

Abstract
Hydrogen sulfide (H2S) is an endogenous gaseous transmitter synthesized in various cell types. It is well established that H2S functions in many physiological processes, including the relaxation of vascular smooth muscle, mediation of neurotransmission, regulation of inflammation, and modulation of insulin signaling. In recent years, it has been revealed that polysulfides, substances with a varying number of sulfur atoms (H2Sn), are generated endogenously from H2S in the presence of oxygen. A series of studies describes that sulfane sulfur has the unique ability to bind reversibly to other sulfur atoms to form hydropersulfides and polysulfides, and that polysulfides activate ion channels and promote calcium influx. Furthermore, polysulfides regulate tumor suppressor activity, promote the activation of transcription factors targeting antioxidant genes and regulate blood pressure by vascular smooth muscle relaxation. Insulin secretion from pancreatic β cells plays a critical role in response to increased blood glucose concentration. H2S has emerged as an important regulator of glycemic control and exhibits characteristic regulation of glucose homeostasis. However, the effects of polysulfides on glucose-stimulated insulin secretion (GSIS) are largely unknown. In this study, we demonstrated that pharmacological polysulfide salts including Na2S2, Na2S3, and Na2S4 considerably inhibit GSIS in mouse and rat pancreatic β-cell-derived MIN6 and INS-1 cell lines, and that the effect is dependent on the activation of ATP-sensitive potassium channels. In addition, we demonstrated that a mixture of Na2S and diethylamine NONOate inhibits GSIS in a similar way to the pharmacological administration of polysulfide salts.
Subject terms: Mechanisms of disease, Diabetes complications
Introduction
Hydrogen sulfide (H2S) is synthesized endogenously in various cell types, and functions as an endogenous gaseous transmitter. H2S function is well established in many physiological processes, including the relaxation of vascular smooth muscle, mediation of neurotransmission, regulation of inflammation, and modulation of insulin signaling1–3. In recent years, it has been revealed that polysulfides, substances with a varying number of sulfur atoms (H2Sn), are generated from H2S in the presence of oxygen1,4,5 and by 3-mercaptopyruvate sulfurtransferase (3MTS)6. In addition, polysulfides are also generated by the interaction between H2S and nitric oxide (NO)7. Polysulfides contain sulfane sulfur, which is present in various proteins and functions as a potential intracellular H2S store that releases H2S under reducing conditions. A series of studies describe the unique ability of sulfane sulfur to bind reversibly to other sulfur atoms to form hydropersulfides (R-S-SH) and polysulfides (-S-Sn-S-)1. Polysulfides have been shown to activate ion channels and promote calcium influx, as well as to regulate tumor suppressor activity, promote the activation of transcription factors targeting antioxidant genes, and modulate blood pressure by vascular smooth muscle relaxation8. H2S and sulfane sulfur constitutively coexist, and recent work suggests that sulfane sulfur species may act as signaling molecules in at least some biological processes9. It is also clear that many of the effects of H2S are mediated through reactions with cysteine sulfur on regulatory proteins and most of these are not mediated directly by H2S but require prior oxidation of H2S and the formation of H2Sn10.
H2S has also emerged as an important molecule in glycemic control, exhibiting characteristic regulation of glucose homeostasis. Specifically, H2S stimulates glucose production via activation of gluconeogenesis and glycogenolysis in hepatocytes, yet inhibits lipolysis in adipocytes and insulin secretion from pancreatic β cells11–13. Currently, however, the effects of polysulfides on glucose-stimulated insulin secretion (GSIS) are largely unknown.
In this study, we investigated the effect of pharmacological polysulfide salts including sodium disulfide (Na2S2), sodium trisulfide (Na2S3), and sodium tetrasulfide (Na2S4) on insulin secretion under low and high glucose conditions using cell biological and electrophysiological methods. We examined the effects of polysulfides at basal conditions and during GSIS and demonstrated that polysulfides activate ATP-sensitive potassium (KATP) channels, suppressing GSIS in mouse insulinoma 6 (MIN6) cells, rat insulinoma 1 (INS-1) cells, and mouse pancreatic β-cells/islets.
Results
Polysulfide salts inhibit glucose-stimulated insulin secretion in MIN6 cells
First, insulin secretion in response to changes in extracellular glucose concentration was examined in MIN6 cells. Cells were pre-cultured under 2 mM glucose conditions for 0.5 h. Then, the cells were exposed to a range of glucose concentrations (5, 10, and 20 mM) for 1 h, and the concentration of insulin in the culture media was assayed (Supplementary Fig. 1a). Next, the time-dependent GSIS profile was investigated. To do this, cells were exposed to 20 mM glucose and insulin concentration was assayed at 0, 20, 40, 60, and 120 min (Supplementary Fig. 1b). Glucose-stimulated insulin secretion was clearly observed in MIN6 cells.
Next, the effect of the polysulfide salt Na2S4 on GSIS was investigated. MIN6 cells were exposed to 1.5–50 µM Na2S4 for 4 h under 2 mM glucose conditions and then the insulin concentration in the culture media was assayed. Na2S4 at concentrations exceeding 3.1 µM significantly inhibited basal insulin secretion in MIN6 cells under 2 mM glucose conditions (Fig. 1a). MIN6 cells were then exposed to Na2S4 for 4 h under 2 mM glucose conditions and then 20 mM glucose conditions for 1 h. Here, Na2S4 concentrations exceeding 1.5 µM significantly inhibited GSIS in MIN6 cells (Fig. 1b). The insulin secreted in response to the polysaccharides sucrose and maltose was also assessed. Only glucose elicited insulin secretion, indicating that other polysaccharides do not largely impact the secretion of insulin (Fig. 1c). Subsequently, the effect of changing the Na2S4 incubation time was investigated. MIN6 cells were preincubated with Na2S4 for 1 h or 4 h and GSIS was measured. No significant differences were detected, but only under 25 µM (Fig. 1d). Finally, the reversibility of the inhibitory effect of Na2S4 was investigated. MIN6 cells were either exposed to Na2S4 for 4 h and then to 20 mM glucose, or incubated for 6 h without Na2S4 and then exposed to 20 mM glucose. The inhibitory effect of Na2S4 was completely recovered after 6 h of incubation (Fig. 1e).
