

Abstract
Background:
Farnesoid X receptor (FXR) agonist, obeticholic acid (OCA), increases total and low-density lipoprotein cholesterol (LDL-C) in patients with nonalcoholic steatohepatitis (NASH). The present study evaluated the impact of OCA therapy on lipoprotein sub-particles.
Method
The study included 196 patients (99 OCA group and 97 placebo) who were enrolled in the FLINT trial and had samples available for lipid analysis and liver biopsies at enrollment and end-of-treatment (EOT) at 72 weeks. Very low-density lipoprotein (VLDL), low density lipoprotein (LDL), and high-density lipoprotein (HDL) particles were evaluated at baseline, 12 and 72 weeks after randomization, and 24 weeks following EOT.
Results
Baseline lipoprotein profiles were similar among OCA and placebo. OCA did not impact total VLDL particle concentrations, but OCA vs placebo treatment was associated with decreased large VLDL particle concentration at 12 weeks (baseline adjusted mean: 6.8 vs. 8.9 nmol/L; P=0.002), mirrored by an increase in less-atherogenic small VLDL particle concentration (33.9 vs. 28.0 nmol/L; P=0.02). After 12 weeks, total LDL particle concentration was higher among OCA vs. placebo (1667 vs. 1329 nmol/L; P<0.0001), characterized by corresponding increases in both less-atherogenic, large-buoyant LDL (475 vs. 308 nmol/L; P=<0.001) and more-atherogenic small-dense LDL particles (1015 vs. 872 nmol/L; P=0.002). The changes in LDL particle concentrations were similar between treatment groups (OCA and placebo) 24 weeks following EOT due to improvement in OCA cohort. Compared to placebo, a reduction in total HDL-particle concentration, particularly large and medium HDL-particles, was noted in the OCA treated patients but this resolved after drug discontinuation.
Conclusion:
OCA therapy is associated with increases at 12 weeks in small VLDL particles and large and small LDL particles and reduction in HDL particles which revert to baseline 24 weeks after drug discontinuation.
Keywords: nonalcoholic steatohepatitis, very low density lipoprotein, low density lipoprotein, high density lipoprotein, lipoproteins, obeticholic acid
Graphical Abstract
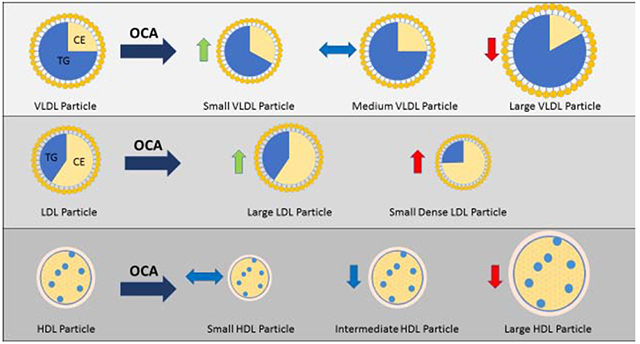
Lay Summary:
Nonalcoholic steatohepatitis is a chronic liver disease that is associated with increased risk of developing cirrhosis and cardiovascular disease. Recently, obeticholic acid (OCA), a farnesoid X receptor agonist, improved liver disease but led to an increase in cholesterol, however, the impact of OCA on cholesterol is not well understood. In the present study, we present data showing that OCA therapy is associated with an increase in lipoprotein levels which improve after drug discontinuation.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common etiology of chronic liver disease affecting nearly a third of the United States population1. Nonalcoholic steatohepatitis (NASH) is the clinically aggressive variant of NAFLD that is characterized histologically by hepatic steatosis along with necro-inflammatory activity2. In the absence of approved pharmacological therapy, NASH is increasingly being targeted for drug development efforts3. Obeticholic acid (OCA) is a farnesoid X receptor (FXR) agonist that was recently shown in a randomized, placebo-controlled clinical trial to improve liver histology in patients with NASH4. FXR is a bile-acid binding transcription factor belonging to the super family of nuclear receptors and, due to its central role in inflammation, glucose and lipid metabolism, FXR agonism is an attractive therapeutic target in NASH.
Mechanistically, FXR activation reduces triglyceride-rich lipoproteins via repression of hepatic sterol responsive element binding protein 1c (SREBP1c), microsomal triglyceride transfer protein (MTTP), and apolipoprotein B (apoB) gene expression5–7. However, it also reduces cholesterol conversion to bile acids which is a major mechanism of cholesterol disposal. FXR agonism via OCA in the Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) trial led to an increase in low-density lipoprotein cholesterol (LDL-C) and reduction in high density lipoprotein cholesterol (HDL-C)4,8. Since cardiovascular disease (CVD) is common among patients with NAFLD and the leading cause of mortality among patients with NAFLD9–11, this rise in serum LDL-C requires a deeper understanding.
The advanced lipoprotein profile consists of measuring a number of lipoprotein particles and sub-particles with varying atherogenic risk that are often better predictors of future CVD events than the traditional lipid panel, especially in subjects with metabolic diseases such as type 2 diabetes12–14. Recent studies have highlighted a close relationship between NAFLD and atherogenic lipoproteins, despite relatively normal traditional lipid panel related parameters15,16. The impact of FXR activation via OCA on atherogenic lipoprotein sub-particles is currently unknown. In order to bridge this knowledge gap, we conducted the current study to evaluate the impact of OCA on atherogenic lipoproteins in patients enrolled in the FLINT trial.
