

Abstract
The use of gaseous allene as an allyl pronucleophile in enantioselective aldehyde reductive coupling is described. Notably, using the same antipode of chiral ligand, (S)-tol-BINAP, an inversion of enantioselectivity is observed for allene vs allyl acetate pronucleophiles. Experimental and computational studies corroborate intervention of diastereomeric π-allyliridium-C,O-benzoate complexes, which arise via allene hydrometalation (from a pentacoordinate iridium hydride) vs allyl acetate ionization (from a square planar iridium species).
Keywords: iridium, allylation, enantioselective, transfer hydrogenation, reductive coupling
Graphical Abstract
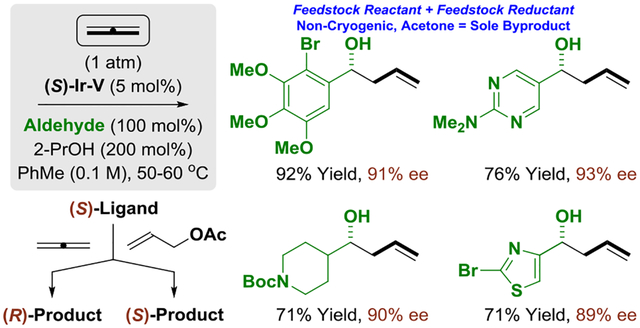
In connection with longstanding efforts to use abundant chemical feedstocks as pronucleophiles in metal-catalyzed carbonyl reductive coupling,1f a diverse suite of enantioselective C-C couplings was developed in our laboratory.1,2 A distinguishing feature of these processes resides in the use of inexpensive terminal reductants (e.g. H2, 2-PrOH) or, more ideally, dual use of alcohols as carbonyl proelectrophiles and reductants in carbonyl addition via hydrogen auto-transfer.1 Given the high occurrence of allenes as constituents in the C3, C4 and C5 petroleum cracking fractions (Figure 1), these patterns of reactivity were applied to the first (2007) allene-carbonyl reductive couplings to furnish homoallylic alcohols (Figure 2).3 Using allene, methylallene and dimethylallene as pronucleophiles, products of carbonyl allylation, crotylation and tert-prenylation were obtained in reactions conducted from either the alcohol or aldehyde oxidation level.3 Shortly thereafter (2009), we reported the first enantioselective reactions of this type using dimethyl allene.4a Following this and other catalytic enantioselective allene-carbonyl additions developed in our laboratory,4,5 Buchwald reported an allene-ketone reductive coupling mediated by (MeO)2MeSiH.6
Figure 1.

Allene feedstocks formed in petroleum cracking.
Figure 2.

Milestones in metal-catalyzed allene-carbonyl reductive coupling and related hydrogen auto-transfer processes.
In this account, we report the first enantioselective allene-aldehyde reductive couplings.7 Additionally, we demonstrate that using the same antipode of chiral ligand, (S)-tol-BINAP, an inversion of enantioselectivity is observed for carbonyl allylations that employ gaseous allene vs allyl acetate as pronucleophile solely in response to stereogenicity at iridium.8,9 Experimental and computational studies corroborate intervention of diastereomeric π-allyliridium-C,O-benzoate complexes, which arise via allene hydrometalation (from a pentacoordinate iridium hydride) vs ionization of allyl acetate (from a square planar iridium species).
An initial series of experiments were conducted in which a sealed reaction vessel back-filled with gaseous allene 1a and charged with aldehyde 2a (100 mol%), 2-propanol (200 mol%), THF (0.4 M) and an iridium catalyst (5 mol%) were heated to 60 °C (Table 1) for 24 hours. It was determined that the (S)-tol-BINAP-modified iridium catalyst, Ir-V, delivered the desired product 3a with the highest levels of enantioselectivity as the (R)-enantiomer, which is the opposite enantiomer observed in corresponding carbonyl allylations mediated by allyl acetate 1b.10 This surprising result compelled us to evaluate the diastereomeric composition of the iridium catalyst Ir-V, which is easily accomplished via LCMS due to the chromatographic stability of π-allyliridium-C,O-benzoates (Figure 3). The catalyst prepared from allyl acetate is enriched in diastereomer D, which, as indicated by experiment and computation, is the thermodynamically most stable isomer. In contrast, the catalyst recovered from the reaction mixture using allene or prepared from allene itself is enriched in diastereomer C. It was posited that use of Ir-V derived from allene would improve enantioselectivity in the allene-mediated allylation of aldehyde 2a. Indeed, an increase from 86% to 92% ee in the formation of homoallylic alcohol 3a was observed (Table 1).
