

Summary
Lactate produced by muscle during high-intensity activity is an important end product of glycolysis that supports whole body metabolism. The lactate shuttle model suggested that lactate produced by glycolytic muscle fibers is utilized by oxidative fibers. MCT4 is a proton coupled monocarboxylate transporter preferentially expressed in glycolytic muscle fibers and facilitates the lactate efflux. Here we investigated the exercise capacity of mice with disrupted lactate shuttle due to global deletion of MCT4 (MCT4−/−) or muscle-specific deletion of the accessory protein Basigin (iMSBsg−/−). Although MCT4−/− and iMSBsg−/− mice have normal muscle morphology and contractility, only MCT4−/− mice exhibit an exercise intolerant phenotype. In vivo measurements of compound muscle action potentials showed a decrement in the evoked response in the MCT4−/− mice. This was accompanied by a significant structural degeneration of the neuromuscular junctions (NMJs). We propose that disruption of the lactate shuttle impacts motor function and destabilizes the motor unit.
Subject Areas: Musculoskeletal Medicine, Genetics, Neuroscience
Graphical Abstract
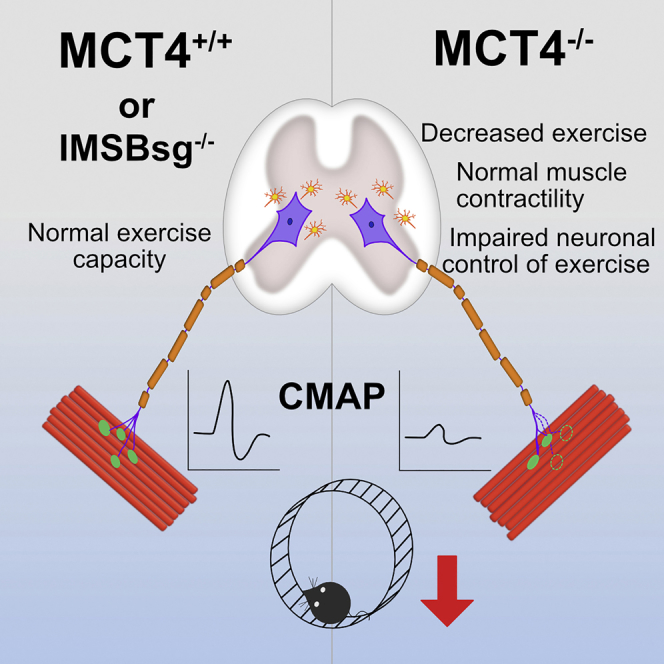
Highlights
-
•
MCT4 facilitates lactate efflux from the cells and is highly expressed in muscles fibers
-
•
MCT4−/− mice are exercise intolerant but there is no impact on muscle physiology
-
•
Action Potential conduction is impaired in the MCT4−/− mice and NMJs are disrupted
-
•
MCT4 and lactate efflux play a role in the maintenance of a functional motor unit
Musculoskeletal Medicine; Genetics; Neuroscience
Introduction
A hallmark of high-intensity exercise is the increased production of lactate by muscles, which is coupled with an increase in lactate in circulation. Lactate was once considered a waste product of anaerobic glycolysis and the cause of muscle fatigue and decreased force generation (Hill et al., 1924). In the late 1980s, George Brooks (Brooks, 1986) proposed that lactate produced by fast-twitch muscle fibers through both aerobic and anaerobic glycolysis (Connett et al., 1990) is shuttled to oxidative fibers and to other organs via circulation, to fuel oxidative metabolism during exercise.
Lactate is transported across the plasma membrane by monocarboxylate transporters (MCTs), encoded by the SLC16A gene family (Adijanto and Philp, 2012). MCT1 (SLC16A1) and MCT4 (SLC16A3) are H+-coupled lactate transporters found in the sarcolemma of skeletal muscle (Bonen, 2001, Wilson et al., 1998). MCT1 has higher affinity for lactate (Poole and Halestrap, 1993) and is predominantly expressed in slow-twitch muscle fibers, where it facilitates the influx of lactate. MCT4 has lower affinity but higher capacity for lactate transport (Dimmer et al., 2000) and is expressed in fast twitch where it mediates lactate efflux. The direction of the flux is primarily controlled by the gradient of lactate and protons (Juel, 1997). To be trafficked to the plasma membrane, MCT1 and MCT4 need to assemble in the ER with an accessory protein called Basigin (Bsg or CD147) (Kirk et al., 2000), a member of the immunoglobulin superfamily. In Bsg knockout mice, MCTs are not trafficked to the plasma membrane but are targeted for degradation (Philp et al., 2003).
The model of the lactate shuttle as a mechanism to distribute high-energy metabolites between glycolytic and oxidative cells has been applied beyond skeletal muscle and exercise. It has been suggested that lactate is shuttled between astrocytes and neurons during neuronal stimulation (Pellerin et al., 1998). In the brain, it is not yet clear if the direction of the lactate shuttle is from astrocytes to neurons or vice versa. Multiple studies have tried to address the question, but the results are still inconclusive as discordant findings have been published (Díaz-García et al., 2017, Mangia et al., 2009).
In this study, we have examined the contribution of the lactate shuttle to exercise capacity of mice using a global Slc16a3 knockout (MCT4−/−) and an inducible muscle specific Bsg knockout mouse (iMSBsg−/−) models. We performed in vivo and ex vivo analyses of exercise capacity, neuromuscular function, and morphometry in these mouse models. Surprisingly, our results revealed that disruption of lactate transport decreased exercise performance without altering muscle structure/function and was accompanied by a progressive structural and functional impairment of the neuronal component of the motor unit.