METHODS
Study Design
The data obtained for this study were from participants of the FLINT trial, which was an adult treatment trial conducted by the Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN)4. Briefly, 283 patients with biopsy proven NASH were randomized to 72 weeks of OCA 25mg once-daily treatment versus placebo. The primary outcome was defined as improvement in liver histology evidenced by a decrease in NAFLD Activity Score (NAS) by at least 2 points without worsening of fibrosis from baseline to end-of-treatment (EOT)4. Liver biopsies in the last 64 patients were not performed after interim analysis showed 43% of patients met the primary outcome with OCA treatment compared to 21% on placebo (P=0.0024) and treatment was also discontinued at that time related to concerns about increases in circulating cholesterol levels in patients taking OCA.
Liver Histology
All liver biopsies were evaluated centrally by the NASH CRN Pathology Committee members as a group for consensus and graded according to the NASH CRN criteria2. Hepatic steatosis was graded on an ordinal scale from 0-3 [grade 0 <5% steatosis; grade 1 = 5-33% steatosis, grade 2= 34-66% steatosis; grade 3 = ≥67% steatosis]. Cytologic ballooning was graded on a scale of 0-2 [grade 0 = none; grade 1 = few ballooned hepatocytes; and grade 2 = many ballooned hepatocytes]. Lobular inflammation was graded on a scale from 0-3 [grade 0 = none; grade 1 = <2 foci/20x hpf; grade 2 = 2-4 foci/20x hpf; and grade 3 = >4 foci/20x hpf]. Hepatic fibrosis was quantified from stages 0-4 and for the purpose of this analysis fibrosis stages 1a, 1b, and 1c were considered to be stage 12. Study pathologists were blinded to treatment assignment and whether biopsies were before or after treatment.
Study Visit and Procedures:
All patients were evaluated at their respective medical center and protocol driven anthropometric measurements, blood tests and patient assessments were collected. All blood was collected after an overnight fast. Fasting traditional lipid profile consisted of serum total cholesterol, HDL-C, LDL-C and triglycerides. There were no pre-specified rules regarding the treatment of dyslipidemia during the clinical trial and the decision to initiate and escalate lipid lowering therapy was left to the discretion of the treating physician.
Lipoprotein Profile
A comprehensive lipoprotein profile analysis was performed on stored EDTA plasma samples collected at 0 weeks, 12 weeks, 72 weeks, and 96 weeks. Nuclear magnetic resonance spectroscopy (NMR) spectra were collected on a Vantera Clinical Analyzer from EDTA plasma samples as previously described17,18. Concentrations for the lipoprotein classes (very low-density lipoprotein [VLDL], LDL, and HDL particle numbers) and subclasses (small, medium and large) were calculated using the LP3 deconvolution algorithm (LabCorp, Morrisville, NC). The LP3 algorithm reports lipoprotein particles of the following diameter ranges (nanometer, nm): large VLDL (>60 nm), medium VLDL (42-60 nm), small VLDL (29-42 nm), IDL (23-29 nm), large LDL (20.5-23 nm), small LDL (18-20.5 nm), large HDL (9.4-14 nm) medium HDL (8.2-9.4 nm) and small HDL (7.3-8.2 nm)18,19. The calculated mean VLDL, LDL, and HDL particle sizes were weighted averages derived from the sum of the diameter of each subclass multiplied by its relative mass percentage17.
Statistical Analysis
The current analysis includes the subset of FLINT patients who had both a baseline and EOT liver biopsy. Summary statistics includes means, standard deviations and percentages. To evaluate the impact of OCA therapy on lipoproteins, the standard lipid profiles and NMR lipoprotein profiles were evaluated at baseline, 12 weeks, and 72 weeks (EOT) on therapy and 24 weeks off-treatment (96 weeks). Treatment group differences at baseline were compared using analysis of variance (ANOVA), while group differences at follow up were compared using analysis of covariance (ANCOVA) adjusting for baseline value. Sensitivity analysis included the testing of the OCA treatment effect by statin use (i.e., effect modification) at each time point. FLINT trial was not designed to evaluate the impact of statin therapy on lipoproteins and as such the methodology did not standardize an approach to lipid management resulting in initiation and titration of statin at the discretion of the treating physician. As such, in the present study we present empiric data regarding statin use. Framingham risk score (FRS), a validated cardiovascular risk score, was used to evaluate the impact of OCA on future CVD risk20. P-values are nominal and have not been adjusted for multiple comparisons, multiple looks or multiple outcomes. Analyses were conducted using SAS (Version 9.3 of the SAS System for Windows, Cary, NC: SAS Institute Inc., 2002-2004) and Stata (StataCorp. 2015. Stata Statistical Software: Release 14. College Station, TX: StataCorp LP).
RESULTS
Patient Characteristics
A total of 196 patients (99 OCA and 97 placebo) who had liver biopsies and lipid profiles performed at baseline and EOT were included in the current analysis. Baseline demographic characteristics of the two groups were similar (Table 1). Overall mean (SD) age was 51 (12) years and the majority of the patients were female (67%) and white (82%). The serum aminotransferase levels and distribution of liver histological features, including steatosis, lobular inflammation, and cytological ballooning, were similar between the placebo and OCA-treated groups. At baseline, the distribution of diabetes, hypertension and dyslipidemia were similar across the two groups. Finally, 52% of the OCA-treated group was on statin therapy at baseline compared to 44% of the placebo (P=0.32). The FRS categories were similar and OCA therapy did impact the FRS at end of study (week 72) and follow up (week 96) (Appendix Table 1).
Table 1.