Figure 1.

Dose- and time-dependent effects of polysulfide salt, sodium tetrasulfide (Na2S4) on glucose-stimulated insulin secretion in MIN6 cells. (a,b) Mouse MIN6 cells were exposed to Na2S4 (0, 1.5, 3.1, 6.2, 12.5, 25 and 50 µM) for 4 h under 2 mM glucose conditions, and insulin secretion was determined in the presence of 2 mM (a) or 20 mM glucose (b) (n = 5). (c) MIN6 cells were treated with 20 mM glucose (glu), sucrose (suc) or maltose (mal) for 1 h (n = 8). (d) MIN6 cells were exposed to Na2S4 (0, 1.5, 3.1, 6.2, 25, and 50 µM) for 1 h or 4 h and then insulin secretion was determined under 20 mM glucose conditions. (n = 5) (e) MIN6 cells were exposed to Na2S4 for 4 h and then to 20 mM glucose or incubated for 6 h without Na2S4 and then insulin secretion was determined in 20 mM glucose conditions (n = 3). Data are presented as mean ± SD. Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared with the control: (Na2S4 0 µM: a and b). *P < 0.05 for comparison of the indicated groups (c, d, and e).
The effect of other sulfane sulfur donors on GSIS, including Na2S2 and Na2S3, was also examined in MIN6 cells. Na2S2 or Na2S3 both inhibited GSIS within 4 h of their addition to the media (Fig. 2a). Remarkably, the inhibitory effect of Na2S2 (IC50 > 250 µM) was weaker than that of Na2S3 (IC50 = 39.9 µM) and the effect of Na2S3 was weaker than that of Na2S4 (IC50 = 9.5 µM). Finally, the effect of H2S was investigated. In contrast to sulfane sulfur donors, Na2S did not significantly affect GSIS at any concentration up to 50 µM (Fig. 2a).
Figure 2.

Effects of hydrogen sulfide donor and polysulfide salts on glucose-stimulated insulin secretion in MIN6 cells and its dependency of S-S bonds. (a) Mouse MIN6 cells were exposed to Na2S, Na2S2, or Na2S3 (0, 6.2,12.5, 25 and 50 µM) for 1 h in the presence of 2 mM glucose and then insulin secretion was determined in 20 mM glucose. (n = 3) (b) MIN6 cells were exposed to Na2S or DEA/NO in 2 mM or 20 mM glucose. (n = 3) (c) MIN6 cells were exposed to Na2S or Na2S4 in 20 mM glucose. (n = 3) (d,e) MIN6 cells were exposed to 50 µM Na2S or 50 µM Na2S4 with 250 µM tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (d) or Na2S or Na2S4 treated by immobilized-TCEP (iTCEP). (n = 3) (e) for 1 hour and then insulin secretion was determined under 20 mM glucose conditions. (n = 3) Data are presented as mean ± SD (n = 3). Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared with the control (0 µM Na2S4: 20 mM or 10 mM glucose)
The inhibition of GSIS by polysulfides generated by the interaction of H2S with NO
The oxidation of H2S generates H2Sn, and the interaction of H2S with S-nitroso cysteine generates cysteine persulfide. In fact, the interaction of H2S with NO directly produces H2Sn7. Here, MIN6 cells were exposed to a mixture of Na2S, a sodium salt of sulfide, and diethylamine NONOate (DEA/NO), a donor of NO that generates polysulfides. Neither 50 µM Na2S nor 50 µM DEA/NO inhibited GSIS in MIN6 cells (Fig. 2b). On the other hand, a mixture of 50 µM Na2S and 50 µM DEA/NO significantly inhibited GSIS, as did 12.5 µM Na2S4 (Fig. 2b). We investigated the synergistic role of polysulfides (Na2S4) and H2S (Na2S) on GSIS, and determined that the combination of Na2S4 and Na2S resulted in an additive effect (Fig. 2c).
Pretreatment with reducing agent abolishes the inhibitory effect of Na2S4
In this study, we have shown that the potency of inhibition is correlated with the number of disulfide bonds, the effect of a reducing agent (tris (2-carboxyethyl) phosphine hydrochloride (TCEP)) that cleaves disulfide bonds was assessed. First, TCEP was added directly into the culture media, and measured insulin levels remained very low (Fig. 2d). However, we also found that TCEP abolishes the immunogenicity of insulin by cleaving the S-S bond in insulin (Supplementary Fig. 2). Therefore, Na2S4 was pretreated with immobilized-TCEP (iTCEP) and then administered to cells. The pretreated Na2S4 did not affect the immunogenicity of insulin (Supplementary Fig. 2) but displayed reduced inhibitory potency in terms of GSIS (Fig. 2e). These results indicate that disulfide bonds in polysulfides are essential for their inhibitory effects on GSIS.
Polysulfide salts inhibit GSIS in INS-1 cells and mouse pancreatic β-cells/islets
To investigate whether the suppressive effect of polysulfides can be observed in other cell types, we used the rat insulinoma INS-1 cell line and mouse β-cells/islets. The effect of Na2S4 on GSIS was investigated in INS-1 cells. As in the case of MIN6 cells, Na2S4 inhibited GSIS in a dose-dependent manner (Fig. 3a,b). Na2S2 and Na2S3 also inhibited GSIS (Fig. 3c). β-cells/islets were incubated with 50 µM of Na2S4 in 2 mM glucose from 30 min to 1 h, and the insulin concentration was assessed. The insulin concentration of β-cells/islets, were compensated with total protein weight. As with MIN6 and INS-1 cells, 50 µM Na2S4 significantly suppressed IS after incubation for 4 h (Fig. 3d). When cells were exposed to 20 mM glucose, 50 µM Na2S4 significantly suppressed GSIS after 1 h of incubation. Thus, the inhibitory effect of polysulfides on GSIS is not only observed in mouse MIN6 cells, but also in rat INS-1 cells and β-cells/islets, suggesting that this phenomenon may be universal.
