

Abstract
Detection of factor VIII (FVIII) in cells by flow cytometry is controversial, and no monoclonal fluorescent antibody is commercially available. In this study, we optimized such an assay and successfully used it as a platform to study the functional properties of phosphoglycerate kinase (PGK)-FVIII lentiviral vector-transduced cells by directly visualizing FVIII in cells after different gene transfer conditions. We could measure cellular stress parameters after transduction by correlating gene expression and protein accumulation data. Flow cytometry performed on transduced cell lines showed that increasing MOI rates resulted in increased protein levels, plateauing after an MOI of 30. We speculated that, at higher MOI, FVIII production could be impaired by a limiting factor required for proper folding. To test this hypothesis, we interfered with the unfolded protein response by blocking proteasomal degradation and measured the accumulation of intracellular misfolded protein. Interestingly, at higher MOIs the cells displayed signs of toxicity with reactive oxygen species accumulation. This suggests the need for identifying a safe window of transduction dose to avoid consequent cell toxicity. Herein, we show that our flow cytometry platform for intracytoplasmic FVIII protein detection is a reliable method for optimizing gene therapy protocols in hemophilia A by shedding light on the functional status of cells after gene transfer.
Keywords: FVIII, Hemophilia A, HA Gene Therapy, FVIII FACS, FVIII flow cytometry, Factor VIII, F8, PBMCs FVIII, FVIII in blood cells, Lentiviral vector, Cellular stress
Graphical Abstract
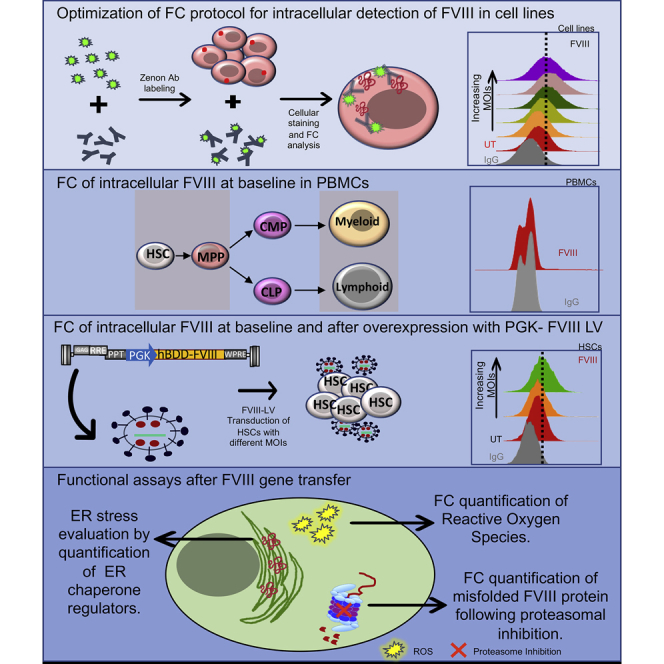
Introduction
Hemophilia A (HA) is a monogenic bleeding disorder caused by defective or absent FVIII. The available current treatment is FVIII replacement therapy from either recombinant or plasma-derived sources. Roughly 30%–40% of the patients develop anti-FVIII alloantibodies that render the replacement therapy ineffective.1 Even achieving subphysiological FVIII levels can alleviate the hemophilic phenotype.2 This made gene therapy (GT) an attractive strategy to treat this disease; accordingly, several clinical trials are currently recruiting patients,3 and promising clinical results were shown after intravenous administration of AAV5-FVIII therapeutic viral vector.4
Gene therapy strategies targeted on CD34+ hematopoietic stem cells (HSCs) have been proposed for clinical trials to develop corrected downstream lineages, including not only megakaryocytes,5 but also myeloid/monocytic cell lines, both proven to be a good source of FVIII.6 While FVIII is widely measured intracellularly with immunohistochemistry staining methods, a reliable protocol for flow cytometry (FC) staining is still not available. Few publications are describing controversial results on FC detection of FVIII in blood cells,7,8 with the clearest results only shown in platelets, where membrane unspecific binding of antibodies (Abs) is not a relevant issue.5
To facilitate the evaluation of FVIII gene modification protocols, we optimized a FC assay to measure and functionally test intracellular FVIII in human cell lines, CD34+ HSCs, and peripheral blood mononuclear cells (PBMCs).
Such an assay could also potentially be of use in the diagnosis of hemophilia cases, especially in clinical cases where a functional protein is present but not secreted, and in cases where an intracellular evaluation is needed.
Results
Choice of Cell Lines
FVIII protein is naturally produced by endothelial cells,9,10 hepatocytes,9 and megakaryocytes.11 In addition, a wide number of cells contain FVIII mRNA, and might produce small amounts of protein, for biological reasons that remain undefined.12
To choose reliable FVIII-producing cell lines, we selected HECV endothelial cells, as “professional” FVIII producer cells, and the HeLa cancer cell line, as, being derived from cervical tissue, it expresses high levels of FVIII mRNA.12 We also included U937 cells, a pre-monocytic cell line, representative of myeloid blood cell capacity for FVIII assembly and production.13
We confirmed the mRNA content of the selected cell lines by qRT-PCR (Figure 1A) and their FVIII protein secretion capacity by an AlphaLISA assay (Figure 1B).
Figure 1.

FVIII mRNA Expression and Protein Secretion in Different Cell Lines
(A) FVIII mRNA expression in HECV, HeLa, and U937 cell lines measured by qPCR over GAPDH mRNA expression; one experiment was run in triplicate (n = 1, mean ± SD). (B) FVIII protein measured in the supernatant of HECV, HeLa, and U937 cell lines (n = 2, ±SD) by AlphaLISA assay; see the Materials and Methods section for details.