Characteristics of the study population at baseline
| N (%) or Mean (SD) | |||
|---|---|---|---|
| Characteristic | OCA (n=99) | Placebo (n=97) | P-value* |
| Demographic | |||
| Age –yrs | 52 (11) | 50 (12) | 0.20 |
| Sex - % male | 30 (30%) | 35 (36%) | 0.45 |
| Race - % white | 84 (85%) | 77 (79%) | 0.35 |
| Anthropometric | |||
| Body mass index – kg/m2 | 35 (6) | 34 (6) | 0.76 |
| Liver tests | |||
| ALT – U/L | 80 (46) | 82 (49) | 0.77 |
| AST – U/L | 62 (39) | 56 (31) | 0.25 |
| GGT – U/L | 74 (80) | 67 (54) | 0.47 |
| Alkaline phosphatase – U/L | 82 (28) | 81 (25) | 0.76 |
| Lipids | |||
| Total cholesterol – mg/dL | 191 (44) | 190 (48) | 0.99 |
| Triglycerides - mg/dL | 189 (109) | 182 (178) | 0.73 |
| HDL – mg/dL | 43 (11) | 45 (14) | 0.23 |
| LDL – mg/dL | 111 (39) | 114 (41) | 0.59 |
| Co-morbidities | |||
| Diabetes | 52 (52%) | 52 (54%) | 0.89 |
| Hypertension | 64 (64%) | 57 (59%) | 0.46 |
| Hyperlipidemia | 62 (63%) | 62 (64%) | 0.88 |
| Concomitant medications | |||
| Statins | 51 (52%) | 43 (44%) | 0.32 |
| Histology | |||
| Steatosis grade | 2.1 (0.8) | 2.1 (0.8) | 0.88 |
| Lobular inflammation grade | 1.8 (0.7) | 1.8 (0.7) | 0.89 |
| Ballooning grade | 1.4 (1.3) | 1.3 (1.2) | 0.37 |
| NAFLD Activity Score (NAS) | 5.4 (1.3) | 5.3 (1.3) | 0.50 |
| Fibrosis stage | 1.8 (1.0) | 1.8 (1.0) | 0.66 |
Derived from Fisher’s exact test for categorical variables and t-test for unequal variance for continuous variables
Lipids
The baseline lipid panel consisting of LDL-C, HDL-C, total cholesterol and triglyceride results were similar between the two groups (Table 1). After 12 weeks of therapy, the baseline adjusted mean difference between OCA-treated and placebo groups was 167 mg/dL vs. 173 mg/dL (P=0.53) for triglycerides, 206 mg/dL vs. 182 mg/dL (P<0.0001) for total cholesterol, 39.3 mg/dL vs. 43.3 mg/dL (P<0.0001) for HDL-C and 135 mg/dL vs. 107 mg/dL (<0.0001) for LDL-C. The treatment group differences in serum triglycerides were not statistically different at week 72 and 96. At 72 weeks, the OCA-group had higher total serum cholesterol (196 vs. 181 mg/dL; P=0.0009) and LDL-C (120 vs. 103 mg/dL; P<0.0001), but the total cholesterol and LDL-C were decreased after OCA discontinuation and were similar to placebo at 96 weeks. As compared to placebo, serum HDL-C levels were lower at 72 weeks in the OCA group (42.5 vs. 44.7 mg/dL; P=0.01) but were similar after treatment discontinuation (96 weeks follow up).
Very Low-Density Lipoprotein Sub-particles
The distribution of total, large, medium and small VLDL particles were similar between OCA-treated and placebo groups at baseline (Table 2). No difference in baseline adjusted mean total VLDL particle concentration was noted at 12, 72 and 96 weeks between the OCA-treated and placebo groups. The baseline adjusted mean large VLDL concentration at 12 weeks in OCA-treated group was lower at 6.8 nmol/L when compared to 8.9 nmol/L in the placebo group (P=0.002). The reduction at 12 weeks in large VLDL particle concentration in the OCA-treated group was accompanied by an increase in small VLDL particle concentration (33.9 vs. 28.0 nmol/L; P=0.02). However, the differences in large and small VLDL particle concentrations were no longer significant at 72 or 96 weeks. The medium VLDL particle concentration was not statistically different between OCA-treated and placebo groups at 12, 72 or 96 weeks.
Table 2.
Change in lipids over time by treatment group
| OCA (n=99) | Placebo (n=97) | Treatment Effect‡ | ||||||
|---|---|---|---|---|---|---|---|---|
| Serum-lipids | Visit | Mean (SD) | Change† from BL | Mean (SD) | Change† from BL | Mean | 95% CI | P-value* |
| Total cholesterol – mg/dL | BL | 191 (44) | 190 (48) | 0 | −13, 13 | 0.99 | ||
| F12 | 206 | 8% | 183 | −4% | 23 | 13, 33 | <0.0001 | |
| F72 | 197 | 3% | 185 | −3% | 12 | 2, 22 | 0.02 | |
| F96 | 176 | −8% | 179 | −6% | −3 | −12, 7 | 0.57 | |
| Triglycerides – mg/dL | BL | 219 (312) | 182 (178) | 37 | −35, 108 | 0.31 | ||
| F12 | 160 | −27% | 177 | −3% | −17 | −40, 5 | 0.13 | |
| F72 | 176 | −20% | 176 | 3% | 0 | −34, 34 | 1.00 | |
| F96 | 193 | −12% | 181 | 0% | 12 | −18, 41 | 0.43 | |
| HDL – mg/dL | BL | 42.6 (11.1) | 44.8 (13.9) | −2.2 | −5.7, 1.4 | 0.23 | ||
| F12 | 39.6 | −7% | 43.7 | −2% | −4.1 | −5.9, −2.3 | <0.0001 | |
| F72 | 42.6 | 0% | 45.1 | 1% | −2.6 | −4.6, −0.6 | 0.01 | |
