

Abstract
Objective
Enhanced glucagon signaling and hepatic glucose production (HGP) can account for hyperglycemia in patients with obesity and type 2 diabetes. However, the detailed molecular mechanisms underlying the enhanced HGP in these patients are not fully understood. Here, we identify Purβ as a positive regulator of HGP and study its molecular mechanisms in the regulation of HGP both in vivo and in vitro.
Methods
Adenovirus-mediated knockdown or overexpression of Purβ was performed in either primary hepatocytes or the livers of db/db mice. Glucose metabolism, insulin sensitivity, and HGP were determined by glucose, insulin, and lactate tolerance tests, respectively. Purβ/ADCY6 protein levels, glucagon signaling (p-CREB/CREB), and insulin signaling (p-Akt/Akt) were measured by immunoblotting. Gene expression was measured by RNA-seq and real-time quantitative polymerase chain reaction. Luciferase reporter and chromatin immunoprecipitation assays were used to study the interaction between Purβ and the Adcy6 promoter.
Results
Purβ was abnormally elevated in obese mice and was also increased under fasting conditions or via the glucagon signaling pathway, which promoted HGP by increasing Adcy6 expression. Liver-specific knockdown of Purβ in db/db mice significantly ameliorated hyperglycemia and glucose intolerance by suppressing the glucagon/ADCY6/cAMP/PKA/CREB signaling pathway. Consistent with this observation, the knockdown of Purβ also inhibited glucose production in isolated primary hepatocytes by inhibiting the glucagon/ADCY6/cAMP/PKA/CREB signaling pathway, whereas the overexpression of Purβ promoted glucose production by activating this signaling pathway. Mechanistically, Purβ directly binds to the promoter of the Adcy6 gene and thereby promotes its transcription.
Conclusions
Taken together, these results illustrate a new model in which Purβ functions to regulate the glucagon/ADCY6/cAMP/PKA/CREB signaling pathway to help maintain glucose homeostasis.
Keywords: Purβ, Liver, Gluconeogenesis, Glucagon signaling, ADCY6, Obesity
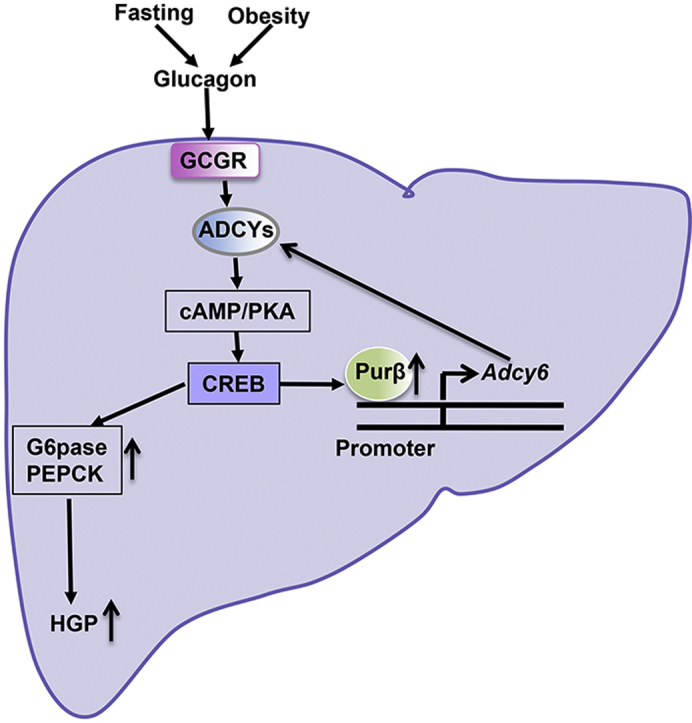
Graphical abstract
Highlights
-
•
Purβ was identified as a novel positive regulator of hepatic glucose production.
-
•
Purβ directly binds to the promoter of the Adcy6 gene, inducing its expression and activating the cAMP/PKA/CREB signaling pathway.
-
•
Liver-specific knockdown of Purβ in db/db mice significantly ameliorates hyperglycemia and glucose intolerance by suppressing the ADCY6/cAMP/PKA/CREB signaling pathway.
1. Introduction
The liver is the main organ producing glucose via glycogenolysis and gluconeogenesis in response to fasting. Under fasting conditions, hepatic glucose production (HGP) is increased in response to elevated blood glucagon levels [1,2]. Conversely, under fed conditions, HGP is suppressed by increased plasma insulin levels [1]. Thus, the HGP rate is determined by a balance between glucagon and insulin. Glucagon, via its G protein–coupled receptors, stimulates cAMP-mediated activation of protein kinase A (PKA), and PKA phosphorylates cAMP response element-binding protein (CREB) on Ser133 [3,4], which in turn activates the transcription of phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase), both key gluconeogenic genes [3,4]. Insulin inhibits HGP by suppressing the expression of PEPCK and G6Pase [1,5]. In patients with obesity and type 2 diabetes, HGP is abnormally elevated, thus contributing to hyperglycemia and glucose intolerance [2,5]. Enhancement of the glucagon signaling pathway and hepatic insulin resistance are two major causes of enhanced HGP [2]. However, the detailed molecular mechanisms underlying this enhanced HGP in patients with obesity are not fully understood.
Purine-rich element binding proteins, such as Purα, Purβ, and Purγ, which form the evolutionarily conserved Pur family, are able to bind to purine-rich single- or double-stranded DNA or RNA [6]. Pur proteins play important roles in development, especially in the development of brain [7,8] and myeloid cells [8], and they are also involved in many human diseases, including cancer [[9], [10], [11]] and fragile-X mental retardation syndrome [7,8,12,13]. Recently, Purβ has been shown to bind to lncRNA (lnc-HOXA1) and mediate repression of Hoxa1 transcription [14]. In addition, Purβ binds to DNA and negatively or positively regulates gene expression in a cell type–specific manner. For example, Purβ serves as a repressor that inhibits the expression of smooth muscle α-actin in fibroblasts and vascular smooth muscles [15,16], whereas it also acts as a transcription factor that increases mTOR and SREBP1 expression in mammary epithelial cells [17]. However, whether Purβ regulates HGP is largely unknown.
