performed RNA sequencing (RNA-seq) analysis on IM and TAM populations sorted from KCKO-luc tumors. Ingenuity pathway analysis of differentially expressed genes (DEGs; versus unirradiated/empty MS; −1.5 < z < 1.5) demonstrated the upregulation of activation pathways (eicosanoid and inducible nitric oxide synthase [iNOS]) accompanied by the downregulation of immunosuppressive pathways (sphingosine-1, P2Y purinergic receptor, thrombin, and STAT3) across both populations (Norris and Dennis, 2014; MacMicking et al., 1997; Park et al., 2014; Barberà-Cremades et al., 2016; White and Gomer, 2015; Mu et al., 2018). Interestingly, only SBRT/IL-12 MS treatment resulted in the differential regulation of metabolic pathways in IMs involving glycolysis (pro-activation, upregulated), and cholesterol biosynthesis (pro-suppression, downregulated) (Figure S4C) (Freemerman et al., 2014; Wei et al., 2015). In both IMs and TAMs, IFNγ activation was identified as a top upstream regulator of differential expression upon SBRT/IL-12 MS treatment (Tables S1 and S2).
We next analyzed expression patterns at the individual gene level by dichotomizing DEGs (|log2[fold-change]| > 0.5 versus unirradiated/empty MS; p < 0.05) into monocyte, M1, and M2 macrophage subsets by using immunologic gene sets from the Molecular Signatures Database (MSigDB) provided by the Broad Institute (MSigDB: GSE5099). Using this classification strategy, we identified an M1-skewed gene upregulation after SBRT/IL-12 MS treatment in both myeloid cell types (IM: monocyte → M1 = 31, monocyte → M2 = 7; TAM: M1 = 13, M2 = 3), in addition to the predominant downregulation of M2-like genes (IM: monocyte → M2 = 9 genes, monocyte → M1 = 4 genes; TAM: M2 = 4, M1 = 1) (Figures 4D and 4E, respectively). Monotherapy treatment with SBRT or IL-12 MS was insufficient to activate comparable levels of differential gene expression in IMs or TAMs (Figures S4D–S4G).
To assess the functional impact of myeloid polarization on KCKO-luc tumors, we performed IM/TAM transplants of SBRT/IL-12 MS-treated populations (Figure 4F). Briefly, IMs and TAMs were sorted from KCKO-luc tumors, and pooled together with freshly cultured KCKO-luc cells. Cell mixtures from each SBRT/IL-12 MS treatment group were orthotopically implanted into naive hosts that received no further treatment. Compared to control KCKO-luc-only tumors, the addition of untreated IM/TAM pools promoted significant increases in relative tumor growth, demonstrating the well-documented protumor capacities of myeloid suppressors. Conversely, the transplantation of SBRT/IL-12 MS-treated IM/TAM pools significantly suppressed tumor outgrowth (Figure 4G). Altogether, these findings demonstrate that SBRT/IL-12 MS treatment induces a cumulative repolarization of the intratumoral myeloid compartment toward an activated, antitumor state.
IFNγ Production Is Necessary to Drive Antitumor T Cell Ratios
To validate our human PDA data demonstrating an increased intratumoral T cell infiltrate following SBRT therapy (Figure 1), we performed flow cytometric analysis on KCKO-luc tumors by using a comprehensive lymphoid marker panel (Figure 5A). Analysis of tumors at day 11 demonstrated modest increases in the percentage of CD8 T cells following SBRT and SBRT/IL-12 treatments; however, by day 14, SBRT-dependent CD8 increases were more pronounced, reaching significance in the SBRT/IL-12 MS group (Figure 5B, top panels). CD4 T cells were significantly increased by SBRT treatment at day 11, and interestingly, the effect was abrogated by the addition of IL-12 MS. Conversely, by day 14, SBRT/IL-12 MS combination had elicited a significant increase in the CD4 compartment (Figure 5B, bottom panels). Antigen-presenting cells (APCs) and other lymphocyte lineages, including B, natural killer (NK), and CD8+NK1.1+ cells, were found to be unchanged or decreased following SBRT/IL-12 MS treatments (Figures S5B–S5D).
Figure 5. SBRT/IL-12 MS Therapeutic Efficacy Is Dependent upon IFNγ-Driven Antitumor T Cell Ratios and Robust CD8 T Cell Activation.

SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors were harvested on days 11 and 14 and digested into single cell suspensions for flow cytometric analysis (n = 4–5). (A) Representative flow cytometry gating strategy used to identify CD8 and CD4 T cell, and Treg populations.
(B) CD8 and CD4 T cell population densities were assessed on days 11 (left panels) and 14 (right panels) by using flow cytometry and are presented as a percentage of total CD45+ cells identified; Holm-Sidak tests, representative of at least two independent experiments.
(C) Treg cell percentages (top panels) and CD8:Treg ratios (bottom panels) were analyzed on day 11 in tumors grown in WT (left panels) and Ifng−/− (right panels) mice; Holm-Sidak tests, representative of at least two independent experiments.
(D) SBRT/IL-12 MS-treated KCKO-luc orthotopic tumors (n = 4) were administered immunoglobulin G (IgG) control or anti-CD8- or anti-CD4-depleting antibodies every three days between days 5 and 20 post-implantations. Tumor size was measured over time using IVIS bioluminescent imaging. Values are presented as the geometric mean of maximum photon emissions within ROIs; Holm-Sidak test, significance relative to UI/empty MS group, pooled data from two independent experiments.
(E) CD8+ T cells were flow sorted prior to RNA-seq analysis. SBRT/empty MS, UI/IL-12 MS, and SBRT/IL-12 MS DEGs (versus UI/empty MS controls) were compared to naive (Tnaive), effector (Teff), effector-memory (Tem), and exhausted (Tex) T cell genesets from the Broad Institute MSigDB (MSigDB: GSE1000002), and gene matches are presented in heatmaps. Blue, downregulated; red, upregulated. Representative of one experiment.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figures S5 and S6 and Table S3.
Much like the M1/M2 paradigm, IFNγ drives CD4 T cells toward a proinflammatory T helper type 1 (Th1) program (Zhou et al., 2009). To assess the inflammatory status of intratumoral CD4 T cells at day 11, we stained for the Treg transcription factor FOXP3 and observed a significant increase in CD4+/FOXP3+ Treg cells after SBRT treatment. Interestingly, the addition of IL-12 MS to SBRT resulted in a reduced percentage of Treg cells, and repeating this experiment in Ifng−/− mice confirmed the dependence of this effect on this proinflammatory cytokine (Figure 5C, top panels). Interestingly, an analysis of day 14 KCKO-luc tumors demonstrated a significant rebound in Treg cells with SBRT/IL-12 MS treatment, suggesting that CD4 reprogramming was a transient event (Figure S5E). Combining CD8 T cell and Treg cell distributions to assess the ratio of activated T cells, we observed a significant increase in the CD8/Treg ratio only in the SBRT/IL-12 MS treatment group. Similar to the Treg effects, the proinflammatory ratio was lost in the Ifng−/− host background (Figure 5C, bottom panels). These findings suggest that SBRT and IL-12 MS treatments cooperatively increase antitumor T cell ratios in KCKO-luc tumors by promoting CD8 accumulation and subsequently eliminating CD4 immunosuppressive regulatory programming.