Figure 3.

Dose- and time-dependent effects of polysulfide salts on glucose-stimulated insulin secretion in INS-1 cells and mouse pancreatic β-cells/islets. (a,b) Rat insulinoma INS-1 cells were exposed to Na2S4 (0, 0.05, 0.5, 5, or 50 µM) for 1 h in the presence of 2 mM glucose and then insulin secretion was determined under 2 mM (a) or 10 mM (b) glucose conditions. (n = 3) (c) INS-1 cells were exposed to 50 µM Na2S, Na2S2, Na2S3, or Na2S4 for 1 h in 2 mM glucose and then insulin secretion was determined under 20 mM glucose conditions (n = 3). (d) Mouse pancreatic β-cells/islets were exposed to 50 µM Na2S4 for 1 h in 2 mM or 20 mM glucose (n = 3). In the case of β-cells/islets insulin concentrations were compensated with total protein weight. Data are presented as mean ± SD (n = 3). Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared with the control (Na2S3 or Na2S4 0 µM).
Effect of polysulfides on death of MIN6 cells
Cell death was assayed by flow cytometry and the trypan blue exclusion method. 4 mM lidocaine treatment induced cell death, as detected by propidium iodide (PI) or Annexin V staining. However, neither 50 µM nor 100 µM Na2S4 induced cell death within 1 h (Fig. 4a). In the case of the flow cytometry assessment method, neither 50 µM nor 100 µM Na2S4 induced cell death within 1 h, whereas lidocaine induced widespread cell death (Fig. 4b, Supplementary Fig. 3).
Figure 4.

Impact of Na2S4 on death of mouse MIN6 cells. (a,b) MIN6 cells were exposed to Na2S4 at doses from 0 µM to 100 µM and to lidocaine as positive control at 10 mM and cultured for periods ranging from 0 h to 4 h prior to cell viability evaluation. Cell death was also assayed by flow cytometry (a) and trypan blue exclusion method (b). Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared to the control cell population (Na2S4 ;0 µM, lidocaine; 0 mM treatment).
Our findings show that polysulfides at 50 µM did not cause MIN6 cell death and indicate that the observed suppression of GSIS is not due to cell toxicity caused by polysulfides.
Effects of polysulfide salts on cellular energy metabolism in MIN6 cells
Intracellular ATP concentration plays a crucial role in GSIS. High extracellular glucose increases ATP concentration in pancreatic β-cells14,15. Here, in agreement with previous studies, we observed an Na2S4 dependent effect on intracellular ATP concentaration upon exposure to 2 mM or 20 mM glucose at the timepoints 1 and 4 h. 20 mM glucose increased ATP concentration compared to 2 mM glucose. However, 25 µM Na2S4 did not affect ATP concentration at all (at all timepoints) in cells treated with 20 mM glucose (Fig. 5a).
Figure 5.

Effects of Na2S4 on cellular energy metabolism. (a) Mouse MIN6 cells were cultured from 1 h to 4 h with 25 µM Na2S4 prior to determination of the cellular ATP level (n = 3) under 2 mM or 20 mM glucose conditions. (b,c) Mouse MIN6 cells were exposed to Na2S4 (0, 1.5, 3.1, 6.2, 12.5, 25 and 50 µM) for a period of 4 h, followed by oxygen consumption rate (OCR) assay (b) and extracellular acidification rate (ECAR) assay (c) (n = 9). Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared to the control cell population (Na2S4 0 µM, glucose 20 mM). *P < 0.05 for comparison of the indicated groups.
It has been reported that glucose increases oxygen consumption in mitochondria and stimulates oxidative phosphorylation (OXPHOS) in mitochondria16,17. We therefore investigated the effect of Na2S4 on oxygen metabolism in MIN6 cells (Supplementary Fig. 4a). High-glucose stimulation activated mitochondrial respiration in MIN6 cells (Supplementary Fig. 4b). The oxygen consumption rate (OCR) was increased in 20 mM glucose conditions (Supplementary Fig. 4b). The extracellular acidification rate (ECAR) also increased in 20 mM glucose conditions (Supplementary Fig. 4c). A concentration of 1.5–25 µM Na2S4 did not affect OCR or ECAR under 20 mM glucose conditions within the 4 h measurement period (Fig. 5b,c). However, 50 µM Na2S4 suppressed OCR in response to exposure to high glucose. Together, these results show that polysulfides applied to cells at concentrations up to 25 µM did not affect cellular energy or oxygen metabolism.
Effects of polysulfides on insulin secretion induced by glibenclamide
There is a consensus that the elevation of intracellular ATP/ADP ratio in response to high glucose conditions results in the closure of ATP-sensitive potassium (KATP) channels, and depolarizes the plasma membrane18. Glibenclamide, a drug that acts as a channel closer, has been reported to facilitate insulin secretion in pancreatic β-cells even under low-glucose conditions, by binding to and inhibiting the KATP channel inhibitory regulatory subunit sulfonylurea receptor 1 (SUR1)14. The effects of Na2S3 and Na2S4 treatment on glibenclamide-induced insulin secretion were examined in MIN6 cells. Here, we found that even upon addition of 2 mM glucose, 10 µM glibenclamide elicited insulin secretion. Such secretion was then suppressed by treatment with more than 6.2 µM Na2S4 (Fig. 6a). Under 20 mM glucose conditions, Na2S4 at concentrations exceeding 6.2 µM suppressed glibenclamide-induced insulin secretion (Fig. 6a). As was the case for Na2S4, high concentrations (>25 µM) of Na2S3 did suppress glibenclamide-induced insulin secretion in a dose-dependent manner in cells treated with 2 mM glucose conditions (Fig. 6b). In addition, we examined the effect of polysulfide salt on Ca2+-triggered insulin secretion by stimulation with 60 mM K+. Stimulation with 60 mM K+ induced insulin secretion. Insulin secretion was suppressed with 50 µM Na2S4, but not with 25 µM Na2S4, which suppressed GSIS (Fig. 6c). These results suggest that polysulfide donors affect cellular processes distal to KATP channels.