Here we identify Purβ as a positive regulator of HGP. Purβ, induced by fasting or glucagon, promotes HGP by increasing Adcy6 transcription, leading to cAMP accumulation, increased PKA activity, CREB activation, and increased transcription of PEPCK and G6Pase, both key gluconeogenic genes. Purβ is abnormally elevated in mice with obesity, and liver-specific knockdown of Purβ in db/db mice significantly ameliorates hyperglycemia and glucose intolerance by suppressing the glucagon/ADCY6/cAMP/PKA/CREB signaling pathway. Consistent with this observation, in isolated primary hepatocytes, knockdown of Purβ also inhibits glucose production by inhibiting this signaling pathway. Moreover, Purβ binds to the promoter of the Adcy6 gene, promoting its expression and activating the cAMP/PKA/CREB signaling pathway. These results support a new model in which Purβ regulates the glucagon/ADCY6/cAMP/PKA/CREB signaling pathway to help maintain glucose homeostasis, indicating that Purβ/ADCY6 may serve as a promising drug target for the treatment of hyperglycemia in patients with obesity.
2. Materials and methods
2.1. Animals
C57BL/6 and db/db mice were purchased from GemPharmatech (Nanjing, China). Mice were housed on a 12-h light/12-h dark cycle and fed either a normal chow or a high-fat diet with free access to water. All animal procedures described in this study were performed in adherence with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA) and with the approval by the Institutional Animal Care and Use Committee of Harbin Institute of Technology.
Liver-specific Purβ knockdown db/db mice were generated via tail vain injection of a purified adenovirus expressing shPURB1 for 8–12 days, and the experiments were conducted in conscious mice. In glucose tolerance tests, insulin tolerance tests, glucagon tolerance tests, and lactate tolerance tests, mice were fasted 6 h (8:00 AM–2:00 PM) and intraperitoneally injected with glucose (0.5 g/kg body weight), insulin (4 U/kg body weight), glucagon (6 μg/kg body weight), and sodium lactate (0.5 g/kg body weight), respectively. Blood glucose was monitored after injection. Blood samples were collected from tail veins, and blood glucose was measured as described previously [18,19]. Plasma insulin was measured using insulin enzyme-linked immunosorbent assay (ELISA) kits (MS100, EZassay). Liver triacylglycerol (TAG) levels were measured using Free Glycerol Reagent (Sigma).
2.2. Primary hepatocyte cell cultures, adenoviral infection, and HGP assays
Primary hepatocytes were isolated from C57BL/6 mice by liver perfusion with type II collagenase (Worthington Biochem, Lakewood, NJ) and cultured at 37 °C and 5% CO2 in DMEM medium supplemented with 5% fetal bovine serum. For Purβ overexpression experiments, primary hepatocytes were infected with βGal or Purβ adenoviruses overnight. For knockdown of Purβ, two target sequences (shPURB1: GTCGGTATGCAGATGAAATGA and shPURB2: GATGAAATGAAAGAGATCCAG) were selected. For Purβ knockdown experiments, primary hepatocytes were infected with Scramble, shPURB1, or shPURB2 adenoviruses for 30 h. These hepatocytes were used for immunoblotting, quantitative polymerase chain reaction, RNA sequencing, cAMP production, PKA activity, and HGP assays. For HGP assays, these hepatocytes were incubated in Hank's Balanced Salt Solution supplemented with 10 mM lactate and 1 mM pyruvate in the presence or absence of glucagon (100 nM) for 4 h. Glucose in the medium was measured and normalized to total protein levels.
2.3. Immunoblotting
For insulin signaling assays in vivo, mice were fasted for 20–24 h and administrated insulin (4 units/kg body weight) via the inferior vena cava for 5 min. For insulin signaling assays in vitro, hepatocytes were treated with insulin at different doses for 5 min. For glucagon signaling assays, hepatocytes were treated with glucagon for 10 min. Total proteins were extracted from livers or hepatocytes in a lysis buffer (50 mM Tris HCl, pH 7.5, 1.0% NP-40, 150 mM NaCl, 2 mM EGTA, 1 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) as described previously [20,21]. Extracts were then immunoblotted with antibodies against phospho-Akt (pSer473 and Thr308 from CST), total Akt (CST), phospho-CREB (pSer133) (CST), total CREB (Proteintech), Purβ (Proteintech), ADCY6 (Proteintech), Flag (Sigma), or Tubulin (Santa Cruz).
