

Abstract
The development of novel inhibitors of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) represents a viable approach to alleviate Alzheimer’s disease. Thirty-six halogenated 2-hydroxy-N-phenylbenzamides (salicylanilides) with various substitution patterns and their esters with phosphorus-based acids were synthesized in yields of 72% to 92% and characterized. They were evaluated for in vitro inhibition of AChE from electric eel and BuChE from equine serum using modified Ellman’s spectrophotometric method. The benzamides exhibited a moderate inhibition of AChE with IC50 values in a narrow concentration range from 33.1 to 85.8 µM. IC50 values for BuChE were higher (53.5–228.4 µM). The majority of derivatives inhibit AChE more efficiently than BuChE and are comparable or superior to rivastigmine—an established cholinesterases inhibitor used in the treatment of Alzheimer’s disease. Phosphorus-based esters especially improved the activity against BuChE with 5-chloro-2-{[4-(trifluoromethyl)phenyl]carbamoyl}phenyl diethyl phosphite 5c superiority (IC50 = 2.4 µM). This derivative was also the most selective inhibitor of BuChE. It caused a mixed inhibition of both cholinesterases and acted as a pseudo-irreversible inhibitor. Several structure-activity relationships were identified, e.g., favouring esters and benzamides obtained from 5-halogenosalicylic acids and polyhalogenated anilines. Both 2-hydroxy-N-phenylbenzamides and esters share convenient physicochemical properties for blood-brain-barrier penetration and thus central nervous system delivery.
Keywords: acetylcholinesterase, benzamides, butyrylcholinesterase, enzyme inhibition, esters, in vitro inhibition, phosphorus derivatives, salicylanilides
1. Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease and the most common cause of dementia, with heavy social and economic costs and problems, which gradually worsen over a number of years [1,2]. Fifty million people worldwide are living with dementia and the World Alzheimer report 2018 estimates an incensement to more than 152 million cases by 2050 [3]. The disease is associated with a loss of cholinergic neurons in the brain and decreased levels of neurotransmitter acetylcholine (ACh). Enzymes called cholinesterases terminate its action. The brain contains two major forms of cholinesterases: acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). Both enzymes may have roles in the aetiology and progression of AD beyond regulation of synaptic ACh levels. They differ genetically, structurally and in their kinetics. The enzyme acetylcholinesterase (a serine hydrolase found at neuromuscular junctions and cholinergic brain synapses) is involved in the termination of impulse transmission by rapid hydrolysis of ACh in numerous cholinergic pathways in the central and peripheral nervous systems. Butyrylcholinesterase serves as a co-regulator of AChE activity [4]. While the overall amount of AChE in the brain decreases with the progression of AD, the enzymatic activity of BuChE remains unchanged or even increases and can compensate the loss of AChE [5]. The presence of AChE in the patients’ brains is associated with β-amyloid plaques and neurofibrillary tangles (NFT), which are the hallmarks of the AD pathogenesis. Increased AChE levels have been discovered around amyloid plaques and NFT [6]. Analogously, overexpression of BuChE is involved in the formation of β-amyloid protein and senile plaques. Therefore, inhibiting BuChE activity could prove beneficial as a hypothetically disease-modifying approach to AD; BuChE is considered as an adjunctive therapeutic target in later stages of AD [7].
Based on these facts, the main medicines used in the treatment of AD are AChE inhibitors. In addition to potent enzyme inhibition, these molecules must cross the blood-brain barrier (BBB) to be sufficiently available in the central nervous system (CNS). According to the mode of action, AChE inhibitors are irreversible or reversible. Reversible inhibitors, competitive or non-competitive (such as the current AD drugs donepezil and galantamine) and pseudo-irreversible inhibitor rivastigmine, a unique dual inhibitor of AChE and BuChE [8], have therapeutic applications, while toxic effects are normally associated with irreversible AChE inhibitors such as organophosphate pesticides and nerve warfare agents.
Moreover, inhibitors of cholinesterases can be used in the therapy of other diseases than AD, such as other dementias, sleep behaviour disorder, myasthenia gravis, glaucoma, postoperative ileus, bladder distention and intoxication of anticholinergic agents [9,10,11]. Cholinergic hypothesis was the first comprehensive paradigm to explain the memory deficit and cognitive impairment in AD. After that, the loss of cholinergic neurons has been associated with other neurodegenerative disorders. Later, the cholinergic hypothesis in dementia has lost some interest, in particular because of the limited clinical efficacy of ACh augmentation therapy and the rising highlight of the role of Aβ and tau protein pathology. Recently, the cholinergic hypothesis has been “revisited” due to many reasons [12]. In addition to this approach, other hypotheses have been formulated, e.g., amyloid cascade, hyperphosphorylation of tau protein, oxidative stress, metal ions hypothesis, or inflammatory hypothesis [13].
Current treatment cannot stop the disease from progressing; it can only temporarily improve symptoms or slow down the progress at best. Thus, a worldwide effort is needed to find better ways to treat the Alzheimer’s disease, delay its onset, and prevent it from developing. Despite multiple features of AD pathogenesis and a wide range of proposed potential targets, no better therapeutic interventions than inhibitors of cholinesterases and NMDA-receptor antagonist memantine have been introduced. AChE inhibition may decrease Aβ plaques and NFT formation. Despite moderate clinical efficacy of cholinesterases inhibitors, AChE is one of the most investigated targets for AD [6]. Thus, AChE- and BuChE-oriented inhibitors-related research can still be considered useful. In particular, these dual inhibitors modulating also other pathogenesis pathway(s) are up-to-date.
2-Hydroxy-N-phenylbenzamides (salicylanilides or 2-hydroxybenzanilides) are weakly acidic phenolic compounds. Depending on their substitution, they can be divided into different groups, including halogenated (mono-, di- and polyhalogenated compounds) and nonhalogenated derivatives, O-esters including carbamates with antibacterial, mainly antimycobacterial and antifungal activity [14,15,16,17,18]. Several halogenated salicylanilides are widely used as veterinary drugs against helminths and ectoparasites [19]. Commercially available closantel, niclosamide, oxyclozanide, rafoxanide and resorantel are also potent against Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pyogenes or Propionibacterium acnes [20]. Niclosamide and N-(3,5-dichlorophenyl)-5-chloro-2-hydroxybenzamide exhibited anti-filamentation and anti-biofilm activity against fungus Candida albicans, and multidrug-resistant yeast Candida auris [21].
We have found salicylanilide O-substituted derivatives with significant activity against AChE and BuChE. Diethyl phosphates act as pseudo-irreversible cholinesterases inhibitors [22], N-phenethyl [23] and N,N-diphenyl carbamates [24] produced clearly favourable BuChE inhibition when compared to other analogues. In addition, salicylanilide diethyl thiophosphates were active, especially against AChE [25]—but there is no study dealing with the inhibition property of parent benzamides with free phenolic group. That is why we initiated this systematic study dealing with the halogenated salicylanilides as potential inhibitors of both cholinesterases and their phosphorus analogues. The halogenation ensures sufficient lipophilicity, which should contribute to the passive targeting of CNS. The current study should answer whether parent salicylanilides are able to inhibit AChE and BuChE, how selective and if the intrinsic activity of this scaffold could participate in the inhibition previously described for salicylanilide carbamates and (thio)phosphates [22,23,24,25]. The second goal is focused on the influence of esterification by phosphorus-based acids on enzyme inhibition and selectivity. The compounds with low IC50 values and dual inhibition of AChE and BuChE with optimal physicochemical properties to penetrate BBB were aimed at in this study.