| F96 | 43.9 | 3% | 44.2 | −1% | −0.2 | −2.3, 1.8 | 0.82 | |
| LDL – mg/dL | BL | 111 (39) | 114 (41) | −3 | −15, 8 | 0.59 | ||
| F12 | 137 | 23% | 108 | −5% | 30 | 22, 38 | <0.0001 | |
| F72 | 122 | 10% | 107 | −6% | 15 | 6, 24 | 0.001 | |
| F96 | 100 | −10% | 101 | −11% | −1 | −9, 8 | 0.89 | |
| NMR-VLDL | ||||||||
| Total – nmol/L | BL | 68.6 (38.4) | 59.6 (34.1) | 9.1 | −1.3, 19.2 | 0.09 | ||
| F12 | 61.5 | −10% | 63.3 | 6% | −1.8 | −8.3, 4.7 | 0.59 | |
| F72 | 59.3 | −14% | 58.3 | −2% | 1.1 | −5.6, 7.7 | 0.75 | |
| F96 | 60.0 | −13% | 60.6 | 2% | −0.6 | −7.6, 6.4 | 0.87 | |
| Large – nmol/L | BL | 9.8 (8.2) | 8.9 (8.6) | 0.9 | −1.5, 3.3 | 0.47 | ||
| F12 | 6.7 | −32% | 8.9 | 0% | −2.2 | −3.5, −0.9 | 0.001 | |
| F72 | 8.3 | −15% | 8.2 | −8% | 0.1 | −1.3, 1.6 | 0.85 | |
| F96 | 9.4 | −4% | 9.3 | 4% | 0.1 | −1.8, 1.9 | 0.93 | |
| Medium – nmol/L | BL | 27.3 (25.2) | 22.8 (23.5) | 4.5 | −2.4, 11.4 | 0.20 | ||
| F12 | 21.3 | −22% | 25.7 | 13% | −1.9 | −8.9, 0.2 | 0.06 | |
| F72 | 20.5 | −25% | 23.0 | 1% | −2.5 | −7.3, 2.2 | 0.29 | |
| F96 | 22.4 | −18% | 23.9 | 5% | −1.5 | −5.9, 3.0 | 0.52 | |
| Small – nmol/L | BL | 31.6 (20.3) | 28.0 (15.5) | 3.6 | −1.5, 8.7 | 0.16 | ||
| F12 | 34.2 | 8% | 28.1 | 0% | 6.0 | 1.1, 11.0 | 0.02 | |
| F72 | 30.7 | −3% | 26.8 | −4% | 3.9 | −0.6, 8.4 | 0.09 | |
| F96 | 28.5 | −10% | 26.8 | −4% | 1.9 | −3.0, 6.7 | 0.45 | |
| NMR-LDL | ||||||||
| Total – nmol/L | BL | 1390 (463) | 1424 (476) | −34 | −166, 98 | 0.61 | ||
| F12 | 1668 | 20% | 1335 | −6% | 333 | 230, 437 | <0.0001 | |
| F72 | 1522 | 9% | 1336 | −6% | 187 | 79, 294 | 0.0007 | |
| F96 | 1281 | −8% | 1278 | −10% | 3 | −89, 95 | 0.95 | |
| Large – nmol/L | BL | 288 (237) | 342 (273) | −54 | −126, 18 | 0.14 | ||
| F12 | 477 | 66% | 313 | −8% | 164 | 104, 225 | <0.0001 | |
| F72 | 399 | 39% | 336 | −2% | 63 | 6, 120 | 0.03 | |
| F96 | 272 | −6% | 287 | −16% | −15 | −66, 36 | 0.57 | |
| IDL – nmol/L | BL | 157 (125) | 165 (135) | −8 | −45, 29 | 0.66 | ||
| F12 | 176 | 12% | 151 | −8% | 25 | −11, 60 | 0.17 | |
| F72 | 168 | 7% | 162 | −2% | 6 | −31, 43 | 0.77 | |
| F96 | 129 | −18% | 155 | −6% | −26 | −56, 4 | 0.08 | |
| Small – nmol/L | BL | 946 (418) | 917 (390) | 28 | −85, 142 | 0.62 | ||
| F12 | 1013 | 6% | 872 | −5% | 141 | 51, 232 | 0.002 | |
| F72 | 955 | 1% | 838 | −9% | 117 | 22, 212 | 0.02 | |
| F96 | 878 | −7% | 839 | −9% | 39 | −45, 123 | 0.36 | |
| NMR-HDL | ||||||||
| Total – μmol/L | BL | 34.8 (6.2) | 34.4 (6.8) | 0.4 | −1.4, 2.2 | 0.68 | ||
| F12 | 30.0 | −14% | 34.9 | 1% | −4.8 | −6.0, −3.6 | <0.0001 | |
| F72 | 32.0 | −8% | 34.6 | 1% | −2.6 | −4.0, −1.2 | 0.0003 | |
| F96 | 33.4 | −4% | 35.2 | 2% | −1.8 | −3.2, −0.3 | 0.02 | |
| Large - μmol/L | BL | 6.1 (2.8) | 6.3 (3.4) | −0.2 | −1.1, 0.7 | 0.67 | ||
| F12 | 4.4 | −28% | 6.3 | 0% | −1.8 | −2.3, −1.3 | <0.0001 | |
| F72 | 4.9 | −20% | 6.1 | −3% | −1.2 | −1.8, −0.7 | <0.0001 | |
| F96 | 5.8 | −5% | 5.8 | −8% | −0.1 | −0.6, 0.5 | 0.82 | |
| Medium - μmol/L | BL | 6.5 (4.7) | 7.3 (5.4) | −0.7 | −2.1, 0.7 | 0.33 | ||
| F12 | 5.5 | −15% | 8.0 | 10% | −2.5 | −3.7, −1.2 | 0.0001 | |
| F72 | 5.6 | −15% | 7.2 | −1% | −1.6 | −2.9, −0.3 | 0.02 | |
| F96 | 7.1 | 9% | 8.0 | 10% | −0.8 | −2.1, 0.4 | 0.18 | |
| Small - μmol/L | BL | 22.1 (6.2) | 20.8 (5.7) | 1.3 | −0.4, 3.0 | 0.13 | ||
| F12 | 20.0 | −10% | 20.7 | 0% | −0.7 | −2.2, 0.8 | 0.35 | |
| F72 | 21.6 | −2% | 21.4 | 3% | 0.2 | −1.3, 1.8 | 0.78 | |
| F96 | 20.5 | −7% | 21.4 | 3% | −0.9 | −2.2, 0.5 | 0.20 | |
Based on ANOVA for differences at baseline and ANCOVA for differences during follow-up adjusting for baseline value
100*[(Mean at FU – Mean at BL) / Mean at BL]
Treatment effect is treatment group difference in change from baseline adjusted for value at baseline
Low Density Lipoprotein Sub-particles