Tumoricidal Effect of SBRT/IL-12 MS Therapy Is Dependent upon Activated CD8 T Cells
To determine if CD8 T and/or CD4 Th1 cells were necessary for therapeutic efficacy, we administered CD8− or CD4-depleting antibodies one day prior to SBRT treatment (day 5) of KCKO-luc tumors and repeated dosing every three days for two weeks. Strikingly, IVIS bioluminescent imaging demonstrated the complete abrogation of antitumor effects upon CD8+ depletion, whereas CD4+ depletion showed no effect (Figure 5D). To assess CD8 T cell activation status, we performed flow cytometric and Luminex analyses of day 11 KCKO-luc tumors. Following SBRT/IL-12 MS, we observed an upregulated expression of the CD44 activation marker and a greater percentage of degranulating CD107a+ cells (Figure S6A, far-left and mid-left panels, respectively). Corroborating increased CD107a degranulation, heightened intratumoral levels of granzyme B (GZMB) were observed in SBRT and SBRT/IL-12 MS groups (Figure S6B). CD8 T cells did not demonstrate increased levels of the exhaustion markers CTLA4 and PD1 on a per cell basis; however, there was a greater percentage of cells expressing these markers, suggesting a greater overall number of activated, but not exhausted, CD8 T cells (Figure S6A, mid-right and far-right panels, respectively).
For further assessment of the T cell activation status in our KCKO-luc orthotopic tumor model, we performed RNA-seq analysis on sorted CD8 cells from each of our four experimental groups. Ingenuity pathway analysis of DEGs (versus unirradiated/empty MS; −1.5 < z < 1.5) identified the activation of proliferative functions, including S-phase entry and cyclin regulation, alongside the deactivation of G2/M checkpoint regulation in IL-12 MS and SBRT/IL-12 MS groups. The SBRT/IL-12 MS group demonstrated the upregulation of protein translation (tRNA charging and pyridoxal 5′-phosphate salvage) and nucleotide biosynthesis pathways (pyrimidine salvage and pyrimidine de novo biosynthesis), which are chief to clonal expansion and effector and memory differentiation of CD8+ T cells (Figure S6C) (Quéméneur et al., 2004). At the individual gene level, we again classified differential expressers (|log2[fold-change]| > 0.5 versus unirradiated/empty MS; p < 0.05) into subsets using the MSigDB (MSigDB: GSE1000002). Upon sorting DEGs into naive (Tnaive), effector (Teff), effector-memory (Tem), and exhausted (Tex) T cell groups, we observed an overwhelming downregulation of naive (26 down, 2 up) and upregulation of effector genes (76 up, 1 down) with SBRT/IL-12 MS treatment. Furthermore, 70% of differentially expressed effector-memory genes were upregulated, and only 6 of 13 exhaustion transcripts identified were augmented. SBRT and IL-12 MS monotherapies demonstrated similar patterns in differential expression; however, the quantity of DEGs was greatly reduced compared to SBRT/IL-12 MS (Figure 5E). Interferon γ was not identified as a top upstream regulator of differential expression in any treatment group, emphasizing its indirect effects by repolarization of suppressor cells (Table S3). Collectively, these findings demonstrate the augmentation of intratumoral T cell activation and memory formation elicited by SBRT/IL-12 MS combination treatment and illustrate the significance of this process for therapeutic response.
SBRT/IL-12 MS Therapy Generates Systemic Antitumor Immunity That Drives an Abscopal Effect
SBRT/IL-12 MS treatment of KCKO-luc tumors lead to long-term survival in 100% of mice. Accordingly, we hypothesized that immunological memory had been established. To test for long-term immunity, we first rechallenged SBRT/IL-12 MS-cured mice with metachronous KCKO-luc tumors approximately 6 months after the treatment of primary tumors. The hemi-spleen tumor model recapitulates metastatic tumor formation in the liver, the most common site of PDA dissemination. Tumor cells were injected into the spleen where they passively diffused to the liver by the hepatic portal vein. Hemi-splenectomy was performed post-implantation to prevent non-specific tumor formation. Three days following rechallenge, we observed decreased KCKO-luc seeding in SBRT/IL-12 MS-cured mice relative to age-matched naive controls, as measured by tumor bioluminescence. By day 7 post-implantations, SBRT/IL-12 MS-cured mice demonstrated no evidence of liver tumor burden, which was corroborated by a significant survival benefit (Figures 6A and 6B).
Figure 6. SBRT/IL-12 MS Therapy Generates Systemic Antitumor Immunity That Drives an Abscopal Effect.

(A and B) Mice cured of primary KCKO-luc tumors by SBRT/IL-12 MS treatment (n = 5) were rechallenged after 6 months by delivering KCKO-luc cells to the liver by using the hemi-spleen metastatic model. Rechallenged mice did not receive any second-line therapy. Liver tumors were followed over time using IVIS bioluminescent imaging to measure growth (A), as well as survival analysis (B). For IVIS imaging analysis, the treatment-naive line plot represents the geometric mean of 5 individual mice, while SBRT/IL-12 MS line plots represent individual mice. Representative of 2 independent experiments.
(C and D) Mice that survived hemi-spleen rechallenge, in addition to age-matched tumor or treatment-naive donors (n = 5), were sacrificed after 3.5 months, and CD8+ T cells from the spleen and lymph nodes were isolated using negative selection. Donor CD8 T cells were transplanted 1:1 into naive recipients 16 h prior to KCKO-luc implantation. Tumor-bearing CD8 T cell recipient mice did not receive any additional treatment, and tumors were followed over time by using IVIS bioluminescent imaging (C) to measure growth. The transferal of partial (>10-fold decrease in bioluminescent tumor volume) and full (unidentifiable tumor by manual palpation) immunity is represented in a pie chart (D) as a percentage of total SBRT/IL-12 MS-cured donors. For IVIS imaging analysis, the treatment-naive line plot represents the geometric mean of 5 individual mice, while the SBRT/IL-12 MS line plots represent individual mice. Representative of one experiment.
(E–G) KCKO-luc cells were implanted on day 0 (n = 5) into the liverby using the hemi-spleen model (a), whereas KCKO cells were simultaneously injected into the pancreas (b). The SBRT/IL-12 MS treatment paradigm (C and D) was followed for the treatment of primary pancreas tumors only (E). KCKO-luc liver metastases were tracked over time by sing IVIS bioluminescent imaging to measure metastatic growth (F), as well as survival analysis (G). For IVIS imaging analysis, values are presented as the geometric mean of maximum photon emissions within ROIs; Holm-Sidak tests, representative of two independent experiments. For each survival analysis, Grehan-Breslow-Wilcoxon tests were performed. All significance relative to UI/empty MS group.
**p < 0.01.
See also Figure S7.
For additional confirmation of long-term antitumor immunity, we next transferred CD8 T cells from rechallenged mice into naive mice, hypothesizing that cells from SBRT/IL-12 MS-cured donor mice would protect naive recipients during tumor challenge. Nine months following primary tumor eradication, CD8 T cells were purified from the remaining spleen and lymph nodes of SBRT/IL-12 MS-treated mice. Donor mice were not primed in any way prior to T cell isolation, and naive donor controls were age-matched. T cells were intravenously injected into recipient mice 16 h prior to orthotopic KCKO-luc implantation. As early as day 5 post-implantations, we observed reduced tumor seeding in recipient mice infused with CD8 T cells from SBRT/IL-12 MS-cured donors, and by day 24, antitumor responses were evident in all 5 mice, as demonstrated by IVIS bioluminescent analysis (Figure 6C). Subsequent analyses at day 40 revealed no evidence of tumor (by palpation) in 60% of mice infused with CD8 T cells from the SBRT/IL-12 MS-cured group, indicating the transferal of full antitumor immunity to naive recipients (Figure 6D). Comprehensively, these results demonstrate the formation of a targeted immune response against KCKO-luc tumors upon SBRT/IL-12 MS treatment that generates tumor-specific memory CD8 T cells.