2. Materials and Methods
2.1. Chemistry
2.1.1. General Methods
All the reagents and solvents were purchased from Sigma-Aldrich (Darmstadt, Germany) or Penta Chemicals (Prague, Czech Republic) and they were used as received. The reactions and the purity of the products were monitored by thin-layer chromatography using a mixture with a ratio of toluene to ethyl acetate of 4:1 or 9:1 (v/v) as the eluent. Plates were coated with 0.2 mm Merck 60 F254 silica gel (Merck Millipore, Darmstadt, Germany) and were visualized by UV irradiation (254 nm). The melting points were determined on a Büchi Melting Point B-540 apparatus (BÜCHI, Flawil, Switzerland) using open capillaries. The reported values are uncorrected. Infrared spectra were recorded on a FT-IR spectrometer using ATR-Ge method (Nicolet 6700 FT-IR, Thermo Fisher Scientific, Waltham, MA, USA) in the range 650–4000 cm−1. The NMR spectra were measured in DMSO-d6, CDCl3 or acetone-d6 at ambient temperature using a Varian V NMR S500 instrument (500 MHz for 1H and 126 MHz for 13C; Varian Comp. Palo Alto, CA, USA). The chemical shifts δ are given in ppm with respect to tetramethylsilane as an internal standard. The coupling constants (J) are reported in Hz. Elemental analysis (C, H, N) was performed on an automatic microanalyser CHNS-O CE instrument (FISONS EA 1110, Milano, Italy). Flash chromatography was performed on a CombiFlash Rf 200 automated chromatograph (Teledyne Isco, Lincoln, NE, USA) using columns filled with Kieselgel 60, 0.040–0.063 mm (Merck, Germany) and a detection wavelength of 280 nm.
2.1.2. Synthesis of 2-Hydroxy-N-phenylbenzamides 1–4
The microwave-assisted synthesis of salicylanilides 1–4 was described previously by our group (e.g., [26]). Briefly, appropriate salicylic acid (1 mmol) was suspended in 10 mL of chlorobenzene, then substituted aniline (1 mmol) was added, followed by phosphorus trichloride (0.5 mmol). The reaction mixture was irradiated in microwave reactor (530 W, 25 min; MicroSYNTH Milestone Ethos 1600 (Milestone, Bergamo, Italy)). The reaction mixture was filtered while hot and was then let to stand at +4 °C for 12 h. The resulted precipitate was filtered off and crystallized from ethanol/water to obtain pure benzamides.
Some of the compounds 1–4 were synthesized and reported previously by our group. For this study, we adopted the salicylanilides from two works: Krátký et al. [26] (compounds 1, 2a–2c, 2f–2k, 3a–3c, 3f–3k, and 4k) and Paraskevopoulos et al. [15] (derivatives 2l, 3l, 4c, 4g, and 4l). Bromosalicylanilides (4a, 4b, 4f, 4h–4j) were prepared as synthetic precursors of salicylanilide diethyl phosphates [17].
The identity of the known compounds was established using NMR and IR spectroscopy by the comparison with previously reported data. Additionally, their purity was checked by melting points measurement and elemental analysis.
5-Chloro-N-(3,5-dichlorophenyl)-2-hydroxybenzamide (2d). White solid; yield 82%; mp 246–248 °C (247–249 °C [27]). IR: 3324 (N-H), 1630 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.54 (1H, s, OH), 10.54 (1H, s, NH), 8.09 (2H, d, J = 2.3 Hz, H2′, H6’), 7.85 (1H, d, J = 2.7 Hz, H6), 7.45 (1H, dd, J = 8.8, 2.7 Hz, H4), 7.37 (1H, t, J = 2.3 Hz, H4′), 7.01 (1H, d, J = 8.8 Hz, H3). 13C NMR (126 MHz, DMSO): δ 165.2, 156.7, 139.8, 133.3, 130.6, 128.7, 124.1, 123.0, 120.3, 120.2, 119.2. Anal. C 49.41, H 2.31, N 4.51%, calculated for C13H8Cl3NO2 (316.56), C 49.32, H 2.55, N 4.42%.
5-Chloro-N-(3,4,5-trichlorophenyl)-2-hydroxybenzamide (2e). White solid; yield 79%; mp 286–288.5 °C (287–290 °C [27]). IR: 3323 (N-H), 1628 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.53 (1H, s, OH), 10.63 (1H, s, NH), 8.04 (2H, s, H2′, H6′), 7.83 (1H, d, J = 2.7 Hz, H6), 7.45 (1H, dd, J = 8.6, 2.5 Hz, H4), 7.05 (1H, d, J = 8.8 Hz, H3). 13C NMR (126 MHz, DMSO): δ 165.8, 157.4, 149.8, 139.0, 134.6, 132.2, 129.4, 123.0, 120.7, 119.2, 113.5. Anal. C 44.34, H 1.94, N 4.00%, calculated for C13H7Cl4NO2 (351.00), C 44.48, H 2.01, N 3.99%.
4-Chloro-N-(3,5-dichlorophenyl)-2-hydroxybenzamide (3d). White solid; yield 80%; mp 257–259 °C (255–256 °C [28]). IR: 3310 (N-H), 1605 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.70 (1H, s, OH), 10.49 (1H, s, NH), 7.84–7.78 (3H, m, H6, H2′, H6′), 7.31 (1H, t, J = 1.9 Hz, H4′), 7.05–6.99 (2H, m, H3, H5). 13C NMR (126 MHz, DMSO): δ 166.2, 159.0, 140.9, 137.7, 133.7, 131.4, 122.3, 120.1, 118.8, 118.2, 116.4. Anal. C 49.60, H 2.41, N 4.70%, calculated for C13H8Cl3NO2 (316.56), C 49.32, H 2.55, N 4.42%.
4-Chloro-N-(3,4,5-trichlorophenyl)-2-hydroxybenzamide (3e). White solid; yield 76%; mp 260–262 °C (261–262 °C [29]). IR (ATR): 3320 (N-H), 1609 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.75 (1H, s, OH), 10.55 (1H, s, NH), 8.03 (2H, s, H2′, H6′), 7.83 (1H, d, J = 8.5 Hz, H6), 7.06 (1H, d, J = 2.1 Hz, H3), 7.02 (1H, dd, J = 8.5, 2.1 Hz, H5). 13C NMR (126 MHz, DMSO): δ 165.6, 158.3, 138.6, 136.5, 133.0, 131.4, 120.6, 119.6, 119.2, 116.9, 114.0. Anal. C 44.40, H 1.97, N 4.10%, calculated for C13H7Cl4NO2 (351.00), C 44.48, H 2.01, N 3.99%.
5-Bromo-N-(3,5-dichlorophenyl)-2-hydroxybenzamide (4d). White solid; yield 81%; mp 244–245.5 °C (243–245 °C [28]). IR: 3327 (N-H), 1628 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.47 (1H, s, OH), 10.54 (1H, s, NH), 7.93 (1H, d, J = 2.5 Hz, H6), 7.81 (2H, d, J = 1.9 Hz, H2′, H6′), 7.56 (1H, dd, J = 8.8, 2.6 Hz, H4), 7.33 (1H, t, J = 1.9 Hz, H4′), 6.95 (1H, d, J = 8.8 Hz, H3). 13C NMR (126 MHz, DMSO): δ 165.2, 156.8, 140.8, 136.1, 134.2, 131.6, 123.5, 121.1, 119.6, 118.4, 110.4. Anal. C 43.40, H 2.27, N 3.75%, calculated for C13H8BrCl2NO2 (361.02), C 43.25, H 2.23, N 3.88%.
5-Bromo-N-(3,4,5-trichlorophenyl)-2-hydroxybenzamide (4e). White solid; yield 78%; mp 297–298.5 °C (297–299 °C [28]). IR: 3322 (N-H), 1634 (C=O) cm−1. 1H-NMR (500 MHz, DMSO): δ 11.46 (1H, s, OH), 10.57 (1H, s, NH), 8.01 (2H, s, H2′, H6′), 7.91 (1H, d, J = 2.6 Hz, H6), 7.56 (1H, dd, J = 8.8, 2.6 Hz, H4), 6.95 (1H, d, J = 8.8 Hz, H3). 13C NMR (126 MHz, DMSO): δ 165.2, 156.7, 138.6, 136.2, 133.5, 131.7, 124.4, 121.0, 120.6, 119.6, 110.4. Anal. C 39.41, H 1.91, N 3.55%, calculated for C13H7BrCl3NO2 (395.46), C 39.48, H 1.78, N 3.54%.
