

Abstract
Bone marrow aspirate (BMA) differential cell counts (DCCs) are critical for the classification of hematologic disorders. While manual counts are considered the gold standard, they are labor intensive, time consuming, and subject to bias. A reliable automated counter has yet to be developed, largely due to the inherent complexity of bone marrow specimens. Digital pathology imaging coupled with machine learning algorithms represents a highly promising emerging technology for this purpose. Yet, training datasets for BMA cellular constituents, critical for building and validating machine learning algorithms, are lacking. Herein, we report our experience creating and employing such datasets to develop a machine learning algorithm to detect and classify BMA cells. Utilizing a web-based system that we developed for annotating and managing digital pathology images, over 10,000 cells from scanned whole slide images of BMA smears were manually annotated, including all classes that comprise the standard clinical DCC. We implemented a two-stage, detection and classification approach that allows design flexibility and improved classification accuracy. In a 6-fold cross-validation, our algorithms achieved high overall accuracy in detection (0.959 +/− 0.008 precision-recall AUC) and classification (0.982 +/− 0.03 ROC AUC) using non-neoplastic samples. Testing on a small set of acute myeloid leukemia and multiple myeloma samples demonstrated similar detection and classification performance. In summary, our algorithms showed promising early results and represent an important initial step in the effort to devise a reliable, objective method to automate DCCs. With further development to include formal clinical validation, such a system has the potential to assist in disease diagnosis and prognosis, and significantly impact clinical practice.
Examination of the bone marrow is an essential part of the hematologic work-up for many blood and bone marrow diseases and a common laboratory procedure 1. As part of this examination, a nucleated differential cell count (DCC) is obtained by microscopy on Wright-stained bone marrow aspirate (BMA) smears. This procedure entails quantification of cells of different lineages to determine the proportions of each, the findings of which aid in the classification of numerous benign and malignant hematologic disorders. In fact, disease defining criteria are based on cutoff percentages of myeloblasts for myeloid malignancies, such as acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), and the percentage of plasma cells for plasma cell neoplasms, such as monoclonal gammopathy of undetermined significance (MGUS) and smoldering myeloma 2.
Several factors render manual DCC analysis suboptimal, as currently performed in clinical laboratories 3. First, DCCs are labor intensive and time consuming. Second, inter- and intra-observer variability in terms of cell identification and choice of cells for counting represent ongoing sources of error. Third, there is inherent statistical imprecision due to the relatively small number of cells generally counted. If successfully developed, automation of DCCs could obviate most of these concerns.
Traditional automated hematology analyzers that do not employ digital images have been explored for performing DCCs. Major problems with this approach included failure to count nucleated red blood cells and to differentiate stages of cell development, as well as interference by bone marrow lipid 4, 5. These issues are perhaps unsurprising given the complex nature of bone marrow compared to blood for which these instruments were designed. However, a computerized method using digital pathology images could potentially perform DCCs on all pertinent bone marrow cells on a smear. Aside from increasing throughput and reducing labor costs, such an approach could potentially improve accuracy, reproducibility, and objectivity and provide much needed standardization for DCCs.
Cell detection and classification are perhaps the most widely studied problems in computational pathology, with most efforts focused on the analysis of hematoxylin and eosin stained solid tumor sections. While commercial blood analyzers have begun utilizing automated image analysis of Wright stained smears 6, their accuracy largely depends on precise control of pre-analytical variables to minimize staining variations and cell crowding while maximizing preservation of cytologic details. Detection and classification in BMA smears is significantly more challenging due to the high density of touching and overlapping cells, as well as the greater diversity and complexity of cell morphologies. Cell and nuclei detection algorithms often rely on circular or axial symmetry and may fail to detect cells with irregular or multi-lobed nuclei or may incorrectly interpret these as multiple cells. Classification is difficult without accurate detection of cells and localization of cell boundaries (using image segmentation algorithms), and is further compounded by the subtlety of differences in cytologic characteristics used to distinguish many cell types found in bone marrow.
Machine learning approaches have emerged as the dominant paradigm in analyzing histology images7–12. Whereas traditional image analysis methods are engineered using domain knowledge or mathematical models, machine learning algorithms that utilize neural networks are adaptive and can learn from data in an unbiased manner 13. While neural networks typically exhibit superior performance in tasks like detection and classification, realizing these benefits can require thousands of labeled examples for training algorithms to recognize variations in staining and morphology and to reach diagnostically-meaningful accuracy. This demand for labeled data places significant emphasis on the process of image annotation, with efficient protocols and software interfaces being key additional ingredients for developing highly accurate, deep learning algorithms. Current literature on image analysis of BMA smears has not adequately addressed the detection of cells, a particularly challenging problem in BMA smears, and has demonstrated success with only a few cytological classes, limiting potential clinical use 14, 15.
In this paper, we describe our initial steps toward the development of a machine learning digital pathology system to perform DCCs and describe promising initial results in detecting and classifying all non-neoplastic bone marrow cellular constituents of the DCC and neoplastic cells in a small set of AML and multiple myeloma (MM) test cases. Our software prototype achieves a high degree of accuracy in cell detection and classification tasks, using a two-stage system, based on convolutional neural networks. This system is, moreover, able to reliably localize closely packed cells and classify diverse cytomorphologies. A large-scale annotation effort to produce data for training and validation was critical in achieving these results. This study outlines a promising prototype system for automating bone marrow DCCs and provides a basis for further development and eventual clinical validation studies that will include a comprehensive array of bone marrow neoplasms. A glossary of technical terms used in this paper is presented in Table S1.