2.4. Quantitative real-time PCR (RT-qPCR) analysis
qPCR analysis was performed as previously described [18,19,22]. Briefly, total RNAs were extracted from indicated hepatocytes or livers using TriPure Isolation Reagent (Roche, Mannheim, Germany). The first-strand cDNAs were synthesized using random primers and M-MLV reverse transcriptase (Promega, Madison, WI). RNA abundance was measured using the Roche LightCycler 480 real-time PCR system (Roche, Mannheim, Germany). The expression of individual genes was normalized to the expression of 36B4, a housekeeping gene. Primers for real-time qPCR are listed below: PEPCK-F: 5′-ATCATCTTTGGTGGCCGTAG-3′, PEPCK-R: 5′-ATCTTGCCCTTGTGTTCTGC-3′; G6Pase-F: 5′-CCGGTGTTTGAACGTCATCT-3′, G6Pase-R: 5′-CAATGCCTGACAAGACTCCA-3′; 36B4–F: 5′-AAGCGCGTCCTGGCATTGTCT-3′, 36B4-R: 5′-CCGCAGGGGCAGCAGTGGT-3′; PURB-F: 5′-TGCAACAAGTACGGGGTGTT-3′, PURB-R: 5′-TCAATCCTCA TCCACTTCCT C-3′; Adcy6-F: 5′-GCATCCTGTTTGCGGACATT-3′, Adcy6-R: 5′-ACAGTGATTCTCCCTCACCG-3′; IL1β-F: 5′-GCCTTGGGCCTCAAAGGAAAGAATC-3′, IL1β-R: 5′-GGAAGACACGGATTCCATGGTGAAG-3′; IL6-F: 5′-AGCCAGAGTCCTTCAGA-3′, IL6-R: 5′-GGTCCTTAGCCACTCCT-3′; TNFα-F: 5′-CATCTTCTCAAAATTCGAGTGACAA-3′, TNFα-R: 5′-TGGGAGTAGACAAGGTACAACCC-3′.
2.5. Nuclear run-on RT-qPCR
Nascent mRNA levels were measured by nuclear run-on RT-qPCR following a published protocol [23]. Briefly, primary hepatocytes were infected with βGal or Purβ adenoviruses overnight. Nuclei were extracted in NP-40 lysis buffer (10 mM Tris HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.5% NP-40) and then incubated with BrUTP and unlabeled ribonucleotides in a transcription reaction buffer supplemented with 100U RNase OUT, 0.5 mM BrUTP, 1 mM ATP, 1 mM GTP, 1 mM CTP, and 0.5 mM UTP at 30 °C for 30 min. Nuclear RNA was extracted using TriPure Isolation Reagent. Labeled and unlabeled nuclear RNA samples were immunoprecipitated with anti-BrdU antibodies. Nascent Adcy6 mRNA levels were then measured by RT-qPCR and normalized by 36B4.
2.6. cAMP and PKA activity assays
Mice were fasted for 20–24 h, and livers were harvested for cAMP and PKA activity assays. Primary hepatocytes were infected with indicated adenoviruses and then treated with 100 nM glucagon for 10 min. cAMP was measured using an ELISA kit (H164-1-2, Nanjing Jiancheng).
For PKA activity assays, livers and hepatocytes were lysed in buffer containing 20 mM MOPS, 50 mM β-glycerolphosphate, 50 mM sodium fluoride, 1 mM DTT, 1 mM benzamidine, 1 mM PMSF, 10 μg/ml leupeptin and aprotinin. PKA activity assays were performed following the manufacturer's protocol (ab9435, Abcam).
2.7. RNA sequencing
Total RNAs were extracted from hepatocytes using TriPure Isolation Reagent (Roche, Mannheim, Germany). RNA-seq was performed by using the Illumina NovaSeq 6000 platform. Paired-end clean reads were aligned to the mouse reference genome (Ensemble_GRCm38.96) with TopHat (version 2.0.12), and the aligned reads were used to quantify mRNA expression by using HTSeq-count (version 0.6.1) as described previously [19]. RNA-seq data that support the findings of this study have been deposited in GEO under accession code GSE136728.
2.8. Luciferase reporter assays
Mouse Adcy6 promoter (−2001 to −1 or −1001 to −1) luciferase reporter plasmids and β-galactosidase reporter plasmids were transiently cotransfected with Purβ expression plasmids into HEK293T cells using polyethylenimine reagents. Cells were collected 24 h after transfection, and luciferase activity was measured using a luciferase assay system (Promega Corporation). Luciferase activity was normalized to β-galactosidase levels.
2.9. Chromatin immunoprecipitation (ChIP) assays
Primary hepatocytes were isolated from C57BL/6 mice and infected with βGal or Flag-Purβ adenoviruses overnight. These hepatocytes were washed with cold phosphate-buffered saline and fixed with 1% formaldehyde for 10 min at 37 °C. Their nuclei were isolated and subjected to sonication (M220 Focused-ultrasonicator; Covaris) to break genomic DNA into 500- to 1000-bp fragments using a chromatin shearing kit (520127 truChIP Chromatin Shearing Kit, Covaris). The samples were immunoprecipitated with Flag beads (A2220, Sigma). DNA was extracted and used for qPCR analysis. Primers for qPCR were as follows: Adcy6 promoter −74 to −147: 5′-TCATGACATTTCTCTTCCGCCT-3′ (forward) and 5′-AGTGGTAGTGGTGGCGAGAT-3′ (reverse); Adcy6 promoter −288 to −387: 5′-GACTCCCCAAGGGGATAACT-3′ (forward) and 5′-GGAGCCCTGTGAGTCCTTTAG-3′ (reverse); Adcy6 promoter −572 to −798: 5′-ATACAACCAGCTCCCACAACC-3′ (forward) and 5′-TCATTTTGCCAACAAGGGCA-3′ (reverse); Adcy6 promoter −1060 to −1211: 5′-GGGAGACACAGGTACCGAAAG-3′ (forward) and 5′-CAATGCCTACTTCCCCAAGGC-3′ (reverse); Adcy6 promoter −1366 to −1543: 5′-TCTGGGCAAGCCTGAAAACT-3′ (forward) and 5′-CAGCGGAGTCCCAAGAGTTG-3′ (reverse); Adcy6 promoter −1558 to −1850: 5′-GATCCCCCACGCTTACCTG-3′ (forward) and 5′-ACAAAAGGAGCTTGTGCCT-3′ (reverse).
2.10. Statistical analysis
Data were presented as means ± SEM. Differences between groups were analyzed by two-tailed Student's t tests. P < 0.05 was considered statistically significant.