Results
MCT4−/− Mice Develop Exercise Intolerance
MCT4−/− mice were purchased from Taconic Biosciences. The animals were born at normal Mendelian ratio, with no apparent defects at birth. There were no developmental delays noted in MCT4−/− mice, as confirmed by normal growth curves and comparable body (Figure 1A) and muscle (Figure 1B) weights between MCT4+/+ and MCT4−/− mice.
Figure 1.

MCT4−/− Mice Have Normal Development
(A and B) (A) Weight gain over 40 weeks of age and (B) muscle weight at 3 months of age in MCT4+/+ and MCT4−/− mice (n = 15 per group). Values are mean and SEM.
(C) Western blot analysis of MCT4 and MCT1 expression in skeletal muscle and cardiac muscle. EDL, extensor digitorum longus; TA, tibialis anterior; Sol, soleus; Quad, quadricep; Gastr, gastrocnemius; Diaph, diaphragm.
(D) Representative immunostaining of EDL frozen sections from MCT4−/− and MCT4+/+ mice labeled with WGA (left, red) and antibodies against MCT4 (middle, green) and MCT1 (right, green).
(E) Representative immunolabeling of EDL frozen sections with MCT1 (blue) and antibodies against type IIb (red) and IIa (green) fibers. Scale bar, 50 μm.
MCT4 was previously shown to be expressed in fast-twitch muscle fibers (Bonen, 2001). In agreement with this we found by western blot analysis of muscle lysates from MCT4+/+ mice that MCT4 was mainly expressed by glycolytic fast-twitch (EDL, TA, and quadriceps) and mixed (gastrocnemius) muscles. MCT4 was completely absent in muscles lysates prepared from MCT4−/− mice (Figure 1C). MCT1, on the other hand, was expressed in all muscle types, but at higher levels in oxidative slow-twitch muscles including soleus, diaphragm, and heart (Figure 1C). We performed immunofluorescent labeling on frozen sections of the extensor digitorum longus (EDL) and found MCT4 was localized at the sarcolemma in all the fibers of MCT4+/+ EDL muscles, where it co-localized with wheat germ agglutinin (WGA). No MCT4 labeling was detected in frozen sections of EDLs from MCT4−/− mice (Figure 1D). Because the MCTs are bidirectional and can function for influx or efflux of lactate, we examined whether MCT1 expression was increased in the EDL for compensation. MCT1 immunofluorescence on EDLs cryosections showed that the transporter has a similar expression pattern from both MCT4+/+ and MCT4−/− mice, where it was preferentially expressed in oxidative fiber (smaller sized) (Figure 1D). Immunofluorescence labeling of EDL cross sections labeled with antibodies detecting MCT1, type IIa MyHC (oxidative fibers), and type IIb MyHC (glycolytic fibers) confirmed that the transporter is primarily expressed in IIa and IIx (identified as non-positive for either IIa or IIb MyHC) fibers (Figure 1E).
To investigate whether alterations of lactate transport in MCT4−/− mice could affect normal exercise performance, we tested MCT4−/− and MCT4+/+ mice with voluntary wheel running and forced endurance treadmill. Voluntary wheel activity was comparable between the two groups at 3 months of age but was reduced by 2.5-fold in 12-month-old MCT4−/− mice (Figure 2A).
Figure 2.

MCT4−/− Are Exercise Intolerant
(A) Voluntary wheel running, cumulative number of revolutions over 60 h of MCT4−/− and MCT4+/+ mice, at 3 and 12 months of age (n = 3 each group, One way-ANOVA test, ****p < 0.0001).
(B) Treadmill protocol for endurance test at 5° incline.
(C) Total distance reached in the endurance treadmill test by MCT4+/+ and MCT4−/− mice at 3, 6, and 18 months of age (n = 10 MCT4+/+ and 23 MCT4−/− at 3 months, n = 10 MCT4+/+ and 15 MCT4−/− at 6 months, n = 7 MCT4+/+ and 8 MCT4−/− at 18 months, three independent groups. Student t test, *p < 0.05, **p < 0.001).
(D) Representative traces of cumulative number of shocks over time during treadmill test of MCT4−/− and MCT4+/+ mice at 6 months of age. All the data are rapresented as mean and SEM.
See also Figure S1.
The endurance treadmill protocol was designed to gradually increase the running speed over time, up to 26 m/min, a speed at which oxygen consumption in C57BL/6 mice reaches approximately 80% of VO2max (high-intensity exercise) (Desai and Bernstein, 2001) (Figure 2B). At high-intensity, most of the energy to support muscle activity is derived from carbohydrates, resulting in the production and release of lactate from muscle fibers and the consequent increase in blood lactate. Consistently, blood glucose decreased by ∼30% after treadmill (Figure S2). Treadmill performance declined with age in both genotypes and, consistent with the wheel running results, MCT4−/− mice ran less compared with controls (Figure 2C). MCT4−/− mice could reach the maximum speed but, unlike MCT4+/+, could not endure the high-intensity performance, as indicated by the quicker accumulation of electric shocks over time (Figure 2D). This suggests that, indeed, lack of MCT4 and disruption of muscle lactate efflux cause an age-dependent and exercise-intensity-dependent impairment of exercise capacity.