2.1.3. Synthesis of Phosphorus-Based Derivatives 5
The syntheses of diethyl phosphate 5a and thiophosphate 5b were reported previously by our group [17,25]. It is based on the reaction of appropriate salicylanilide with a mild excess of diethyl chlorophosphate or diethyl chlorothiophosphate in the presence of a tertiary base.
Diethyl phosphite 5c was obtained by the following procedure: 1 mmol of the salicylanilide 3k was suspended in 10 mL of dichloromethane (DCM) and treated by triethylamine (1.5 of equivalents). After a complete dissolution, diethyl chlorophosphite was added in one portion (1.5 of equivalents). The reaction mixture was stirred for 24 h at room temperature. Then, it was evaporated to provide a viscous liquid. Ethyl acetate was added, and the insoluble portion was filtered off. The filtrate was washed sequentially by sodium carbonate solution (10%), hydrochloric acid (0.1 M) and saturated brine. The organic phase was dried using sodium sulphate and evaporated to provide a viscous oily liquid, which was purified using flash chromatography (mobile phase composition was with a ratio of ethyl acetate to hexane of 1:3 (v/v)).
Cyclic analogues 5d and 5e were synthesized by modified procedure adopted from [30]. 1 mmol of the salicylanilide 3k was suspended in 10 mL of dichloromethane (DCM) and treated by triethylamine (3 of equivalents). After a complete dissolution, phenylphosphonic dichloride (or phenylphosphonothioic dichloride) was added in one portion (1.5 of equivalents). The reaction mixture was heated for 4 h and then stirred at room temperature. After 20 h, it was evaporated to dryness. The resulting crystals were suspended in ethyl acetate. The insoluble portion was filtered off and the filtrate was washed sequentially by sodium carbonate solution (10%), hydrochloric acid (0.1 M) and saturated brine. The organic phase was dried using sodium sulphate and the crystallization was initiated by the addition of n-hexane. The reaction mixture was stored for 24 h at +4 °C. Then, the resulting precipitate was filtered off and the crystals were recrystallized from ethyl acetate/hexane mixture.
5-Chloro-2-{[4-(trifluoromethyl)phenyl]carbamoyl}phenyl diethyl phosphite (5c). Brownish viscous oil; yield 28%. IR: 3310 (N-H), 1684 (C=O), 1601, 1323, 1292, 1165, 1118, 1066, 1033, 964, 845, 766, 689 cm−1. 1H-NMR (500 MHz, acetone-d6): δ 9.99 (1H, s, NH), 8.06-8.02 (2H, m, H3′, H5′), 7.80 (1H, dd, J = 8.3, 1.1 Hz, H3), 7.76–7.72 (2H, m, H2′, H6′), 7.52–7.51 (1H, m, H6), 7.44 (1H, ddd, J = 8.3, 2.0, 0.9 Hz, H4), 4.28-4.20 (4H, m, CH2), 1.29 (6H, td, J = 7.1 Hz, J = 1.1 Hz, CH3). 13C NMR (126 MHz, CDCl3): δ 162.0, 147.5 (d, J = 7.1 Hz), 141.3, 138.1 (d, J = 2.1 Hz), 132.9 (d, J = 1.0 Hz), 126.2 (d, J = 1.5 Hz), 126.0 (d, J = 1.7 Hz), 126.2 (q, J = 3.8 Hz), 126.2 (q, J = 32.6 Hz), 124.1 (q, J = 270.0 Hz), 121.7 (d, J = 3.0 Hz), 119.6, 64.1 (d, J = 6.0 Hz), 14.0 (d, J = 6.8 Hz). Anal. C 49.68, H 4.29, N 3.09%, calculated for C18H18ClF3NO4P (435.76), C 49.61, H 4.16, N 3.21%.
(RS)-7-Chloro-2-phenyl-3-[4-(trifluoromethyl)phenyl]-3-hydrobenzo[e][1,3,2]oxazaphosphinin-4-one 2-oxide (5d). White solid; yield 76%; mp 185–186 °C. IR: 3337, 1656 (C=O), 1575, 1564, 1330, 1219, 1181, 1118, 1070, 715 cm−1. 1H-NMR (500 MHz, CDCl3): δ 8.16 (1H, d, J = 8.5 Hz, H5), 7.77–7.72 (2H, m, H3′, H5′), 7.66–7.56 (3H, m, H2′, H6′, H4′′), 7.50-7.44 (2H, m, H3′′, H5′′), 7.39-7.28 (4H, m, H6, H8, H2′′, H6′′). 13C NMR (126 MHz, CDCl3): δ 161.1 (d, J = 4.3 Hz), 151.3 (d, J = 7.9 Hz), 141.9, 136.6, 134.4 (d, J = 3.2 Hz), 132.3 (d, J = 11.2 Hz), 131.5, 131.0 (q, J = 32.7 Hz), 130.2 (d, J = 2.4 Hz), 129.0 (d, J = 16.1 Hz), 126.5 (q, J = 3.9 Hz), 125.9, 125.5 (d, J = 182.6 Hz), 123.5 (q, J = 272.6 Hz), 119.4 (d, J = 9.2 Hz), 115.8 (d, J = 2.0 Hz). Anal. C 54.65, H 2.69, N 3.29%, calculated for C20H12ClF3NO3P (437.74), C 54.88, H 2.76, N 3.20%.
(RS)-7-Chloro-2-phenyl-3-[4-(trifluoromethyl)phenyl]-3-hydrobenzo[e][1,3,2]oxazaphosphinin-4-one 2-sulfide (5e). Yellow solid; yield 52%; mp 136.5–139 °C. IR: 3076, 1675 (C=O), 1610, 1417, 1330, 1315, 1299, 1130, 1069, 949, 750, 692 cm−1. 1H-NMR (500 MHz, CDCl3): δ 8.13 (1H, d, J = 8.4 Hz, H5), 7.94-7.88 (2H, m, H3′, H5′), 7.64–7.59 (1H, m, H4′′), 7.53 (2H, d, J = 8.3 Hz, H2′, H6′), 7.50–7.44 (2H, m, H3′′, H5′′), 7.34 (1H, td, J = 8.5, 1.3 Hz, H6), 7.30–7.24 (3H, m, H8, H2′′, H6′′). 13C NMR (126 MHz, CDCl3): δ 161.3 (d, J = 3.7 Hz), 151.5 (d, J = 8.7 Hz), 141.7, 134.4 (d, J = 3.3 Hz), 132.2 (d, J = 12.9 Hz), 131.4, 130.6 (d, J = 2.4 Hz), 130.8 (q, J = 32.6 Hz), 129.6 (d, J = 139.3 Hz), 128.7 (d, J = 15.8 Hz), 126.1 (q, J = 3.8 Hz), 125.8, 123.5 (q, J = 272.5 Hz), 120.9, 119.9 (d, J = 8.4 Hz), 116.6 (d, J = 3.3 Hz). Anal. C 53.11, H 2.77, N 2.94%, calculated for C20H12ClF3NO2PS (453.80), C 52.94, H 2.67, N 3.09%.
2.1.4. Determination of Physicochemical Parameters
The molecular weights, logP values and topological polar surface areas (tPSA) were calculated using the CS ChemOffice Ultra program (version 18.0, CambridgeSoft, Cambridge, MA, USA).
2.2. Determination of Cholinesterases Inhibition
The IC50 values were determined using the spectrophotometric Ellman’s method modified according to Zdražilová et al. [31], which is a simple, rapid and direct method to determine the SH and -S-S- group content in proteins. This method is widely used for the screening of the efficiency of cholinesterases inhibitors. The enzymatic activity is measured indirectly by quantifying the concentration of the 5-thio-2-nitrobenzoic acid ion formed in the reaction between the thiol reagent 5,5′-dithio-bis(2-nitrobenzoic acid) and thiocholine, a product of substrate hydrolysis (i.e., acetylthiocholine; ATCH) by cholinesterases [32].