MATERIALS AND METHODS
Bone marrow aspirate smears.
Wright-stained BMA smears, made for routine patient care from 17 patients, were de-identified and scanned at 0.25 μm/pixel (40X objective) using an Aperio AT2 scanner™ to generate whole-slide images. The smears were uniformly prepared in the bone marrow laboratory at Emory University Hospital from June to August 2015 using the same procedure and reagent vendors. Smears were selected at random from a set of cases, previously studied in manual DCC analyses 3, provided they included cellular particles with at least 500 bone marrow hematopoietic cells, displayed reference range DCCs, minimal cellular degeneration, and a paucity of smearing artifacts. Moreover, complete pathologic investigation in all cases failed to disclose morphologic, immunophenotypic, or genetic abnormalities. In addition, whole-slide images of BMA smears were similarly prepared from materials from 3 AML and 2 MM patients. These disease cases were selected based on having high malignant cell content of 30–50% for AML and 20–30% for MM cases. The 17 non-neoplastic cases were used to develop and validate software algorithms. The additional 3 AML and 2 MM cases were used to measure the performance of these algorithms on an initial set of disease samples. This study was approved by the Institutional Review Board.
Cell annotation.
Whole-slide images were uploaded to a Digital Slide Archive (DSA) server for visualization and annotation. The DSA enables web-based viewing, allowing users to pan and zoom through large whole-slide images, and features a collection of annotation tools for marking and labeling regions and structures 16. The annotation interface is shown in Figure 1A. Regions-of-interest (ROIs) for annotation were first selected using the rectangle or polygon tool and included non-hemodilute areas of the smears adjacent to bone marrow spicules where the cells are mostly evenly distributed, cytologically intact, visually distinguishable, and best represent the spectrum of hematopoiesis. Within these ROIs, other regions were selected using the polygon tool to exclude erythrocytes and non-counted cells (macrophages, stromal cells, mast cells, etc.), that typically would not be included in DCCs (not shown). Individual cells for the DCC were annotated using the point annotation tool by placing a single point at the cell center-of-mass (including nucleus and cytoplasm) and assigning each cell to one of the 13 classes shown in Table 1. The cells within each region were exhaustively annotated to enable accurate assessment of cell detection algorithms. Cells of uncertain class, such as those with suboptimal cytologic preservation including smudged and/or naked nuclei, were assigned to an “unknown” class. Megakaryocytes were also annotated, but not included in the cell detection or classification analyses since they are relatively few and not typically included in DCCs.
Figure 1. The Digital Slide Archive (DSA) annotation interface.
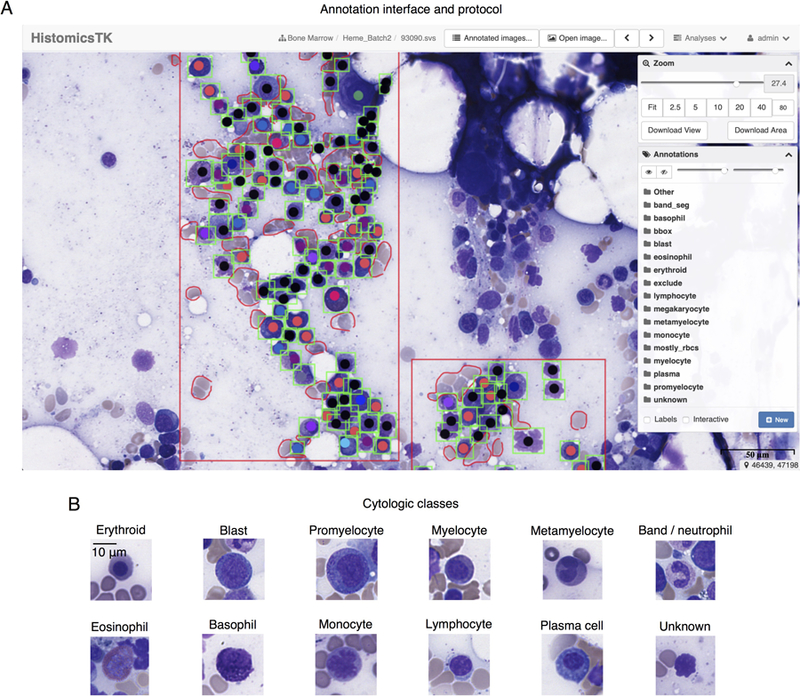
(A) ROIs were defined using the rectangle draw tool, shown in red. Cells within these regions were then annotated exhaustively using the point tool to indicate cytologic class. Finally, bounding boxes, shown in green, were drawn around each annotated cell to delineate the cell boundary for detection algorithm training. The annotations are organized in layers in the Annotation menu, at right, where colors, transparency, and visibility of the annotation markers can be controlled. In addition to the layers for cytological classes, layers are also provided for the region-of-interest (“Other”), artifacts (“exclude”), and regions containing mostly red blood cells (“mostly_rbcs”). (B) Thumbnail images, representing examples of the 11 cell classes encountered in generating DCCs plus an unknown class, are presented.
Table 1. Counts of annotated cells by cytological class.
Annotations outside ROIs were performed to increase counts for rare classes.
| Cytologic class | Total annotated | Inside ROI | Outside ROI |
|---|---|---|---|
| Erythroid | 1526 | 1396 | 130 |
| Blast | 571 | 288 | 283 |
| Promyelocyte | 295 | 112 | 183 |
| Myelocyte | 613 | 414 | 199 |
| Metamyelocyte | 547 | 443 | 104 |
| Band/neutrophil | 1036 | 1005 | 31 |
| Eosinophil | 412 | 156 | 256 |
| Basophil | 62 | 21 | 41 |
| Monocyte | 178 | 109 | 69 |
| Lymphocyte | 544 | 363 | 181 |
| Plasma cell | 283 | 86 | 197 |
| Megakaryocyte* | 39 | 14 | 25 |
| Unknown | 3163 | 3162 | 1 |
| Total | 9269 | 7569 | 1700 |
ROI: Region-of-interest.