Lack of MCT4 Does Not Affect the Intrinsic Properties of the EDL Muscle
To determine if the exercise intolerance was due to structural alterations of MCT4−/− muscle fibers, we performed a histological analysis of EDL cross sections, looking for signs of degeneration, fibrosis, or fiber type switch. We focused on EDL muscles for a variety of reasons: first, EDL is highly engaged during locomotion in rodents; second, it expresses high levels of MCT4 and its fiber type composition is primarily fast; and finally, it can be easily and consistently dissected for functional studies.
H&E staining showed normal fiber morphology with no signs of immune infiltrates, fibrotic tissue, or central nucleation (Figure 3A). Consistent with the normal muscle mass (Figure 1B), fiber size measured as minimum Feret diameter was comparable between genotypes both at 3 and 12 months of age (Figures 3B and 3C).
Figure 3.

Skeletal Muscle Histology Is Not Affected by the Lack of MCT4
(A) Representative H&E staining of EDL cryosections from MCT4+/+ and MCT4−/− mice; scale bar, 50 μm.
(B and C) Frequency histograms of fiber size of EDLs measured as minimum Feret diameter from MCT4+/+ and MCT4−/− at (B) 3 and (C) 12 months (n = 5 each group, counting all the fibers in the entire muscle section).
(D) Representative immunolabeling of MCT4+/+ and MCT4−/− EDL sections with antibodies against type IIB and IIa fibers and laminin for sarcolemma.
(E and F) Percentage of IIb and IIa fiber types in EDLs from MCT4+/+ and MCT4−/− at (E) 3 and (F) 12 months of age (n = 5 each group, two-way ANOVA test). All the data are rapresented as mean and SEM.
Changes in muscle metabolism are often correlated with changes in the proportion of glycolytic and oxidative fibers. To address this possibility, we immunolabeled EDL frozen sections with antibodies against the myosin heavy chain (MHC) isoforms (Figure 3D). The relative abundance of each fiber type was similar between genotypes at 3 and 12 months of age (Figures 3E and 3F).
To determine if the exercise intolerance in MCT4−/− mice could be caused by defects in muscle contractility, we performed an ex vivo assessment of strength and fatigability in isolated EDL muscles using field depolarization. This technique bypasses the neuronal and neuromuscular junction (NMJ) components and focuses specifically on measuring the contractile and metabolic properties of the muscle. Isometric twitch and tetanic forces, both absolute (Table S1) and after normalization by cross-sectional area (CSA) (Figures 4A and 4B), were not different between MCT4−/− and MCT4+/+. Similar optimal length (L0), mass, and CSA were consistent with the tissue weight measurements (Figure 1B) and the histomorphometric analyses (Figures 3A–3C). EDL muscles from MCT4+/+ and MCT4−/− mice had similar fatigability profiles (Figure 4C), as determined by administering repeating submaximal tetanic contractions every second for 8 min. The same trend was found in younger (3 months old) and older (18 months old) mice (data not shown). This was despite significant differences in the accumulation of intramuscular lactate, which was higher (∼50%) in MCT4−/− stimulated muscles compared with the unstimulated and MCT4+/+ conditions (Figure 4D). Therefore, intramuscular accumulation of lactate due to lack of MCT4 did not have an effect on muscle contractility. Because MCT4 is a proton-linked lactate transporter and contributes to the control of the intracellular pH, we tested whether other pH modulators like carbonic anhydrases 2, 3, and 4 (Car2, Car3, Car4) or proton exchangers such as Mct1 and Na+/H+ exchanger (Nhe1) were overexpressed in the MCT4−/− muscle as a compensatory response. qPCR analysis of TA muscles from 6-month-old mice showed no difference in the expression of these genes (Figure 4E). Together, these data suggest that lactate accumulation in muscle does not alter muscle contractility or fatigability and therefore is not the main cause of the reduced exercise capacity of MCT4−/− mice.
Figure 4.

Lack of MCT4 Does Not Impair the Contractile Properties of the Skeletal Muscles
(A and B) (A) EDL twitch and (B) tetanic forces generated ex vivo (n = 4 each group).
(C) EDL fatigue traces (n = 4 each group).
(D) Intramuscular lactate in unstimulated and stimulated gastrocnemius muscles (n = 4 each group, two-way ANOVA test, interaction: p < 0.01, stimulation: ***p < 0.001, genotype: *p < 0.05).
(E) qPCR analysis of genes involved in pH control in TA muscles of 6-month-old MCT4+/+ and MCT4−/− mice (n = 3 MCT4+/+ and 5 MCT4−/−). All the data are rapresented as mean and SEM.
iMSBsg−/− Mice Have Normal Exercise Capacity
To strengthen our finding and further investigate whether deletion of MCT4 in muscle could have any effect on exercise capacity, we generated mice with a skeletal muscle-specific deletion of Basigin (BSG). BSG is the accessory protein required for maturation and trafficking of both MCT1 and MCT4 to the sarcolemma. When Bsg is knocked out, MCT1 and MCT4 fail to traffic to the plasma membrane and are targeted for degradation. Specific deletion of Bsg in skeletal muscle was achieved by crossing BsgFlox/Flox mice with the tamoxifen-inducible skeletal muscle-specific cre recombinase mice, Acta1-cre mice. The cre recombinases was induced when the mice reached 12 weeks of age to avoid possible developmental effects of the lack of BSG. Western blot analysis confirmed that Bsg was ablated from skeletal muscle of iMSBsg−/− mice and that the cre-recombinase driven by Acta1 was not active in the cardiac muscle (Figure 5A). BSG, MCT1, and MCT4 were undetectable in lysates prepared from skeletal but not cardiac muscle.
Figure 5.