The total LDL particle, intermediate density lipoprotein (IDL), large LDL, and small LDL particle concentrations were similar across the two groups at baseline. The baseline adjusted mean LDL particle concentrations in the OCA-treated group were elevated at 12 and 72 weeks at 1667 nmol/L and 1511 nmol/L, respectively, compared to the LDL particle concentrations of 1329 nmol/L (P<0.0001) and 1331 nmol/L (P=0.001) at 12 and 72 weeks of the placebo group. The increase in LDL particles was mirrored by a similar increase in large-buoyant LDL and small-dense LDL particle concentrations at 12 and 72 weeks. The large LDL particle concentration was 475 nmol/L in the OCA-treated group and 308 nmol/L in the placebo group at 12 weeks (P<0.0001) and 391 nmol/L in OCA group compared to 332 nmol/L in the placebo group at 72 weeks (P=0.04). Similarly, baseline adjusted mean small LDL particle concentration was higher in OCA vs placebo groups at 12 weeks (1015 vs. 872 nmol/L; P=0.002) and 72 weeks (959 nmol/L vs. 838 nmol/L; P=0.01). The total, large, and small LDL particle concentrations were similar between placebo and OCA-treated groups at 96 weeks. Finally, IDL concentrations at 12, 72 and 96 weeks were not different in OCA treatment compared to placebo.
High Density Lipoprotein Sub-Particles
The total HDL and HDL sub-particle (small, medium, and large) concentrations were similar between the treatment groups at baseline. The baseline adjusted mean total HDL concentrations of OCA-treated groups were 30.2 μmol/L at 12 weeks, 32.1 μmol/L at 72 weeks and 33.4 μmol/L compared to 35.0 μmol at 12 weeks (P<0.0001), 34.8 μmol/L at 72 weeks (P=<0.001) and 35.3 μmol/L at 96 weeks (P=0.01) in the placebo group. These changes were driven by reduction in large and medium HDL sub-particles in the OCA treated groups compared to placebo. OCA therapy did not affect small HDL sub-particles which were comparable to placebo group throughout therapy and after drug discontinuation.
Patterns of Lipid Lowering Therapy During the Study
Statin use was similar between the treatment groups at baseline (p=0.60) (Table 3). In the OCA group with complete statin use data, 36% (32/90) of patients were not on statin therapy at enrollment, 12 weeks and EOT; 46% (41/90) were on statin therapy at enrollment, 12 weeks and EOT; and 18% (17/90) patients transiently required statin therapy during the duration of the trial. In the placebo group with complete statin use data, 42% (39/92) of patients were not on statin therapy at enrollment, 12 weeks and EOT; 39% (36/92) were on statin therapy at enrollment, 12 weeks and EOT; and 19% (17/92) on intermittent use of statin therapy during the duration of the trial. There were no significant interaction effects of the OCA vs placebo differences by time-dependent statin use for any lipoprotein particle at baseline or baseline-adjusted change in any of the lipoprotein particles at 12 weeks, 72 weeks or 96 weeks (Appendix Table 2). When treatment groups were stratified according statin use, patients on OCA had increased total LDL concentrations, small LDL, and a reduction in large HDL at week 12 (Appendix Table 3). In the OCA group, 6 patients were on ezetimibe at baseline and 2 additional patients were started on ezetimibe during follow up. In the placebo cohort, 6 patients were on ezetimibe at baseline, 1 stopped during follow up and ezetimibe was not initiated in any patients during the study course. No patients took clofibrate during the course of the study.
Table 3.
Statin use by treatment group
| Statin use history | OCA (n=98)* | Placebo (n=97) | P-value |
|---|---|---|---|
| 0.60 | |||
| Not taking at baseline, week 12 or 72 | 33 (34%) | 39 (40%) | |
| Taking at baseline, week 12 and F72 | 46 (47%) | 39 (40%) | |
| Transient use at baseline, week 12 and 72 | 19 (19%) | 19 (20%) |
Excludes one patient with missing statin use data at F12 and F72
Cardiovascular Events in OCA Treatment Arm
There were 14 (14%) OCA patients who developed an adverse event related to cardio-vascular disease (CVD) during the trial. There were no significant differences between those developing vs not developing CVD adverse event in baseline-adjusted changes in any lipoprotein particle at 12 weeks, 72 weeks or 96 weeks (Appendix Table 3).
DISCUSSION
The activation of FXR by OCA led to an increase in LDL-C during the FLINT trial4 and in the present study we further explored this observation by demonstrating the impact of OCA therapy on the lipoprotein subfractions. The relationship of elevated LDL-C on CVD risk is well established21 and the impact of an increase in LDL-C with OCA therapy compared to observed histological benefit regarding the long-term outcome remains undefined. The findings from the FLINT trial have served a major impetus for concomitant use of statin therapy22 and a priori guidelines with medications targeting the bile acid pathway for treatment of NAFLD.