Annotated but not used in the cell detection or classification analyses.
Following point annotations, rectangular bounding boxes were drawn to demarcate the extent of each cell (RC, NK). These bounding boxes are required to train and validate the detection algorithm. Additional point annotations were generated outside ROIs to augment the number of examples of cell types that inherently occur less frequently in bone marrow, such as basophils (Table 1). These latter annotations were utilized only during classifier training and neither for classifier validation, nor for training and validation of the cell detection algorithm. Cell annotations were based on well-established cytomorphological criteria used for the microscopic identification of each cell type 17.
Subsequently, all annotated cells were examined for cytologic quality and appropriateness of classification through a consensus review by 3 pathologists (AAA, BRD, DLJ). To accomplish this review, the DSA application-programming interface was used to extract a 96×96 pixel thumbnail image of each annotated cell. These thumbnail images were next organized into folders by assigned cell type. The few initially misclassified cells were identified, and corrections were made to the annotation database. Representative examples of the cytologic classes used in our analysis are displayed in Figure 1B.
Cell detection algorithm.
Our cell detection algorithm is based on the Faster Region-Based Convolutional Network (Faster-RCNN) 18. This network combines bounding box regression for predicting bounding box locations, region pooling, and a residual convolutional network for extracting feature maps from the input images. Detection was approached by treating all cells as a single ‘object’ class, without regard to actual cytomorphologic class. The residual network was trained using two equally weighted loss functions: 1. A cross entropy loss for object classification and 2. An L1 loss on the bounding box coordinates and sizes. Proposed regions were then pooled for computational efficiency, since many proposals are generated for each object. A pre-trained model was used to initialize the residual convolutional net 19, where the remaining network components were random normal initialized (zero mean, variance 1e-4). The entire network was trained for 500 epochs, where an epoch represents one training pass through all training instances. Training employed a momentum-based gradient optimization with momentum 0.9, learning rate 3e-4, weight decay 5e-4, and dropout fraction 0.2. Non-max suppression with a threshold of 0.5 was applied to reduce duplicate proposals.
Cell classification algorithm.
Cell classes were predicted using the VGG16 convolutional network 20. Cell images, sized at 96×96 pixels, were cropped from the center of each manually-generated bounding box. These bounding boxes were mapped to the point annotations using the Hungarian algorithm applied to the pairwise distances between each box and each point. These images were unit normalized to the range [0, 1]. A cross entropy loss for the 12 classes (including “unknown” and excluding megakaryocytes) was used for network optimization. A pre-trained network was used for initialization and then trained using the gradient descent optimizer with 100 cell batches and a learning rate of 1e-4 for 500 epochs. Dropout fraction 0.3 was applied to the fully connected layers.
Data augmentation.
Augmentation techniques were utilized in cell detection and classification to improve prediction accuracy. For cell detection, we generated randomly cropped 600 × 600 pixel regions from the ROIs and randomly flipped these horizontally and vertically. For cell classification, we applied standard augmentation techniques to manipulate the orientation, brightness, and contrast of the cropped cell images. Each cell image was randomly mirror-flipped along the horizontal and vertical axes, rotated by an increment of 90 degrees, brightness adjusted (random_brightness, delta=0.25), and contrast adjusted (random_contrast, range [0.9, 1.4]). To simulate errors in the detection algorithm, we performed a random translation of up to 5 pixels horizontally and vertically. Testing time augmentation was also performed to improve classification performance. During inference, 16 augmented instances of each cell were generated. The softmax values for these augmented versions were then aggregated to generate a single prediction for each cell.
Detection and classification validation.
Non-neoplastic cases were used to perform a 6-fold cross-validation to measure the prediction accuracy of our cell detection and classification methods. Each training set was used to develop a cell detection and a cell classification model. These models were evaluated on the validation test slides, yielding six total measurements of detection accuracy and of classification accuracy in non-neoplastic samples. To test performance in the AML and MM samples, we combined data from all 17 non-neoplastic slides to generate a detection model and a classification model. These models were then applied to the AML and MM samples to assess their performance on a small set of neoplastic test cases.
Cells and ROIs from each training slide set were used to train the detection and classification models using the manual point and bounding box annotations. These models were applied to the validation test slides as follows: 1. The detection model was applied to the test slides to generate prediction bounding boxes and their probabilities and the detection accuracy was measured (see details next paragraph). 2. For detections regarded as true positives, cell images were cropped and centered at the predicted bounding box locations. These cells were then used to evaluate the accuracy of the cell classification model.