Optimization of Intracellular FVIII FC Staining Protocol
To visualize FVIII in cells by FC, we concurrently compared different methods. Since no commercial fluorescent mAb is available, we custom-labeled Abs (Green Mountain Abs [GMA]) anti-FVIII Abs against different protein domains with the staining kit DyLight 650 N-hydroxysuccinimide (NHS) ester, or with Zenon mouse labeling kit, based on Fc-Ab fragment staining.
The first series of experiments with DyLight-labeled Ab showed a high non-specific binding of anti-FVIII Ab to the surface and intracellular compartments of PBMCs. To dissect the cause of this non-specificity, we co-incubated anti-FVIII Abs with FVIII pure protein before labeling, expecting a decreased staining due to the engagement of Ag-specific binding sites by free FVIII. On the contrary, the staining was enhanced, suggesting that FVIII protein itself contributes to additional fluorescent signals (Figure S1).
Therefore, in order to optimize a reliable staining method, we critically considered the biological features of FVIII protein contributing to staining non-specificity.
First, addition of a high-concentration (10%) mouse serum blocking step and a stringent, mathematical gating strategy (illustrated in Figure 3 and in the Materials and Methods section) were essential to decrease aspecific labeling. Mouse serum was found to give better results than Fc blocking alone for this purpose both on human peripheral CD14+ cells (Figure S2) and cell lines (data not shown), and the use of immunoglobulin (Ig)G1 isotype control completely corrected the gating offset, compared to IgG2. This IgG1 correction was noted only for DyLight-labeled Abs, while the difference was negligible for Zenon technology-labeled Abs. GMA Abs were titrated with both labeling methods (data not shown), and a final concentration of 10 μg/mL was chosen.
Figure 3.

Gating Strategy Illustration
Intracellular staining of FVIII on U937 cells, Zenon labeling. (A–C) Logical gating strategies are shown on (A) forward light scatter (FSC)/side light scatter (SSC) to define the populations and to exclude (B) doublet cells and (C) death cells/debris. (D and E) FVIII positivity (E) is then compared to the appropriate IgG isotype (D). Superimposed plots and histograms include the following: unstained cells (black) and cells stained with only fixable live marker (light gray) superimposed to FVIII-positive cells (blue) and relative IgG (light blue). (F–H) Different domains in HeLa and U937 cells (F and G) are visualized with different intensities (H) (mean ± SD, n = 2 on cells after FVIII transduction).
In its active form, FVIII protein interacts through its C2 domain with phosphatidylserine (PS) molecules,14 naturally exposed on the surface of dead, damaged cell membranes,15 or activated cells.16
While this is an important property of FVIII physiological coagulation activity, at the same time, it leads to binding of FVIII protein to surrounding non-FVIII-producing cells (bystander cells and cell debris), therefore hampering the identification of FVIII real producer cells. In order to mitigate this effect, we added a fixable viability marker consistently in every FC evaluation. Lactadherin is a milk protein that competes with FVIII protein for PS binding sites through its C2 domains.17 In order to increase the staining specificity, we exposed cells overnight to lactadherin before FVIII Ab labeling, and we then measured FVIII surface and intracellular protein. Surface non-specific protein binding on U937 cells was greatly reduced (Figures 2A and 2C), while FVIII cell internal content was not impacted (Figure 2B). This result suggests that the addition of non-specific competitors for FVIII surface binding, such as lactadherin, could be helpful in FVIII surface detection experiments, such as investigation of FVIII-producing cell populations in a tissue-derived complex cell set or in PBMCs. The addition of lactadherin allows for a more specific definition of cells that are secreting FVIII as opposed to those cells that acquire it by binding secreted FVIII from the milieu.
Figure 2.

Lactadherin Competition Assay
FC analysis showing FVIII expression after in vitro lactadherin overnight incubation with U937 cells. Experiments were performed in parallel conditions with/without lactadherin. (A) FVIII surface staining; indicated are means ± SD of n = 4, p = 0.0039 (paired t test). (B) FVIII intracellular staining; indicated are means ± SD of n = 3, in triplicate, p = 0.69. Percentage of IgG background staining (here and in the figures below, if not otherwise specified) is subtracted from the rate of positive cells. (C) Representative dot plots of surface staining after competition assay of lactadherin.
After protocol optimization, FVIII protein detection by FC in both U937 cell line and PBMCs (data not shown) was detectable as an overall shift in the mean fluorescent intensity of the cell population, rather than as a distinct population of cells, requiring a negative control with the relevant IgG fluorescent background staining to measure the positive fraction present (Figures 3A–3G). A1, A2, A3, and LC domains are separately visible by FC with different percentages (Figure 3H).
Validation of FVIII FC Staining
A definitive validation step for a FC staining would be a species-specific FVIII negative control. We were unable to find PBMCs or human cell lines totally negative for FVIII mRNA, and therefore we sought to generate a FVIII knockout human cell line using the CRISPR-Cas9 technique to induce an indel frameshift.18,19 Exon 4 of the FVIII gene was targeted in HeLa, HECV, and U937 cells using 5′- ATACTAGTAGGGCTCCAATG-3′ as the target sequence.
Despite the successful introduction of a frameshift mutation on both alleles, as illustrated in Figure S3, all of the cell lines continued to translate FVIII protein in a form visible by western blot (WB) and FC intracellular analyses. This is probably the result of an alternative splicing mechanisms of FVIII,20,21 or of a translation at alternate open reading frames downstream of an edited gene segment. The presence of FVIII in this form and its continued functionality post-editing do, however, pose other intriguing research questions that fall outside the scope of this work.