The enzyme activity in final reaction mixture (2000 µL) was 0.2 U/mL, concentration of acetylthiocholine (or butyrylthiocholine, BTCH) 40 µM and concentration of 5,5′-dithio-bis(2-nitrobenzoic acid) 0.1 mM for all reactions. The tested compounds were dissolved in DMSO and then diluted in demineralized water to the concentration of 1 µM. For all tested compounds and standard (rivastigmine), five different concentrations of inhibitor in final reaction mixture were typically used. All measurements were carried in triplicate and the average values of reaction rate (v0-uninhibited reaction, vi-inhibited reaction) were used for construction of the dependence v0/vi vs. concentration of inhibitor. From obtained equation of regression curve, the value of IC50 was calculated (based on the definition of IC50).
The determination of mechanism of enzyme inhibition was described in detail previously [22]. The inhibitor 5c was evaluated at three different concentrations for each enzyme.
For the determination of the type of inhibition Lineweaver-Burk plot [33] was used and the measuring procedure was similar to that for the determination of IC50 (modified Ellman’s method was used). The enzyme activity in the final reaction mixture (2000 µL) was 0.2 U/mL, a concentration of ATCH or BTCH 20–80 µM and a concentration of 5,5′-dithio-bis(2-nitrobenzoic acid) 0.1 mM. For each of the substrate concentrations, four different concentrations of inhibitor were chosen. The dependence absorbance vs. time was observed and the reaction rate was calculated. All measurements were carried in duplicate and the average values of reaction rate were used for the construction of the Lineweaver-Burk plot. From obtained equations of regression curves, the values of KM (Michaelis constant) and Vm (maximum velocity) were calculated and the type of inhibition was evaluated.
Acetylcholinesterase was obtained from electric eel (Electrophorus electricus L.) and butyrylcholinesterase was from an equine serum. Rivastigmine was used as a reference drug. All of the enzymes and rivastigmine were purchased from Sigma-Aldrich (Prague, Czech Republic).
3. Results and Discussion
3.1. Chemistry
2-Hydroxy-N-phenylbenzamides 1–4l were synthesized from substituted salicylic acids and anilines using phosphorus trichloride (PCl3) under microwave (MW) irradiation (Scheme 1). When compared to analogous reaction performed under refluxing for 6 h, this MW-mediated synthesis offers shorter reaction time and higher yields (72–92%) [26]. The majority of these benzamides have been reported in our previous studies [15,17,26]; for details, see Section 2.1.2.
Scheme 1.

Synthesis of salicylanilides 1-4 (R1 = H, 5-Cl, 4-Cl, 5-Br; R2 = H, 3-F, 3-Cl, 3-Br, 3-CF3, 4-F, 4-Cl, 4-Br, 4-CF3, 3,4-diCl, 3,5-diCl, 3,4,5-triCl, 3,5-bis-CF3; Ph-Cl: chlorobenzene).
The selected benzamide 3k was esterified using corresponding chlorides of phosphorus-based acids. It was converted by triethylamine (Et3N; 1.5 of equivalents for 5a–5c, 3 of equivalents for 5d and 5e) in situ into triethylammonium salts in dichloromethane (DCM) and then a mild excess (1.5 of equivalents) of chlorides were added [17,25]. After 24 h of stirring, the final esters 5 (Scheme 2) were obtained in yields of 28% to 93%. The preparation of the cyclic analogues 5d and 5e involved also initial refluxing for 4 h.
Scheme 2.

Synthesis of the esters 5 (X = O, S, or is missing; Y = O, S; DCM: dichloromethane).
The compounds were characterised by melting points, IR, and NMR spectra. Their purity was checked by thin-layer chromatography and elemental analysis.
The structures of all the compounds are reported in Table 1.
Table 1.
Structures of 2-hydroxybenzamides and IC50 for acetylcholinesterase and butyrylcholinesterase.
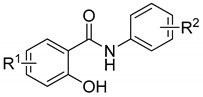
| |||||
|---|---|---|---|---|---|
| Code | R1 | R2 | IC50 (EeAChE) [µM] | IC50 (EqBuChE) [µM] | Selectivity AChE/BuChE |
| 1 | H | H | 49.71 ± 3.73 | 205.73 ± 12.15 | 0.24 |
| 2a | 5-Cl | 3-Cl | 59.50 ± 3.80 | 228.42 ± 11.29 | 0.26 |
| 2b | 5-Cl | 4-Cl | 76.53 ± 2.17 | 169.60 ± 3.09 | 0.45 |
| 2c | 5-Cl | 3,4-diCl | 60.79 ± 6.88 | 186.47 ± 15.69 | 0.33 |
| 2d | 5-Cl | 3,5-diCl | 46.21 ± 0.63 | 111.59 ± 7.98 | 0.41 |
| 2e | 5-Cl | 3,4,5-triCl | 51.06 ± 0.49 | 102.72 ± 0.97 | 0.50 |
| 2f | 5-Cl | 3-Br | 50.03 ± 0.15 | 141.04 ± 1.13 | 0.35 |
| 2g | 5-Cl | 4-Br | 58.25 ± 4.87 | 118.74 ± 6.65 | 0.49 |
| 2h | 5-Cl | 3-F | 58.65 ± 3.98 | 154.40 ± 8.38 | 0.38 |
| 2i | 5-Cl | 4-F | 48.27 ± 8.90 | 132.99 ± 15.81 | 0.36 |
| 2j | 5-Cl | 3-CF3 | 54.41 ± 0.25 | 120.30 ± 3.02 | 0.45 |
| 2k | 5-Cl | 4-CF3 | 58.47 ± 2.80 | 154.65 ± 3.58 | 0.38 |
| 2l | 5-Cl | 3,5-bis-CF3 | 50.18 ± 0.88 | 64.44 ± 1.34 | 0.78 |
| 3a | 4-Cl | 3-Cl | 58.08 ± 1.20 | 134.29 ± 0.24 | 0.43 |
| 3b | 4-Cl | 4-Cl | 56.19 ± 3.23 | 152.00 ± 3.13 | 0.37 |
| 3c | 4-Cl | 3,4-diCl | 46.26 ± 0.56 | 100.27 ± 1.15 | 0.46 |
| 3d | 4-Cl | 3,5-diCl | 50.15 ± 0.26 | 141.10 ± 2.80 | 0.36 |
| 3e | 4-Cl | 3,4,5-triCl | 57.78 ± 4.05 | 132.36 ± 4.31 | 0.44 |
| 3f | 4-Cl | 3-Br | 60.03 ± 0.42 | 167.80 ± 2.06 | 0.36 |
| 3g | 4-Cl | 4-Br | 58.32 ± 1.57 | 173.99 ± 4.02 | 0.34 |
| 3h | 4-Cl | 3-F | 60.79 ± 1.74 | 145.66 ± 4.07 | 0.42 |
| 3i | 4-Cl | 4-F | 62.85 ± 1.53 | 151.76 ± 7.58 | 0.41 |
| 3j | 4-Cl | 3-CF3 | 68.28 ± 3.78 | 163.18 ± 11.68 | 0.42 |
| 3k | 4-Cl | 4-CF3 | 60.29 ± 2.39 | 199.53 ± 2.02 | 0.30 |
| 3l | 4-Cl | 3,5-bis-CF3 | 54.72 ± 2.05 | 150.50 ± 0.87 | 0.36 |
| 4a | 5-Br | 3-Cl | 62.04 ± 6.48 | 134.00 ± 1.63 | 0.46 |
| 4b | 5-Br | 4-Cl | 85.75 ± 6.10 | 130.61 ± 3.99 | 0.66 |
| 4c | 5-Br | 3,4-diCl | 60.83 ± 3.65 | 122.60 ± 7.20 | 0.50 |
| 4d | 5-Br | 3,5-diCl | 33.13 ± 0.47 | 135.92 ± 0.14 | 0.24 |
| 4e | 5-Br | 3,4,5-triCl | 42.08 ± 2.41 | 140.07 ± 6.20 | 0.30 |
| 4f | 5-Br | 3-Br | 69.72 ± 9.97 | 145.78 ± 3.53 | 0.48 |
| 4g | 5-Br | 4-Br | 70.30 ± 2.85 | 112.16 ± 1.79 | 0.63 |
| 4h | 5-Br | 3-F | 54.44 ± 0.73 | 119.24 ± 2.53 | 0.46 |
| 4i | 5-Br | 4-F | 61.22 ± 1.08 | 136.57 ± 1.47 | 0.45 |
| 4j | 5-Br | 3-CF3 | 42.67 ± 0.18 | 116.91 ± 2.23 | 0.36 |
| 4k | 5-Br | 4-CF3 | 44.79 ± 3.44 | 134.81 ± 0.86 | 0.33 |
| 4l | 5-Br | 3,5-bis-CF3 | 48.46 ± 1.07 | 53.46 ± 3.20 | 0.91 |
| Rivastigmine | 56.10 ± 1.41 | 38.40 ± 1.97 | 1.46 | ||
AChE and BuChE inhibition are expressed as the mean ± SD (n = three experiments). The two or three lowest IC50 values for each enzyme are shown in bold.