The findings of the current study must be evaluated in the context of published literature that have demonstrated the relationship between atherogenic dyslipidemia and NASH15,16. NASH is associated with upregulation of hepatic synthesis of triglyceride and cholesterol, which are transported out of the liver in large VLDL particles16. These large triacylglycerol-rich VLDL particles, which are slowly metabolized in the periphery, are subject to a cholesteryl ester transfer protein (CETP)-mediated exchange process that removes cholesteryl ester from the particle core and replaces it with triacylglycerol23. LDL, so altered, is a potential substrate for hepatic lipase which can then result in the generation of smaller, more atherogenic lipoproteins often called small-dense LDL24,25. In contrast, metabolism of cholesteryl ester enriched small VLDL particles results in the formation of large-buoyant LDL particles, which are thought to be less atherogenic26. In the present study, OCA treatment resulted in a reduction in large VLDL particles and an increase in small VLDL particles, while total VLDL lipoprotein particle concentration was not affected by OCA therapy. These findings are consistent with published literature showing FXR reduces triglyceride-rich VLDL lipoproteins by repressing hepatic SREBP1c,MTTP and apoB expression5–7,27. Thus, the net effect of FXR agonism is a shift towards small VLDL particles, which are a reflection of a less atherogenic lipoprotein profile.
The VLDL particle is hydrolyzed by lipoprotein lipase and slow metabolism is a key prerequisite for formation of atherogenic small-dense LDL particles26. FXR activation amplifies effects of lipoprotein lipase by increasing expression of apoCII, an activator of lipoprotein lipase, and reducing expression of apoCIII, an inhibitor of lipoprotein lipase, thereby promoting intravascular VLDL lipolysis and generation of LDL28,29. Furthermore, since FXR inhibits bile acid synthesis from cholesterol, it leads to increased intrahepatic cholesterol content27. Increased intrahepatic cholesterol reduces LDL-receptor activity and concentration. Thus, increased lipoprotein lipase activity, coupled with reduction in the LDL-receptor, results in reduced LDL clearance and elevated concentrations of circulating LDL particles and LDL-C30. In the present study, we observed an increase in LDL-C as early as 12 weeks of therapy which persisted for the duration of therapy and returned to baseline after OCA discontinuation. Expectedly, we also observed an increase in both large-buoyant LDL and small-dense LDL particles while the patients were on OCA therapy and an improvement with OCA discontinuation. We also note that no statistically significant changes in atherogenic IDL were observed with OCA therapy, although there was a non-significant increase in IDL particles with OCA therapy. The exact mechanism underlying these changes is not forthcoming from the present study, but may be potentially related increased lipoprotein lipase activity30.
HDL particles also play a key role in atherogenesis and CVD and are better predictors of cardiovascular risk than HDL-C19,31,32 FXR agonism has been shown to increase HDL-C clearance by increasing CETP expression and hepatic scavenger receptor-B1 and decreasing HDL production by decreasing apolipoprotein A-I30,33–36. In the present study, OCA treatment led to a reduction in HDL-C, along with a reduction in total, large and medium HDL particle concentrations, particularly after 12 weeks of therapy, and the effect persisted until EOT. As with VLDL and LDL, HDL-C and HDL particle concentrations improved after OCA cessation.
The current study provides greater granularity regarding OCA and dyslipidemia using the FLINT trial, however, the findings should be evaluated in the context of study limitations. The data presented show correlative relationships but do not explore the key mechanisms responsible for the dyslipidemia associated with OCA use. In the FLINT trial, the rise in total cholesterol and LDL-C was not fully anticipated, thus a standardized lipid management plan was not part of the clinical trial design. In addition, the dose of statin therapy was not collected. Within this limitation, it is difficult to ascertain how statin therapy might have impacted the observed lipid changes with OCA therapy and the results presented with regards to statin use on lipoprotein profile must be interpreted with caution. Future mechanistic studies incorporating statin therapy as clinical designs are necessary to evaluate this relationship further. While data linking atherogenic lipoprotein sub-particles to CVD events exists in the cardiovascular literature, similar data is limited in patients with NASH and the current study provides the framework to evaluate this association further. Well designed prospective studies with standardized lipid lowering therapeutic regimens are essential to evaluate not only the impact of lipoproteins on cardiovascular events but also the impact of lipid lowering therapy on lipoproteins in patients with NASH. Furthermore, ongoing clinical trials with FXR agonists and fibroblast growth factor (FGF19) analogues should provide additional data on the interactions between bile acid and lipid metabolism. Since diet data was not collected as part of the FLINT trial, it is difficult to ascertain how the diet might have impacted lipoprotein profile in the present study.
In summary, OCA treatment was associated with an increase in LDL-C and total cholesterol which was largely due to an increase in less-atherogenic small VLDL and large-buoyant LDL particles. Co-administration of statin therapy with FXR agonist may potentially blunt the increases in lipoprotein sub-particles associated with FXR agonism, however, this strategy requires further validation.
Supplementary Material
Figure 1:

The change in lipoproteins in patients treated with Obeticholic acid and placebo over the study duration.
HDL; high-density lipoprotein, LDL; low density lipoprotein, VLDL; very low density lipoprotein
Highlights:
Obeticholic acid (OCA) has shown promise as a potential treatment of NASH
OCA is associated with increase in LDL-cholesterol, triglycerides, total cholesterol and reduction in HDL-cholesterol
Deep lipid analysis showed increase in atherogenic (i.e. small dense LDL-cholesterol) and less atherogenic lipoproteins (i.e. large LDL particles) after 12 weeks of therapy which improve after discontinuation of OCA
OCA therapy also lead to reduction in large and medium HDL sub-particles which remained significantly lower than placebo treated group even after drug discontinuation
Acknowledgments
FUNDING INFORMATION: The Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grants U01DK061718, U01DK061728, U01DK061731, U01DK061732, U01DK061734, U01DK061737, U01DK061738, U01DK061730, U01DK061713). Additional support is received from the National Center for Advancing Translational Sciences (NCATS) (grants UL1TR000439, UL1TR000436, UL1TR000006, UL1TR000448, UL1TR000100, UL1TR000004, UL1TR000423, UL1TR000058).