In the absence of a species-specific negative control, to ensure that detection of FVIII was truly due to the presence of the protein, and not to a fluorescence artifact, we double-stained cells with two antibodies targeting different FVIII domains, each labeled with a different dye. This staining approach has been pioneered in the study of the HIV reservoir by Chomont et al.22 Following co-staining on HECV cells, both Abs bound to the same target cells, with this colocalization confirming the specificity of staining (Figures 4A–4C). Intriguingly, the rate of FVIII double fluorescence was lower than the expected cumulative value, as visualized by single staining. Steric hindrance of Abs is unlikely to explain this decrease, since simultaneous binding of multiple anti-fVIII mAbs (mAbs) to the C2 domain of FVIII was shown in previous studies.23,24 Either the presence of incomplete FVIII fragments, or still a margin of staining unspecificity despite the extensive controls, might explain this finding (Figure 4D).
Figure 4.

Double Staining of A2 and C1 Domains of FVIII
GMA® 8012 IgG1 anti A2 domain and GMA® 8011 IgG2a anti C1 domain were used with IgG1 and IgG2a as controls for a comparison of single and double staining on HECV un-transduced cells (A), HECV cells transduced at MOI 30 (B) and 40 (C) with PGK-FVIII-LVV. The average of 2 experiments performed in duplicate is summarized in the graph, with IgG subtracted values (D).
In summary, we were able to reliably detect FVIII protein by FC collectively in HECV, HeLa, and U937 cell lines as well as in PBMCs (Figures 5A and 5B). PBMC staining was further validated by comparison to WB, where it was observed that the sensitivity detection by WB required at least 2 × 10e6 cells/test in order to detect FVIII (Figure 5C).
Figure 5.

FVIII Expression in Cell Lines and in PBMCs
(A) FVIII percentage by FC after IgG subtracted in different cell lines (HeLa, n = 4; HECV, n = 2; U937, n = 6) and PBMCs (n = 3) (mean ± SD). (B) FVIII intracellular staining by FC; total PBMCs show 4% of FVIII-positive population. (C) FVIII A2 detection by WB at different cell numbers of PBMCs.
The optimized staining protocol is summarized in Table 1.
Table 1.
Optimized Protocol of FVIII Staining for FC Visualization with Zenon and with Custom-Labeled Abs
| Step | Name of Step | Details | Interval |
|---|---|---|---|
| 1 | harvesting of cells | wash cells once with PBS (centrifuge); harvest cells and count using trypan blue and hemocytometer, and divide cells as 1 million cells per condition | centrifuge at 300 × g for 5 min at RT |
| 2 | viability staining | in 1 mL of PBS, add 1 μL of viability dye (incubate), according to the manufacturer’s protocol (LIVE/DEAD Fixable Dead Cell Stain Viability Kit, Molecular Probes) | incubate for 15 min in the dark at RT |
| 3 | staining buffer wash | wash cells in 3 mL of staining buffer: 0.1% BSA-PBS solution (centrifuge) | centrifuge at 300 × g for 5 min at RT |
| 4 | CD markers or FVIII (surface staining) | cells are re-suspended in X μL of staining buffer for surface staining; desired labeled surface Abs are added (total volume for surface staining should be 100 μL) (incubate) | incubate for 30 min in the dark at RT |
| 5 | staining buffer wash | wash cells in 3 mL of staining buffer: 0.1% BSA-PBS solution (centrifuge) | centrifuge at 300 × g for 5 min at RT |
| 6 | cell fixation | re-suspend cell pellet in 100 μL of fixing buffer (incubate) according to the manufacturer’s protocol (Invitrogen FIX & PERM cell permeabilization kit). | incubate for 15 min at RT |
| 7 | staining buffer wash | wash cells in 3 mL of staining buffer: 0.1% BSA-PBS solution (centrifuge) | centrifuge at 300 × g for 5 min at RT |
| 8 | cell permeabilization | re-suspend the cells in 100 μL of perm buffer; add 10% final concentration of mouse serum (incubate) | incubate for 15 min at RT |
| 9 | FVIII (intracellular staining) | – Zenon staining: label anti-FVIII Abs and IgG isotype control with Zenon labeling dye (according to manufacturer’s protocol for Zenon Ab labeling kits; Thermo Fisher) and add the labeled anti-FVIII Ab to the cells (incubate) | incubate for 20 min in the dark at RT |
| – stably labeled anti-FVIII Abs: add to cells the labeled Abs at titrated concentration (incubate) | |||
| 10 | staining buffer wash | wash cells in 3 mL of staining buffer: 0.1% BSA-PBS solution (centrifuge) | centrifuge at 300 × g for 5 min at RT |
| 11 | final cell fixation | re-suspend the cells in 200 μL of 1% paraformaldehyde-PBS (this step is important to minimize the off-rate of Abs, and to stabilize the Zenon complex) | |
| 12 | data acquisition | acquire the results on FC instrument |
For non-Zenon custom-labeled Abs, use of the IgG1 isotype is highly recommended. RT, room temperature.
FVIII Transgene Visualization by FC
When tested by FC, transduced U937 cells showed a rate of FVIII proportional to the rate of transduction, performed with increasing MOIs (Figure 6A). This indicates that by FC the transgene is not distinguishable from the native protein and is displayed in a cumulative proportion, and it is correlated with an increasing cell vector copy number (VCN) (Figure 6B). This was mirrored by the respective FVIII mRNA content and protein secretion and was verified by confocal imaging on HECV cells with the same Zenon technology used for FC (Figures 6C–6E) and through WB imaging (Figure 7).
Figure 6.