3. Results
3.1. Identification of Purβ as a positive regulator of HGP
HGP is increased during fasting via enhanced glucagon/cAMP/CREB signaling. Thus, fasting-induced genes may regulate HGP. We first measured the levels of the Purβ mRNA and protein in the livers of fasted mice via qPCR and immunoblotting analyses, respectively. As shown in Figure 1A,B, fasting increased the levels of both Purβ mRNA and protein. As fasting increases glucagon secretion and its signaling pathway in the liver, these observations raise the possibility that glucagon may increase the levels of hepatic Purβ in primary hepatocytes. To test this hypothesis, primary hepatocytes were isolated from C57BL/6 mice and treated with glucagon. As shown in Figure 1C,D, glucagon treatment increased both the Purβ mRNA and protein levels.
Figure 1.

Identification of Purβ as a positive regulator of hepatic glucose production. (A) C57BL/6 male mice (8 weeks) were fasted for 24 h. Liver Purβ mRNA abundance was measured by qPCR and normalized to 36B4 expression. Randomly fed: n = 6, fasted: n = 6. B. C57BL/6 male mice were fasted as described above. Liver extracts were immunoblotted with antibodies against Purβ or Tubulin. (C–D) Primary hepatocytes were prepared from C57BL/6 males (8–10 weeks), grown overnight, and then treated with 0 or 100 nM glucagon for 4 h. Purβ mRNA and protein levels were measured by qPCR and immunoblotting analysis (n = 4). (E–F) Primary hepatocytes were infected with Scramble, shPURB1, or shPURB2 adenovirus for 30 h, then Purβ protein levels were measured by immunoblotting assays (E), and HGP assays were performed (F) (n = 4). (G–H) Primary hepatocytes were infected with βGal or Purβ adenovirus overnight, and Flag-Purβ protein levels were then measured by immunoblotting assays (G) and HGP assays were also performed (H) (n = 4). *p < 0.05. **p < 0.01.
To determine whether Purβ was essential for HGP, primary hepatocytes from C57BL/6 mice were infected with an Ad-shPURB1 or Ad-shPURB2 adenovirus to knockdown Purβ expression, and HGP assays were then performed. Adenovirus-mediated expression of shPURB1 or shPURB2 in primary hepatocytes led to a significant reduction in Purβ (Figure 1E), which resulted in a decrease in HGP under both basal and glucagon-treated conditions (Figure 1F).
To determine whether Purβ promotes HGP, primary hepatocyte cultures were prepared from C57BL/6 mice and infected with Purβ or βGal adenoviruses. The results indicated that recombinant Purβ was dramatically increased in Purβ adenovirus-infected hepatocytes (Figure 1G). These infected hepatocytes were then treated with or without glucagon and subjected to HGP assays. The results demonstrated that Purβ overexpression could significantly enhance HGP under both basal and glucagon-treated conditions (Figure 1H). These results demonstrated that Purβ could function to promote HGP.
3.2. Liver-specific knockdown of Purβ protects against hyperglycemia and glucose intolerance in obesity
Enhanced HGP contributes to hyperglycemia in both patients and rodents with obesity. To determine whether Purβ expression is abnormally increased under such conditions, both Purβ mRNA and protein levels were measured using qPCR and western blot analysis, respectively, in two obese mouse models (high-fat-diet–induced obesity and leptin receptor deficiency [db/db] mice). Both Purβ mRNA and protein levels were significantly increased in the livers of these models (Figure 2A,B), and these data indicate that Purβ might function to regulate hepatic glucose metabolism in both patients and rodents with obesity.
Figure 2.

Liver-specific knockdown of Purβ protects against hyperglycemia and glucose intolerance in obesity. (A–B) C57BL/6 male mice (8 weeks) were fed a regular chow diet or HFD for 12 weeks; wild-type and db/db mice (11 weeks) were fed a regular chow diet. Purβ mRNA and protein levels were measured by qPCR and immunoblotting analysis. (C) Liver-specific Purβ knockdown db/db mice were generated via tail-vain injection of a purified adenovirus expressing shPURB1. At the same time, db/db mice were injected with the same amount of purified Scramble adenovirus as their control. Liver extracts were immunoblotted with antibodies against Purβ or Tubulin. (D) Body weight of Purβ-KD and control db/db mice. (E) Fasting blood glucose levels. (F) Fed blood glucose levels. (G) Plasma insulin levels. (H–I) Glucose tolerance tests and AUC. (J) Insulin tolerance tests. n = 7–8. *p < 0.05. **p < 0.01.
To determine whether Purβ could regulate hepatic glucose metabolism in obese mice, we generated liver-specific Purβ knockdown db/db mice via tail-vain injection of a purified adenovirus expressing shPURB1 and measured the blood glucose and glucose tolerance in these mice. The expression of Purβ was significantly decreased in the liver of Purβ-KD db/db mice compared with control db/db mice (Figure 2C), but body weight was similar between Purβ-KD and control db/db mice (Figure 2D). However, fasting blood glucose was 42.8% lower in Purβ-KD than in the control db/db mice (Figure 2E), and blood glucose levels were also reduced by 36.8% in the former mice group under fed conditions (Figure 2F). Plasma insulin levels were also significantly decreased in Purβ-KD db/db mice (Figure 2G). Glucose tolerance in Purβ-KD db/db mice was significantly improved (Figure 2H). The area under the curve (AUC) was decreased by 62.9% in Purβ-KD db/db mice (Figure 2I). To further assess insulin sensitivity in these mice, insulin tolerance tests were performed. Surprisingly, exogenous insulin reduced blood glucose in these two groups to a similar extent (Figure 2J). These data suggest that the knockdown of Purβ can ameliorate hyperglycemia and glucose intolerance in db/db mice independent of insulin sensitivity.