Greater than 50% of locally advanced pancreatic malignancies present with metastatic disease that precludes patients from surgery (Hidalgo et al., 2015). It has been postulated that the abscopal effect induced by RT is driven by the activation of a systemic immune response, characteristic of an in situ tumor vaccination (de la Cruz-Merino et al., 2014). Unfortunately, the abscopal effect following RT monotherapy is rarely observed clinically, suggesting the need for optimization by targeting the immune system (e.g., IL-12 MS). Having shown that SBRT/IL-12 MS elicited potent local effects on orthotopic pancreas tumors, we next tested whether combination therapy could elicit an abscopal effect on a synchronous secondary tumor. Primary KCKO (luciferase null) tumors were injected in the pancreas alongside simultaneous implantation of secondary KCKO-luc metastases in the liver by using the hemi-spleen technique (Figure 6E). SBRT and IL-12 MS therapeutic scheduling was not modified, and treatments were delivered only to the primary pancreas tumor. IVIS bioluminescent imaging was used to track luciferase-expressing liver metastases, and although untreated and monotherapy-treated controls developed aggressive metastatic disease (Figures S7A and 6F), SBRT/IL-12 MS treatment resulted in the elimination of established liver metastases (Figure 6F) and significantly improved survival (Figure 6G). These experiments also demonstrated the therapeutic potency of SBRT/IL-12 MS in aged mice (30 weeks old); however, similar results were observed when the experiment was repeated in 6- to 8-week-old mice (Figure S7B). Additionally, the abscopal effect elicited by SBRT/IL-12 MS was not limited to the liver. We also used a distant metastasis model in which KCKO-luc tumor cells were synchronously implanted in the pancreas (primary lesion) and leg muscle (secondary lesion) and both were measured using IVIS imaging (Figure S7C). Although no difference in the therapeutic response of primary tumors was observed, both bioluminescent and caliper measurements confirmed a significant size reduction of untreated leg tumors upon SBRT/IL-12 MS treatment of the pancreas, resulting in 60% of mice being tumor-free at 25 days post-implantation (Figures S7D and S7E, respectively). By monitoring plasma IL-12 concentrations, we could determine if systemic increases were generating the observed abscopal effect. Plasma IL-12 levels were found to be uniformly upregulated in both IL-12 MS and SBRT/IL-12 MS groups following treatment (Figure S7F); however, the abscopal effect was only observed in the SBRT/IL-12 MS treatment group. These data suggest that although systemic increases in IL-12 may contribute to the therapeutic effect on secondary lesions, only the combination of SBRT with IL-12 MS generates a systemic antitumor effect that is capable of destroying established metastases.
DISCUSSION
The development of conRT for PDA has lost initiative in recent years after clinical trials demonstrated ineffectual overall survival and local tumor control outcomes (Neoptolemos et al., 2004; Rich et al., 2004; Hammel et al., 2016). Studies over the last decade have characterized the importance of an intact immune response for tumor resolution following radiotherapy, and immune attenuation could be a chief contributor to these clinical shortcomings. This work examined two emerging therapeutic strategies for PDA, SBRT and immunotherapy, hypothesizing that the combination would stimulate an immunogenic response capable of downsizing locally advanced lesions. The PDA tumor microenvironment (TME) is highlighted by a profoundly immunosuppressive stroma that accompanies PDA transformation and prevents the establishment of CD8 T effector responses (Feig et al., 2012). Furthermore, commonly utilized conRT scheduling can also impart lymphopenia in addition to promoting an immunosuppressive cellular milieu (Schrek, 1961; Rech et al., 2018; Xu et al., 2013). Our immunohistochemistry (IHC) findings (Figure 1) highlighted the core benefit of SBRT, namely, increased intratumoral CD8 T cell load, and accordingly, we approached SBRT scheduling as a means of delivering acute tumor damage to amplify the antigen-specific adaptive immune response. Conversely, we observed abundant myeloid suppressor cell densities in both untreated and SBRT-treated patient samples, suggesting that the presence of immunosuppressor cells remained a central therapeutic obstacle. These results prompted us to develop an immunotherapy combination (IL-12 MS) that could both stimulate the activation of recruited CD8 T cells, as well as reprogram the abundance of immunosuppressor cells throughout the tumor.
PDA is underscored by a diverse protumor landscape that includes a predominance of Treg and myeloid suppressor cells, the exclusion or exhaustion of cytotoxic T cells, and a desmoplastic and/or avascular extracellular matrix. Each of these characteristics contribute to an immune imbalance favoring the immunosuppression inherent to PDA (Feig et al., 2012). The development of an antitumor immune response in this setting requires a multifaceted intervention that comprehensively repolarizes the stromal component. For this reason, the pleiotropic activity of IL-12 made it an attractive candidate for intervention. Our work demonstrates a robust antitumor effect in recalcitrant PDA tumors by using SBRT/IL-12 MS treatment. Furthermore, we show therapeutic efficacy across three aggressive preclinical models, with the KCKO-luc orthotopic model resulting in 100% cures (Figure 2). The ability of SBRT/IL-12 MS therapy to reprogram and commandeer the diverse immunosuppressive stroma in PDA may explain why we see such powerful immune responses across characteristically cold tumor models.
The synergistic IFNγ induction following SBRT/IL-12 MS combination is our most striking finding, and we speculate that SBRT-driven increases in IFNγ-producing cells is a crucial precursory event. Although NK cells and APCs are common contributors to IFNγ production, we found their relative abundances to be unaffected by SBRT treatment alone (Figures S5A and S5C). Rather, we observed a SBRT/IL-12 MS-treated CD8 T cell transcriptome that was primed for IFNγ production (Figure 5E). Surprisingly, coordinate studies investigating intratumoral IFNγ levels under CD8+ T cell depletion demonstrated increased cytokine amounts with SBRT/IL-12 MS treatment, underscoring both CD4 T cell and myeloid contributions and compensatory potential (data not shown). Although we observed robust IFNγ induction, the expression was transient (similar to the profile of IL-12 release; Figure 3), and the mechanism by which acute IFNγ priming can initiate a sustained antitumor response is not completely understood. One contributing factor may be the coordination of IFNγ and tumor necrosis factor α (TNFα) stimulation. Yarilina et al. (2008) has demonstrated that synchronous high-level IFNγ/TNFα signaling can initiate an autocrine loop of proinflammatory signaling, leading to the stable reprogramming of myeloid suppressors.
IFNγ has been shown to elicit transcriptional feedback on IL-12 through IFN consensus sequence binding protein (ICSBP) activation, driving a feedforward response (Wang et al., 2000). Although this signaling architecture can produce powerful proinflammatory events, persistent interferon stimulation has been shown to drive epigenetic changes that promote multiple T cell exhaustion paradigms (Benci et al., 2016). Our intratumoral Luminex profiling (Figure 3) suggests that feedforward IL-12/IFNγ production occurs following SBRT/IL-12 MS treatment in our KCKO-luc model (Figure S5A). Importantly, during peak IL-12/IFNγ signaling (day 11), we did not observe marked CD8 T cell exhaustion, and heightened cell numbers at day 14 suggest sustained proliferation for multiple days following IL-12 MS delivery (Figures 5B and 5E). In addition to IM/TAM repolarization events, the CD8 T cell response was likely supported by acute reductions in Treg density mediated by IFNγ; however, Treg restoration approximately 96-h post-treatment may be of similar therapeutic importance (Figure S5E). IFNγ-dependent indoleamine 2,3-dioxygenase (IDO) induction has been found to enhance Treg rebound following cytotoxic events, and work by Kalia et al. (2015) would suggest that Treg signaling through CTLA4 is necessary for the formation of fit and functional Tmem populations. The ability of SBRT/IL-12 MS to evoke a dynamic proinflammatory stimulus followed by standard immunomodulatory feedback and memory formation may be essential for therapeutic efficacy. Maintaining an invigorated repertoire of tumor-specific T cells following the SBRT/IL-12 MS response dramatically improves the potential for successful second-round treatment with a tertiary immunotherapy or repeated IL-12 MS administration.