iMSBsg−/− Mice Do Not Show Exercise Intolerance
(A) Representative western blot analysis of BSG, MCT1, and MCT4 in skeletal muscles and cardiac muscle of iMSBsg−/− and iMSBsg+/+ mice after tamoxifen or vehicle treatment.
(B) Total distance reached during the treadmill test by iMSBsg−/− and iMSBsg+/+ at 6 months of age (n = 5 each group). Ex vivo EDL (C) twitch force, (D) tetanic force, and (E) fatigue traces (n = 3 each group). All the data are rapresented as mean and SEM.
See also Figure S2
Since tamoxifen can acutely affect cardiac muscle contractility (Asp et al., 2013), after the induction of the cre recombinase, we waited 3 weeks for the tamoxifen to be completely cleared from the system before testing the animals for exercise capacity. Using the forced treadmill test, we found that there was no significant difference in the total distance run by the iMSBsg−/− mice compared with controls (Figure 5B). We then tested whether the complete disruption of the lactate shuttle had an effect on the contractile properties of the muscles by performing ex vivo measurements of force and fatigability in isolated EDLs. As for the MCT4−/− muscles, we did not detect any difference in maximal isometric force (Figures 5C and 5D) and fatigability (Figure 5E) between iMSBsg−/− and control mice. Since the MCT4−/− presented a decrease in exercise capacity already at 3 months and we did not observe any difference between controls and iMBsg−/−, we did not investigate the performance of these animals at older ages. Overall, the studies on iMSBsg−/− mice confirmed that the absence of MCT1 and MCT4 in the sarcolemma of skeletal muscle does not affect exercise capacity and muscle ex vivo contractile properties.
Loss of MCT4 Leads to Degeneration of the NMJ and Decreased Compound Muscle Action Potential Amplitude
In many neuropathies (Hill, 2003), e.g., Charcot-Marie-Tooth disease (CMT) and Amyotrophic Lateral Sclerosis (ALS), a progressive decline in muscle strength and exercise capacity is caused by pathologies of the motoneuron, nerves, and/or the NMJ rather than a primary defect in the muscle. We therefore investigated whether ablation of MCT4 could impact the structure and function of the motor unit components upstream of muscle.
It was previously reported that MCT4 is expressed in astrocytes in the Central Nervous System (CNS), particularly in the cortex and hippocampus of both rats and mice. qPCR analysis of Mct4 (Slc16a3) mRNA in spinal cord samples demonstrated the presence of the transcript, although the levels were 100 times lower than in skeletal muscle (Figure 6A). To determine the cellular distribution of MCT4-expressing cells in the spinal cord, we performed fluorescence in situ hybridization (FISH) on frozen sections from the lumbar spinal cord of MCT4+/+ mice and co-labeled with nuclear staining and GFAP (Glial fibrillary acidic protein), a marker for glial cells. The in situ hybridization showed widespread expression of Mct4 in both the gray and white matter (Figure 6B). Since we detected Mct4 expression in the spinal cord, we sought to test whether the lack of MCT4 affected the motor unit, beginning with the motoneuron. We used cresyl violet staining in lumbar spinal cord cryosections to detect the Nissl substance in neurons (Figure 6C). We did not detect any difference in the spinal cord histomorphological features between genotypes, with normal boundaries between white and gray matter. Motor neurons were identified as Nissl-positive cells with a clearly visible nucleus and a soma larger than 100 μm and were counted in three separate sections of the lumbar spinal cord. The number of motoneurons was not different between MCT4−/− and MCT4+/+ (Figure 6D), suggesting that lack of MCT4 does not induce neuronal cell death.
Figure 6.

Lack of MCT4 Does Not Affect the Morphology of Motoneurons
(A) mRNA expression of Mct4 in spinal cord and skeletal muscle. Values are normalized to Rplp0 housekeeping gene.
(B) In situ hybridization of Mct4 mRNA (red) in lumbar spinal cord cryosections (10 μm thickness). Sections were co-labeled with nuclear DAPI staining (blue) and an antibody against GFAP (green). Scale bar, 200 μm; insert scale bar, 50 μm.
(C) Representative cresyl violet staining of lumbar spinal cord cryosections. Arrowheads in the enlarged insert indicate motoneurons in the ventral horn. Scale bar, 400 μm; insert scale bar, 200 μm.
(D) Quantification of the number of motoneurons in lumbar spinal cord sections (25μm) of MCT4+/+ and MCT4−/− mice (n = 3 animals each group, three sections per animal evaluated).
(E) Examples of toluidine blue staining of semithin sections of sciatic nerves (0.5 μm thickness). Scale bar, 200 μm.
(F) Morphometric analysis of axon diameter and g-ratio in MCT4−/− and MCT4+/+ sciatic nerves (n = 3 each group, entire section was counted). *p < 0.05. All the data are rapresented as mean and SEM.
We then performed a morphometric analysis of the sciatic nerve of 6-month-old MCT4+/+ and MCT4−/− mice (Figure 6E) to determine if the lack of MCT4 altered the number, size, and/or myelination of axons. Toluidine blue staining of semithin sciatic nerve sections revealed no differences in the density of myelinated axons; however, we found a small but significant increase in axons diameters in MCT4−/− samples, driven by the increased occurrence of large axons (Figure 6F). The g-ratio (a measure of myelin thickness) was similar between genotypes.