Table 1.
Selected optimization experiments in the enantioselective reductive coupling of allene 1 with aldehyde 2a and divergent enantioselectivity observed upon use of allyl acetate vs allene pronucleophiles.a
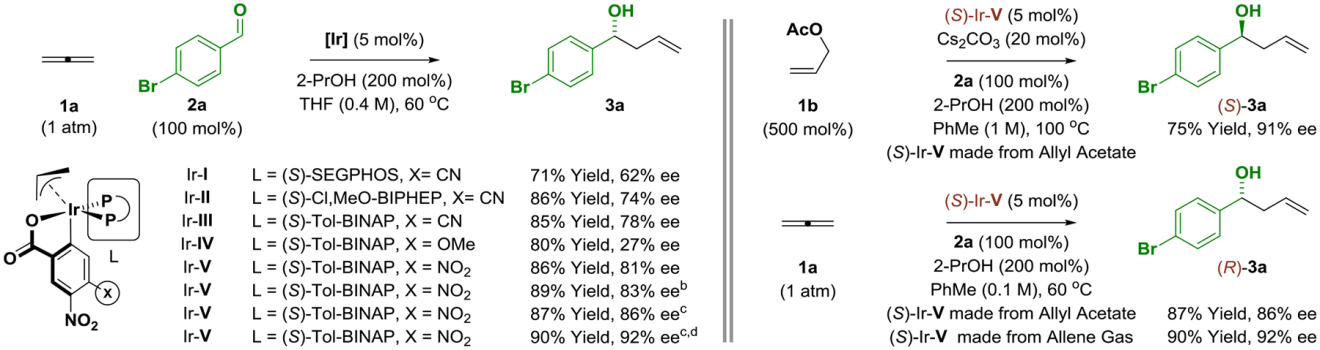
|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis.
PhMe (0.4 M).
PhMe (0.1 M).
(S)-Ir-V derived from allene. See Supporting Information for experimental details.
Figure 3.

Diastereomeric composition of the (S)-Ir-V and calculated thermodynamic stabilities.
Using catalyst (S)-Ir-V derived from allene, the scope of the allene-mediated reductive aldehyde allylation mediated by 2-propanol was explored (Table 2). Diverse aryl aldehydes 2a-2i and heteroaryl aldehydes 2j-2p were converted to the corresponding homoallylic alcohols 3a-3p in good yield with uniformly high levels of enantioselectivity. As illustrated by the formation of adduct 3d, due to the mild reaction conditions, sensitive functional groups such as pinacol boronates are tolerated. The formation of 3l, which incorporates an unprotected indole nitrogen, also is notable. The α,β-unsaturated aldehyde 2q, as well as linear and branched aliphatic aldehydes 2r and 2s also participate in highly enantioselective allylation. Allene-mediated allylation of 2a mediated by d8-2-propanol delivers deuterio-3a (eq. 1).11 The pattern of deuterium incorporation corroborates reversible allene hydrometalation with incomplete regiocontrol. The relatively low levels of deuterium incorporation are attributed to reversibility of the hydrometalation event and H/D-exchange with adventitious water.12
Table 2.