Our work has uncovered a multifaceted mechanism, illustrated in Figure 7, through which SBRT/IL-12 MS elicits antitumor effects. PDA tumorigenesis is typically highlighted by marked infiltration of immunosuppressive Treg cells, IMs that seed TAM populations, and a paucity of CD8 T cells in the lesion periphery (Figure 7A). An antitumor immune response is initiated by SBRT, which likely induces immunogenic tumor cell death producing tumor antigen and presentation, both of which are necessary for Teff formation in the draining lymph node (DLN) (green) (Figure 7B). However, these increases in intratumoral CD8 Teff cells have modest antitumor effects due to the ancillary recruitment of Treg and IM/TAM suppressors. To overcome this obstacle, local IL-12 MS treatment stimulates intratumoral T effectors to produce IFNγ, which repolarizes both lymphoid and myeloid suppressors (Figure 7C). The resolution of PDA tumors is highlighted by Treg rebound and Tmem formation, resulting in lasting tumor-specific immune memory that can control and/or eliminate distal metastases (Figure 7D). This proposed summary likely represents the oversimplification of a much more complex antitumor mechanism involving a multitude of cells and pathways not yet defined. For example, the IL-12/IFNγ axis is capable of eliciting effects on non-immune targets, such as tumor cells (increased MHCI expression and cytostatic and/or cytotoxic effects) and endothelial cells (release of antiangiogenic and immune adhesion molecules), that may also contribute to immune-mediated antitumor effects (Suzuki et al., 1998; Strasly et al., 2001).
Figure 7. Schematic of SBRT/IL-12 MS Therapeutic Mechanism in PDA.

(A) PDA tumorigenesis is highlighted by marked infiltration of immunosuppressive Treg cells, IMs that seed TAM populations, and a paucity of CD8 T cells in the lesion periphery.
(B) SBRT initiates necrotic cell death that produces tumor antigen necessary for Teff formation in the DLN (green). Increases in intratumoral CD8 Teff cells have modest antitumor effects due to the ancillary recruitment of Treg and IM/TAM suppressors.
(C) IL-12 MS treatment stimulates intratumoral T effectors to produce IFNγ, which initiates Th1 repolarization of Tregs, activation of IMs (adding to IFNγ pools) and M1 reprogramming of TAMs. The actuation of Teff cell number and function elicits marked tumor cell apoptosis.
(D) The resolution of PDA tumors is highlighted by Treg rebound and Tmem formation, resulting in lasting tumor-specific immune memory.
Intratumoral IL-12 administration was used to mitigate the systemic toxicity of intravenous administration observed in some clinical studies (Jenks, 1996). Importantly, no mice experienced adverse events or exhibited signs of immune reaction to IL-12 MS treatment (data not shown), and a phase I clinical trial of SBRT/IL-12 MS in locally advanced PDA is in preparation. Serial surgeries were used to deliver IL-12 in PDA mouse models (Figure 2); however, translating this approach to the clinic would likely involve endoscopic ultrasound-guided (EUS) techniques. EUS intervention is currently used for PDA diagnosis and staging as well as fiducial marker placement for radiotherapy and would be a safe, minimally invasive approach for i.t. IL-12 MS delivery (Al-Haddad and Eloubeidi, 2010). We also postulate that MS packaging provides an added level of IL-12 intratumoral sequestration due to the enhanced permeability and retention (EPR) effect. The encapsulation of IL-12 in a 1- to 5-μm polymer coating likely prevents passive clearance by weakened lymphatic drainage while protecting the cytokine from proteolytic degradation or phosphatidylserine capture in the TME (Sevenich and Joyce, 2014; Oyler-Yaniv et al., 2017). Apart from supporting its sustenance in the TME, MS technology may also affect the cellular uptake and trafficking of IL-12, as work by Champion et al. (2008) demonstrated the phagocytosis and internalization of 2- to 3-μm polystyrene MSs by rat alveolar macrophages. We observed similar engulfment following i.p. injection of AF594-labeled MS (Figures S2C and S2D). These findings evoke compelling questions surrounding intracellular IL-12 signaling mechanisms, the role of phagocytosis in myeloid reprogramming, and the active cellular transport of IL-12 MS to sites of PDA dissemination, such as the liver- and tumor-draining lymph nodes.
Approximately 60% of locally advanced PDA malignancies present with metastatic disease, and furthermore, this value excludes additional cases with undetectable micrometastases (Gillen et al., 2010). A therapy that harnesses the capacity for both local and distal tumor control would dramatically increase the number of patients eligible for neoadjuvant intervention and, potentially, surgical candidacy. Similar to other works investigating SBRT and immunotherapy combination (Yasmin-Karim et al., 2018), our evaluation of systemic immune memory following SBRT/IL-12 MS therapy (Figure 6) confirmed its capacity for abscopal control of an untreated synchronous lesion, suppression of outgrowth upon metachronous rechallenge, and transferal of protection to naive recipients. Comprehensively, these findings strongly suggest that SBRT/IL-12 MS treatment initiated a potent in situ vaccination. From this perspective, rather than classifying IL-12 MS as an adjuvant to radiation, SBRT may also be viewed as a tool for producing tumor a neoantigen that primes the IL-12 MS-driven immune response. The capacity of this combination therapy to potentiate a robust systemic immune response expands interventional opportunity beyond the scope of borderline resectable lesions to include advanced metastatic disease. Alongside the potency and durability of SBRT/IL-12 MS in preclinical models of advanced disease, these features strongly advocate the continued clinical translation of this therapeutic approach for PDA.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILIBILITY
This study did not generate new reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott A. Gerber (scott_gerber@urmc.rochester.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo Animal Studies
All experiments were approved by the University Committee on Animal Resources, and were performed in compliance with the NIH and University of Rochester approved guidelines for the care and use of animals. Six- to eight-week old C57BL/6J and B6.129S7-Ifngtm1Ts/J (IFNγ−/−) mice were obtained from The Jackson Laboratory. P48-Cre+/−; Tp53L/L and LSL-KrasG12D mice were obtained from Dr. Aram Hezel, and crossed to obtain the P48-Cre; LSL-KrasG12D; Tp53L/L (KPC) genotype (Hezel et al., 2012). For orthotopic modeling, age-matched female mice were used; for KPC GEMM studies male and female mice were randomly assigned to experimental groups. All mice were maintained on a 12-hour light/dark cycle and were kept in individually ventilated cages with bedding and nesting material. Health monitoring was performed according to the FELASA Guideline 2014.
Human Studies
All human protocols were reviewed and approved by the University of Rochester institutional review board. Informed consent was obtained from all subjects involved in this study. For immunohistochemistry analyses, de-identified sections were obtained from the University of Rochester Department of Surgical Pathology. All specimens included in the study were curated from patients diagnosed with resectable PDA, recruited at the University of Rochester Medical Center. No information on the sex/age of human subjects is available.