The synapse between the motoneurons and the skeletal muscle is called neuromuscular junction (NMJ). Alterations in the NMJ are associated with poor motor performance and exercise intolerance. We then sought to understand if the lack of MCT4 affects the integrity of the NMJ. We assessed the morphology of the NMJ in EDL and soleus muscles by performing whole-mount immunolabeling of the pre-synaptic axons (SMI32 ab) and vesicles (SV2), as well as of the post-synaptic nicotinic acetylcholine receptors (alpha-bungarotoxin) (Figure 7A). Although we did not detect any significant difference in NMJ morphology in muscles from 6-month-old mice, the incidence of disrupted NMJs in EDL of 12-month-old MCT4−/− mice was significantly higher (p < 0.0001, Chi-square test), with signs of denervation and re-innervation (Figure 7A). In 12-month-old MCT4+/+ muscles, only a minimum number of NMJs showed signs of partial denervation. Among the NMJs analyzed in MCT4−/− mice, 15% were partially or total denervated and 8% had terminal sprouting (Figures 7A and 7B). The analysis of NMJs in the soleus muscles showed no difference between MCT4−/− and MCT4+/+ mice (Figure S3A), suggesting that the effects of MCT4 ablation are more evident in fast motor units. Analysis of the density of the acetylcholine receptors (AChRs) at the post-synaptic endplates showed a similar result, with an increase in the fragmentation of the NMJs of the 12-month-old MCT4−/− mice compared with the MCT4+/+ (Figures S3B and S3C). No difference in the integrity of the NMJs was detected in the 6-month-old mice.
Figure 7.

NMJ Morphology and CMAPs Are Impaired in MCT4−/− Mice
(A) Representative images of whole-mount immunohistochemistry of EDL muscles labeling motor axons (SMI32, green), pre-synaptic nerve terminals (SV2, green) and post-synaptic endplate (rhodamine-conjugated α-bungarotoxin, red). I., intact; P.D., partially denervated; F.D., fully denervated; M.I., multiple innervated.
(B) Quantification of intact and disrupted NMJs in EDLs of 12-month-old MCT4−/− and MCT4+/+ (n = 3 each group, Chi-square, p < 0.0001).
(C) Representative CMAP trace showing the electromyographic signal recorded from gastrocnemius muscle (black trace) upon stimulation of the sciatic nerve (green trace). Amplitude and RMS are indicated.
(D) CMAPs RMS (Root-Mean-Square) recorded at 20 Hz in 6-month-old MCT4−/− and MCT4+/+ mice (n = 4 each group, Student t test, *p < 0.05). All the data are rapresented as mean and SEM.
See also Figure S3
To assess the functional activity of the motor unit, we performed in vivo compound muscle action potential (CMAP) studies on 6-month-old MCT4+/+ and MCT4−/− mice. We applied repeated sciatic nerve stimulations with increasing frequencies and recorded the evoked electromyographic (EMG) signal in the gastrocnemius muscle (Figure 7C). No difference was found in the amplitude decrement after 30 s of repetitive stimulations (data not shown). CMAP amplitude, however, expressed as root mean squared (RMS), was significantly lower in MCT4−/− at all frequencies tested (Figure 7D). Interestingly, loss of NMJ integrity and decreased CMAP amplitude have been described in patients with CMT (Elbracht et al., 2014) (Spaulding et al., 2016) (Cipriani et al., 2018). Together, these data suggest that ablation of MCT4 had a negative effect on the structural and functional stability of the motor unit.
Discussion
In the current study, we investigated whether disruption of the lactate shuttle had an effect on exercise capacity, by characterizing the phenotype of the MCT4−/− mouse. The results of our study showed that mice with a global knockout of MCT4 had defects in locomotion both when tested for spontaneous in-cage activity (wheel running) and forced treadmill exercise (Figure 2). The decrement in motor function was more evident in the spontaneous activity after the mice reached the adult age, suggesting that the phenotype is progressive, and exacerbated during forced endurance test (Figure 2C). By combining the results from the MCT4−/− mice with those obtained from the iMSBsg−/−, we can conclude that the deletion of lactate transporters from the skeletal muscles is not detrimental for the muscle itself. Instead, the exercise intolerance observed is accompanied by a functional and structural impairment of the neuronal component of the motor unit. Although we cannot exclude that lack of MCT4 in the CNS affects behavior, the integration of in vivo and ex vivo functional assessments with morphological analyses suggests that α motor neurons and NMJs are mostly affected.
For decades, lactate was considered the cause of fatigue in exercising muscles based on experiments conducted in the 1920s on non-oxygenated frog muscles (Hill et al., 1924). The lactate shuttle hypothesis was described over 30 years ago by Brooks (Brooks, 1986), who suggested that lactate is a substrate exchanged between glycolytic and oxidative tissues/cells, allowing for the control of its cytosolic concentration. During the past 20 years, researchers in the muscle and exercise physiology fields have worked to understand the real effects of lactate and H+ during muscle activity (Allen et al., 2008, Azevedo et al., 2007). It has now been established that lactate is not cause of fatigue (Posterino et al., 2001). The fate of the H+ ions, which are co-transported together with lactate during fatiguing muscle contraction (Kemp et al., 2006, Robergs et al., 2004, Robergs, 2017), however, is still not clear. In our study we showed that in ex vivo electrically stimulated MCT4−/− muscles, despite a 2-fold accumulation of intramuscular lactate, there was no decrement in force generation or fatigability (Figure 4), supporting the fact that increased lactate was not the cause of fatigue. We also tested whether addition of exogenous L-lactate in the bath during the fatigue test could alter muscle performance, but no difference was detected (data not shown).