FVIII Transgene Visualization in U937 Cells
(A) FC analysis of four replicate experiments, after U937 transduction with PGK-FVIII LV (one-way ANOVA test, p = 0.0048; conditions significant against UT are labeled with stars). (B) VCN was calculated in parallel with FC by interpolating the CT values of unknowns on two curves: one curve of the CT values of titrated FVIII transgene plasmid (representing the number of copies), and the other curve of the CT values for the GAPDH housekeeping gene of titrated cell numbers. (C) FVIII RNA expression after U937 transduction with PGK-FVIII LV (one-way ANOVA test, p = 0.0107; conditions significant against UT are labeled with stars). (D) FVIII protein was measured by AlphaLISA in the supernatant of UT U937 and transduced at MOIs of 10 and 30 (mean ± SD). (E) Confocal images of FVIII Zenon TSA-enhanced immunostaining in UT HECV cells and transduced at an MOI of 40 against IgG isotype, at different magnifications (upper panel: original magnification, ×63; lower panel: original magnification, ×20, zoomed); FVIII in green (FITC) and nucleus in blue (DAPI). The right bar graph shows the representative mean fluorescent intensity (MFI) of the same cells, calculated from three individual fields of view. Of note, the confocal imaging sensitivity was not sufficient to significantly capture FVIII positivity in UT cells, whereas the FC technique was able to detect FVIII positivity even with minimal differences in expression.
Figure 7.

Protein Measurement by WB and FC
FVIII expression in HECV, HeLa, and U937 cell lines was tested by WB and FC with GMA anti-FVIII Ab GMA012 (suited for WB analyses) in a parallel FC experiment with Zenon technology. FC is shown on the left, and WB is shown on the right. The FVIII 43-kDa, A2 domain band (http://greenmoab.com/product/gma-012/) and the 55-kDa α-tubulin (DM1A clone, Sigma-Aldrich) are shown. The ratio of FVIII over tubulin is shown in the y axes of the bars.
We then transduced cord blood CD34+ cells at increasing MOIs and measured FVIII by FC after 1 week of transduction. Remarkably, CD34+ cells express FVIII physiologically (confirming previous data)13,25 and overexpress it after transduction to a level detectable also by ELISA (Figure S4C). As functional readout, we performed colony-forming unit (CFU) colonies, showing comparable results through different MOIs with a lower trend at increasing MOIs (Figure S4D).
Interestingly, FVIII protein expressed after transduction with higher MOIs, did not increase linearly, in the cell lines transduction plateaued around an MOI of 30 (Figures 6A and 6C). We wondered if this result was due to Ab saturation or possibly caused by the saturation of the cellular protein folding/secretion machinery. Indeed, with FVIII being a large glycoprotein, overexpression could potentially limit the protein-building capacity of cells.
FC Functional Studies on FVIII Transgene
The first cellular response to protein accumulation is endoplasmic reticulum (ER) stress. This is associated with the ubiquitin-proteasome system (UPS) and the unfolded protein response (UPR) pathways, leading eventually to cell autophagy if the ER stress is prolonged.26
We first studied the effect of high VCN on the initiation of ER stress and consequent UPR by measuring the mRNA level of binding immunoglobulin protein (BiP)/Grp78 in U937 cells transduced at different MOIs with phosphoglycerate kinase (PGK)-FVIII lentiviral vector (LV). BiP/Grp78 gave comparable results across the conditions with a (nonsignificant) trend of increased values at MOIs of 20 and 30 (Figure 8A).
Figure 8.

Functional Studies of FVIII-Transduced Cells
(A–C) Initiation of UPR in the U937 cell line after different transduction platforms, measured by BiP/Grp78 mRNA levels (A), ROS accumulation by FC (B), and FVIII misfolded protein accumulation by FC in U937 and HECV cells with and without proteasome inhibition by lactacystin (C). Mean ± SD is visualized. p = not significant (NS).
Reactive oxygen species (ROS) are also generated during ER stress, and their unbalanced accumulation contributes to drift UPS and UPR roles from being cytoprotective to cytotoxic.27 ROS levels, measured by FC, increased at MOIs of 30 and higher, indicating a potential toxic effect, resulting from overexpression of FVIII transgene (Figure 8B).
To further explore the effects of FVIII accumulation, we interfered with the proteasomal degradation pathway by blocking it through incubation in the presence of the proteasome inhibitor lactacystin, before FC FVIII intracellular measurement. Proteasome inhibition blocks the displacement of misfolded protein physiologically routed for intracellular degradation and forces protein accumulation in the cell.28 FVIII accumulated misfolded protein was still recognizable by FC (Figure 8C). As previously seen, the transgene increased with MOI. After proteasome inhibition, FVIII accumulated in the cells, revealing how the cell machinery is actively engaged in clearing misfolded FVIII proteins. Noticeably, this also occurs in untransduced cells and in both non-professional and professional FVIII producers, such as U937 and HECV cells, reinforcing the need to monitor FVIII production and to ensure that a safe upper limit of transduction is applied in order to avoid cytotoxicity when designing potential gene therapeutic approaches.
Discussion
FC detection of intracellular FVIII in the literature is still controversial and is not currently performed in clinical practice. FVIII protein levels are routinely measured in plasma via functional rather than quantitative assays in order to diagnose and evaluate the severity of HA. In this study, we have optimized a robust reproducible protocol to visualize and quantify intracellular FVIII protein by FC. The protocol has been optimized in human cell lines of different tissue origin and in PBMCs, suggesting that it can further be applied to any cell type in order to evaluate the presence of intracellular FVIII. Due to the lack of commercial labeled mAbs against FVIII FACS staining, the available data in the literature mainly describe results obtained with polyclonal Abs or fluorescent secondary Abs.7,29, 30, 31 Since secondary staining in intracellular compartments adds further to the level of non-specific staining, we chose to work with reliable primary Abs, labeled in-house with fluorescent dyes or with Zenon technology. With our work, we achieved a comprehensive and systematic optimization of the current available methods, adding functional insights to FC analyses.