3.2. In Vitro Inhibition of Acetylcholinesterase and Butyrylcholinesterase
Thirty-six benzamides 1–4l were evaluated as potential inhibitors of AChE from electric eel (EeAChE) and BuChE from equine serum (EqBuChE) using Ellman’s spectrophotometric method modified according to Zdražilová et al. [31]. Their efficacy is expressed as IC50, i.e., the concentration required for 50% inhibition of the enzymatic activity. Based on inhibition of both cholinesterases, we calculated selectivity indexes (SI) as the ratio of IC50 value for AChE/IC50 value for BuChE to express the selectivity quantitatively (Table 1). The results were compared with those obtained for rivastigmine, an established carbamate drug used in the therapy of Alzheimer’s and Parkinson’s dementias. Rivastigmine is a dual acylating pseudo-irreversible inhibitor of both AChE and BuChE.
Focusing on inhibition of AChE, IC50 values were in a close range 33.13 (4d)–85.75 (4b) μM. These results are fully comparable to those obtained for phenolic carbamate drug rivastigmine (56.10 μM). Interestingly, unsubstituted salicylanilide 1 produced a comparatively better inhibition (49.71 μM) than a majority of salicylanilides, i.e., the halogenation of salicylanilide moiety is not essential for inhibition of AChE. However, the bromination of salicylic acid in combination with a polyhalogenated aniline improved the activity (1 vs. 4d, 4e, 4j, and 4k) and provided the three most active compounds in this study, 5-bromo-2-hydroxy-N-(3,5-dichlorophenyl)benzamide 4d (33.13 μM), followed by 5-bromo-2-hydroxy-N-(3,4,5-trichlorophenyl)benzamide 4e (42.08 μM) and 5-bromo-2-hydroxy-N-[3-(trifluoromethyl)phenyl]benzamide 4j (42.67 μM).
Additionally, several general structure-activity relationships were identified:
Slightly better inhibition is associated with derivatives of 5-substituted salicylic acid (series 2 and 4).
The substitution of the position 3 at the aniline ring led to improved activity when compared to the position 4 (majority of the series 3 and the pair 2h and 2i are partial exceptions).
The order of preferred aniline substituents in the case of monosubstitution is as follows: CF3 > F > Br > Cl (series 4); Cl > Br > F > CF3 (series 3); F > Br > CF3 > Cl (series 2).
The di- and tri-substitution of aniline (both by chlorine c–e and trifluoromethyl group l) resulted predominantly in significantly more potent inhibition of AChE when compared to monosubstituted analogues.
Regarding polychlorinated anilides, the derivatives of 3,5-dichloroaniline d are superior to 3,4-dichloroanilides c and also to 3,4,5-trichloroanilides e; the additional substitution of the 3,5-dichloro compounds d by 4-chlorine (i.e., providing trichloro structures e) is detrimental but it is still superior to 3,4-dichloroanilides c. Series 3 is a partial exception.
In contrast to AChE, somewhat different results were obtained for BuChE. This enzyme was inhibited at higher concentrations and in a broader concentration range starting from 53.46 μM (4l) up to 228.42 μM (2a); all the compounds are less potent than rivastigmine (38.40 μM). The halogenation of the salicylanilide core is essential and it improves IC50 values of the salicylanilide 1. Two amides share significantly low IC50 values: 5-bromo- and 5-chloro-2-hydroxy-N-[3,5-bis(trifluoromethyl)phenyl]benzamides 2l and 4l (64.44 and 53.46 μM, respectively). The following SAR were revealed:
The most potent inhibition was exhibited by the derivatives of 5-bromosalicylic acid (series 4) followed by 5-chlorosalicylic acid (2).
The order of preferred atoms on monosubstituted aniline ring is as follows: Br > CF3 > F > Cl (series 2); Cl > F > Br > CF3 (series 3); CF3 > F > Br > Cl (series 4; i.e., the identical results as obtained for AChE). There was no sharp difference in the activity of 3- and 4-substituted anilines.
The presence of di- and tri-substituted anilines increases the BuChE inhibition predominantly, especially for trifluoromethyl derivatives.
Regarding polychlorinated anilides within the series 3 and 4, the derivatives of 3,4-dichloroaniline c are superior to 3,5-dichloroanilides d and also to 3,4,5-trichloroanilides e. However, the reverse SAR was found in the series 2.
Regarding selectivity to both cholinesterases, it can be distinguished based on the values of SI. SI lower than 1 means selectivity for AChE. On the other hand, compounds with SI higher than 1 tends to the selectivity for BuChE. The salicylanilides 1–4 share an obvious tendency to inhibit AChE more efficiently (SI within the range of 0.24 to 0.91; Table 1). Only the bis-trifluoromethyl derivative 4l exhibited almost identical inhibition of both enzymes.
To investigate the influence of phosphorus-based substituents, a known scaffold for the inhibition of cholinesterases [8,22,25], of phenolic group on inhibition, we prepared several derivatives of 4-chloro-2-hydroxy-N-[4-(trifluoromethyl)phenyl]benzamide 3k, i.e., the salicylanilide with a moderate activity against AChE and low activity against BuChE. Thus, if the presence of phosphorus-based moiety was beneficial, a drop in IC50 values would be more significant.
Previously, we synthesized diethyl phosphate 5a [17,22] and diethyl thiophosphate 5b [25]. Both derivatives share an enhanced inhibition of BuChE (up to 20.3 times) and moreover, thiophosphate 5b was 6.6 times more effective AChE inhibitor (Table 2). Then, we prepared analogous diethyl phosphite 5c. Regarding BuChE, this modification was the most successful one with IC50 value of 2.37 µM (84.2 times more potent than the parent 3k) and highly selective for BuChE (SI 28.0), but still remained the dual inhibitor of both cholinesterases. The removal of oxygen from phosphate 5a to phosphite 5c improved the activity especially against BuChE (4.2 times). Then, we prepared two cyclic analogues, 7-chloro-2-phenyl-3-[4-(trifluoromethyl)phenyl]-3-hydrobenzo[e][1,3,2]oxazaphosphinin-4-one 2-oxide (5d)/2-sulfide (5e). The first derivative showed an enhanced inhibitory potency against both enzymes when compared to parent salicylanilide and, regarding AChE, also when compared to the majority of the compounds involved in this study (IC50 48.13 µM). However, the isosteric replacement of oxygen by sulphur (5d→5e) resulted in a substantial lower activity comparable to parent amide 3k; it contrasts with the pair 5a and 5b, where this switch led to a sharply stronger AChE inhibition. The activity of [1,3,2]oxazaphosphinine-4-ones 5d and 5e suggests that the amidic hydrogen is not essential for the existence of biological activity, but its presence contributes to more potent enzyme inhibition.