Detection accuracy was measured using precision-recall and intersection-over-union (IoU) analysis. IoU is defined for any pair of predicted and manually-annotated bounding boxes, the latter representing the ground truth (gold standard) bounding box, as the area of box intersection over the area of box union. This reaches 1 for perfect overlap and 0 for non-overlapping boxes. The following definitions were used for precision-recall analysis: 1. True positive (TP) where a manually-annotated box has a corresponding predicted box meeting the IoU threshold. 2. False negative (FN) where a manually-annotated box has no predicted box meeting the IoU threshold. 3. False positive (FP) where a predicted box does not have a corresponding manually-annotated box meeting the IoU threshold. The Hungarian algorithm was used to generate a correspondence between manually-annotated and predicted boxes that maximizes the sum of IoUs to avoid double counting of manually-annotated boxes in accuracy calculations. Each predicted bounding box has an associated confidence score and so a precision-recall curve is generated using TP, FN, and FP for the range of detection confidence thresholds from 0 to 1. The area under this precision recall curve measures detection accuracy over a broad range of detection sensitivities 21. In addition, we measured error in the positioning of predicted bounding boxes as the difference in location between the predicted box centers and the matched manually-annotated box centers. Using the TP correspondence from above, we calculated the Euclidean distance between box centers and normalized by the manually-annotated bounding box size (using half the length of the annotated box diagonal).
Classification accuracy was measured using receiver-operating characteristic (ROC) analysis. For each classification model, we measured the sensitivity and specificity of a binary classifier for each cell type (this cell type versus all others) to generate an ROC curve. The area under the ROC curve (AUC) was measured for each cell type, along with the macro average (average performance over all classes, not weighted by class prevalence) to measure class specific and overall accuracy 22.
Execution time analysis.
Analysis of execution times was accomplished using the python time module. Times were measured for loading the ROI file from disk, performing detection on the ROI, cropping images for detected cells from the ROI, and performing cell classification. Execution times were measured for each ROI in 10 trials. Regression analysis was performed to predict execution time from ROI size and number of detected cells using least squares. Variables in this analysis were 1, the number of detected cells, 2, the square root of the number of pixels in the ROI, and 3, a constant bias term. To extrapolate this model to an ROI with 500 detected cells, we trained a second regression model to relate ROI size and the logarithm of the number of detected cells.
Software and hardware.
We employed the following software tools: Tensorflow 1.8 served as the basic framework for the entire system. Luminoth v0.2.0 is Tensorflow-based and provided the detection framework. The Hungarian algorithm was implemented in Scipy v1.1.0. The OpenCV 3.1.0 library was used to handle png/jpeg images. All experiments were run on a dual-socket server equipped with Intel Xeon CPUs, 128GB RAM, and two NVIDIA Tesla P100 cards.
RESULTS
Study overview.
An overview of our approach is presented in Figure 2. Data for training and validating algorithms were generated using a web-based DSA annotation system (Figure 1A). Our cell detection and classification analyses included 11 cytological classes that constitute those of standard DCCs and an unknown class (Figure 1B). These annotations were used to train a two-stage pipeline consisting of cell detection (Figure 2A) and cell classification algorithms (Figure 2B), both based on convolutional networks. The accuracy of these algorithms was evaluated through a 6-fold cross-validation on non-neoplastic samples (Figure 2C). In addition, the cell detection and cell classification algorithms were then trained on all non-neoplastic samples, and tested on a small set of AML and MM samples to assess their potential application to neoplastic cells.
Figure 2. Computational detection and classification of cells in bone marrow aspirate smears.

(A) Cell detection was performed using a Faster R-CNN network built on the resnet101 fully convolutional network. (B) Following cell detection, a separate convolutional network was used to classify the detected cells into 12 cytological classes. (C) Detection and classification accuracy were evaluated through 6-fold cross-validation to measure detection and classification accuracy using human annotations of cytological class and bounding box location. Cross-validation was performed at the case level, so that annotated cells from each case were assigned entirely to either the training or testing set.
Large-scale annotation.
Convolutional networks can deliver outstanding performance given large training datasets of thousands of examples that represent the morphological and staining variations observed in clinical practice 7. To generate annotations at sufficient scale, we developed a protocol using the DSA 16. A screenshot of the DSA annotation interface is presented in Figure 1A with example ROIs, cell annotations, and bounding boxes. We used the DSA and the annotation protocol to annotate 9269 non-neoplastic cells that are specified in Table 1, and included those within ROIs and a smaller subset outside ROIs. The latter subset increased the representation of less common cell classes. Annotation efficiency was improved using a tiered approach that took into account the expertise and availability of annotators, and the effort involved. Labeling cell types requires expertise in the cytomorphology of bone marrow cells. A simple and efficient point and click method utilizing a mouse was developed for cell type labeling by pathologists. Since point annotations alone are not adequate for training detection algorithms, students performed the more laborious task of placing rectangular bounding boxes around the pre-identified cells. Although the bounding boxes alone could be utilized for both localization and cell type labeling, this tiered approach proved more efficient and allowed us to generate a much larger number of annotations.
Cell detection with region-proposal networks.
Detection results for one representative non-neoplastic ROI are presented in Figure 3A. Cells that were missed often had a corresponding detection bounding box that was close, but did not have adequate overlap, based on IoU analysis, to be called a match (Figure 3A, subpanels 1–4). A number of false positives correspond to cells that were mistakenly not annotated by our human observers (Figure 3A, subpanels 5 and 6). A precision-recall analysis was performed to evaluate detection performance from the most sensitive to the most specific tuning of the detection algorithm threshold. The detectors generated in cross-validation simultaneously achieved high precision and recall with only minor variation in performance from fold to fold, as displayed in Figure 3B. The median area under precision-recall curves, observed in cross-validation, was 0.959 +/− 0.008 (see Table S2). In addition to these discrete detection errors, we also measured the positional errors in the placement of predicted bounding boxes. Correct bounding box placement is critical for the classification stage, since the center of the predicted boxes is used to extract cell images for classification. Since the error tolerance for bounding box placement is higher when detecting larger cells, we developed a relative error measure that considers both cell size and predicted bounding box position (see Figure S1). The median relative placement error observed in cross-validation was 6% (Figure 3C), indicating good coincidence between the centers of predicted and actual cell centers.