In our hands, the Zenon staining method was the most reproducible, and, when combined with 10% mouse serum blocking, it generated the lowest background levels. However, the use of IgG isotypes was still found to be necessary as a negative control in order to distinguish the positive population.
Due to the absence of a proper species-specific negative control, we initially sought to create FVIII knockout cell lines through CRISPR/Cas9 gene editing, but despite successful editing of the FVIII gene, we failed to knock out the protein translation. Instead, we demonstrated the specificity of staining by alternate means. First, cell lines transduced with increasing FVIII viral load showed progressive amounts of FVIII intracellular protein, and this scale of protein content was mirrored in the WB data. Additionally, when cells were co-stained with two mAbs specific for different domains of FVIII, a method shown to increase sensitivity in intracellular staining for HIV-1 protein p24 gag, both of these Abs were found to bind to the same cells in a linear fashion.
HA is a monogenic disorder for which gene therapy is a potentially effective treatment option, and both preclinical and clinical studies have recently shown remarkable success in trials targeting liver with FVIII-AAV.3 However, some limitations have been identified in these studies, including the potential for high immunogenicity, leading to the need to pursue alternative strategies, both in terms of the viral vectors used and the identification of different target cells. Among them there are CD34+ HSCs targeted with a myeloid promoter,6 megakaryocytes with a promoter of genes specifically expressed in platelets,5,32 and liver sinusoidal endothelial cells (LSECs)9,10 as the main natural producer of FVIII in the human body.
Selecting candidate target cells for FVIII gene therapy necessitates robust analysis to determine cell eligibility criteria. A long life-span of the target cell is not the only suitable feature. Cell machinery has to be adequate to produce the chosen transgene and be sufficient to cope with the levels of protein produced without triggering cellular UPRs, or worse, uncontrolled cell death. Indeed, over-accumulation of misfolded protein in cells, if not properly compensated by a UPR, may also trigger immunogenicity,33 with its downstream concatenate effects that could affect the survival of the whole population of transduced cells in the body. This is why we sought to add FC-based toxicity assessments as a novel method to evaluate the functional status of FVIII-producing cells.
Moreover, a FC analysis will also serve as an effective readout tool to analyze the efficiency both of gene therapy and during follow-up after treatment. Interestingly, no clinical platform based on FC is available for detection of intracellular FVIII, and this assay will open novel diagnostic possibilities as in lectin mannose binding protein 1 (LMAN1) and multiple coagulation factor deficiency protein 2 (MCFD2), where FVIII is produced but cannot be excreted from cells due to ER-Golgi transporter protein defects.34 Beyond diagnosis, future observational studies might be able to associate FVIII intracellular protein abundance with pathological events, as bleedings, and propose FC as a prediction/prevention assay.
Also, FVIII mRNA in different isoforms has been found to be transcribed in almost all human cells without clear data on the translated protein, its abundance, and its role in different cells.12 Therefore, FC analysis could allow for the stratification of different cells according to their ability to translate FVIII mRNA into actual protein.
Finally, we used FVIII FC as a tool for the evaluation of cytotoxicity induction through the UPS system following saturation with lactacystin. Even following the accumulation of misfolded protein forms, after proteosomal inhibition, FC was still capable of measuring protein abundance, and to provide an assessment of cellular toxicity, which correlated with other parameters of cell toxicity, such as ROS, and BiP/Grp78 expression. Defining a toxicity threshold could help guide reaching a compromise between increasing protein expression via gene therapy and induction of toxicity. The combination of different toxicity evaluation tools in a FC assay would give an enhanced overall view about the cell status of gene therapy target cells.
In summary, in our study we have developed a FC platform to quantify the cell productivity of FVIII and to measure the influence of its overexpression on cell status.
Materials and Methods
Cell Culture and Cell Line Maintenance
HECV (human vascular endothelial cells) and U937 (pre-monocytic cells) cell lines were kindly provided by Antonia Follenzi (UPO, Italy). HeLa (cervical cancer cells) were purchased from ATCC. PBMCs were purchased from AllCells (Alameda, CA, USA). Cells were cultured in Iscove’s modified Dulbecco's medium (IMDM; Gibco) and RPMI 1640 (Fisher Scientific, Pittsburgh, PA, USA) media supplemented with 10% (v/v) fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA).
FC Staining
For FC staining experiments, 1 million cells/condition were tested. Cells were washed with PBS for viability staining using LIVE/DEAD Fixable Violet Dead Cell Stain (Molecular Probes/Life Technologies, Carlsbad, CA, USA). FC experiments were done based on the manufacturer’s procedures for the FIX & PERM cell permeabilization kit (Life Technologies, Carlsbad, CA, USA). Surface staining for PBMCs was performed with anti-CD19, anti-CD14, and anti-CD33 (BioLegend, San Diego, CA, USA) Abs. Anti-hFVIII mAbs used were as follows: A1 domain (GMA8002), A2 domain (GMA012, GMA8024), A3 domain (GMA8001), light chain domain (GMA8041), C1 domain (GMA8011) (Green Mountain Abs, San Francisco, CA, USA), and IgG isotype controls IgG2a and IgG1 (SouthernBiotech, Birmingham, AL, USA). Since the FVIII Ab-stained population shows a positive gradient with increased antigen intensity rather than a distinct population, a gate is set mathematically on the IgG isotype control calculated via the following formula: mean fluorescence intensity (MFI) + 3 × robust SD (rSD) of fluorescence intensity. Then, the same gate is applied to the FVIII-stained population. This allows us to avoid setting arbitrary gates, overlapping between positive and negative populations.