Table 2.
Phosphorus-bases inhibitors of AChE and BuChE.
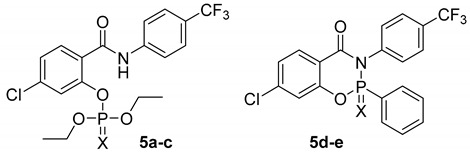
| ||||
|---|---|---|---|---|
| Code | X | IC50 (EeAChE) [µM] | IC50 (EqBuChE) [µM] | Selectivity AChE/BuChE |
| 5a [22] | O | 86.3 ± 4.9 | 9.84 ± 0.06 | 8.77 |
| 5b [25] | S | 9.16 ± 0.17 | 18.3 ± 1.0 | 0.50 |
| 5c | − | 66.37 ± 1.14 | 2.37 ± 0.01 | 28.00 |
| 5d | O | 48.13 ± 1.70 | 25.00 ± 0.30 | 1.93 |
| 5e | S | 63.48 ± 1.41 | 170.10 ± 13.58 | 0.37 |
| 3k | 60.29 ± 2.39 | 199.53 ± 2.02 | 0.30 | |
| Rivastigmine | 56.10 ± 1.41 | 38.40 ± 1.97 | 1.46 | |
AChE and BuChE inhibition are expressed as the mean ± SD (n = three experiments). The lowest IC50 value for each enzyme is shown in bold.
Four phosphorus-based esters (5a–5d) were superior to rivastigmine against BuChE and two of them against AChE (5b, 5d). Interestingly, the type of phosphorus functional group can modulate the AChE/BuChE selectivity. The presence of only oxygen(s) leads to preferential inhibition of BuChE (SI of 1.93–28.0; 5a, 5c and 5d) while the incorporation of sulphur increased the relative affinity to AChE (SI 0.37 and 0.50 for 5e and 5b, respectively).
We have demonstrated unequivocally that both salicylanilides and their phosphorus-based analogues act as dual AChE-BuChE inhibitors. Based on anti-AD drug rivastigmine, it was shown that this dual inhibition might be beneficial for the therapy of dementia [34]. The parent 2-hydroxybenzamides inhibit AChE preferentially and their modification did not improve the activity predominantly. On the other hand, the esterification of phenolic hydroxyl led to a significantly enhanced inhibition of BuChE. Regarding the structure of phosphorus part of the molecules, it is possible to modulate selectivity to each cholinesterase.
Mechanism and Type of Inhibition
Previously, we identified salicylanilide diethyl phosphates as pseudo-irreversible inhibitors of both cholinesterases [22]. That is why we also investigated the mechanism of inhibition of the most potent BuChE inhibitor 5c.
Based on the changes of enzyme activity in the presence of tested inhibitor during prolonged incubation, it is possible to distinguish reversible, pseudo-irreversible and irreversible inhibition [35,36]. In the case of reversible inhibition, the inhibitor is bound to the enzyme for a short period of time, the activity of the enzyme goes down immediately and, after dissociation of complex enzyme-inhibitor, the enzyme activity is restored. During pseudo-irreversible inhibition, the inhibitor is bound to the enzyme molecule covalently, but the bond is slowly broken down and the enzyme activity returns to the initial state. For irreversible inhibition, covalent and permanent bond between enzyme and inhibitor is typical and therefore the enzyme becomes inactivated. Irreversible inhibitor decreases enzyme activity successively and the dependence log% A vs. time in the presence of inhibitor is linear [37,38].
The procedure of measurement and data evaluation were in detail described previously in the reference [22]. From the dependence log% A vs. time the mechanism of action of tested inhibitor can be deduced. Obtained results assert the pseudo-irreversible inhibition of both cholinesterase enzymes (Figure 1 for AChE, Figure 2 for BuChE).
Figure 1.

The dependence log% A vs. time. Enzyme: acetylcholinesterase, derivative 5c, concentration of inhibitor: 10 µM (rhomb), 30 µM (square), 90 µM (triangle).
Figure 2.

The dependence log% A vs. time. Enzyme: butyrylcholinesterase, derivative 5c, concentration of inhibitor: 3 µM (rhomb), 6 µM (square), 9 µM (triangle).
The enzyme inhibitors may bind to the active site or at some other sites. Based on this fact, they can be divided into four categories: competitive, non-competitive, uncompetitive or mixed. Competitive inhibitors compete with the substrate for the same binding site on the enzyme, the so-called active site. Non-competitive, uncompetitive and mixed inhibitors do not bind at the active site but at some other [39].
The type of inhibition could be distinguished based on the Lineweaver-Burk plot [33] and the comparison of the kinetic parameters KM and Vm of uninhibited and inhibited reactions. The diagnostic criterion for competitive inhibition is that Vm is unaffected by inhibitor and KM for inhibited reaction is increased. All lines in Lineweaver-Burk plot share a common y-intercept. For non-competitive inhibitor KM is unchanged, but Vm of inhibited reaction is decreased. All lines in Lineweaver-Burk plot share a common x-intercept. Uncompetitive inhibitor decreases KM and Vm, but KM/Vm remains the same as for the uninhibited reaction. The pattern obtained in Lineweaver-Burk plot is a set of parallel lines. For mixed inhibition KM, Vm and KM/Vm are altered. The lines in Lineweaver-Burk plot intercept in the quadrant II or III [33].
From obtained results of KM and Vm and Lineweaver-Burk plots presented in Figure 3 (AChE) and Figure 4 (BuChE), it is possible to conclude, that the inhibitor 5c acts via mixed inhibitory mechanism.
Figure 3.

Lineweaver-Burk plot for AChE inhibition (ATCH = acetylthiocholine).
Figure 4.

Lineweaver-Burk plot for BuChE inhibition (BTCH = butyrylthiocholine).
3.3. Prediction of Physicochemical Parameters, Drug-Likeness and CNS Delivery
We determined physicochemical parameters that are important for the prediction of drug-likeness and also CNS availability (Table 3). Lipinski’s rule of five defines four physicochemical parameters that are associated with a favorable profile for oral administration. It postulates that the molecular weight (MW) should not be higher than 500, logP over five, number of hydrogen bond donors and acceptors should be of ≤5 and ≤10, respectively [40]. All of the derivatives 1–5 meet the criteria for MW and the number of hydrogen bond donors and acceptors. One salicylanilide (4l) and four phosphoric derivatives (5a, 5b, 5d, and 5e) share slightly higher logP values. However, the escalated lipophilicity may help to overcome bioavailability-related problems of the parent compounds 1–4, which is known substantially low for salicylanilides, despite generally meeting Lipinski’s rule [14,41]. Increased lipophilicity should also improve crossing biological barriers including BBB by a non-specific passive diffusion.
Table 3.