Figure 6.
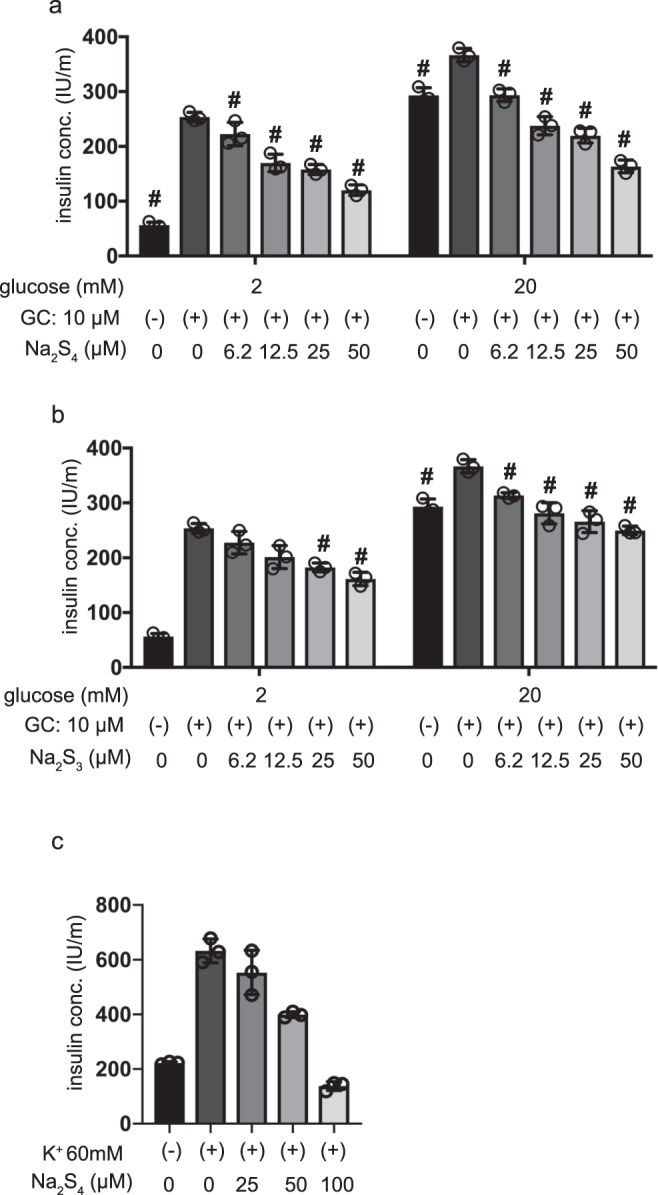
Effects of polysulfide salts on insulin secretion induced by glibenclamide. (a,b) Mouse MIN6 cells were exposed to Na2S4 (a) or Na2S3 (b) (0, 6.2, 12.5, 25, or 50 µM) for 4 h without or with glibenclamide (10 µM) under 2 mM or 20 mM glucose conditions (n = 3). (c) MIN6 cells were exposed to 60 mM K+ with Na2S4 (n = 3). Insulin secretion was determined as described in Materials and Methods. Data are presented as the mean ± SD (n = 4). Differences between treatments were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons; #P < 0.05, as compared with the control (glucose = 2 mM, without glibenclamide or without diazoxide treatment or without high K+ treatment).
Effects of Na2S4 on the membrane potential of MIN6 cells
The expression of Glut2 (Slc2a2), Cav1.2 (Cacna1c), Kv2.1 (Kcnb1), Kv2.2 (Kcnb2), Kir6.2 (Kcnj11), SUR1 (Abcc8), insulin (Ins1), and 18 S ribosomal RNA (Rn18s) was examined by RT-PCR (Supplementary Fig. 5). Glut2, Cav1.2, Kv2.1, Kv2.2, Kir6.2, SUR1, or insulin expressions were not affected within 4 h of glucose or Na2S4 treatment (Supplementary Fig. 5). Results from qRT-PCR indicated that the expression of KATP channel interacting intermediates Kir6.2 and SUR1 was not affected by Na2S4 treatment (Fig. 7a). Kir6.2 expression increased in response to exposure to 20 mM glucose, but was not affected by Na2S4 treatment (Fig. 7a). These results demonstrate that the expression of molecules which play critical roles in glucose uptake or membrane depolarization was not affected by Na2S4.
Figure 7.

Effects of Na2S4 on the membrane potential of mouse MIN6 cells. (a) Mouse MIN6 cells were exposed to 50 µM Na2S with 20 mM glucose and harvested. Then the mRNA levels of Kir6.2, and SUR1 were assayed by qRT-PCR. Data are presented as mean ± SD (n = 3). #P < 0.05, as compared with the control (Na2S4 0 µM). (b) Na2S4 (50 μM) induced hyperpolarization of membrane potential with gramicidin-perforated patch. Glibenclamide (Glb; 100 μM) induced depolarization. These traces are representative of six glucose-responsive cells. (c) Representative current–voltage relationships for the whole-cell currents with gramicidin-perforated patch. The current was elicited by a voltage ramp from −123 to −43 mV with a rate of 0.2 V/s. Na2S4 (50 μM) increased the current in the KRBH buffer contained 10 mM glucose (C; control). Thereafter, the current induced by Na2S4 decreased by glibenclamide (Glb, 10 μM). (d) Summary of the effects of Na2S4 and glibenclamide on the conductance (n = 9). *P < 0.05 for comparison of the indicated groups.