Mouse serum (Life Technologies, Carlsbad, CA, USA) was added at 10% v/v directly with FVIII Abs in PBMCs and cell lines, and FC block was performed at different concentrations (Figure S2) only for PBMC staining before the surface staining step. A Zenon Alexa Fluor mouse labeling kit (Invitrogen, Carlsbad, CA, USA) and DyLight 650 NHS ester (Life Technologies, Rockford, IL, USA) were used according to the manufacturers’ instructions. For the lactadherin competition assay, cells were incubated overnight with 4 nM lactadherin (Avivasysbio, San Diego, CA, USA) prior to FC staining. Lactacystin 26S proteasome inhibitor (Sigma-Aldrich, St. Louis, MO, USA) was used for FVIII intracellular accumulation analysis at a concentration of 50 μM for 4 h prior to FC analysis. CellROX Green reagent (Thermo Fisher Scientific, Eugene, OR, USA) was used according to the manufacturer’s protocol at a concentration of 5 μM in co-incubation with cells for 30 min at 37°C.
FC acquisition was performed through ACEA Biosciences NovoCyte and BD Biosciences LSRFortessa flow cytometers. Detailed steps of the optimized protocol for FC analysis are shown in Table 1. FC data were analyzed using FlowJo version 8.5 and Beckman Coulter Kaluza software. The gate was set on the corresponding IgG isotype control then applied to the condition according to the equation (fluorescence mean + scalar × fluorescence rSD). The minimum value of the scalar is 4.
LV Transduction
Cell lines were transduced with a self-inactivating (SIN) third-generation LV hPGK.hFVIIIBDD, kindly provided by Antonia Follenzi (UPO, Italy) at MOIs of 5–40.
Transduction was performed in 24-well plates at 500,000 cells per well for 24 h. Cells were then washed to remove the vector and further cultured at least for 5 days before FVIII assessment analyses.
RNA Extraction and qPCR
Isolation of mRNA from cells was performed using a RNA extraction kit from Thermo Fisher Scientific (Pittsburgh, PA, USA) according to the manufacturer’s procedures. Synthesis of cDNA from RNA was achieved by using a GoScript reverse transcription kit (Promega, Madison, WI, USA). Furthermore, qPCR was performed using GoTaq qPCR Master Mix (Promega, Madison, WI, USA) and relevant primers.
FVIII-specific primers (forward, 5′-CCAGAGTTCCAAGCCTCCAACA-3′; reverse, 5′-GGAAGTCAGTCTGTGCTCCAATG) were used for evaluating the FVIII transcripts.5 For the UPR qPCR, the following primers were used: human BiP/Grp78-specific primers (forward, 5′-AAGGAGCGCATTGATACTAGA-3′; reverse, 5′-AGGGCCTGCACTTCCATAGAG-3′) and GAPDH (forward, 5′-AACGTGTCAGTGGTGGACCTG-3′; reverse, 5′-AGTGGGTGTCGCTGTTGAAGT-3′). Gene expression relative to the GAPDH housekeeping gene was calculated with relevant negative controls and in triplicates.
AlphaLISA for FVIII Quantification
Quantification of FVIII concentration was measured using the AlphaLISA FVIII detection kit (PerkinElmer, Waltham, MA, USA), which is an ELISA system commercially available for highly sensitive and specific quantitative measurement of FVIII protein levels in the supernatants of cells and human plasma. For protein detection experiments, cells were cultured in amounts of 300,000–500,000 cells in 24-well plates in 300–500 μL of X-VIVO 15 serum-free/phenol red-free cell medium (Lonza, MD, USA) for overnight incubation.
Supernatants were collected after culture and centrifugation at 2,500 rpm, placed on ice, and processed with an AlphaLISA assay according to the manufacturer’s procedures. Data were acquired with EnSight multimode plate reader (PerkinElmer, Waltham, MA, USA)
Western Blotting for FVIII Detection
Cells were collected and lysed with CytoBuster cell lysis buffer (Merck, Darmstadt, Germany). After a 30-min incubation on ice and spinning down at 2,500 rpm, subsequently, supernatants containing FVIII protein were collected. Protein quantification was performed according to the manufacturer’s protocol with a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Pittsburgh, PA, USA).
Samples were electrophoresed at 100 V for 1 h 30 min through an in-house pre-prepared polyacrylamide gel (separating gels with stacking gels). Gels were transferred onto semi-dry blots (Bio-Rad Laboratories, Hercules, CA, USA) transfer unit at 1.0 A, 25 V for 30 min. Proper blocking was performed in milk/PBS-Tween 20 (PBS-T) for 30 min, followed by multiple PBS-T washes. Overnight incubation was performed with primary Abs at 4°C, followed by PBS-T multiple washes (10 min per wash). Secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse polyclonal Ab was then added to the blot for 1 h. After several washing steps, the western HRP substrate was added for blot development. Blots were read on the ChemiDoc MP imaging system (Bio-Rad Laboratories, Hercules, CA, USA). 55-kDa α-tubulin (DM1A clone, Sigma-Aldrich) Ab was used as loading control as relevant. Protein measurement analysis was done by ImageJ software and MS Excel.
VCN Quantification
Quantification of the VCN was performed using qPCR on genomic DNA of U937 cells according to the protocol previously described.35 Briefly, the VCN of each sample has been calculated by comparing the two standard curves, one curve of the CT values of titrated FVIII transgene Wpre-dNEF (forward, 5′-TGGATTCTGCGCGGGACGTC-3′; reverse 5′-GGCTAAGATCTACAGCTGCCTTG-3′), and the other curve of the CT values for GAPDH (forward, 5′-AACGTGTCAGTGGTGGACCTG-3′; reverse, 5′-AGTGGGTGTCGCTGTTGAAGT-3′).