Figure 3. Cell detection results.

(A) Sample detection result on cross-validation test ROI. Here, green boxes indicate true-positive detections, red boxes false negatives missed by the detector, blue boxes false positives where a detection does not match ground truth, and orange boxes the ground-truth annotations that best correspond to false positives. In many cases (examples in panels 1–4), false positives were due to insufficient overlap with a ground truth annotation (intersection-over-union at least 0.5). Some false positives correspond to cells correctly detected by the algorithm but that were missed during the annotation process (examples in panels 5 and 6). (B) Precision-recall of detection algorithm for cross-validation test sets. Shaded region indicates standard deviation of precision-recall over the six cross-validation sets. (C) Histogram of cross-validation bounding box placement error. This error measures the distance between predicted and actual bounding box centers relative to actual bounding box size.
Cell classification and augmentation strategies.
Classification results for one representative non-neoplastic ROI are presented in Figure 4A. In this example, two unknowns were misclassified as erythroid precursors, as shown in subpanels 1 and 2. Classifier accuracy was evaluated using a one-versus-all classifier for each cytologic class. The classification threshold was varied from the most sensitive to the most specific, generating an ROC profile and AUC measurement for each class (see Figure 4B). The median total AUC for non-neoplastic cells (all classes weighted equally) observed in cross-validation was 0.982 +/− 0.03, while the median AUC for each class ranged from 0.960 (monocyte) to 1.00 (basophil). All AUCs from cross-validation cases are displayed in Tables S3A and B. A confusion matrix describing the cross-validation misclassifications is presented in Figure 4C. As shown, the most common specific classification errors were encountered for defined cell types, particularly monocytes (14%) and lymphocytes (21%), which were predicted to be unknown cell types. Other common errors included adjacent cell classes in the myeloid series: blasts being misclassified as promyelocytes (10%), myelocytes being predicted to be promyelocytes (8%), and promyelocytes being predicted to be blasts (7%). Lastly, lymphocytes were predicted to be erythroid cells (6%) and monocytes were misclassified as metamyelocytes (7%) or myelocytes (6%).
Figure 4. Cell classification results.

(A) Sample classification result on test ROI. Predicted class and location of detected cells is indicated with color-code dots. Classification errors are indicated with a bounding box in sub panels 1 and 2, colored to indicate the annotated cell class. (B) Classification area-under-curve on cross-validation test sets. Each point represents the AUC of one class in one testing fold. Total AUC was calculated as the average AUC over all classes (unweighted by class proportions). (C) Cross-validation confusion matrix indicating the classification errors that were made for each class. Rows display the true cell class while columns indicate the cell type predicted by the classifier. Values are normalized as percentages across rows. The diagonal shows the proportion of TPs for each cell class. Values outside of the diagonal represent misclassification rates. Results presented are aggregated over all cross-validation folds.
We next estimated how these misclassifications might affect key cell types in the DCC, namely plasma cells and blasts. The misclassification rates from the confusion matrix were used to analyze manual DCCs from 5 patient samples that represent a clinically relevant spectrum of plasma cells (N=2) and blasts (N=3). We calculated upper and lower estimates for plasma cell and blast percentages, given the importance of these cell types in disease classification. The projected percentages with lower and upper error estimates, in parentheses, include 8.6% (7.7%, 8.8%) and 57.2% (50.9%, 57.3%) for plasma cells, and 6.4% (5.4%, 7.4%), 9.6% (8.2%, 12.1%) and 26% (22.1%, 28.0%) for blasts (see Table S4 for complete DCCs).
The aforementioned results are based on non-neoplastic cells. Since the cytomorphologies of neoplastic cells can differ, to varying degrees, from their non-neoplastic counterparts, we explored the feasibility of employing the algorithms, developed using non-neoplastic cells, for detecting and classifying neoplastic cells. We studied a small set of AML and MM test cases selected for neoplastic cell content of 20–50%. In aggregate, 1373 cells were annotated which included 223 AML blasts and 76 malignant plasma cells (see Table S5 for all annotated cells). The detection AUC for all cell classes was 0.970 for AML cases and 0.979 for MM cases (see Table S2). For AML blasts, the classification AUC was 0.893 and that for MM plasma cells was 0.970 (see Table S3B). Confusion matrix analysis for the AML cases suggested an overall predication accuracy of 87.2% for blasts. Interestingly, a subset of blasts was classified by the algorithm as “unknown”, something that is often done clinically with neoplastic cells at the time of manual DCC, until the neoplastic cell lineage is determined by ancillary studies, then such cells are generally re-categorized into their correct class (e.g. blasts). If blasts that were counted as unknowns are included as true positives here, the prediction accuracy increases to 93.3% (see Table S6). Lastly, confusion matrix analysis for MM cases suggested a prediction accuracy for neoplastic plasma cells of 96.5%. This improves to 98.8% if plasma cells that were classified as unknowns are likewise counted as true positives (See Table S6).
Handling detection and classification with two separate networks provided more flexibility in network design, and enabled us to employ advanced strategies for data augmentation that had a significant positive impact on classification accuracy. During class inference, each detected cell was augmented to generate 16 versions with different orientations and intensity transformations, as displayed in Figure 5A. The classification network was applied to these augmented versions, and the predicted class probabilities were aggregated. This procedure improved the total AUC an average of 5.0% over all cross-validation folds as displayed in Figure 5B (see also Tables S3A and B). This increase in classification accuracy was statistically significant (p=3.12e-2, Wilcoxon signed rank).