Lipoprotein samples were tested free of charge at LipoScience (now LabCorp) and the agreement was between LabCorp and VCU (Arun Sanyal) for NMR testing. the analysis, interpretation and manuscript preparation was done by the authors and independent on LabCorp.
FINANCIAL DISCLOSURE:
| Mohammad Siddiqui: | Advisory board member with Pfizer |
| Mark Van Natta | None |
| Margery Connelly | Employed by LabCorp® |
| Raj Vuppalanchi | |
| Brent Tetri | Consulting or advising relationships with Allergan, Arrowhead, ARTham, Blade, Boehringer Ingleheim, BMS, Coherus, Consynance, Durect, Enanta, Gelesis, Gilead, Intercept, Lipocine, Madrigal, Medimmune, Merck, Metacrine, Mundipharma, NGM, pH-Pharma, Prometheus, Siemens |
| James Tonascia | None |
| Cynthia Guy | consulting agreements with several pharmaceutical companies including Immuron, Taiwan J, Madrigal, CymaBay, and NGM related to NASH and other chronic liver diseases, and her institution receives or has received research grants on her behalf from some these entities. |
| Rohit Loomba | consultant or advisory board member for Arrowhead Pharmaceuticals, AstraZeneca, Bird Rock Bio, Boehringer Ingelheim, Bristol-Myer Squibb, Celgene, Cirius, CohBar, Conatus, Eli Lilly, Galmed, Gemphire, Gilead, Glympse bio, GNI, GRI Bio, Intercept, Ionis, Janssen Inc., Merck, Metacrine, Inc., NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Prometheus, Sanofi, Siemens, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Boehringer-Ingelheim, Bristol-Myers Squibb, Cirius, Eli Lilly and Company, Galectin Therapeutics, Galmed Pharmaceuticals, GE, Genfit, Gilead, Intercept, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, NuSirt, Pfizer, Prometheus, and Siemens. He is also co-founder of Liponexus, Inc. |
| Srinivasan Dasarathy | None |
| Julia Wattacheril | Paid consulting activities with Astra Zeneca and has received research support from Janssen, Genfit, Intercept, Galectin, Gilead, Zydus, Conatus, Shire. |
| Naga Chalasani: | Ongoing consulting activities (or had in preceding 12 months) with NuSirt, Abbvie, Afimmune (DS Biopharma), Axovant, Allergan (Tobira), Madrigal, Shire, Coherus, Siemens, Centurion, and Genentech. These consulting activities are generally in the areas of nonalcoholic fatty liver disease and drug hepatotoxicity. Dr. Chalasani receives research grant support from Exact Sciences, Intercept, Lilly, Galectin Therapeutics and Cumberland where his institution receives the funding. Over the last decade, Dr. Chalasani has served as a paid consultant to more than 30 pharmaceutical companies and these outside activities have regularly been disclosed to his institutional authorities. |
| Arun J. Sanyal | Conatus, GenFit, Gilead, Mallinckrodt, Pfizer, Salix, Boehringer Ingelhiem, Novartis, Nimbus, Merck, Hemoshear, Sequana, Lilly, Novo Nordisk, Fractyl, Durect, Indalo, Allergan, Chemomab, Affimmune, Ardelyx, Teva, Terns, Enyo, Birdrock, Albrieo, Sanofi, Jannsen, Takeda, Zydus, BASF, Amra, Perspectum, OWL, Poxel, Servier, Second Genome, General Electric, 89 Bio, Immuron, Intercept. Stock in Exhalenz, Akarna, GenFit, Hemoshear, Durect, indalo, Tiziana. Owner of Sanyal Bio. Research Grants: Conatus, Echosens-Sandhill, Gilead, Mallinckrodt, Salix, Novartis, Galectin, Bristol Myers, Sequana. Royalties: Elsevier, UpToDate |
FLINT:
The FLINT trial was conducted by the NASH CRN and supported in part by a Collaborative Research and Development Agreement (CRADA) between NIDDK and Intercept Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ClinicalTrials.gov numbers:
REFERENCES CITED
- 1.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–1395. doi: 10.1002/hep.20466 [DOI] [PubMed] [Google Scholar]
- 2.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701 [DOI] [PubMed] [Google Scholar]
- 3.Shadab Siddiqui M, Harrison SA, Abdelmalek MF, et al. Case definitions for inclusion and analysis of endpoints in clinical trials for NASH through the lens of regulatory science. Hepatology. October 2017. doi: 10.1002/hep.29607 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113(10):1408–1418. doi: 10.1172/JCI21025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. 2009;284(15):10023–10033. doi: 10.1074/jbc.M808818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokane H, Nakahara M, Tachibana S, Shimizu M, Sato R. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J Biol Chem. 2004;279(44):45685–45692. doi: 10.1074/jbc.M404255200 [DOI] [PubMed] [Google Scholar]
- 8.Hameed B, Terrault NA, Gill RM, et al. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2018;47(5):645–656. doi: 10.1111/apt.14492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekstedt M, Franzen LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–873. doi: 10.1002/hep.21327 [DOI] [PubMed] [Google Scholar]