Cell lines
KCKO and Pan02 cell lines stably expressing firefly luciferase (KCKO-luc, Pan02-luc) were obtained from Dr. David DeNardo (Zhu et al., 2014). Both cell lines were syngeneic to the C57BL/6J background. Cultures were maintained in DMEM/F-12 (GIBCO) supplemented with 10% fetal bovine serum (GIBCO) and 1% penicillin/streptomycin (ThermoFisher Scientific) at 37°C and 5% CO2.
METHOD DETAILS
Orthotopic Tumor Implantation
Mice were anesthetized using an isoflurane anesthetic vaporizer (Scivena Scientific) and a 10-mm laparotomy incision was made to expose the spleen and pancreas. Cell lines were detached with 0.25% trypsin/EDTA (GIBCO) and resuspended in a 1:1 PBS:Matrigel (BD Biosciences) solution. One-hundred-thousand cells (50 μL) were injected into the pancreatic tail, and two 4-mm titanium fiducial markers (Horizon) to assist in SBRT targeting were implanted adjacent to the tumor bubble. For one minute immediately following tumor cell injection, a cotton swab was placed over the injection site to prevent peritoneal leakage. IVIS bioluminescent imaging verified successful implantations with no peritoneal studding and provided baseline measurements for standardizing pretreatment groups.
Radiation Therapy
All radiation was delivered using the Small Animal Radiation Research Platform (SARRP, XStrahl) with a 5-mm collimator. Mice were anaesthetized with vaporized isoflurane during all radiation treatments. Tumor-bearing mice that underwent conventional radiotherapy (conRT) were dosed with 2 Gy in 15 fractions yielding a biologically effective dose (BED = nd(1 + d /(α/β))) of 36 for tumor and 50 for normal tissue (assuming an α/β ratio of 10 for tumor and 3 for normal). Stereotactic body radiation therapy (SBRT) was administered to tumor-bearing mice following a schedule of 6 Gy radiation in 4 fractions on days 6–9 post-implantation, yielding a BED of 38.4 for tumor and 72 for normal tissue. Localized delivery was targeted using previously mentioned titanium fiducial markers; markers were visualized with pretreatment computed tomography (CT) scans. The dosing isocenter was positioned using a beam angle designed to circumvent major organs. In each case, a dose volume histogram (DVH) was generated to confirm full dose deposition to the tumor and negligible amounts to surrounding organs (e.g., liver).
Bioluminescent Imaging
In vivo tumor growth was measured using the IVIS Spectrum Imaging System (IVIS, PerkinElmer). Mice were anesthetized by vaporized isoflurane and injected subcutaneously (s.c.) with D-luciferin (2.5 mg, Invitrogen) in 100 μL PBS vehicle. While in the right lateral recumbent position, a series of images were taken at 2-minute intervals for 24 min and photon emissions were collected. Bioluminescence (p/sec/cm2/sr) was calculated within matching (circular) regions of interest (ROIs) manually placed over tumors. Peak intensity was recorded for each tumor upon two sequential measurements demonstrating signal decay.
Immunohistochemistry
All human SBRT-treated specimens were acquired 10–14 days following neoadjuvant intervention; mouse tumors were harvested on day 11. All PDA tissue samples were fixed in 10% neutral buffered formalin. Fixed human tissue was processed (paraffin embedded), and sectioned by the University of Rochester Department of Surgical Pathology. Fixed mouse tissue was processed (paraffin embedded), and sectioned by the University of Rochester Center for Tumor Immunology Research Histology Core. For IHC analyses, serial sections were stained overnight at 4°C with anti-CD8 (C8/144B, 1:100, Thermo Scientific MS-457–57), and anti-CD68 (KP1, 1:200, Thermo Scientific MS-397-P0) antibodies. A polymer-based system was used for detection (GBI Broad Spectrum Polink 2 Plus (GBI D22), DAB chromogen (GBI CO2–12) incubation). Slides were counterstained with Mayer’s hematoxylin. Staining was completely absent in identical tissue sections in which a universal negative control solution was used (Enzo ADI-950–231-0025). Whole tissue sections were digitized and registered at 20× magnification and registered. Regions of interest were defined by a licensed pathologist (blinded) as follows: “margin” - area defined by 500 μm within and outside of a manually drawn line at tumor/stroma interface; “center” - area defined by 1000 μm within the invasive front, mucosal surface, or tissue edge. Areas of tissue loss/artifacts were excluded from analysis. Random forest classification was used to enumerate the number of events, and margin index (ratio of center:margin) was calculated for entire ROIs of each case individually.
Microsphere Injection
Polylactic acid microspheres were created using phase inversion phenomena as previously described (Mathiowitz et al., 2000; Egilmez et al., 2000). Lyophilized microspheres were resuspended in PBS (20 μL per mouse) prior to i.t. delivery. Twenty-four hours following the final SBRT fraction (day 10), mice were anesthetized with vaporized isoflurane and a 10-mm laparotomy incision was made to expose pancreas tumors. Empty MS control (2 mg beads) or IL-12 MS (2 mg beads containing 0.5 μg recombinant IL-12) were injected i.t. using an 18-guage Hamilton syringe.
Luminex Analyte Assay
Following sacrifice, mouse tumors were dissociated with a tissue homogenizer in 100 μL 0.5× Cell Lysis Buffer 2 (R&D Systems, diluted in PBS), containing 1× Halt Protease Inhibitor Cocktail and 1x Halt Phosphatase Inhibitor Cocktail (ThermoFisher Scientific). Tissues were lysed at room temperature for 30 min with gentle agitation. Magnetic Luminex Assays were performed using a Mouse Premixed Cytokine/Chemokine Multi-Analyte Kit (R&D Systems). Assay procedures were carried out following manufacturer’s instructions. Microplates were run on a Bio-Plex 200 system (Bio-Rad) collecting 50–100 beads per target with less than 20% aggregate. Pierce BCA Protein Assays (ThermoFisher Scientific) were performed on remaining lysates following manufacturer’s instructions. Total protein concentrations for each sample were used for analyte normalization into pg/mg protein values.
Flow Cytometry
Following sacrifice, mouse tumors were mechanically dissociated followed by digestion with 30% collagenase (30 mins, 37°C, Sigma-Aldrich). Homogenates were then passed through 40 μm filters and cells were resuspended in PAB (1L PBS, 1g sodium azide, 10 g BSA) at approximately 1 × 106 cells/reaction. The following conjugated antibodies were used for staining: CD45-PerCP-Cy5.5 (30-F11, BD Biosciences), CD8-eFluor 450 (53–6.7, eBioscience), CD4-APC-Cy7 (GK1.5, BD PharMingen), NK-1.1-PE-CF594 (PK136, BD Biosciences), CD11b-eFluor 450 (M1/70, Invitrogen), Ly-6C-APC-Cy7 (AL-21, BD Biosciences), Ly-6G-BV605 (1A8, BioLegend), F4/80-APC (BM8, eBioscience), I-A/I-E-PE (M5/114.15.2, BD Biosciences), FOXP3-APC (FJK-16 s, eBioscience), IFN-gamma-PE (XMG1.2, BD Biosciences), CD11c-PE-Cy7 (HL3, BD Biosciences), CD19-BV510 (1D3, BD Biosciences), CD44-BV510 (IM7, BioLegend), CD107a-PE-Cy7 (1D4B, BD Biosciences), CD152-PE-CF594 (UC10–4F10–11, BD Biosciences), CD279-APC-R700 (J43, BD Biosciences). Cell surface antigens were stained for 30 min at 4°C in the dark. Samples were then washed with PAB and fixed overnight using the FOXP3 fixation/permeabilization kit (eBioscience) following manufacturer’s instructions. The following day, cells were washed with FOXP3 Permeabilization Buffer (eBioscience) and stained for intracellular targets for 30 min at 4°C in the dark. FMO controls were utilized for intracellular activation markers. Cells were washed and resuspended in PAB and run on a LSRII Fortessa (BD Biosciences). Fifty to one hundred thousand events/sample were collected and analyzed using FlowJo software (FlowJo). The same procedure was followed for imaging flow cytometry, and samples were run on an Amnis ImageStream GenX (Luminex Corporation).