CRISPR/Cas9 Edition
Guide RNA (gRNA) synthesis was designed with the GeneArt CRISPR gRNA design tool, and the one validated targeting exon 4 was targeting the sequence 5′-ATACTAGTAGGGCTCCAATG-3′. TrueCut Cas9 protein v2, GeneArt CRISPR gRNA design tool, GeneArt precision gRNA synthesis kit, GeneArt genomic cleavage detection kit, Lipofectamine CRISPRMAX Cas9 transfection reagent, MeltDoctor HRM Master Mix, and a Neon transfection system 10-μL kit (Thermo Fisher Scientific, Pittsburgh, PA, USA) were used per the manufacturers’ recommendations. Briefly, design gRNAs were produced in vitro and transfected to the cells either by lipid transfection (Lipofectamine) or electroporation. Genomic cleavage detection was used to assess the cutting efficiency using the gRNA. Clones were made using single-cell sorting by FC on a SORP FACSAria3 (BD Biosciences). Data were processed with FACSDiva 6.3 software (BD Biosciences). A high-throughput screening for genome edition of the clones was done via high-resolution melting (HRM) as previously described.36,37 Selected clones were further validated by Sanger sequencing (ABI 3500 sequencer). Sanger sequence analysis was performed using two online tools: CRISP-ID38 and Synthego.39 The selected clones had similar results in each of the software analyses.
Confocal Imaging
HECV cells were fixed, permeabilized, and stained for confocal imaging in well glass-bottomed chamber sides (Nunc Lab-Tek II). Staining was performed with FVIII Zenon tyramide signal amplification (TSA)-enhanced immunostaining (fluorescence in situ hybridization [FITC]), and nuclear staining was conducted with DAPI (Thermo Fisher). Imaging was conducted on a Zeiss 880 inverted microscope with Airyscan. Images were acquired with Plan-Apochromat 10×/0.45 numerical aperture (NA), Plan-Apochromat 20×/0.8 NA, and Plan-Apochromat 63×/1.4 NA oil immersion objectives. Cellular identification was performed using the Imaris (Bitplane) surface creation utility. FVIII expression was calculated from mean fluorescence intensity in the FITC channel per surface object.
Statistical Analysis
GraphPad Prism and Microsoft Excel software were used for data analyses. Data are represented as mean ± SD. Significance analysis of the results was done using appropriate t tests or one-way ANOVA to compare means, as specified in the figure legends.
Author Contributions
M.E: Conducted conceived and planned experiments, wrote the manuscript; A.A: Performed experiments, wrote the manuscript; D.K, CM.R, S.A, G.G, I.P., A.S., N.V.P: Performed experiments; C.B: planned research; A.F: planned and supervised research; JC.G: planned research, supervised experiments; S.D: Planned research, directed, supervised the project and wrote the manuscript.
Acknowledgments
We would like to acknowledge the continued support of Simone Merlin, Valentina Bruscaggin, and Silvia Pignani from the Histology Department, Faculty of Medicine at Università del Piemonte Orientale, Novara, Italy.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2019.11.003.
Supplemental Information
References
- 1.Dimichele D. Inhibitors: resolving diagnostic and therapeutic dilemmas. Haemophilia. 2002;8:280–287. doi: 10.1046/j.1365-2516.2002.00626.x. [DOI] [PubMed] [Google Scholar]
- 2.Kay M.A., High K. Gene therapy for the hemophilias. Proc. Natl. Acad. Sci. USA. 1999;96:9973–9975. doi: 10.1073/pnas.96.18.9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doshi B.S., Arruda V.R. Gene therapy for hemophilia: what does the future hold? Ther. Adv. Hematol. 2018;9:273–293. doi: 10.1177/2040620718791933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5–factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 5.Wang X., Shin S.C., Chiang A.F., Khan I., Pan D., Rawlings D.J., Miao C.H. Intraosseous delivery of lentiviral vectors targeting factor VIII expression in platelets corrects murine hemophilia A. Mol. Ther. 2015;23:617–626. doi: 10.1038/mt.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doering C.B., Denning G., Shields J.E., Fine E.J., Parker E.T., Srivastava A., Lollar P., Spencer H.T. Preclinical development of a hematopoietic stem and progenitor cell bioengineered factor VIII lentiviral vector gene therapy for hemophilia A. Hum. Gene Ther. 2018;29:1183–1201. doi: 10.1089/hum.2018.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandey G.S., Tseng S.C., Howard T.E., Sauna Z.E. Detection of intracellular factor VIII protein in peripheral blood mononuclear cells by flow cytometry. BioMed Res. Int. 2013;2013:793502. doi: 10.1155/2013/793502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishaque A., Thrift J., Murphy J.E., Konstantinov K. Cell surface staining of recombinant factor VIII is reduced in apoptosis resistant BHK-21 cells. J. Biotechnol. 2008;137:20–27. doi: 10.1016/j.jbiotec.2008.07.1856. [DOI] [PubMed] [Google Scholar]
- 9.Do H., Healey J.F., Waller E.K., Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J. Biol. Chem. 1999;274:19587–19592. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 10.Shahani T., Covens K., Lavend’homme R., Jazouli N., Sokal E., Peerlinck K., Jacquemin M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014;12:36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- 11.Sadelain M., Chang A., Lisowski L. Supplying clotting factors from hematopoietic stem cell-derived erythroid and megakaryocytic lineage cells. Mol. Ther. 2009;17:1994–1999. doi: 10.1038/mt.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanolini D., Merlin S., Feola M., Ranaldo G., Amoruso A., Gaidano G., Zaffaroni M., Ferrero A., Brunelleschi S., Valente G. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100:881–892. doi: 10.3324/haematol.2014.123117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert G.E., Drinkwater D. Specific membrane binding of factor VIII is mediated by O-phospho-l-serine, a moiety of phosphatidylserine. Biochemistry. 1993;32:9577–9585. doi: 10.1021/bi00088a009. [DOI] [PubMed] [Google Scholar]