To identify the ion channels affected by Na2S4, we measured the membrane potential of MIN6 cells using the gramicidin-perforated patch technique (Fig. 7b). Na2S4 (50 µM) hyperpolarized the membrane potential induced by 10 mM glucose (n = 6). Washing out of Na2S4 (50 µM) promptly restored the membrane potential depolarization induced by 10 mM glucose, to its original level. Following perfusion with standard 2 mM glucose solution, the firing of action potentials induced by 10 mM glucose disappeared. Glibenclamide (100 µM) addition also induced the firing of action potentials. These results indicated that Na2S4 activated KATP channels reversibly.
We measured whole-cell currents with the gramicidin-perforated patch technique and specifically measured KATP currents in MIN6 cells bathed in a 10 mM glucose solution. Na2S4 at 50 µM facilitated inwardly rectifying K+ currents (n = 9, Fig. 7c). Measured conductance was 0.11 ± 0.22 nS/pF in 10 mM glucose solution (Fig. 7d). Na2S4 significantly increased the conductance to 0.27 ± 0.08 nS/pF. The addition of 10 µM glibenclamide significantly decreased the conductance to 0.17 ± 0.06 nS/pF (n = 9, Fig. 7d).
Na2S4 facilitated the inwardly rectifying K+ conductance in a dose-dependent manner with an intracellularly standard K+ pipette solution that included 0.01 mM ATP, measured with the whole-cell patch-clamp configuration (Fig. 8a). The half maximal effective concentration (EC50) of Na2S4 was estimated at 16.7 ± 3.1 μM with a Hill coefficient of 5.7 ± 1.7 (n = 5, Fig. 8b). However, the effect of Na2S4 was suppressed with a standard K+ solution containing 1 mM ATP (n = 6, Fig. 8b). Stromatoxin-1 (100 nM, STx), an inhibitor of Kv 2.1 channels, did not decrease the conductance (n = 3, Supplementary Fig. 6). These results together suggest that Na2S4 affects KATP channels in MIN6 cells.
Figure 8.

Effect of Na2S4 on voltage-dependent outward potassium currents in mouse MIN6 cells. (a) Current–voltage relationships of whole-cell currents with varying concentrations of Na2S4 (in 0, 5, 25, 50, or 500 μM). Na2S4 increased inwardly rectifying potassium currents in a dose-dependent manner (n = 5). (b) Semi-logarithmic plot of the conductance between −108 and −68 mV by concentration of Na2S4 with the standard K+ pipette solution included ATP at 0.01 mM (closed circles) and 1 mM (open circles).
Discussion
In this study, we demonstrated that pharmacological polysulfide salts including Na2S2, Na2S3, and Na2S4 significantly inhibited GSIS in mouse and rat pancreatic β-cell-derived MIN6 and INS-1 cell lines, and that the effect is dependent on activation of the KATP channel. Moreover, a mixture of Na2S and diethylamine NONOate (DEA/NO), which generates polysulfides in cells, also had comparative suppressive effects on GSIS.
Polysulfides, a mixture of substances with varying numbers of sulfurs (H2Sn) is endogenously generated from H2S in the presence of oxygen6,7,19,20. Polysulfides contain sulfane sulfur, which is maintained in various proteins as a potential intracellular H2S store, which releases H2S under reduced conditions. It has also been reported that polysulfides are enzymatically biosynthesized by reacting with cysteine19. A mixture of Na2S (a sodium salt of sulfide) and diethylamine NONOate (DEA/NO, a NO donor), has been reported to generate H2S2 and H2S37,21,22. In fact, we demonstrated here that Na2S and DEA/NO efficiently inhibited GSIS in MIN6 cells.
Polysulfides, and not H2S, have previously been reported to be entities that bind within sulfhydrate or sulfurate proteins19,20,23. Polysulfides regulate the function of proteins by modulating cysteine residues in target protein to form -SSH moieties, thereby causing structural change of the protein1. In this study, we demonstrated that in fact polysulfides suppressed GSIS far more efficiently than Na2S. Na2S at a concentration of 50 µM inhibited GSIS but the potency of the inhibition was reduced compared to polysulfides. Notably the potency of the inhibition after treatment with 1.5 µM Na2S4 was stronger than 50 µM Na2S. In fact, the IC50 of Na2S4 was calculated to be 9.5 µM. However, Na2S in the presence of an NO donor results in an inhibitory effect similar to the use of polysulfides4. Another interesting finding is that the suppressive effect of a polysulfide is related to the number of disulfide bonds present. The suppressive effect of Na2S4 is stronger than that of Na2S3 and Na2S4. In addition, TCEP treatment counteracted the suppressive effect observed in response to polysulfides24.
These factors suggest that polysulfide acts as an intermediate species during H2S signaling. The present study demonstrates that polysulfides are more potent GSIS suppressors than H2S. Polysulfides are known to promote protein sulfhydration more efficiently than H2S25.
It has also been reported that H2S acts as a regulator of O2 consumption of mitochondria and intracellular O2 metabolism in mammalian cells. In HeLa cells, 100 µM H2S was shown to significantly suppress oxygen consumption and affect intracellular ATP concentration26. Contrary to this, our experimental results indicate that even 50 µM Na2S4 failed to significantly influence oxygen consumption or intracellular ATP concentration in MIN6 cells.