We used Bsg floxed mice to indirectly knock out both transporters in adult muscle, with the goal of testing whether it would have a detrimental impact on exercise capacity. Mice ubiquitously lacking Bsg are reported to be sterile and with memory and learning deficits (Naruhashi et al., 1997), but their exercise capacity has not been investigated. To our surprise, there was no difference between controls and iMSBsg−/− in terms of running capacity or ex vivo contractility. This result is in contrast to the lactate shuttle hypothesis and more recent work, which suggest that lactate derived from skeletal muscle is a key metabolite utilized by different tissues as energy substrate (Hui et al., 2017). We cannot exclude that the lack of both transporters and their binding partner, BSG, may have caused a metabolic reprogramming in the skeletal muscle to limit lactate production and utilization. Taken together, the results from the two mouse models show that the systemic redistribution of muscle-derived lactate, operated by MCT1 and MCT4, can be bypassed during exercise.
Since its conception, the lactate shuttle theory has been applied to other systems such as cancer and neuroenergetics. Pellerin and Magistretti (Pellerin et al., 1998) presented the astrocytes-neurons lactate shuttle (ANLS) hypothesis where, in situations of high energy demand, astrocytes are a source of lactate and neurons are lactate consumers. Several studies have shown the expression of MCT4 in astrocytes in brain cortex and hippocampus (Bergersen et al., 2001) (Rafiki et al., 2003). In this study, we investigated the presence of MCT4 in the spinal cord and the potential role of lactate in supporting motor neuron activity. The in situ hybridization data (Figure 6B) showed that MCT4 is expressed by multiple cell types, but we did not confirm the expression in any particular cell type. Based on information available from the Allen Brain Atlas, as well as the topology of our in situ mRNA staining, we suggest that motoneurons are among the cells expressing MCT4. This is surprisingly new since previous reports indicated that MCT4 is expressed only in astrocytes. The neuronal MCT4 expression, in contrast with the ANLS hypothesis, is, however, supported by several publications suggesting that neurons rely on glycolytic metabolism for energy production, when stimulated (Gjedde and Marrett, 2001, Simpson et al., 2007) (Mangia et al., 2009) (Díaz-García et al., 2017). Future investigations will need to address if MCT4 and lactate have roles in the viability of the motor neuron and whether, as seen in several cancer studies (Marchiq et al., 2015), the blockade of lactate flux induces senescence and impairs neuronal signaling.
By performing CMAP measurements on MCT4−/− mice, we found a decrement in the amplitude of the EMG signal upon direct stimulation (Figure 7). Lower CMAP amplitude is a hallmark of axonal neuropathies such as CMT, multiple sclerosis (MS), and other pathologies of the NMJ. Despite the clear functional decline of the motor capacity (Figure 2) and the lower CMAPs amplitude in young adult mice, we did not detect any evident structural change in the sciatic nerve (Figure 6). This is consistent with the normal latency estimated during our CMAP experiments in both young adult and old mice (Manganelli et al., 2016) (Cornett et al., 2016).
Structural disruption of NMJs was evident in older mice (Figure 7), when the decline in spontaneous activity is also more pronounced. The results suggested that MCT4 and lactate may have an effect on the long-term integrity of the motor unit and NMJs. It is interesting to note that the degeneration of the NMJs was observed only in EDL muscles, which express MCT4 at the sarcolemma, and not in soleus muscles, which express mainly MCT1. Based on our results, we speculate that lactate released by the skeletal muscle may be necessary for the stability of the NMJs either functioning as metabolic substrate or as signaling molecule. A variety of factors as myokines and neurotrophic factors are important for the structural and functional integrity of the NMJ (Lin et al., 2008, Lin et al., 2001). Indeed, exercise has been shown to induce remodeling and adaptation of the NMJs (Deschenes et al., 2019), and lack or reduced muscle activity can have a detrimental effect on NMJ structure and function as seen during aging (Gonzalez-Freire, 2014).
It has been reported that MCT4 is expressed by Schwann cells of the peripheral nervous system (PNS) (Domènech-Estévez et al., 2015), where it might have a role in the maintenance of axon myelination. In our model, we did not detect the expression of MCT4 mRNA or protein in the PNS and we also did not observe any defect in axons myelination (Figure 6).
In conclusion, the results of our study show that the exercise intolerance observed in the MCT4−/− mice is not attributable to disruption of the lactate shuttle between fast-twitch and slow-twitch muscle fibers. Rather, in the MCT4−/− mice there is an instability of the motor unit and, in particular, a progressive degeneration of the NMJs. We propose that the lack of MCT4 from motoneurons decreases their activity possibly by altering the coordinate action of the metabolic pathways responsible for ATP production. This may in turn affect the release of neurotransmitters at the NMJs, hence the decreased amplitude of the CMAPs recorded. Therefore, the decreased overall activity of the MCT4−/− mice is associated with instability and premature degeneration of the NMJs. We suggest that the phenotype of MCT4−/− mice is reminiscent of neurodegenerative diseases such as Charcot-Marie-Tooth, Multiple Sclerosis, or ALS, and therefore it would be interesting to evaluate the role of MCT4 in these pathologies.