RNA-Seq
Following sacrifice, 2 mouse tumors were pooled per treatment group. Tissues were mechanically dissociated followed by digestion with 30% collagenase (30 mins, 37°C, Sigma-Aldrich). Homogenates were passed through 40 μm filters and cells were resuspended in PAB (1L PBS, 1g sodium azide, 10 g BSA). Between 2 × 106 and 4 × 106 cells from each treatment group were stained for cell surface antigens for 30 min at 4°C in the dark. CD8+ T cell, IM, and TAM populations were sorted on a FACSAria II cell sorter (BD Biosciences) using a 100 μm nozzle. Cells were immediately lysed in Buffer RLT (containing β-mercaptoethanol), homogenized with QIAShredder spin columns, and RNA was purified using the RNeasy Micro Kit (QIAGEN) following manufacturer’s instructions. RNA sequencing and analysis was performed by the University of Rochester Genomics Research Center. RNA quality was assessed using an Agilent Bioanalyzer (Agilent), and all samples analyzed demonstrated RNA integrity values > 5. CDNA libraries were constructed using the TruSeq RNA Sample Preparation Kit V2 (Illumina) following manufacturer’s instructions, and sequencing was performed on an Illumina high-throughput HiSeqTM 2500 platform (Illumina). Genes differentially-expressed in treatment groups relative to unirradiated + empty MS controls were analyzed using Ingenuity Pathway Analysis (IPA) software (QIAGEN).
Myeloid Transplant
Following sacrifice on day 11, KCKO-luc tumors were harvested and prepared for flow sorting following the procedure used for RNA-seq analysis. IM, and TAM populations were sorted on a FACS Aria II cell sorter (BD Biosciences) using a 100 μm nozzle into DMEM/F-12 supplemented with 10% fetal bovine serum. Sorted IMs and TAMs were counted and pooled with freshly cultured KCKO-luc cells in a 1:1:2 ratio, respectively. Cell mixtures were resuspended in a 1:1 PBS:Matrigel (BD Biosciences) solution, and 100,000 cells (50 μL) were injected into the pancreatic tail of naive mice following the standard orthotopic implantation procedure. No further treatments were administered, and IVIS bioluminescent imaging was used to measure tumor outgrowth.
Antibody Depletion
Following KCKO-luc tumor implantation on day 0, 200 μg depleting antibody (resuspended to 100 μL in PBS) was injected in mice s.c. every 3 days between days 5 and 20 (6 doses). The antibodies (Bio X Cell) delivered were rat isotype control (IgG2a, C1.18.4), rat anti-mouse CD8α (IgG2a, 53–6.7), and rat anti-mouse CD4 (IgG2a, GK1.5).
Hemi-Spleen Tumor Implantation
Mice cured of primary KCKO-luc tumors were rechallenged approximately 6 months following initial orthotopic tumor implantation. Vaporized isoflurane was used to anesthetize mice prior to ten-mm laparotomy incision to expose the spleen. Six-mm titanium clips (Horizon) were placed adjacent to the hepatic portal vein, and a hemisecting cut was made between ligations. KCKO-luc cells were detached with 0.25% trypsin/EDTA, resuspended in PBS, and passed through a 40 μm filter to achieve a single cell suspension. The hemi-spleen segment connected to the hepatic portal vein was injected with 5 × 105 cells (in 100 μL PBS) slowly, over the course of 1 min. Following implantation, a third titanium clip was used to ligate the hepatic portal vein immediately adjacent to the spleen, and the injected splenic segment was resected and distal vasculature was cauterized. For bioluminescent imaging, mice were placed in the supine position.
T Cell Transplants
Mice with no evidence of tumor burden following primary orthotopic implantation and hemi-spleen rechallenge were sacrificed approximately 9 months following initial tumor challenge. Additionally, 5 age-matched tumor-naive mice were also sacrificed for controls. Spleens as well as draining, axillary, inguinal, and iliac lymph nodes were harvested and mechanically dissociated. CD8+ T cells were isolated by negative selection using the EasySep Mouse CD8+ T Cell Isolation Kit (STEMCELL Technologies), following manufacturer’s instructions. Approximately 4 × 106 CD8+ T cells per donor were purified from an input of 5 × 107 cells per donor. CD8+ T cells were resuspended in 100 μL PBS and delivered to recipient mice via tail vein injection 16 h prior to KCKO-luc orthotopic tumor implantation (1:1 donor:recipient transferal). IVIS tumor burden measurements were used to classify the transferal of immunological memory (> 10-fold decrease in tumor volume), and blinded manual palpation at day 40 was used to confirm full immunological memory (unidentifiable tumor).
Abscopal Studies
Prior to standard pancreatic orthotopic implantation on day 0, 5 × 104 cells (suspended in 100 μL PBS) or 5 × 105 cells (suspended in 100 μL PBS) were intramuscularly injected into the left hindlimb or seeded in the liver using the hemi-spleen technique, respectively. To prevent interference with SBRT targeting, Vicryl suture (Ethicon) was used for spleen and portal vein ligations. Following SBRT treatment of the primary pancreatic tumor, dose volume histograms were generated to confirm full dose deposition into the primary tumor without spillover into the secondary leg or liver tumors. For IVIS imaging, mice were administered 2.5 mg D-luciferin s.c. (in 100 μL PBS), and both primary and secondary tumor ROIs were designated for separate bioluminescent measurements. Digital calipers were used to measure leg tumor diameters in two perpendicular dimensions, and volumes were calculated using the formula: d1 × d22 × 0.52. Blinded caliper measurements were used to identify tumor-free mice.
QUANTIFICATION AND STATISTICAL ANALYSES
Prism 7 software (GraphPad) was used for all statistical analyses, and p values of < 0.05 were considered significant. Unless stated otherwise, all summary values represent the geometric mean/standard deviation of individual data points. One or two way ANOVAs were used as appropriate for the dataset analyzed. For IHC analyses, independent t tests were used to compare the mean values of untreated to SBRT-treated tumors. Laevene’s test was used to evaluate equality of variances. Holm-Sidak tests were performed on tumor bioluminescence measurements post-treatment and prior to recurrence (i.e., days 13–20). Tumor growth/survival analyses compared the curves of each treatment group to UI/Empty MS controls, while all other multiple comparisons additionally analyzed SBRT/Empty MS to SBRT/IL-12 MS and UI/IL-12 MS to SBRT/IL-12 MS. All flow cytometry gating was done using FlowJo 10 software (FlowJo). Ingenuity Pathway Analysis (IPA) software (QIAGEN) was used for all RNA-seq differentially expressed gene analyses. All diagrammatic figures were created with BioRender.