Treatment with 50 µM Na2S4 did not affect the expression of GLUT2, Cav1.2, Kir6.2, or SUR1, nor did it affect cell growth and death. ATP content, OCR, or ECAR, a surrogate marker of glycolysis, was not significantly affected by Na2S4. The evidence strongly suggests that glucose intake was not affected by polysulfides. In addition, polysulfide treatment did not affect glibenclamide-induced insulin secretion. Taken together, the evidence prompted us to focus on KATP channels. The KATP channel is a potassium channel composed of Kir6.x: Kir6.1 and Kir6.2 (a K+ channel member belonging to the ‘inward rectifier’ subclass) and sulfonylurea receptor (SUR; SUR1, SUR2A, and SUR2B) subunits27. It is sensitive to adenosine triphosphate (ATP), and various subtypes of KATP channel are expressed in a wide range of tissues28,29. In pancreatic β cells, including MIN6 cells and INS-1 cells, KATP channels are comprised of SUR1 and Kir6.228. Mutations of SUR1 or Kir6.2 genes are known to cause familial hypoglycemia associated with unregulated insulin secretion14,15,30. It is reported that H2S stimulates KATP channels in vascular smooth muscle cells and that H2S also functions as an endogenous opener of KATP channels in INS-1E cells. A series of studies has indicated that H2S stimulates a variety of ion channels such as TRPA1, TRPV1, T-type Ca2+ channels, and KATP channels2,3,7,31–33. The evidence strongly suggests therefore that polysulfides would also affects these ion channels.
In line with this logic, we demonstrated the polysulfides activate KATP channels in MIN6 cells derived from mouse insulinoma. First, Na2S4 reversibly hyperpolarized the membrane potential after induction by 10 mM glucose. Second, Na2S4 facilitated conductance of inwardly rectifying K+ currents in a dose-dependent manner upon addition of a standard K+ pipette solution that included ATP. Another series of structural studies indicated that Kir6.x subunits sense changes in intracellular ATP concentration34,35. In this study, we demonstrated that Na2S4 facilitated the inwardly rectifying K+ conductance in a dose-dependent manner with an intracellularly standard K+ pipette solution with 0.01 mM ATP. However, the effect of Na2S4 was suppressed with a K+ pipette solution comprising 1 mM ATP. Conversely, we also demonstrated that 10 µM of glibenclamide significantly decreased inwardly rectifying K+ currents elicited by polysulfides. Taking into account existing evidence that the direct target of glibenclamide is SUR, it is reasonable to suggest that polysulfides affect Kir6.2, SUR1, or the interaction between them.
H2S is sequentially oxidized to form polysulfides with a varying number of sulfur atoms, until the number of sulfur atoms reaches eight. At that point, sulfur molecules cyclize and separate from polysulfides. Polysulfides also sulfurate cysteine residues in TRPA1 channels to modify their activity7,31. Polysulfides sulfurate Cys422 and Cys622 in TRPA1 channels to activate them, and it is reported that vascular KATP channels are inhibited during oxidative stress caused by S-glutathionylation; the Kir6.1 subunit specifically has been reported to be responsible for oxidant sensitivity in these channels2,36. Systematic mutational analysis revealed three cysteine residues in the N terminal and transmembrane domains of Kir6.1: namely Cys43, Cys120, and Cys176. Among them, Cys176 contributed to >80% of the oxidant sensitivity of the protein36. In contrast to Cys176, Cys43 exhibited only a modest contribution to S-glutathionylation, and Cys120 was modulated by extracellular oxidants but not by intracellular glutathione disulfide36. In Kir6.2 channels, Cys166 (corresponding to Cys176 in Kir6.1) is suggested to be involved in intrinsic channel gating. As in the case of Kir6.1, several cysteine residues are well conserved between species (Supplementary Fig. 8). In addition to Kir6.x, SUR1 also have well-conserved cysteine residues (Supplementary Fig. 7). Notably, the inhibitory effects of polysulfides on GSIS were demonstrated to be reversible in this study. In addition, Na2S4 activated KATP channels in MIN6 cells reversibly. Together, the evidence presented here suggests that polysulfides modulate reactive cysteine residues.
There is a limitation of this study. Because we focused on the mechanism of the suppression of insulin secretion by pharmacological polysulfides in vitro, we did not perform in vivo experiments. In vivo experiments using mice may warrant the impact of polysulfides on systemic insulin secretion and glucose metabolism.
Materials and Methods
Cell culture
Mouse insulinoma MIN6 cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) containing 450 mg/dl glucose. Rat INS-1 cells were cultured in RPMI1640 (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 50 μM β-mercaptoethanol, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Culture conditions used replicated those reported in the literature for these cells37,38.
Reagents
Details of reagents used in this study are described in Table S1.
Isolation of mouse pancreatic islets
Male C57BL/6JJcl mice (8–10 weeks old, n = 8) were sacrificed by cervical dislocation in accordance with protocols approved by the Animal Experimentation Committee, Kansai Medical University (#19–088). Pancreatic islets were isolated from the pancreas by enzymatic digestion of the tissue, using a slight modification to a protocol described by Lacy et al.39. The pancreas was removed and digested with collagenase (Type IV, 195 U/ml; Worthington Biochemical, Lakewood, NJ, USA) in a solution containing 2 mM glucose and trypsin inhibitor (0.01%; Sigma-Aldrich, St Louis, MO, USA) at 37 °C for 30 min with vigorous shaking. The pancreatic tissue was triturated with a pipette and washed two times with an enzyme-free solution. Islets were selected with a glass micropipette under a stereomicroscope. Batches of ten islets were used to measure insulin concentration.
Measurement of insulin concentration
Insulin concentration in the culture medium of MIN6, INS-1 cells, and pancreatic β cells/ islets was assayed using a Mouse/Rat Insulin H-type™ enzyme-linked immunosorbent assay kit (Shibayagi Co. Ltd., Shibukawa, Japan), following the manufacturer’s instructions40. Detailed protocols are available in the provided supplementary information and at protocols.io (10.17504/protocols.io.v63e9gn).