Limitations
In the current study we found that the lack of MCT4 negatively affects the exercise capacity of the mice but it had no impact on ex vivo muscle physiology. MCT4−/− mice did exhibit a decrease in amplitude of the CMAPs accompanied by structural changes at the levels of the NMJs. Although our data suggest that the exercise intolerance is due to instability of the motor unit, our approach did not have sufficient resolution to determine whether the functional impairment of the NMJs is a cause or a consequence of exercise intolerance. In addition, we could only detect MCT4 transcripts in the spinal cord since the immunostaining to demonstrate the presence of the protein was not reliable because was giving a high non-specific signal also in the MCT4−/− samples. We could not ascertain whether these effects resulted from lack of MCT4 expression in the motoneurons or astrocytes. In this study, owing to limited technology, we could not show whether the lack of MCT4 had a direct effect on neurotransmitter release from the pre-synapses or impaired activation of the motoneurons. Future studies using motoneurons and/or astrocytes MCT4 conditional knockout mice will help elucidate this important point.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the Bioimaging Shared Resource of the Sidney Kimmel Cancer Center (NCI 5 P30 CA-56036) and the Penn Muscle Institute Muscle Physiology Assessment Core for muscle contraction study. This work was primarily supported by the US National Institute of Health grants R01EY012042 to N.J.P. and R01NS079702 and R01NS110385 to A.C.L.
Author Contributions
S.B, E.L., and N.J.P. designed and developed project and wrote the manuscript. S.B. and E.L. performed experiments and analyzed data. M.C.W. performed NMJ analysis. R.A.N. provided Bsgflox/flox mice. A.C.L. and T.S.K. contributed with reagents and discussion. All the authors read and reviewed the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: December 20, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.11.041.
Contributor Information
Emanuele Loro, Email: eloro@pennmedicine.upenn.edu.
Nancy J. Philp, Email: nancy.philp@jefferson.edu.
Supplemental Information
References
- Adijanto J., Philp N.J. The SLC16A family of monocarboxylate transporters (MCTs)–physiology and function in cellular metabolism, pH homeostasis, and fluid transport. Curr. Top. Membr. 2012;70:275–311. doi: 10.1016/B978-0-12-394316-3.00009-0. [DOI] [PubMed] [Google Scholar]
- Allen D.G., Lamb G.D., Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Asp M.L., Martindale J.J., Metzger J.M. Direct, differential effects of tamoxifen, 4-hydroxytamoxifen, and raloxifene on cardiac myocyte contractility and calcium handling. PLoS One. 2013;8:e78768. doi: 10.1371/journal.pone.0078768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo J.L., Tietz E., Two-Feathers T., Paull J., Chapman K. Lactate, fructose and glucose oxidation profiles in sports drinks and the effect on exercise performance. PLoS One. 2007;2:e927. doi: 10.1371/journal.pone.0000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergersen L., Waerhaug O., Helm J., Thomas M., Laake P., Davies A.J., Wilson M.C., Halestrap A.P., Ottersen O.P. A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp. Brain Res. 2001;136:523–534. doi: 10.1007/s002210000600. [DOI] [PubMed] [Google Scholar]
- Bonen A. The expression of lactate transporters (MCT1 and MCT4) in heart and muscle. Eur. J. Appl. Physiol. 2001;86:6–11. doi: 10.1007/s004210100516. [DOI] [PubMed] [Google Scholar]
- Brooks G.A. The lactate shuttle during exercise and recovery. Med. Sci. Sports Exerc. 1986;18:360–368. doi: 10.1249/00005768-198606000-00019. [DOI] [PubMed] [Google Scholar]
- Cipriani S., Phan V., Médard J.-J., Horvath R., Lochmüller H., Chrast R., Roos A., Spendiff S. Neuromuscular junction changes in a mouse model of Charcot-Marie-Tooth disease type 4C. Int. J. Mol. Sci. 2018;19:4072. doi: 10.3390/ijms19124072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connett R.J., Honig C.R., Gayeski T.E., Brooks G.A. Defining hypoxia: a systems view of VO2, glycolysis, energetics, and intracellular PO2. J. Appl. Physiol. 1990;68:833–842. doi: 10.1152/jappl.1990.68.3.833. [DOI] [PubMed] [Google Scholar]
- Cornett K.M.D., Menezes M.P., Bray P., Halaki M., Shy R.R., Yum S.W., Estilow T., Moroni I., Foscan M., Pagliano E. Phenotypic variability of childhood Charcot-Marie-Tooth disease. JAMA Neurol. 2016;73:645–651. doi: 10.1001/jamaneurol.2016.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai K.H., Bernstein D. Exercise and oxygen consumption in the mouse. In: Hoit B.D., Walsh R.A., editors. Vol. 238. Springer; 2001. pp. 277–302. (Cardiovascular Physiology in the Genetically Engineered Mousee. Developments in Cardiovascular Medicine). [Google Scholar]
- Deschenes M.R., Tufts H.L., Noronha A.L., Li S. Both aging and exercise training alter the rate of recovery of neuromuscular performance of male soleus muscles. Biogerontology. 2019;20:213–223. doi: 10.1007/s10522-018-9788-y. [DOI] [PubMed] [Google Scholar]
- Dimmer K.S., Friedrich B., Lang F., Deitmer J.W., Bröer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000;350 Pt 1:219–227. [PMC free article] [PubMed] [Google Scholar]
- Díaz-García C.M., Mongeon R., Lahmann C., Koveal D., Zucker H., Yellen G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017;26:361–374.e4. doi: 10.1016/j.cmet.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domènech-Estévez E., Baloui H., Repond C., Rosafio K., Médard J.-J., Tricaud N., Pellerin L., Chrast R. Distribution of monocarboxylate transporters in the peripheral nervous system suggests putative roles in lactate shuttling and myelination. J. Neurosci. 2015;35:4151–4156. doi: 10.1523/JNEUROSCI.3534-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbracht M., Senderek J., Schara U., Nolte K., Klopstock T., Roos A., Reimann J., Zerres K., Weis J., Rudnik-Schöneborn S. Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot-Marie-Tooth disease type 2E. Clin. Neuropathol. 2014;33:335–343. doi: 10.5414/NP300742. [DOI] [PubMed] [Google Scholar]