Figure 5. Impact of data augmentation on classification performance.

(A) Data augmentation procedure for inference. (A.1) At inference time, for each detected cell we extracted an image centered at the predicted bounding box location. (A.2) This image is transformed using rotations, translations, and pixel intensity transforms to generate an “augmented” set of 16 images for inference. These images are passed through the classification network to generate 16 total predictions of cytologic class. Each prediction describes the probabilities that the image belongs to each of the 12 cytologic classes. These predictions are aggregated to smooth out noise and to improve robustness. (A.3) The cell in question is assigned to the highest-probability cytologic class using the aggregated predictions. (B) This augmentation procedure significantly improved classification accuracy in cross-validation experiments (Wilcoxon signed rank test). Each dot represents the accuracy of one fold in the cross-validation.
We also analyzed the execution time of our algorithms on a high-performance server equipped with graphical processing unit (GPU) accelerators (see Materials and Methods for configuration). Execution of the detection and classification models during inference consumed almost all computation time, with loading and preprocessing consuming only minimal time (Figure S2, Table S7). We modeled total execution time using a regression analysis based on the number of detected cells and ROI size (Figure S3). This model was highly accurate with R2=0.999. We extrapolated this model, as detailed in the Materials and Methods, to predict an average execution time of 162 seconds for an ROI containing 500 cells.
DISCUSSION
BMA DCC is routinely performed to assess hematopoietic activity, to compare the proportions of the different cell lineages with reference ranges, and to quantify abnormal cells when present. It is generally performed by pathologists and/or the laboratory technical staff depending on workflow and the laboratory case volume. While publications vary in the total number of cells recommended for performance of DCCs, they generally fall between 300–500 cells, but can vary based on specific clinical circumstances 2, 23, 24. At the high end, counts of more than 500 cells have been recommended based on theoretical work that considered the odds of unacceptable error in classification when initial counts fall near diagnostic cutoffs for critical cells classes 1, 25. Yet, manual DCCs suffer from being labor intensive with inherent inter- and intra-observer variability in cell classification and choice of cells counted. Automation of the DCC could not only obviate these issues, including the ability to readily analyze the many hundreds to thousands of pertinent cells on a smear, but also could also afford standardization. If successfully developed, such a system could thus have a tremendous impact on the practice of pathology.
One promising method to create an automated DCC system, which we explored in this work, is digital image analysis with machine learning. Images of BMA smears present significant technical challenges for image analysis algorithms. BMA smears contain cells representing diverse cytomorphologies with some cell types exhibiting only subtle differences. A large number of cells in any given smear may be ambiguous and the boundaries between cells indistinguishable, particularly in areas with clumping. Traditional image analysis techniques that rely on models of cell appearance and morphology are difficult to apply in these scenarios, and may fail to accurately detect and distinguish closely packed cells from one another, a process called segmentation. Reliable segmentation is absolutely necessary to extract shape, texture, and color features that are used for classification. Importantly, difficulties in segmentation will often be reflected in poor classification performance. A data-driven approach, based on machine learning with convolutional networks, can perform classification without explicitly segmenting cells by relying on detection algorithms to localize cells. This approach also does not rely on a-priori definitions of cell features for classification, but does require extensive annotations of data for training and validation of the detection and classification networks.
To generate sufficient data for such convolutional network approaches, we developed an efficient tiered annotation protocol using the DSA. This web-based platform facilitated de-centralized annotation and review, and helped to scale our labeled dataset. The tiered protocol utilized experts to classify cells using a simple point annotation tool, and students to do the more laborious work of placing bounding boxes, enabling us to annotate over 10,000 cells from neoplastic and non-neoplastic cases. This large dataset allowed us to engineer an analysis pipeline based on convolutional networks for cell detection and classification. This pipeline achieved high detection accuracy on both non-neoplastic cases in cross-validation (0.959 +/− 0.008 AUC) as well as AML (0.970 AUC) and MM (0.979 AUC) test cases. Classification accuracy for all cell types was also high in non-neoplastic cases (0.982 +/− 0.03 ROC AUC) and largely in AML (0.912 AUC) and MM (0.906 AUC) test cases. Importantly, high classification accuracy of neoplastic cells will be crucial for developing a diagnostic tool for diseases such as AML and MM. In light of the fact that our classifiers were trained entirely on non-neoplastic cases, the classification accuracies achieved in this small test set of AML blasts (0.893 AUC) and MM plasma cells (0.970 AUC) are promising and represent a good starting point for further progress. Of note, these levels approach the performance of a commercially available clinical image analysis system for blood 26, 27. In addition, the estimated effects on blast and plasma cell percentages from DCCs we calculated using the misclassification rates from confusion matrix analysis suggest that reasonable error ranges are likely to be encountered in future clinical validation studies. Decoupling the detection and classification steps provided definite benefits in our system. As we demonstrated, the ability to perform augmentation of detected cells significantly improved classification accuracy, increasing the accuracy from 0.917 +/− 0.027 to 0.982 +/− 0.03 (p = 3.12e-2). Using separate networks for detection and classification also improved flexibility in design. Detection and classification tasks have very different design requirements and creating a single convolutional network to perform both tasks is difficult and will likely result in suboptimal overall performance. Importantly, these two networks will appear seamless to users of the software once fully optimized. Analysis of execution time shows that the expected runtime for an ROI containing 500 cells is less than three minutes. This performance could be significantly improved using additional hardware, and can be largely hidden from the end-user by processing slides offline prior to inspection.