Cell viability assay
Cell viability was assessed using the CellTiter 96™ AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA)41,42. Cell viability was determined by comparing the absorbance values of the treated cells with those of the control cells (MIN6 cells at 24 h of incubation) defined as 100%. All experiments were performed in triplicate or quadruplicate. Detailed protocols are available in the supplementary information and at protocols.io (10.17504/protocols.io.v64e9gw).
ATP assay
Intracellular ATP content was evaluated using the CellTiter-Glo™ luminescent cell viability assay kit (Promega)42. Assays were performed in triplicate and repeated at least twice. Detailed protocols are available in the supplementary information and at protocols.io (10.17504/protocols.io.v7ke9kw).
Measurement of cellular oxygen consumption and extracellular acidification
The cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XFp Extracellular Flux Analyzer™ (Agilent Technologies, Santa Clara, CA)41,43. MIN6 cells were seeded at a density of 1 × 104 cells/well on an XFp Cell Culture microplate. The XF Cell Mito Stress Test™ was performed in glucose-containing XF base medium, following the manufacturer’s protocol with 1 µM oligomycin, 1 µM FCCP, 0.5 µM rotenone, and 0.5 µM antimycin A. Detailed protocols are available at Supplementary information and protocols.io (10.17504/protocols.io.v92e98e).
Quantitative reverse transcriptase-PCR (qRT-PCR) analysis
Total RNA was isolated using RNeasy™ Mini Kit (Qiagen, Valencia, CA, USA). First-strand cDNA synthesis and real-time PCR were performed as described41,43. PCR primers sequences were demonstrated as Table S2 primer sequences for Actb (actin, beta; β-actin):, Abcc8 (ATP binding cassette subfamily C member 8; SUR1):, Abcc9 (ATP binding cassette subfamily C member 9; SUR2), Kcnj11 (potassium inwardly rectifying channel, subfamily J member 11; Kir6.2), Kcnj8 (potassium inwardly rectifying channel, subfamily J, member 8; Kir6.1), Slc2a2 (solute carrier family 2 (facilitated glucose transporter), member 2; Glut2), and Cacna1c (calcium channel, voltage-dependent, L type, alpha 1 C subunit; Cav1.2). Detailed protocols are available at Supplementary information and protocols.io (10.17504/protocols.io.v7ne9me).
Electrophysiological studies
MIN6 cells were incubated in an extracellular bath solution containing 2 mM glucose for 30 min at 37 °C before patch-clamp experiments44–46. Membrane potential measurements and whole-cell current recordings were performed using the EPC 800 patch-clamp amplifier (HEKA Elektronik Inc. Holliston, MA, USA). Experiments were conducted at 23–30 °C. Detailed protocols are available at Supplementary information and protocols.io (10.17504/protocols.io.v68e9hw).
Statistical analysis
Data are presented as means ± SD. Differences between groups were evaluated with one-way analysis of variance (ANOVA) and two-way ANOVA followed by Dunnett’s test for multiple comparisons. Statistical analyses were performed with Prism8™ (GraphPad Software, Inc. La Jolla, CA). Statistical significance was defined by P-values < 0.05.
Supplementary information
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science KAKENHI, Grants JP26670693 and JP24592336 to K.H., JP16K10975 and JP19K09339 to Y.M., and JP18K16501 to A.O. This work was also supported by a research grant from the Kansai Medical University (KMU) research consortium to K.H., the branding program as a world-leading research university on intractable immune and allergic diseases from MEXT Japan, and a research grant from Katano Kai to A.O. and K.H. We would like thank to Editage (www.editage.jp) for English language editing.
Author contributions
T.S., M.H., H.K., Y.M., and K.H. conceived and designed the experiments. T.S., M.H., C.S., M.K., T.U., and Y.M. performed the experiments. T.S., M.H., and K.H. prepared figures and/or tables and wrote the paper with comments from H.K. All authors read and approved the final manuscript.
Data availability
The datasets analyzed in this study are available in the Supplementary Information and the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-55848-7.
References
- 1.Kimura H. Hydrogen Sulfide and Polysulfide Signaling. Antioxid Redox Signal. 2017;27:619–621. doi: 10.1089/ars.2017.7076. [DOI] [PubMed] [Google Scholar]
- 2.Yang W, Yang G, Jia X, Wu L, Wang R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J Physiol. 2005;569:519–531. doi: 10.1113/jphysiol.2005.097642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Untereiner A, Wu L. Hydrogen Sulfide and Glucose Homeostasis: A Tale of Sweet and the Stink. Antioxid Redox Signal. 2018;28:1463–1482. doi: 10.1089/ars.2017.7046. [DOI] [PubMed] [Google Scholar]
- 4.Paul BD, Snyder SH. H(2)S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 5.Lowicka E, Beltowski J. Hydrogen sulfide (H2S) - the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- 6.Kimura Y, et al. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci Rep. 2015;5:14774. doi: 10.1038/srep14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyamoto R, et al. Polysulfides (H2Sn) produced from the interaction of hydrogen sulfide (H2S) and nitric oxide (NO) activate TRPA1 channels. Sci Rep. 2017;7:45995. doi: 10.1038/srep45995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olson KR, et al. Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 2018;15:74–85. doi: 10.1016/j.redox.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanaoka K, et al. Discovery and Mechanistic Characterization of Selective Inhibitors of H2S-producing Enzyme: 3-Mercaptopyruvate Sulfurtransferase (3MST) Targeting Active-site Cysteine Persulfide. Sci Rep. 2017;7:40227. doi: 10.1038/srep40227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolluru GK, Shen X, Bir SC, Kevil CG. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rorsman P. The pancreatic beta-cell as a fuel sensor: an electrophysiologist’s viewpoint. Diabetologia. 1997;40:487–495. doi: 10.1007/s001250050706. [DOI] [PubMed] [Google Scholar]
- 12.Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J Physiol. 1986;374:531–550. doi: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi S, Niki I. Significance of hydrogen sulfide production in the pancreatic beta-cell. J Pharmacol Sci. 2011;116:1–5. doi: 10.1254/jphs.11R01CP. [DOI] [PubMed] [Google Scholar]