- 10.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42(1):132–138. doi:S0168-8278(04)00435-0 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Bhala N, Angulo P, van der Poorten D, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collaborative study. Hepatology. 2011;54(4):1208–1216. doi: 10.1002/hep.24491; 10.1002/hep.24491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cromwell WC, Otvos JD, Keyes MJ, et al. LDL particle number and risk of future cardiovascular disease in the Framingham Offspring Study—Implications for LDL management. J Clin Lipidol. 2007;1(6):583–592. doi: 10.1016/j.jacl.2007.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ai M, Otokozawa S, Asztalos BF, et al. Small Dense LDL Cholesterol and Coronary Heart Disease: Results from the Framingham Offspring Study [Lipids, Lipoproteins, and Cardiovascular Risk Factors]. Clin Chem. 2010. doi: 10.1373/clinchem.2009.137489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koba S, Hirano T, Ito Y, et al. Significance of small dense low-density lipoprotein-cholesterol concentrations in relation to the severity of coronary heart diseases. Atherosclerosis. 2006; 189(1):206–214. doi: 10.1016/j.atherosclerosis.2005.12.002 [DOI] [PubMed] [Google Scholar]
- 15.Siddiqui MS, Sterling RK, Luketic VA, et al. Association Between High-Normal Levels of Alanine Aminotransferase and Risk Factors for Atherogenesis. Gastroenterology. 2013. doi: 10.1053/j.gastro.2013.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siddiqui MS, Fuchs M, Idowu M, et al. Severity of Nonalcoholic Fatty Liver Disease and Progression to Cirrhosis Associate With Atherogenic Lipoprotein Profile. Clin Gastroenterol Hepatol. October 2014. doi:S1542-3565(14)01467-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeyarajah EJ, Cromwell WC, Otvos JD. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin Lab Med. 2006;26(4):847–870. doi: 10.1016/j.cll.2006.07.006 [DOI] [PubMed] [Google Scholar]
- 18.Matyus SP, Braun PJ, Wolak-Dinsmore J, et al. NMR measurement of LDL particle number using the Vantera Clinical Analyzer. Clin Biochem. 2014;47(16-17):203–210. doi: 10.1016/j.clinbiochem.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 19.Matyus SP, Braun PJ, Wolak-Dinsmore J, et al. HDL particle number measured on the Vantera®, the first clinical NMR analyzer. Clin Biochem. 2015;48(3):148–155. doi: 10.1016/j.clinbiochem.2014.11.017 [DOI] [PubMed] [Google Scholar]
- 20.Treeprasertsuk S, Leverage S, Adams LA, Lindor KD, St Sauver J, Angulo P. The Framingham risk score and heart disease in nonalcoholic fatty liver disease. Liver Int. 2012;32(6):945–950. doi: 10.1111/j.1478-3231.2011.02753.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459–2472. doi: 10.1093/eurheartj/ehx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinella ME, Trotter JF, Abdelmalek MF, et al. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol. 2019;70(4):735–744. doi: 10.1016/j.jhep.2018.11.032 [DOI] [PubMed] [Google Scholar]
- 23.Yamada N, Shames DM, Takahashi K, Havel RJ. Metabolism of apolipoprotein B-100 in large very low density lipoproteins of blood plasma. Kinetic studies in normal and Watanabe heritable hyperlipidemic rabbits. J Clin Invest. 1988;82(6):2106–2113. doi: 10.1172/JCI113832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Packard CJ, Demant T, Stewart JP, et al. Apolipoprotein B metabolism and the distribution of VLDL and LDL subfractions. J Lipid Res. 2000;41(2):305–318. [PubMed] [Google Scholar]
- 25.Packard CJ. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem Soc Trans. 2003;31(Pt 5): 1066–1069. doi:10.1042/ [DOI] [PubMed] [Google Scholar]
- 26.Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43(9):1363–1379. [DOI] [PubMed] [Google Scholar]
- 27.Papazyan R, Liu X, Liu J, et al. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J Lipid Res. 2018;59(6):982–993. doi: 10.1194/jlr.M081935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kast HR, Nguyen CM, Sinai CJ, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. 2001;15(10):1720–1728. doi: 10.1210/mend.15.10.0712 [DOI] [PubMed] [Google Scholar]
- 29.Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CM I expression. Gastroenterology. 2003;125(2):544–555. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh Laskar M, Eriksson M, Rudling M, Angelin B. Treatment with the natural FXR agonist chenodeoxycholic acid reduces clearance of plasma LDL whilst decreasing circulating PCSK9, lipoprotein(a) and apolipoprotein C-III. J Intern Med. 2017;281(6):575–585. doi: 10.1111/joim.12594 [DOI] [PubMed] [Google Scholar]
- 31.Williams PT, Feldman DE. Prospective study of coronary heart disease vs. HDL2, HDL3, and other lipoproteins in Gofman’s Livermore Cohort. Atherosclerosis. October 2010. doi: 10.1016/j.atherosclerosis.2010.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharrett AR, Ballantyne CM, Coady SA, et al. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001; 104(10): 1108–1113. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Yin L, Anderson J, et al. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J Biol Chem. 2010;285(5):3035–3043. doi: 10.1074/jbc.M109.083899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gautier T, de Haan W, Grober J, et al. Farnesoid X receptor activation increases cholesteryl ester transfer protein expression in humans and transgenic mice. J Lipid Res. 2013;54(8):2195–2205. doi: 10.1194/jlr.M038141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chao F, Gong W, Zheng Y, et al. Upregulation of scavenger receptor class B type I expression by activation of FXR in hepatocyte. Atherosclerosis. 2010;213(2):443–448. doi: 10.1016/j.atherosclerosis.2010.09.016 [DOI] [PubMed] [Google Scholar]
- 36.Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109(7):961–971. doi: 10.1172/JCI14505 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.