Limited studies have evaluated image analysis in automating BMA DCCs. Choi et al 14 published promising preliminary results using convolutional networks for cell classification in DCCs. Their dataset comprised 2174 cells of non-neoplastic erythroid and myeloid precursors, and did not include other cells types important in DCCs including eosinophils, basophils, monocytes, lymphocytes, and plasma cells. This study focused on classification and did not address detection, utilizing only manually cropped images of cells to develop and validate the classifier. Moreover, noise due to the detection process was not accounted for in their classification. Their reported classification performance was 0.971 precision at 0.971 recall. This is comparable to the overall classification performance for our system that included analysis of all relevant cell types in the DCC. Reta et al 15 developed a cell detection and classification framework for classification of acute leukemia subtypes. Their dataset comprised 633 cells from acute lymphoblastic leukemias and acute myeloid leukemias. They developed an elaborate software pipeline to detect and segment leukocytes in digital images of Wright-stained BMA smears. Detected cells were characterized using a set of features that describe the shape, color, and texture of each cell. These cells were classified individually using basic machine learning algorithms, and the cell classifications were aggregated to provide a single diagnosis for the sample. While their application is narrow and focuses only on a few cell types, their reported segmentation accuracy has a high precision (95.75%) and their subtype classification accuracy ranged from 0.921 to 0.784 ROC AUC. Our findings expand the work in these earlier publications and point to the promise of machine learning approaches towards automation of DCCs.
In many circumstances, manual examination of BMA smears employs a 100x objective (1000x). In this work, we utilized whole slide images collected at a resolution of 0.25 μm/pixel, which approximates a 40x objective (400x). Scanning at 400x offers useful advantages in digital pathology workflow. For example, whole slide scanning beyond 400x magnification is time consuming, and in contrast to scanning at 400x, leads to extremely large file sizes that are impractical to archive 28. And, while scanning beyond 400x could become feasible if limited to smaller ROIs, it requires that these are identified before scanning. Thus, capturing whole slide images will facilitate the automation process by avoiding the introduction of additional human workflow interactions. Moreover, objectives offering magnifications higher than 400x often require oil immersion which can introduce significant challenges, including difficulties in dispensation and containment of oil that can contaminate imaging systems and increase maintenance requirements. We are currently aware of only one slide scanning equipment vendor who is pursuing high-throughput scanning with oil immersion for clinical use. Importantly, our results show that images acquired at a resolution offered by a 40x objective can form the basis of compelling detection and classification algorithms for the specific purpose of cell-type identification for DCCs. The need for higher resolution images will, nonetheless, likely be required for applications aimed at detecting and differentiating more subtle cytomorphologic details such as dysplastic changes, Auer rods, iron particles, and specific intracellular microorganisms.
In this study, we present highly promising preliminary results in developing a computational system for DCC of BMAs. Our approach combines state-of-the-art detection and classification algorithms based on convolutional networks, and achieved excellent performance in detection and classification tasks. This success was enabled primarily by extensive annotation and curation of training and validation data using the DSA. While our results are quite encouraging, this study currently has some important limitations. First, while we evaluated our system on AML and MM cases, we did not include cells from these cases in training, and so the reported classification accuracies for disease cases are likely subject to improvement. Furthermore, the number of these disease cases was limited, and certainly did not represent the full spectrum of hematologic malignancies. Since neoplastic cells often exhibit cytomorphologic differences from benign counterparts, it will be important to include examples of these cells when training algorithms to realize optimal performance on disease cases. Future studies will greatly expand the number of disease cases, will include other diagnostic categories, and will grow the training set to cover the wide spectrum of abnormal cytomorphologies. Second, the small ROIs employed in this study were biased towards better cytologic preservation. The performance on random large ROIs encompassing marrow particles as would typically be analyzed by pathologists has not been assessed. These areas contain more highly dense overlapping marrow cells and stromal cells that will need to be addressed by detection and classification models. Third, exploration of the potential benefits of employing higher resolution scanned images will also be useful as the acquisition and storage of these large digitized images becomes more practical for clinical use. Fourth, we have not established performance criteria for clinical validation of this novel method that is still early in development, but this will certainly be required before deploying for clinical use, as it has for automated analysis of blood smear images 6, 27. Any automated approach will ultimately have to be shown to be at least as reliable and accurate clinically as manual microscopic review of slides and faster than manual DCC performance, even after reclassification by pathologists/technologists of any cells wrongly categorized.
Future annotation efforts will include an inter-observer variability study to better understand the ranges for classification and detection performance of human observers. The final software application will also require a convenient graphical interface that allows users to identify errors and to manually override the algorithm. While our algorithms performed well on samples processed in our lab, variations in pre-analytic factors like smearing and staining quality will impact generalization to other sites, and additional data collection would be required to deploy the system in other labs. Nonetheless, the annotation system and protocols presented here establish a template to generate similar training and validation data, and results. Once the above limitations are addressed, the advances made in this study can be integrated into a practical computerized system with potential to have significant impact on clinical practice.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by the National Cancer Institute Informatics Technology for Cancer Research grants U01CA220401 and U24CA19436201. We thank Drs. Geoffrey Smith and Thomas Durant for helpful suggestions and comments on the manuscript. RC and NK performed this work as part of a senior research course at the Department of Electronic and Telecommunication Engineering, University of Moratuwa, Sri Lanka (instructor Dr. N. W. Nuwan Dayananda, PhD). We thank the University of Moratuwa and Dr. Dayanada for their support.
Funding: This research was supported in part by the National Cancer Institute Informatics Technology for Cancer Research grants U01CA220401 and U24CA19436201.
Footnotes
Disclosure/conflict of interest: Nothing to disclose.