- 15.Fadok V.A., Bratton D.L., Frasch S.C., Warner M.L., Henson P.M. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 16.Fischer K., Voelkl S., Berger J., Andreesen R., Pomorski T., Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108:4094–4101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- 17.Rasmussen J.T. Bioactivity of milk fat globule membrane proteins. Aust. J. Dairy Technol. 2009;64:63–67. [Google Scholar]
- 18.González F., Zhu Z., Shi Z.D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y., Cao J., Xiong M., Petersen A.J., Dong Y., Tao Y., Huang C.T., Du Z., Zhang S.C. Engineering human stem cell lines with inducible gene knockout using CRISPR/Cas9. Cell Stem Cell. 2015;17:233–244. doi: 10.1016/j.stem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalonde S., Stone O.A., Lessard S., Lavertu A., Desjardins J., Beaudoin M., Rivas M., Stainier D.Y.R., Lettre G. Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS ONE. 2017;12:e0178700. doi: 10.1371/journal.pone.0178700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shovlin C.L., Angus G., Manning R.A., Okoli G.N., Govani F.S., Elderfield K., Birdsey G.M., Mollet I.G., Laffan M.A., Mauri F.A. Endothelial cell processing and alternatively spliced transcripts of factor VIII: potential implications for coagulation cascades and pulmonary hypertension. PLoS ONE. 2010;5:e9154. doi: 10.1371/journal.pone.0009154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardons M., Baxter A.E., Massanella M., Pagliuzza A., Fromentin R., Dufour C., Leyre L., Routy J.P., Kaufmann D.E., Chomont N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019;15:e1007619. doi: 10.1371/journal.ppat.1007619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brison C.M., Mullen S.M., Wuerth M.E., Podolsky K., Cook M., Herman J.A., Walter J.D., Meeks S.L., Spiegel P.C. The 1.7 Å X-ray crystal structure of the porcine factor VIII C2 domain and binding analysis to anti-human C2 domain antibodies and phospholipid surfaces. PLoS ONE. 2015;10:e0122447. doi: 10.1371/journal.pone.0122447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walter J.D., Werther R.A., Brison C.M., Cragerud R.K., Healey J.F., Meeks S.L., Lollar P., Spiegel P.C., Jr. Structure of the factor VIII C2 domain in a ternary complex with 2 inhibitor antibodies reveals classical and nonclassical epitopes. Blood. 2013;122:4270–4278. doi: 10.1182/blood-2013-08-519124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Follenzi A., Raut S., Merlin S., Sarkar R., Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119:5532–5542. doi: 10.1182/blood-2011-07-367680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Høyer-Hansen M., Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–1582. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 27.Sies H., de Groot H. Role of reactive oxygen species in cell toxicity. Toxicol. Lett. 1992;64–65:547–551. doi: 10.1016/0378-4274(92)90230-h. [DOI] [PubMed] [Google Scholar]
- 28.Werner E.D., Brodsky J.L., McCracken A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei B., Chen Y., Chen J., Pan C.Q., Murphy J.E. Expression of human coagulation factor VIII in a human hybrid cell line, HKB11. Mol. Biotechnol. 2006;34:165–178. doi: 10.1385/MB:34:2:165. [DOI] [PubMed] [Google Scholar]
- 30.Becker S., Simpson J.C., Pepperkok R., Heinz S., Herder C., Grez M., Seifried E., Tonn T. Confocal microscopy analysis of native, full length and B-domain deleted coagulation factor VIII trafficking in mammalian cells. Thromb. Haemost. 2004;92:23–35. doi: 10.1160/TH03-06-0360. [DOI] [PubMed] [Google Scholar]
- 31.Fomin M.E., Zhou Y., Beyer A.I., Publicover J., Baron J.L., Muench M.O. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS ONE. 2013;8:e77255. doi: 10.1371/journal.pone.0077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Q., Kuether E.L., Chen Y., Schroeder J.A., Fahs S.A., Montgomery R.R. Platelet gene therapy corrects the hemophilic phenotype in immunocompromised hemophilia A mice transplanted with genetically manipulated human cord blood stem cells. Blood. 2014;123:395–403. doi: 10.1182/blood-2013-08-520478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoo Y.S., Han H.G., Jeon Y.J. Unfolded protein response of the endoplasmic reticulum in tumor progression and immunogenicity. Oxid. Med. Cell. Longev. 2017;2017:2969271. doi: 10.1155/2017/2969271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng C., Liu H.H., Yuan S., Zhou J., Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116:5698–5706. doi: 10.1182/blood-2010-04-278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merlin S., Cannizzo E.S., Borroni E., Bruscaggin V., Schinco P., Tulalamba W., Chuah M.K., Arruda V.R., VandenDriessche T., Prat M. A novel platform for immune tolerance induction in hemophilia A mice. Mol. Ther. 2017;25:1815–1830. doi: 10.1016/j.ymthe.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price E.P., Smith H., Huygens F., Giffard P.M. High-resolution DNA melt curve analysis of the clustered, regularly interspaced short-palindromic-repeat locus of Campylobacter jejuni. Appl. Environ. Microbiol. 2007;73:3431–3436. doi: 10.1128/AEM.02702-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas H.R., Percival S.M., Yoder B.K., Parant J.M. High-throughput genome editing and phenotyping facilitated by high resolution melting curve analysis. PLoS ONE. 2014;9:e114632. doi: 10.1371/journal.pone.0114632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dehairs J., Talebi A., Cherifi Y., Swinnen J.V. CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci. Rep. 2016;6:28973. doi: 10.1038/srep28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsiau T. Inference of CRISPR edits from Sanger trace data. bioRxiv. 2018 doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.