3.3. Purβ does not regulate hepatic insulin signaling, steatosis, or inflammation
To further determine whether Purβ regulates glucose metabolism independent of insulin signaling in obesity, we measured insulin-induced phosphorylation of Akt in Purβ-KD db/db mice. The phosphorylation (Ser 473 and Thr 308) of Akt was similar in Purβ-KD and control db/db mice (Figure 3A). Insulin sensitivity was also associated with hepatic steatosis and inflammation. As shown in Figure 3B,C, liver-specific knockdown of Purβ did not alter liver TAG levels or the expression of inflammatory genes such as IL1β, IL6, and TNFα. Furthermore, insulin-induced phosphorylation (Ser 473 and Thr 308) of Akt was not altered by either knockdown or overexpression of Purβ in primary hepatocytes (Figure 3D,E). These data demonstrated that Purβ could regulate hepatic glucose metabolism independent of insulin signaling, hepatic steatosis, and inflammation.
Figure 3.

Purβ does not regulate hepatic insulin signaling, steatosis, or inflammation. (A) Purβ-KD and control db/db mice were fasted for 20–24 h and administered insulin (4 units/kg body weight) via the inferior vena for 5 min. The phosphorylation of Akt (at Ser 473 and Thr 308) was measured by immunoblotting. (B) Liver TAG levels. (C) IL1β, IL6, and TNFα mRNA levels. (D) Primary hepatocytes were infected with Scramble or shPURB1 adenoviruses for 30 h. Insulin-induced phosphorylation (Ser 473 and Thr 308) of Akt was measured by immunoblotting. (E) Primary hepatocytes were infected with βGal or Purβ adenoviruses overnight. Insulin-induced phosphorylation (Ser 473 and Thr 308) of Akt was measured by immunoblotting.
3.4. Liver-specific knockdown of Purβ decreases glucagon sensitivity and gluconeogenesis in obesity
In both patients and rodents with obesity and type 2 diabetes, plasma glucagon levels, glucagon sensitivity, and glucagon/CREB signaling are abnormally increased, contributing to higher HGP and hyperglycemia. To determine whether Purβ regulates glucagon-induced gluconeogenesis, glucagon tolerance tests and lactate tolerance tests were measured in Purβ-KD and control db/db mice. Exogenous glucagon markedly increased blood glucose levels in the control db/db mice; however, its ability to increase blood glucose was severely impaired in Purβ-KD db/db mice (Figure 4A), with the AUC decreased by 46% in Purβ-KD db/db mice (Figure 4B). Hepatic gluconeogenesis, as estimated by lactate tolerance tests, was also significantly lower in Purβ-KD db/db mice, with the AUC reduced by 64.7% (Figure 4C,D).
Figure 4.

Knockdown of Purβ decreases glucagon sensitivity and gluconeogenesis in obesity. (A–B) Purβ-KD and control db/db mice were fasted for 6 h and intraperitoneally injected with glucagon (6 μg/kg body weight). Blood glucose was monitored after injection, and AUC was calculated. (C–D) Hepatic gluconeogenesis was measured by injection of sodium lactate (0.5 g/kg body weight) in Purβ-KD and control db/db mice fasted for 6 h, and AUC was calculated. (E) p-CREB, CREB, Purβ, and Tubulin protein levels were measured by western blot. (F–G) G6Pase and PEPCK mRNA levels were measured by qPCR. n = 7–8. *p < 0.05. **p < 0.01.
To determine whether hepatic Purβ modulates glucagon-stimulated CREB phosphorylation, Purβ-KD and control db/db mice were fasted for ∼20 h, and liver tissue extracts were immunoblotted using a phospho-CREB (pSer133) antibody. This analysis indicated that CREB phosphorylation was decreased by 62% in Purβ-KD db/db mice (Figure 4E). In addition, total CREB levels were slightly increased in Purβ-KD db/db mice compared with the control db/db mice (Figure 4E), possibly compensating for the decreased p-CREB levels. To determine whether Purβ regulates the hepatic gluconeogenic program, liver mRNA was extracted and used to measure the mRNA abundance of key genes via qPCR. The expression of G6Pase and PEPCK was dramatically decreased by 56.7% and 56.2% in Purβ-KD db/db mice, respectively (Figure 4F,G). These data suggest that hepatic knockdown of Purβ can ameliorate hyperglycemia and glucose intolerance, and this most likely occurs via a decrease in HGP in obese mice.
3.5. Purβ promotes the hepatic gluconeogenic program in primary hepatocytes
Knockdown of Purβ decreased HGP, whereas overexpression of Purβ increased HGP in primary hepatocytes (Figure 1E-H), indicating that Purβ could cell-autonomously regulate HGP. To further confirm that Purβ cell-autonomously regulates the hepatic gluconeogenic program, the expression of G6Pase and PEPCK and its upstream regulator p-CREB levels were measured in Purβ knockdown or overexpressing hepatocytes. The expression of G6Pase and PEPCK was significantly reduced in Purβ knockdown hepatocytes (Figure 5A,B), which was most likely due to decreased p-CREB levels (Figure 5C). Conversely, the expression of G6Pase and PEPCK was significantly increased in hepatocytes overexpressing Purβ (Figure 5D,E), likely because of increased p-CREB levels (Figure 5F). However, CREB protein levels did not show any change in Purβ-overexpressing and Purβ KD hepatocytes (Figure 5C, F, and Supplemental Fig. 1), suggesting that Purβ does not regulate CREB expression.
Figure 5.