Lipinski’s rule of five parameters and PSA values of the derivatives 1-5.
| Code | R1 | R2 | MW | LogP | H-bond Donors | H-bond Acceptors | Number of Violations | tPSA [Å2] |
|---|---|---|---|---|---|---|---|---|
| 1 | H | H | 213.24 | 2.45 | 2 | 3 | 0 | 49.33 |
| 2a | 5-Cl | 3-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 2b | 5-Cl | 4-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 2c | 5-Cl | 3,4-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 2d | 5-Cl | 3,5-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 2e | 5-Cl | 3,4,5-triCl | 351.00 | 4.68 | 2 | 3 | 0 | 49.33 |
| 2f | 5-Cl | 3-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 2g | 5-Cl | 4-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 2h | 5-Cl | 3-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 2i | 5-Cl | 4-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 2j | 5-Cl | 3-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 2k | 5-Cl | 4-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 2l | 5-Cl | 3,5-bis-CF3 | 383.67 | 4.85 | 2 | 3 | 0 | 49.33 |
| 3a | 4-Cl | 3-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 3b | 4-Cl | 4-Cl | 282.12 | 3.57 | 2 | 3 | 0 | 49.33 |
| 3c | 4-Cl | 3,4-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 3d | 4-Cl | 3,5-diCl | 316.56 | 4.12 | 2 | 3 | 0 | 49.33 |
| 3e | 4-Cl | 3,4,5-triCl | 351.00 | 4.68 | 2 | 3 | 0 | 49.33 |
| 3f | 4-Cl | 3-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 3g | 4-Cl | 4-Br | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 3h | 4-Cl | 3-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 3i | 4-Cl | 4-F | 265.67 | 3.17 | 2 | 3 | 0 | 49.33 |
| 3j | 4-Cl | 3-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 3k | 4-Cl | 4-CF3 | 315.68 | 3.93 | 2 | 3 | 0 | 49.33 |
| 3l | 4-Cl | 3,5-bis-CF3 | 383.67 | 4.85 | 2 | 3 | 0 | 49.33 |
| 4a | 5-Br | 3-Cl | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 4b | 5-Br | 4-Cl | 326.57 | 3.84 | 2 | 3 | 0 | 49.33 |
| 4c | 5-Br | 3,4-diCl | 361.02 | 4.4 | 2 | 3 | 0 | 49.33 |
| 4d | 5-Br | 3,5-diCl | 361.02 | 4.4 | 2 | 3 | 0 | 49.33 |
| 4e | 5-Br | 3,4,5-triCl | 395.46 | 4.95 | 2 | 3 | 0 | 49.33 |
| 4f | 5-Br | 3-Br | 371.03 | 4.11 | 2 | 3 | 0 | 49.33 |
| 4g | 5-Br | 4-Br | 371.03 | 4.11 | 2 | 3 | 0 | 49.33 |
| 4h | 5-Br | 3-F | 310.12 | 3.44 | 2 | 3 | 0 | 49.33 |
| 4i | 5-Br | 4-F | 310.12 | 3.44 | 2 | 3 | 0 | 49.33 |
| 4j | 5-Br | 3-CF3 | 360.13 | 4.2 | 2 | 3 | 0 | 49.33 |
| 4k | 5-Br | 4-CF3 | 360.13 | 4.2 | 2 | 3 | 0 | 49.33 |
| 4l | 5-Br | 3,5-bis-CF3 | 428.13 | 5.12 | 2 | 3 | 1 | 49.33 |
| 5a | - | - | 451.76 | 5.08 | 1 | 6 | 1 | 73.86 |
| 5b | - | - | 467.82 | 5.82 | 1 | 5 | 1 | 56.79 |
| 5c | - | - | 435.76 | 4.70 | 1 | 5 | 0 | 56.79 |
| 5d | - | - | 437.74 | 5.64 | 0 | 4 | 1 | 46.61 |
| 5e | - | - | 453.80 | 6.37 | 0 | 3 | 1 | 29.54 |
Polar surface area (PSA) was described as another crucial factor for drugs targeting CNS. PSA is calculated as a sum of polar atoms surfaces (i.e., nitrogens, oxygens and their attached hydrogens) [42]. A PSA value of less than 76 Å2 should indicate an increased probability of BBB permeability [43]. Table 3 reports topological polar surface areas (tPSA) calculated using ChemOffice software. All the investigated derivatives exhibited a sufficiently low tPSA values.
Ghose et al. [43] analysed a range of CNS and non-CNS drugs and, in addition to tPSA, they provided additional parameters for designing CNS drugs including: (i) ideally one or two nitrogen atom(s), (ii) fewer than seven (two to four) linear chains outside of rings, (iii) fewer than three (zero or one are optimal) polar hydrogens. Obviously, our compounds meet these guidelines.
4. Conclusions
We evaluated 36 lipophilic halogenated 2-hydroxy-N-phenylbenzamides 1–4 prepared using PCl3 under MW conditions in high yields. The selected salicylanilide was esterified by chlorides of phosphorus-based acids in the presence of a tertiary base. These derivatives were screened in vitro for their activity against acetylcholinesterase and butyrylcholinesterase.
All salicylanilides exhibited moderate inhibition of both cholinesterases in micromolar concentration range starting from 33.1 μM with a clear tendency to affect AChE at lower concentrations and in a closer range of IC50. Some of them were superior to phenolic carbamate rivastigmine against this enzyme. Structure-activity relationships were also identified. Introduction of a phosphorus-based acid fragment (monovalent or bivalent) improved inhibitory potency for BuChE significantly uniformly. Importantly, depending of type of phosphorus containing moiety, it is possible to modulate the selectivity to cholinesterases. For the most active BuChE inhibitor, type and mechanism of inhibition were determined experimentally. Based on in silico prediction/calculation, the investigated compounds meet the criteria for drug-likeness and various parameters for BBB permeability/CNS availability.
Parent 2-hydroxy-N-phenylbenzamides (salicylanilides) with free phenolic group can be considered as convenient compounds (with an intrinsic activity especially against AChE and good physicochemical properties for CNS delivery) for conjugation with another cholinesterase inhibiting scaffolds to obtain drugs that are more active.
Author Contributions
Conceptualization, M.K. and J.V.; methodology, M.K. and Š.Š.; investigation, M.K., N.-H.H., R.V., Š.Š. and K.V.; writing—original draft preparation, M.K. and Š.Š.; writing—review and editing, K.V. and J.V.; supervision, M.K. and J.V.
Funding
This research was funded by the Czech Science Foundation, grant number 17-27514Y, by the Czech Ministry of Education, Youth and Sports, grant number SVV 260 401, and by the project EFSA-CDN [grant No. CZ.02.1.01/0.0/0.0/16_019/0000841] co-funded by ERDF. The authors wish to acknowledge financial support from the University of Pardubice, Faculty of Chemical Technology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bondi M.W., Edmonds E.C., Salmon D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017;23:818–831. doi: 10.1017/S135561771700100X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karlawish J., Jack C.R., Rocca W.A., Snyder H.M., Carrillo M.C. Alzheimer’s disease: The next frontier-Special Report 2017. Alzheimers Dement. 2017;13:374–380. doi: 10.1016/j.jalz.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 3.World Alzheimer Report 2018. [(accessed on 20 June 2019)]; Available online: https://www.alz.co.uk/research/world-report-2018.