Note: This work was presented at the Academy of Clinical Laboratory Physicians and Scientists (ACLPS) 2018 meeting as a platform presentation.
SUPPLEMENTARY TABLES AND FIGURES
Supplementary information is available at Laboratory Investigation’s website.
REFERENCES
- 1.Lee SH, Erber WN, Porwit A, et al. ICSH guidelines for the standardization of bone marrow specimens and reports. Int J Lab Hematol 2008;30(5):349–364. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Vol. 2, 4th edn. Lyon, France: IARC publications, 2017, p. 585. [Google Scholar]
- 3.Abdulrahman AA, Patel KH, Yang T, et al. Is a 500-Cell Count Necessary for Bone Marrow Differentials?: A Proposed Analytical Method for Validating a Lower Cutoff. American journal of clinical pathology 2018;150(1):84–91. [DOI] [PubMed] [Google Scholar]
- 4.d’Onofrio G, Zini G. Analysis of bone marrow aspiration fluid using automated blood cell counters. Clin Lab Med 2015;35(1):25–42. [DOI] [PubMed] [Google Scholar]
- 5.Mori Y, Mizukami T, Hamaguchi Y, et al. Automation of bone marrow aspirate examination using the XE-2100 automated hematology analyzer. Cytometry B Clin Cytom 2004;58(1):25–31. [DOI] [PubMed] [Google Scholar]
- 6.Kratz A, Bengtsson HI, Casey JE, et al. Performance evaluation of the CellaVision DM96 system: WBC differentials by automated digital image analysis supported by an artificial neural network. American journal of clinical pathology 2005;124(5):770–781. [DOI] [PubMed] [Google Scholar]
- 7.Janowczyk A, Madabhushi A. Deep learning for digital pathology image analysis: A comprehensive tutorial with selected use cases. J Pathol Inform 2016;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirinukunwattana K, Ahmed Raza SE, Yee-Wah T, et al. Locality Sensitive Deep Learning for Detection and Classification of Nuclei in Routine Colon Cancer Histology Images. IEEE Trans Med Imaging 2016;35(5):1196–1206. [DOI] [PubMed] [Google Scholar]
- 9.Mobadersany P, Yousefi S, Amgad M, et al. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc Natl Acad Sci U S A 2018;115(13):E2970–E2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saltz J, Gupta R, Hou L, et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep 2018;23(1):181–193 e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bychkov D, Linder N, Turkki R, et al. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci Rep 2018;8(1):3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senaras C, Niazi MKK, Lozanski G, et al. DeepFocus: Detection of out-of-focus regions in whole slide digital images using deep learning. PLoS One 2018;13(10):e0205387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521(7553):436–444. [DOI] [PubMed] [Google Scholar]
- 14.Choi JW, Ku Y, Yoo BW, et al. White blood cell differential count of maturation stages in bone marrow smear using dual-stage convolutional neural networks. Plos One 2017;12(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reta C, Altamirano L, Gonzalez JA, et al. Segmentation and Classification of Bone Marrow Cells Images Using Contextual Information for Medical Diagnosis of Acute Leukemias. PLoS One 2015;10(6):e0130805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutman DA, Khalilia M, Lee S, et al. The Digital Slide Archive: A Software Platform for Management, Integration, and Analysis of Histology for Cancer Research. Cancer Res 2017;77(21):e75–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glassy EF. Color Atlas of Hematology; an illustrated field guide based on proficiency testing Illinois, USA: College of American Pathologists, 1998. [Google Scholar]
- 18.Ren S, He K, Girshick R, et al. Faster r-cnn: Towards real-time object detection with region proposal networks. Advances in neural information processing systems, 2015, p. 91–99. [DOI] [PubMed]
- 19.He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. Proceedings of the IEEE conference on computer vision and pattern recognition, 2016, p. 770–778.
- 20.Simonyan K, Zisserman A. Very deep convolutional networks for large-scale image recognition. arXiv preprint arXiv:14091556 2014.
- 21.Davis J, Goadrich M. The relationship between Precision-Recall and ROC curves. Proceedings of the 23rd international conference on Machine learning: ACM, 2006, p. 233–240. [Google Scholar]
- 22.Sokolova M, Lapalme G. A systematic analysis of performance measures for classification tasks. Information Processing & Management 2009;45(4):427–437. [Google Scholar]
- 23.Bain BJ, Bates I, Laffan M, et al. Dacie and Lewis Practical Hematology London: Churchill Livingstone, 2011. [Google Scholar]
- 24.Ryan DH. Examination of the Marrow. In: Kaushansky K, Lichtman MA, Prchal JT, et al. , editors. Williams Hematology 9th ed. New York, NY: McGraw-Hill Education, 2015. p. 27–40. [Google Scholar]
- 25.Vollmer RT. Blast counts in bone marrow aspirate smears: analysis using the poisson probability function, bayes theorem, and information theory. American journal of clinical pathology 2009;131(2):183–188. [DOI] [PubMed] [Google Scholar]
- 26.Cornet E, Perol JP, Troussard X. Performance evaluation and relevance of the CellaVision DM96 system in routine analysis and in patients with malignant hematological diseases. Int J Lab Hematol 2008;30(6):536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Briggs C, Longair I, Slavik M, et al. Can automated blood film analysis replace the manual differential? An evaluation of the CellaVision DM96 automated image analysis system. Int J Lab Hematol 2009;31(1):48–60. [DOI] [PubMed] [Google Scholar]
- 28.Campanella G, Hanna MG, Geneslaw L, et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nature Medicine 2019. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.