Purβ promotes the hepatic gluconeogenic program in primary hepatocytes. (A–B) Primary hepatocytes were prepared from C57BL/6 males (8–10 weeks) and infected with Scramble, shPURB1, or shPURB2 adenoviruses for 30 h and then treated with or without glucagon for 2 h. The abundance of G6Pase and PEPCK mRNA levels was measured by qPCR (n = 3–4). (C) Primary hepatocytes were infected with Scramble, shPURB1, or shPURB2 adenoviruses for 30 h and then treated with or without glucagon for 30 min. p-CREB, CREB, Purβ, and Tubulin protein levels were measured by immunoblotting. (D–E) Primary hepatocytes were infected with βGal or Purβ adenovirus overnight and then treated with or without glucagon for 2 h. The abundance of G6Pase and PEPCK mRNA levels was measured by qPCR (n = 4). (F) Primary hepatocytes were infected with βGal or Purβ adenovirus overnight and then treated with or without glucagon for 30 min. p-CREB, CREB, Flag, and Tubulin protein levels were measured by immunoblotting. *p < 0.05. **p < 0.01.
3.6. Purβ promotes hepatic cAMP production and PKA activity
Both cAMP and PKA function upstream of the CREB signaling pathway. To determine whether Purβ can regulate hepatic cAMP production and PKA activity, intracellular cAMP levels and PKA activity were measured in Purβ-overexpressing hepatocytes, PurβKD hepatocytes, and livers of PurβKD db/db mice. Overexpression of Purβ in primary hepatocytes increased cAMP levels and PKA activity (Figure 6A,B), whereas knockdown of Purβ decreased cAMP levels and PKA activity both in vitro (Figure 6C,D) and in vivo (Figure 6E,F). These data suggest that Purβ could promote the CREB signaling pathway and hepatic gluconeogenic program by increasing hepatic cAMP production and PKA activity.
Figure 6.

Purβ promotes hepatic cAMP production and PKA activity. (A–B) Primary hepatocytes were infected with βGal or Purβ adenovirus overnight and then treated with glucagon (100 nM) for 10 min. cAMP levels and PKA activity were measured using commercial kits (n = 5–6). (C–D) Primary hepatocytes were prepared from C57BL/6 males (8–10 weeks) and infected with Scramble or shPURB1 adenoviruses for 30 h and then treated with glucagon (100 nM) for 10 min. cAMP levels and PKA activity were measured using commercial kits (n = 5–6). (E–F) cAMP levels and PKA activity in Purβ-KD and control db/db mice were measured using commercial kits (n = 7–8). *p < 0.05. **p < 0.01.
3.7. Purβ promotes Adcy6 expression
Hepatic cAMP homeostasis is controlled by adenylate cyclases (ADCYs) and phosphodiesterases (PDEs), and Purβ promotes hepatic cAMP production either by increasing expression of ADCYs or by decreasing expression of PDEs. To address this possibility, RNA sequencing (RNA-seq) analysis was performed in Purβ-overexpressing and PurβKD hepatocytes. As shown in Figure 7A, the expression of liver-enriched PDEs, such as Pde4b, Pde3b, Pde8a, Pde7a, Pde6d, and Pde5a, did not show any correlated change in Purβ-overexpressing and Purβ KD hepatocytes. However, the expression of liver-enriched Adcy6 was upregulated by overexpression of Purβ and downregulated by knockdown of Purβ (Figure 7B). RT-qPCR and immunoblotting analysis showed that overexpression of Purβ increased both the ADCY6 mRNA and protein levels (Figure 7C), whereas knockdown of Purβ decreased ADCY6 expression (Figure 7D). Furthermore, liver-specific knockdown of Purβ in db/db mice also resulted in a significant decrease in both the ADCY6 mRNA and protein levels (Figure 7E). Interestingly, similar to Purβ, hepatic ADCY6 expression could also be induced by fasting (Figure 7F) and glucagon (Supplemental Fig. 2). Hepatic ADCY6 expression was also abnormally elevated in the two obese mouse models (Figure 7G,H). These data demonstrate that Purβ could promote HGP and lead to hyperglycemia by increasing Adcy6 expression.
Figure 7.

Purβ promotes Adcy6 expression. Primary hepatocytes were infected with Scramble or shPURB1 adenoviruses for 30 h or infected with βGal or Purβ adenovirus overnight. Total RNA was isolated, and RNA-seq was performed. (A) Heat map of PDEs in Purβ-overexpressing and PurβKD hepatocytes. (B) Heat map of ADCYs in Purβ-overexpressing and PurβKD hepatocytes. (C) ADCY6 mRNA and protein levels in Purβ-overexpressing hepatocytes (n = 4). (D) ADCY6 mRNA and protein levels in PurβKD hepatocytes (n = 4). (E) ADCY6 mRNA and protein levels in PurβKD and control db/db mice (n = 7–8). (F) ADCY6 mRNA and protein levels in fasted or fed mice (n = 6). (G–H) ADCY6 mRNA and protein levels in the livers of HFD or ob/ob mice (n = 6–8). *p < 0.05. **p < 0.01.
3.8. Purβ directly binds to the Adcy6 promoter and increases its transcription
To further test whether Purβ promotes Adcy6 expression at the transcriptional level, nascent Adcy6 mRNA levels were measured by performing nuclear run-on RT-qPCR. As shown in Figure 8A, the nascent Adcy6 mRNA level was significantly increased in Purβ-overexpressing hepatocytes, indicating that Purβ promoted Adcy6 transcription. Furthermore, ChIP assays showed that Purβ directly binds to the Adcy6 promoter at −1558 to −1850 (Figure 8B), which is a purine-enriched region. In addition, Purβ increased Adcy6 promoter-controlled luciferase activity in a dose-dependent manner, and deletion of the purine-enriched region abolished this enhancement (Figure 8C). These results demonstrated that Purβ directly binds to the Adcy6 promoter and increases its transcription.
Figure 8.