- Gjedde A., Marrett S. Glycolysis in neurons, not astrocytes, delays oxidative metabolism of human visual cortex during sustained checkerboard stimulation in vivo. J. Cereb. Blood Flow Metab. 2001;21:1384–1392. doi: 10.1097/00004647-200112000-00002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Freire M. The neuromuscular junction: aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014;6:208. doi: 10.3389/fnagi.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A.V., Long C.N.H., Lupton H. Muscular exercise, lactic acid and the supply and utilisation of oxygen. Proc. R. Soc. B: Biol. Sci. 1924;97:155–176. [Google Scholar]
- Hill M. The neuromuscular junction disorders. J. Neurol. Neurosurg. Psychiatry. 2003;74(Suppl 2):ii32–ii37. doi: 10.1136/jnnp.74.suppl_2.ii32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S., Ghergurovich J.M., Morscher R.J., Jang C., Teng X., Lu W., Esparza L.A., Reya T., Le Zhan, Guo J.Y. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juel C. Lactate-proton cotransport in skeletal muscle. Physiol. Rev. 1997;77:321–358. doi: 10.1152/physrev.1997.77.2.321. [DOI] [PubMed] [Google Scholar]
- Kemp G., Böning D., Beneke R., Maassen N. Explaining pH change in exercising muscle: lactic acid, proton consumption, and buffering vs. strong ion difference. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;291:R235–R237. doi: 10.1152/ajpregu.00662.2005. [author reply R238–9] [DOI] [PubMed] [Google Scholar]
- Kirk P., Wilson M.C., Heddle C., Brown M.H., Barclay A.N., Halestrap A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19:3896–3904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Landmann L., Ruegg M.A., Brenner H.R. The role of nerve- versus muscle-derived factors in mammalian neuromuscular junction formation. J. Neurosci. 2008;28:3333–3340. doi: 10.1523/JNEUROSCI.5590-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Burgess R.W., Dominguez B., Pfaff S.L., Sanes J.R., Lee K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Manganelli F., Pisciotta C., Reilly M.M., Tozza S., Schenone A., Fabrizi G.M., Cavallaro T., Vita G., Padua L., Gemignani F. Nerve conduction velocity in CMT1A: what else can we tell? Eur. J. Neurol. 2016;23:1566–1571. doi: 10.1111/ene.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangia S., Simpson I.A., Vannucci S.J., Carruthers A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009;109(Suppl 1):55–62. doi: 10.1111/j.1471-4159.2009.06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiq I., Le Floch R., Roux D., Simon M.-P., Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75:171–180. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- Naruhashi K., Kadomatsu K., Igakura T., Fan Q.-W., Kuno N., Muramatsu H., Miyauchi T., Hasegawa T., Itoh A., Muramatsu T. Abnormalities of sensory and memory functions in mice LackingBsgGene. Biochem. Biophys. Res. Commun. 1997;236:733–737. doi: 10.1006/bbrc.1997.6993. [DOI] [PubMed] [Google Scholar]
- Pellerin L., Pellegri G., Bittar P.G., Charnay Y., Bouras C., Martin J.L., Stella N., Magistretti P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Philp N.J., Ochrietor J.D., Rudoy C., Muramatsu T., Linser P.J. Loss of MCT1, MCT3, and MCT4 expression in the retinal pigment epithelium and neural retina of the 5A11/basigin-null mouse. Invest. Ophthalmol. Vis. Sci. 2003;44:1305–1311. doi: 10.1167/iovs.02-0552. [DOI] [PubMed] [Google Scholar]
- Poole R.C., Halestrap A.P. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. 1993;264:C761–C782. doi: 10.1152/ajpcell.1993.264.4.C761. [DOI] [PubMed] [Google Scholar]
- Posterino G., Dutka T., Lamb G. L (+)-lactate does not affect twitch and tetanic responses in mechanically skinned mammalian muscle fibres. Pflugers Arch. 2001;442:197–203. doi: 10.1007/s004240100528. [DOI] [PubMed] [Google Scholar]
- Rafiki A., Boulland J.L., Halestrap A.P., Ottersen O.P., Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 2003;122:677–688. doi: 10.1016/j.neuroscience.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Robergs R.A., Ghiasvand F., Parker D. Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R502–R516. doi: 10.1152/ajpregu.00114.2004. [DOI] [PubMed] [Google Scholar]
- Robergs R.A. Competitive cation binding computations of proton balance for reactions of the phosphagen and glycolytic energy systems within skeletal muscle. PLoS One. 2017;12:e0189822. doi: 10.1371/journal.pone.0189822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson I.A., Carruthers A., Vannucci S.J. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007;27:1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding E.L., Sleigh J.N., Morelli K.H., Pinter M.J., Burgess R.W., Seburn K.L. Synaptic deficits at neuromuscular junctions in two mouse models of charcot-marie-tooth type 2d. J. Neurosci. 2016;36:3254–3267. doi: 10.1523/JNEUROSCI.1762-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.C., Jackson V.N., Heddle C., Price N.T., Pilegaard H., Juel C., Bonen A., Montgomery I., Hutter O.F., Halestrap A.P. Lactic acid efflux from white skeletal muscle is catalyzed by the monocarboxylate transporter isoform MCT3. J. Biol. Chem. 1998;273:15920–15926. doi: 10.1074/jbc.273.26.15920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.