- 4.Sawatzky E., Wehle S., Kling B., Wendrich J., Bringmann G., Sotriffer C.A., Heilmann J., Decker M. Discovery of Highly Selective and Nanomolar Carbamate-Based Butyrylcholinesterase Inhibitors by Rational Investigation into Their Inhibition Mode. J. Med. Chem. 2016;59:2067–2082. doi: 10.1021/acs.jmedchem.5b01674. [DOI] [PubMed] [Google Scholar]
- 5.Darvesh S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr. Alzheimer Res. 2016;13:1173–1177. doi: 10.2174/1567205013666160404120542. [DOI] [PubMed] [Google Scholar]
- 6.García-Ayllón M.-S., Small D.H., Avila J., Saez-Valero J. Revisiting the Role of Acetylcholinesterase in Alzheimer’s Disease: Cross-Talk with P-tau and β-Amyloid. Front. Molec. Neurosci. 2011;4:22. doi: 10.3389/fnmol.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q., Yang H.Y., Chen Y., Sun H.P. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017;132:294–309. doi: 10.1016/j.ejmech.2017.03.062. [DOI] [PubMed] [Google Scholar]
- 8.Mehta M., Adem A., Sabbagh M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012;2012:728983. doi: 10.1155/2012/728983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colovic M.B., Krstic D.Z., Lazarevic-Pasti T.D., Bondzic A.M., Vasic V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013;11:315–335. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horáková E., Drabina P., Brož B., Štěpánková Š., Vorčáková K., Královec K., Havelek R., Sedlák M. Synthesis, characterization and in vitro evaluation of substituted N-(2-phenylcyclopropyl)carbamates as acetyl- and butyrylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2016;31:173–179. doi: 10.1080/14756366.2016.1212193. [DOI] [PubMed] [Google Scholar]
- 11.Carrarini C., Russo M., Dono F., Di Pietro M., Rispoli M.G., Di Stefano V., Ferri L., Barbone F., Vitale M., Thomas A., et al. A Stage-Based Approach to Therapy in Parkinson’s Disease. Biomolecules. 2019;9:388. doi: 10.3390/biom9080388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohnen N.I., Grothe M.J., Ray N.J., Müller M.L.T.M., Teipel S.J. Recent Advances in Cholinergic Imaging and Cognitive Decline—Revisiting the Cholinergic Hypothesis of Dementia. Curr. Geri. Rep. 2018;7:1–11. doi: 10.1007/s13670-018-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z., Zhang A., Sun H., Han Y., Kong L., Wang X. Two decades of new drug discovery and development for Alzheimer’s disease. RSC Adv. 2017;7:6046–6058. doi: 10.1039/C6RA26737H. [DOI] [Google Scholar]
- 14.Krátký M., Vinšová J. Salicylanilide ester prodrugs as potential antimicrobial agents–a review. Curr. Pharm. Des. 2011;17:3494–3505. doi: 10.2174/138161211798194521. [DOI] [PubMed] [Google Scholar]
- 15.Paraskevopoulos G., Monteiro S., Vosátka R., Krátký M., Navrátilová L., Trejtnar F., Stolaříková J., Vinšová J. Novel salicylanilides from 4,5-dihalogenated salicylic acids: Synthesis, antimicrobial activity and cytotoxicity. Bioorg. Med. Chem. 2017;25:1524–1532. doi: 10.1016/j.bmc.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Krátký M., Vinšová J. Salicylanilide N-monosubstituted carbamates: Synthesis and in vitro antimicrobial activity. Bioorg. Med. Chem. 2016;24:1322–1330. doi: 10.1016/j.bmc.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Vinšová J., Kozic J., Krátký M., Stolaříková J., Mandíková J., Trejtnar F., Buchta V. Salicylanilide diethyl phosphates: Synthesis, antimicrobial activity and cytotoxicity. Bioorg. Med. Chem. 2014;22:728–737. doi: 10.1016/j.bmc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Krátký M., Vinšová J., Stolaříková J. Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains. Molecules. 2012;17:12812–12820. doi: 10.3390/molecules171112812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swan G.E. The pharmacology of halogenated salicylanilides and their anthelmintic use in animals. J. S. Afr. Vet. Assoc. 1999;70:61–70. doi: 10.4102/jsava.v70i2.756. [DOI] [PubMed] [Google Scholar]
- 20.Rajamuthiah R., Fuchs B.B., Conery A.L., Kim W., Jayamani E., Kwon B., Ausubel F.M., Mylonakis E. Repurposing salicylanilide anthelmintic drugs to combat drug resistant Staphylococcus aureus. PLoS ONE. 2015;10:e0124595. doi: 10.1371/journal.pone.0124595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia C., Burgain A., Chaillot J., Pic E., Khemiri I., Sellam A. A phenotypic small-molecule screen identifies halogenated salicylanilides as inhibitors of fungal morphogenesis, biofilm formation and host cell invasion. Sci. Rep. 2018;8:11559. doi: 10.1038/s41598-018-29973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krátký M., Štěpánková Š., Vorčáková K., Vinšová J. Salicylanilide Diethyl Phosphates as Cholinesterases Inhibitors. Bioorg. Med. Chem. 2015;58:48–52. doi: 10.1016/j.bioorg.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Krátký M., Vorčáková K., Vinšová J., Štěpánková Š. Investigation of salicylanilide and 4-chlorophenol-based N-monosubstituted carbamates as potential inhibitors of acetyl- and butyrylcholinesterase. Bioorg. Chem. 2018;80:668–673. doi: 10.1016/j.bioorg.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Krátký M., Štěpánková Š., Vorčáková K., Švarcová M., Vinšová J. Novel Cholinesterases Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules. 2016;21:191. doi: 10.3390/molecules21020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vinšová J., Krátký M., Komlóová M., Dadapeer E., Štěpánková Š., Vorčáková K., Stolaříková J. Diethyl 2-(Phenylcarbamoyl)phenyl Phosphorothioates: Synthesis, Antimycobacterial Activity and Cholinesterase Inhibition. Molecules. 2014;19:7152–7168. doi: 10.3390/molecules19067152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krátký M., Vinšová J. Antifungal Activity of Salicylanilides and Their Esters with 4-(Trifluoromethyl)benzoic Acid. Molecules. 2012;17:9426–9442. doi: 10.3390/molecules17089426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee I.Y., Gruber T.D., Samuels A., Yun M., Nam B., Kang M., Crowley K., Winterroth B., Boshoff H.I., Barry C.E. Structure-activity relationships of antitubercular salicylanilides consistent with disruption of the proton gradient via proton shuttling. Bioorg. Med. Chem. 2013;21:114–126. doi: 10.1016/j.bmc.2012.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Institute of Medicinal Molecular Design, Inc. Muto S., Itai A. Inhibitors against the Production and Release of Inflammatory Cytokines. No. EP1512396, A1. Patent. 2005 Mar 9;
- 29.Geigy A.G., Bindler J., Model E. Poly Halo-Salicylanilides. No. US2703332 (A) Patent. 1955 Mar 1;
- 30.Charles University, Faculty of Pharmacy in Hradec Králové. Vinšová J., Krátký M., Paraskevopoulos G. Substituted derivative of oxyphosphorus acids, its use and pharmaceutical preparation containing it. No. WO 2016095878 A1. Patent Application. 2016 Jun 23;
- 31.Zdrazilova P., Stepankova S., Komers K., Ventura K., Cegan A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. Naturforsch. C. 2004;59:293–296. doi: 10.1515/znc-2004-3-430. [DOI] [PubMed] [Google Scholar]
- 32.Sinko G., Calic M., Bosak A., Kovarik Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007;370:223–227. doi: 10.1016/j.ab.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Lineweaver H., Burk D. The Determination of Enzyme Dissociation Constants. J. Am. Chem. Soc. 1934;56:658–666. doi: 10.1021/ja01318a036. [DOI] [Google Scholar]
- 34.Kandiah N., Pai M.C., Senanarong V., Looi I., Ampil E., Park K.W., Karanam A.K., Christopher S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging. 2017;12:697–707. doi: 10.2147/CIA.S129145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szedlacsek S.E., Duggleby R.G. [6] Kinetics of slow and tight-binding inhibitors. Methods Enzymol. 1995;249:144–180. doi: 10.1016/0076-6879(95)49034-5. [DOI] [PubMed] [Google Scholar]
- 36.Singh J., Petter R.C., Bailli T.A., Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 37.Jann M.W., Shirley K.L., Small G.W. Clinical Pharmacokinetics and Pharmacodynamics of Cholinesterase Inhibitors. Clin. Pharmacokinet. 2002;41:719–739. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- 38.Bartolini M., Cavrini V., Andrisano V. Characterization of reversible and pseudo-irreversible acetylcholinesterase inhibitors by means of an immobilized enzyme reactor. J. Chromatogr. A. 2007;1144:102–110. doi: 10.1016/j.chroma.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 39.Biochemistry. Fourth edition. [(accessed on 20 June 2019)]; Available online: http://gtu.ge/Agro-Lib/Reginald%20H.%20Garrett,%20Charles%20M.%20Grisham%20-%20Biochemistry%20(4th%20ed.)%20-%202010.pdf.
- 40.Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug. Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Zhang Y., Zhang T., Zhang J., Wu B. Significantly enhanced bioavailability of niclosamide through submicron lipid emulsions with or without PEG-lipid: A comparative study. J. Microencapsul. 2015;32:496–502. doi: 10.3109/02652048.2015.1057251. [DOI] [PubMed] [Google Scholar]
- 42.Hitchcock S.A., Pennington L.D. Structure-Brain Exposure Relationships. J. Med. Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 43.Ghose A.K., Herbertz T., Hudkins R.L., Dorsey B.D., Mallamo J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]