Purβ directly binds to the Adcy6 promoter and increases its transcription. (A) Nascent Adcy6 mRNA levels were measured by nuclear run-on RT-qPCR in Purβ-overexpressing hepatocytes (n = 3). (B) ChIP assays showing Purβ occupancy at the promoter of Adcy6 (n = 3). (C) Mouse Adcy6 promoter (−2001 to −1 or −1001 to −1) luciferase reporter plasmids and β-galactosidase reporter plasmids were transiently cotransfected with Purβ expression plasmids into HEK293T cells. Luciferase activity was measured 24 h after transfection and normalized to βGal levels (n = 6). *p < 0.05. **p < 0.01.
4. Discussion
HGP is controlled by glucagon and insulin, and enhanced glucagon signaling in the liver leads to increased HGP, contributing to hyperglycemia in both patients and rodents with obesity and type 2 diabetes [3,4]. The detailed molecular mechanisms underlying this enhanced HGP are not yet fully understood. In this study, Purβ was identified as a positive regulator of HGP and was shown to promote HGP by increasing Adcy6 expression and subsequent activation of the cAMP/PKA/CREB signaling pathway, which contributes to the occurrence of hyperglycemia in obesity.
This study provides several lines of evidence indicating that Purβ acts as a positive regulator of HGP. First, hepatic Purβ expression was increased by fasting and glucagon. Glucagon could activate CREB via the cAMP/PKA signaling pathway. We noted that there are four binding sites of CREB at the promoter (−22 to −29; −98 to −105; −1337 to −1344; and −1349 to −1356) of PURB gene by searching the JASPAR database [24], indicating that glucagon induces Purβ expression through the activation of CREB. Knockdown of Purβ decreased the ability of glucagon to stimulate CREB phosphorylation, the expression of G6Pase and PEPCK, and glucose production in primary hepatocytes, whereas Purβ overexpression had the opposite effects. Purβ did not regulate CREB expression, indicating that Purβ enhances glucagon-induced CREB phosphorylation mainly through the activation of the upstreams of CREB. Mechanistically, Purβ likely increases Adcy6 transcription by directly binding to its promoter, which then leads to increased cAMP production, enhanced PKA activity, and increased HGP. Both Purβ and ADCY6 were induced at a later stage by fasting and then promoted HGP for survival during prolonged fasting.
Second, hepatic Purβ and ADCY6 are abnormally elevated in obesity and type 2 diabetes mouse models. Hepatocyte-specific knockdown of Purβ in db/db mice severely impaired the ability of glucagon to increase blood glucose levels and inhibited CREB phosphorylation and PKA activity (due to decreased cAMP production) by suppressing Adcy6 expression in the liver. These observations indicate that hepatic Purβ/ADCY6 could promote the glucagon signaling pathway, contributing to the occurrence of hyperglycemia in type 2 diabetes.
Surprisingly, Purβ does not regulate insulin signaling, although Purβ-KD db/db mice displayed reduced plasma insulin levels, probably secondary to a decrease in blood glucose in Purβ-KD db/db mice given that low blood glucose stimulates less insulin secretion from β cells. Hepatic deletion of TRAF2 also shows similar phenotypes [20]. Liver-specific knockdown of Purβ in db/db mice did not alter hepatic insulin signaling, steatosis, or inflammation. Exogenous insulin reduced blood glucose to a similar degree in both Purβ-KD and control db/db mice, suggesting that hepatic Purβ does not alter hepatic or systemic insulin sensitivity. Consistently, in isolated primary hepatocytes, neither knockdown nor overexpression of Purβ altered the insulin-induced phosphorylation of Akt, further supporting the conclusion that hepatic Purβ does not regulate insulin sensitivity.
In summary, Purβ was shown to positively regulate HGP by directly binding to the promoter of the Adcy6 gene, increasing its expression and thereby enhancing the glucagon/cAMP/PKA/CREB signaling pathway. Purβ/ADCY6 is abnormally elevated in obese mice and is also increased by fasting or via the glucagon signaling pathway. Hepatic knockdown of Purβ in db/db mice significantly ameliorated hyperglycemia and glucose intolerance by suppressing the ADCY6/cAMP/PKA/CREB signaling pathway. These data suggest that Purβ/ADCY6 might serve as an important drug target for the treatment of hyperglycemia in patients with type 2 diabetes.
Disclosure statement
All authors have nothing to declare.
Author contributions
L.J. performed most of the experiments. Y.J. and X.L. researched data. Z.C. designed the project and wrote the manuscript.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grants 31671225 and 31971083) and the Natural Science Foundation of Heilongjiang Province (YQ2019C011). X.L. is supported by the Chinese Postdoctoral Science Foundation (AUGA4130900619).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.11.008.
conflict of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplemental Figure 1.

Purβ does not regulate CREB expression in primary hepatocytes. (A) Primary hepatocytes were prepared from C57BL/6 males (8 weeks) and infected with Scramble, shPURB1, or shPURB2 adenoviruses for 30 h. CREB, Purβ, and Tubulin protein levels were measured by immunoblotting. (B) Primary hepatocytes were infected with βGal or Purβ adenovirus overnight. CREB, Flag, and Tubulin protein levels were measured by immunoblotting.
Supplemental Figure 2.

The expression of hepatic PURB and Adcy6 was regulated by glucagon and fasting. (A) Primary hepatocytes were prepared from C57BL/6 males (8 weeks) and treated with glucagon for different times (0, 1, 2, 4, and 6 h). The expression of PURB and Adcy6 was measured by RT-qPCR (n = 4-5). (B) C57BL/6 males (8 weeks) were fasted for different times (0, 6, 12, and 24 h). The expression of PURB and Adcy6 was measured by RT-qPCR (n = 5). *p < 0.05.