- 14.Seino S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia. 2012;55:2096–2108. doi: 10.1007/s00125-012-2562-9. [DOI] [PubMed] [Google Scholar]
- 15.Seino S. Physiology and pathophysiology of K(ATP) channels in the pancreas and cardiovascular system: a review. J Diabetes Complications. 2003;17:2–5. doi: 10.1016/S1056-8727(02)00274-X. [DOI] [PubMed] [Google Scholar]
- 16.Sato Y, et al. Cellular hypoxia of pancreatic beta-cells due to high levels of oxygen consumption for insulin secretion in vitro. J Biol Chem. 2011;286:12524–12532. doi: 10.1074/jbc.M110.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurokawa H, et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci Rep. 2015;5:10657. doi: 10.1038/srep10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul Bindu D., Snyder Solomon H. The conversion of H2S to sulfane sulfur: authors' response. Nature Reviews Molecular Cell Biology. 2012;13(12):803–803. doi: 10.1038/nrm3391-c2. [DOI] [PubMed] [Google Scholar]
- 20.Toohey JI. Sulfur signaling: is the agent sulfide or sulfane? Anal Biochem. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 21.Kimura H. Hydrogen polysulfide (H2Sn) signaling along with hydrogen sulfide (H2S) and nitric oxide (NO) J Neural Transm (Vienna) 2016;123:1235–1245. doi: 10.1007/s00702-016-1600-z. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama Y, Takahashi K, Tominaga M, Kimura H, Ohta T. Polysulfide evokes acute pain through the activation of nociceptive TRPA1 in mouse sensory neurons. Mol Pain. 2015;11:24. doi: 10.1186/s12990-015-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mustafa AK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olson KR, et al. Fluorescence quenching by metal centered porphyrins and poryphyrin enzymes. Am J Physiol Regul Integr Comp Physiol. 2017;313:R340–R346. doi: 10.1152/ajpregu.00202.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura H. Hydrogen sulfide and polysulfides as biological mediators. Molecules. 2014;19:16146–16157. doi: 10.3390/molecules191016146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kai S, et al. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxidants & redox signaling. 2012;16:203–216. doi: 10.1089/ars.2011.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gribble FM, Tucker SJ, Seino S, Ashcroft FM. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell K(ATP) channels. Diabetes. 1998;47:1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- 28.Tucker SJ, et al. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heron L, et al. Human alpha-endosulfine, a possible regulator of sulfonylurea-sensitive KATP channel: molecular cloning, expression and biological properties. Proc Natl Acad Sci USA. 1998;95:8387–8391. doi: 10.1073/pnas.95.14.8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miki T, et al. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA. 1998;95:10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura Y, et al. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 2013;27:2451–2457. doi: 10.1096/fj.12-226415. [DOI] [PubMed] [Google Scholar]
- 32.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ujike A, Otsuguro K, Miyamoto R, Yamaguchi S, Ito S. Bidirectional effects of hydrogen sulfide via ATP-sensitive K(+) channels and transient receptor potential A1 channels in RIN14B cells. Eur J Pharmacol. 2015;764:463–470. doi: 10.1016/j.ejphar.2015.07.029. [DOI] [PubMed] [Google Scholar]
- 34.Li N, et al. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell. 2017;168:101–110 e110. doi: 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–294. doi: 10.1016/S0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- 36.Shi Y, Cui N, Shi W, Jiang C. A short motif in Kir6.1 consisting of four phosphorylation repeats underlies the vascular KATP channel inhibition by protein kinase C. J Biol Chem. 2008;283:2488–2494. doi: 10.1074/jbc.M708769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyazaki J, et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 38.Asfari M, et al. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- 39.Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39. doi: 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki K, et al. Volatile anesthetics suppress glucose-stimulated insulin secretion in MIN6 cells by inhibiting glucose-induced activation of hypoxia-inducible factor 1. PeerJ. 2015;3:e1498. doi: 10.7717/peerj.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumi C, et al. Suppression of mitochondrial oxygen metabolism mediated by the transcription factor HIF-1 alleviates propofol-induced cell toxicity. Sci Rep. 2018;8:8987. doi: 10.1038/s41598-018-27220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumi C, et al. Propofol induces a metabolic switch to glycolysis and cell death in a mitochondrial electron transport chain-dependent manner. PLoS One. 2018;13:e0192796. doi: 10.1371/journal.pone.0192796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumi C, et al. Cancerous phenotypes associated with hypoxia-inducible factors are not influenced by the volatile anesthetic isoflurane in renal cell carcinoma. PLoS One. 2019;14:e0215072. doi: 10.1371/journal.pone.0215072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi M, Inagaki A, Novak I, Matsuda H. The adenosine A2B receptor is involved in anion secretion in human pancreatic duct Capan-1 epithelial cells. Pflugers Arch. 2016;468:1171–1181. doi: 10.1007/s00424-016-1806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YZ, Zhang R, Zeng XZ, Song CY. The inhibitory effect of propofol on Kv2.1 potassium channel in rat parietal cortical neurons. Neurosci Lett. 2016;616:93–97. doi: 10.1016/j.neulet.2016.01.058. [DOI] [PubMed] [Google Scholar]
- 46.MacDonald PE, et al. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cells enhances glucose-dependent insulin secretion. J Biol Chem. 2002;277:44938–44945. doi: 10.1074/jbc.M205532200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed in this study are available in the Supplementary Information and the corresponding author upon reasonable request.