DATA AND CODE AVAILABILITY
The accession number for the RNA sequencing data reported in this paper is GEO: GSE136368.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CD8 | Lab Vision | Cat# MS-457-R7; RRID: AB_60754 |
| Mouse monoclonal anti-CD68 | Lab Vision | Cat# MS-397-P0; RRID: AB_720547 |
| Rat monoclonal anti-CD45, PerCP-Cy5.5 conjugated | BD Biosciences | Cat# 550994; RRID: AB_394003 |
| Rat monoclonal anti-CD8, eFluor 450 conjugated | eBioscience | Cat# 48–0081–82; RRID: AB_1272198 |
| Rat monoclonal anti-CD4, APC-Cy7 conjugated | BD Biosciences | Cat# 552051; RRID: AB_394331 |
| Mouse monoclonal anti-NK-1.1, PE-CF594 conjugated | BD Biosciences | Cat# 562864; RRID: AB_2737850 |
| Rat monoclonal anti-CD11b, eFluor 450 conjugated | eBioscience | Cat# 48–0112–82; RRID: AB_1582236 |
| Rat monoclonal anti-Ly-6C, APC-Cy7 conjugated | BD Biosciences | Cat# 560596; RRID: AB_1727555 |
| Rat monoclonal anti-Ly-6G, BV605 conjugated | BioLegend | Cat# 127639; RRID: AB_2565880 |
| Rat monoclonal anti-F4/80, APC conjugated | eBioscience | Cat# 17–4801; RRID: AB_469452 |
| Rat monoclonal anti-I-A/I-E, PE conjugated | BD Biosciences | Cat# 557000; RRID: AB_396546 |
| Rat monoclonal anti-FOXP3, APC conjugated | eBioscience | Cat# 17–5773–80; RRID: AB_469456 |
| Rat monoclonal anti-IFN-gamma, PE conjugated | BD Biosciences | Cat# 554412; RRID: AB_395376 |
| Hamster monoclonal anti-CD11c, PE-Cy7 conjugated | BD Biosciences | Cat# 561022; RRID: AB_2033997 |
| Rat monoclonal anti-CD19, BV510 conjugated | BD Biosciences | Cat# 562956; RRID: AB_2737915 |
| Rat monoclonal anti-CD44, BV510 conjugated | BioLegend | Cat# 103044; RRID: AB_2650923 |
| Rat monoclonal anti-CD107a, PE-Cy7 conjugated | BD Biosciences | Cat# 560647; RRID: AB_1727419 |
| Hamster monoclonal anti-CD152, PE-CF594 conjugated | BD Biosciences | Cat# 564332; RRID: AB_2732917 |
| Hamster monoclonal anti-CD279, APC-R700 conjugated | BD Biosciences | Cat# 565815; RRID: AB_2739366 |
| Mouse monoclonal IgG2a isotype control | Bio X Cell | Cat# BE0085; RRID: AB_1107771 |
| Rat monoclonal anti-CD8α | Bio X Cell | Cat# BE0004–1; RRID: AB_1107671 |
| Rat monoclonal anti-CD4 | Bio X Cell | Cat# BE0003–1; RRID: AB_1107636 |
| Biological Samples | ||
| Human PDA blocks | University of Rochester Department of Surgical Pathology | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Empty PLA microspheres | TherapyX | https://therapyxinc.com |
| IL-12 microspheres | TherapyX | https://therapyxinc.com |
| AF594-BSA microspheres | TherapyX | https://therapyxinc.com |
| Critical Commercial Assays | ||
| Mouse Magnetic Luminex Assay | R&D Systems | Cat# LXSAMSM-08 |
| EasySep Mouse CD8+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19853 |
| Deposited Data | ||
| MSigDB: GSE5099, GSE1000002 | Broad Institute | RRID:SCR_007073 |
| GEO: GSE136368 | NCBI | N/A |
| Experimental Models: Cell Lines | ||
| Mouse: KCKO cells | Zhu et al., 2014 | N/A |
| Mouse: Pan02 cells | Zhu et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S7-Ifngtm1Ts/J | The Jackson Laboratory | JAX: 002287 |
| Mouse: P48-Cre; LSL-KrasG12D; Tp53L/L | Hezel et al., 2012 | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | RRID:SCR_008520 |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653 |
| Prism | GraphPad | RRID:SCR_002798 |
Highlights.
SBRT/IL-12 MS combination immunotherapy eradicates murine orthotopic PDA tumors
IFNγ produced following SBRT/IL-12 MS repolarizes intratumoral myeloid suppressors
Robust CD8 T cell activation and memory formation is initiated by SBRT/IL-12 MS
SBRT/IL-12 MS treatment elicits an abscopal effect, eliminating hepatic metastases
ACKNOWLEDGMENTS
Supported by grants from the NIH (1P50CA196510-01A1 to Washington University in St. Louis School of Medicine, University of Rochester, University of North Carolina at Chapel Hill, and Johns Hopkins Medicine; R01CA230277 to S.A.G.; R01CA168863 to D.C.L.; T90DE021985 to University of Rochester; and S10OD021548-01 to Jacqueline Williams). We additionally thank Mary Georger of the Histology Core, Eric Hernady of the Small Animal Irradiation Core, and Dr. John Ashton in the Genomics Research Core at URMC.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.095.
REFERENCES
- Al-Haddad M, and Eloubeidi MA (2010). Interventional EUS for the diagnosis and treatment of locally advanced pancreatic cancer. JOP 11, 1–7. [PubMed] [Google Scholar]
- Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzone PD, et al. (1997). Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. Cancer Res 3, 409–417. [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Barberà-Cremades M, Baroja-Mazo A, and Pelegrín P (2016). Purinergic signaling during macrophage differentiation results in M2 alternative activated macrophages. J. Leukoc. Biol 99, 289–299. [DOI] [PubMed] [Google Scholar]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Champion JA, Walker A, and Mitragotri S (2008). Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res 25, 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo MP, Vagliani M, Spreafico F, Parenza M, Chiodoni C, Melani C, and Stoppacciaro A (1996). Amount of interleukin 12 available at the tumor site is critical for tumor regression. Cancer Res. 56, 2531–2534. [PubMed] [Google Scholar]
- Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, Lord EM, Linehan DC, and Gerber SA (2016). Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 7, 86522–86535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz-Merino L, Illescas-Vacas A, Grueso-López A, Barco-Sánchez A, and Míguez-Sánchez C; Cancer Immunotherapies Spanish Group (GETICA). (2014). Radiation for Awakening the Dormant Immune System, a Promising Challenge to be Explored. Front. Immunol 5, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplanque G, Shabafrouz K, and Obeid M (2017). Can local radiotherapy and IL-12 synergise to overcome the immunosuppressive tumor microenvironment and allow “in situ tumor vaccination”? Cancer Immunol. Immunother 66, 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, and Bankert RB (2000). In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 60, 3832–3837. [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, and Tuveson DA (2012). The pancreas cancer microenvironment. Clin. Cancer Res 18, 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem 289, 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, and Kleeff J (2010). Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 7, e1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, Borbath I, Bouché O, Shannon J, André T, et al. ; LAP07 Trial Group (2016). Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 315, 1844–1853. [DOI] [PubMed] [Google Scholar]
- Hezel AF, Deshpande V, Zimmerman SM, Contino G, Alagesan B, O’Dell MR, Rivera LB, Harper J, Lonning S, Brekken RA, and Bardeesy N (2012). TGF-β and αvβ6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 72, 4840–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M, Cascinu S, Kleeff J, Labianca R, Löhr JM, Neoptolemos J, Real FX, Van Laethem JL, and Heinemann V (2015). Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology 15, 8–18. [DOI] [PubMed] [Google Scholar]
- Jenkins RW, Barbie DA, and Flaherty KT (2018). Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks S (1996). After initial setback, IL-12 regaining popularity. J. Natl. Cancer Inst 88, 576–577. [DOI] [PubMed] [Google Scholar]
- Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, and Beatty GL (2017). Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res 23, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, and Sarkar S (2015). Quiescence of Memory CD8(+) T Cells Is Mediated by Regulatory T Cells through Inhibitory Receptor CTLA-4. Immunity 42, 1116–1129. [DOI] [PubMed] [Google Scholar]
- Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, et al. (2011). IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Invest 121, 4746–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, and Lord EM (2008). Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol 180, 3132–3139. [DOI] [PubMed] [Google Scholar]
- MacMicking J, Xie QW, and Nathan C (1997). Nitric oxide and macrophage function. Annu. Rev. Immunol 15, 323–350. [DOI] [PubMed] [Google Scholar]
- Mathiowitz E, Chickering Iii D, Jong YS, and Jacob JS (2000). Process for Preparing Microparticles Through Phase Inversion Phenomena. US patent 6,143,211, filed July 3, 1996, and published November 7, 2000.
- Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. (2018). Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, et al. ; European Study Group for Pancreatic Cancer (2004). A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med 350, 1200–1210. [DOI] [PubMed] [Google Scholar]
- Norris PC, and Dennis EA (2014). A lipidomic perspective on inflammatory macrophage eicosanoid signaling. Adv. Biol. Regul 54, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Order SE (1977). The effects of therapeutic irradiation on lymphocytes and immunity. Cancer 39, 737–743. [DOI] [PubMed] [Google Scholar]
- Oun R, Moussa YE, and Wheate NJ (2018). The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 47, 6645–6653. [DOI] [PubMed] [Google Scholar]
- Oyler-Yaniv J, Oyler-Yaniv A, Shakiba M, Min NK, Chen YH, Cheng SY, Krichevsky O, Altan-Bonnet N, and Altan-Bonnet G (2017). Catch and Release of Cytokines Mediated by Tumor Phosphatidylserine Converts Transient Exposure into Long-Lived Inflammation. Mol. Cell 66, 635–647.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee KP, Kang S, Lee J, Sato K, Chung HY, Okajima F, and Im DS (2014). Sphingosine 1-phosphate induced anti-atherogenic and atheroprotective M2 macrophage polarization through IL-4. Cell. Signal 26, 2249–2258. [DOI] [PubMed] [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quéméneur L, Beloeil L, Michallet MC, Angelov G, Tomkowiak M, Revillard JP, and Marvel J (2004). Restriction of de novo nucleotide biosynthesis interferes with clonal expansion and differentiation into effector and memory CD8 T cells. J. Immunol 173, 4945–4952. [DOI] [PubMed] [Google Scholar]
- Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, and Vonderheide RH (2018). Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 78, 4282–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T, Harris J, Abrams R, Erickson B, Doherty M, Paradelo J, Small W Jr., Safran H, and Wanebo HJ (2004). Phase II study of external irradiation and weekly paclitaxel for nonmetastatic, unresectable pancreatic cancer: RTOG-98–12. Am. J. Clin. Oncol 27, 51–56. [DOI] [PubMed] [Google Scholar]
- Schrek R (1961). Qualitative and quantitative reactions of lymphocytes to x rays. Ann. N Y Acad. Sci 95, 839–848. [DOI] [PubMed] [Google Scholar]
- Sedlar A, Kranjc S, Dolinsek T, Cemazar M, Coer A, and Sersa G (2013). Radiosensitizing effect of intratumoral interleukin-12 gene electrotransfer in murine sarcoma. BMC Cancer 13, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, Alqunaibit D, Avanzi A, Daley D, Barilla R, et al. (2016). Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 150, 1659–1672.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L, and Joyce JA (2014). Pericellular proteolysis in cancer. Genes Dev. 28, 2331–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, and Bussolino F (2001). IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J. Immunol 166, 3890–3899. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Umezu Y, Saijo Y, Satoh G, Abe Y, Satoh K, and Nukiwa T (1998). Exogenous recombinant human IL-12 augments MHC class I antigen expression on human cancer cells in vitro. Tohoku J. Exp. Med 185, 223–226. [DOI] [PubMed] [Google Scholar]
- Thind K, Padrnos LJ, Ramanathan RK, and Borad MJ (2017). Immunotherapy in pancreatic cancer treatment: a new frontier. Therap. Adv. Gastroenterol 10, 168–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman RD, Herman J, and Cho LC (2014). Emergence of stereotactic body radiation therapy and its impact on current and future clinical practice. J. Clin. Oncol 32, 2847–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Wysocka M, D’Andrea A, Rengaraju M, Aste-Amezaga M, Kubin M, Valiante NM, and Chehimi J (1992). Natural killer cell stimulatory factor (NKSF) or interleukin-12 is a key regulator of immune response and inflammation. Prog. Growth Factor Res 4, 355–368. [DOI] [PubMed] [Google Scholar]
- Vonderheide RH (2018). The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 33, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walle T, Martinez Monge R, Cerwenka A, Ajona D, Melero I, and Lecanda F (2018). Radiation effects on antitumor immune responses: current perspectives and challenges. Ther. Adv. Med. Oncol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, and Ozato K (2000). An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J. Immunol 165, 271–279. [DOI] [PubMed] [Google Scholar]
- Wang B, Li Q, Qin L, Zhao S, Wang J, and Chen X (2011). Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Tarling EJ, McMillen TS, Tang C, and LeBoeuf RC (2015). ABCG1 regulates mouse adipose tissue macrophage cholesterol levels and ratio of M1 to M2 cells in obesity and caloric restriction. J. Lipid Res 56, 2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, and Gomer RH (2015). Trypsin, Tryptase, and Thrombin Polarize Macrophages towards a Pro-Fibrotic M2a Phenotype. PLoS One 10, e0138748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Tsai YT, Lee IJ, Wu PY, Lu LS, Tsao WS, Huang YJ, Chang CC, Ka SM, and Tao MH (2018). Combination of radiation and interleukin 12 eradicates large orthotopic hepatocellular carcinoma through immunomodulation of tumor microenvironment. OncoImmunology 7, e1477459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, and Wu L (2013). CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 73, 2782–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilina A, Park-Min KH, Antoniv T, Hu X, and Ivashkiv LB (2008). TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol 9, 378–387. [DOI] [PubMed] [Google Scholar]
- Yasmin-Karim S, Bruck PT, Moreau M, Kunjachan S, Chen GZ, Kumar R, Grabow S, Dougan SK, and Ngwa W (2018). Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Front. Immunol 9, 2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeh HJ III, Hurd S, Storkus WJ, and Lotze MT (1993). Interleukin-12 promotes the proliferation and cytolytic maturation of immune effectors: implications for the immunotherapy of cancer. J. Immunother. Emphasis Tumor Immunol 14, 155–161. [DOI] [PubMed] [Google Scholar]
- Zhong J, Patel K, Switchenko J, Cassidy RJ, Hall WA, Gillespie T, Patel PR, Kooby D, and Landry J (2017). Outcomes for patients with locally advanced pancreatic adenocarcinoma treated with stereotactic body radiation therapy versus conventionally fractionated radiation. Cancer 123, 3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chong MM, and Littman DR (2009). Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, and DeNardo DG (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 47, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing data reported in this paper is GEO: GSE136368.