References
- 1.Sharabi K., Tavares C.D., Rines A.K., Puigserver P. Molecular pathophysiology of hepatic glucose production. Molecular Aspects of Medicine. 2015;46:21–33. doi: 10.1016/j.mam.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen M.C., Vatner D.F., Shulman G.I. Regulation of hepatic glucose metabolism in health and disease. Nature Reviews Endocrinology. 2017;13:572. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang G., Zhang B.B. Glucagon and regulation of glucose metabolism. American Journal of Physiology. Endocrinology and Metabolism. 2003;284(4):E671–E678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 4.Ali S., Drucker D.J. Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. American Journal of Physiology. Endocrinology and Metabolism. 2009;296(3):E415–E421. doi: 10.1152/ajpendo.90887.2008. [DOI] [PubMed] [Google Scholar]
- 5.Rines A.K., Sharabi K., Tavares C.D.J., Puigserver P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nature Reviews Drug Discovery. 2016;15:786. doi: 10.1038/nrd.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson E.M., Daniel D.C., Gordon J. The pur protein family: genetic and structural features in development and disease. Journal of Cellular Physiology. 2013;228(5):930–937. doi: 10.1002/jcp.24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hokkanen S., Feldmann H.M., Ding H., Jung C.K., Bojarski L., Renner-Muller I. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Human Molecular Genetics. 2012;21(3):473–484. doi: 10.1093/hmg/ddr476. [DOI] [PubMed] [Google Scholar]
- 8.Khalili K., Del Valle L., Muralidharan V., Gault W.J., Darbinian N., Otte J. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Molecular and Cellular Biology. 2003;23(19):6857–6875. doi: 10.1128/MCB.23.19.6857-6875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darbinian N., Gallia G.L., King J., Del Valle L., Johnson E.M., Khalili K. Growth inhibition of glioblastoma cells by human Pur(alpha) Journal of Cellular Physiology. 2001;189(3):334–340. doi: 10.1002/jcp.10029. [DOI] [PubMed] [Google Scholar]
- 10.Lezon-Geyda K., Najfeld V., Johnson E.M. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia. 2001;15(6):954–962. doi: 10.1038/sj.leu.2402108. [DOI] [PubMed] [Google Scholar]
- 11.Wang L.G., Johnson E.M., Kinoshita Y., Babb J.S., Buckley M.T., Liebes L.F. Androgen receptor overexpression in prostate cancer linked to Pur alpha loss from a novel repressor complex. Cancer Research. 2008;68(8):2678–2688. doi: 10.1158/0008-5472.CAN-07-6017. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi S., Koike K., Omori A., Ichinose S., Ohara S., Kobayashi S. Identification of mRNA/protein (mRNP) complexes containing Puralpha, mStaufen, fragile X protein, and myosin Va and their association with rough endoplasmic reticulum equipped with a kinesin motor. Journal of Biological Chemistry. 2002;277(40):37804–37810. doi: 10.1074/jbc.M203608200. [DOI] [PubMed] [Google Scholar]
- 13.Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T.C. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55(4):556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maamar H., Cabili M.N., Rinn J., Raj A. linc-HOXA1 is a noncoding RNA that represses Hoxa1 transcription in cis. Genes & Development. 2013;27(11):1260–1271. doi: 10.1101/gad.217018.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knapp A.M., Ramsey J.E., Wang S.X., Godburn K.E., Strauch A.R., Kelm R.J., Jr. Nucleoprotein interactions governing cell type-dependent repression of the mouse smooth muscle alpha-actin promoter by single-stranded DNA-binding proteins Pur alpha and Pur beta. Journal of Biological Chemistry. 2006;281(12):7907–7918. doi: 10.1074/jbc.M509682200. [DOI] [PubMed] [Google Scholar]
- 16.Rumora A.E., Wang S.X., Ferris L.A., Everse S.J., Kelm R.J., Jr. Structural basis of multisite single-stranded DNA recognition and ACTA2 repression by purine-rich element binding protein B (Purbeta) Biochemistry. 2013;52(26):4439–4450. doi: 10.1021/bi400283r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huo N., Yu M., Li X., Zhou C., Jin X., Gao X. PURB is a positive regulator of amino acid-induced milk synthesis in bovine mammary epithelial cells. Journal of Cellular Physiology. 2019;234(5):6992–7003. doi: 10.1002/jcp.27452. [DOI] [PubMed] [Google Scholar]
- 18.Ren X., Li X., Jia L., Chen D., Hou H., Rui L. A small-molecule inhibitor of NF-κB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. The FASEB Journal. 2017;31(2):711–718. doi: 10.1096/fj.201600840R. [DOI] [PubMed] [Google Scholar]
- 19.Li X., Jia L., Chen X., Dong Y., Ren X., Dong Y. Islet α-cell inflammation induced by NF-κB inducing kinase (NIK) leads to hypoglycemia, pancreatitis, growth retardation, and postnatal death in mice. Theranostics. 2018;8(21):5960–5971. doi: 10.7150/thno.28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z., Sheng L., Shen H., Zhao Y., Wang S., Brink R. Hepatic TRAF2 regulates glucose metabolism through enhancing glucagon responses. Diabetes. 2012;61(3):566–573. doi: 10.2337/db11-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z., Canet M.J., Sheng L., Jiang L., Xiong Y., Yin L. Hepatocyte TRAF3 promotes insulin resistance and type 2 diabetes in mice with obesity. Molecular Metabolism. 2015;4(12):951–960. doi: 10.1016/j.molmet.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin W., Li X., Xie L., Li S., Liu J., Jia L. A long non-coding RNA, APOA4-AS, regulates APOA4 expression depending on HuR in mice. Nucleic Acids Research. 2016;44(13):6423–6433. doi: 10.1093/nar/gkw341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts T.C., Hart J.R., Kaikkonen M.U., Weinberg M.S., Vogt P.K., Morris K.V. Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nature Protocols. 2015;10(8):1198–1211. doi: 10.1038/nprot.2015.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathelier A., Fornes O., Arenillas D.J., Chen C-y, Denay G., Lee J. Jaspar 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Research. 2015;44(D1):D110–D115. doi: 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]