Enantioselective iridium-catalyzed reductive coupling of gaseous allene 1a with aldehydes 2a-2s mediated by 2-propanol.a
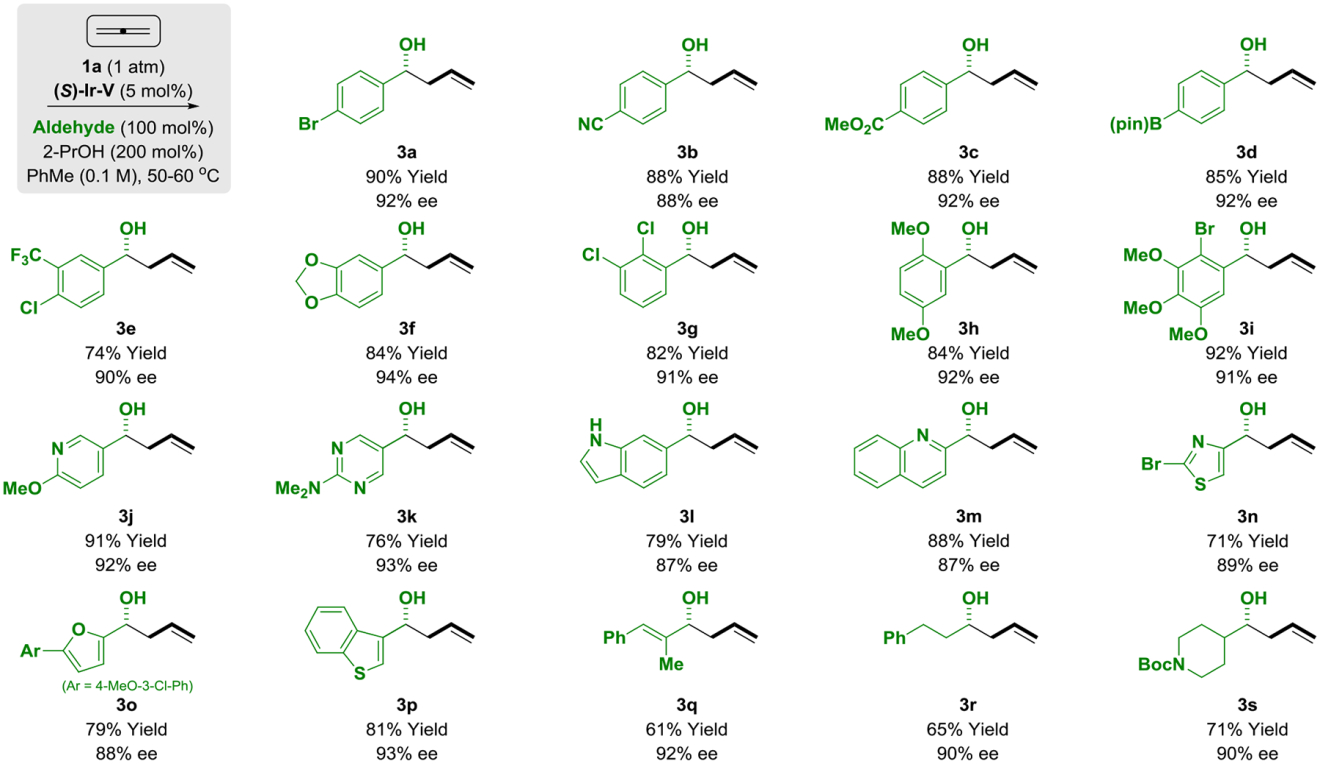
|
Yields are of material isolated by silica gel chromatography. The catalyst (S)-Ir-V prepared from allene was used. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for experimental details.
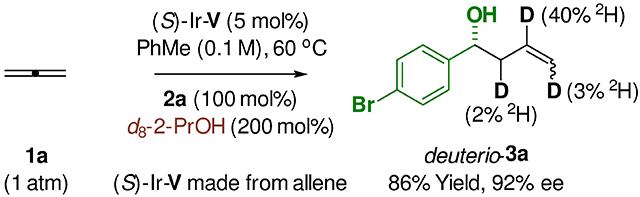 |
(eq. 1) |
The experimental data suggest that the observed divergence in enantioselectivity in reactions of allene vs allyl acetate is due to intervention of diastereomeric π-allyliridium-C,O-benzoate complexes C and D, which arise via allene hydrometalation (from an pentacoordinate iridium hydride) vs ionization of allyl acetate (from a square planar iridium species), respectively (Figure 4).13,14 To challenge the veracity of our hypothesis, we then turned our efforts to density functional theory (DFT) calculations.15 The reaction pathways to form the π-allyliridium complexes under the different experimental conditions were first computed. When allyl acetate pronucleophile is used, the π-allyliridium is formed via coordination of allyl acetate to the C,O-benzoate complex 4 followed by ionization (TS1, Figure 5A). Although the common intermediate 4 can potentially lead to all four diastereomeric π-allyl complexes, formation of D is kinetically and thermodynamically favored (see Figure S2 for less favorable pathways to A and B). The relatively low barrier of TS1D suggests a reversible ionization process. The thermodynamically more stable complex D is operative in the subsequent aldehyde addition step (vide infra). On the other hand, π-allyliridium complexes derived from allene are formed via hydrometalation from a square pyramidal iridium hydride (6C or 6D, Figure 5B). DFT calculations suggest 6C is thermodynamically more stable than 6D, and subsequent allene coordination to form 7C and hydrometalation via TS3C are both very facile. Therefore, π-allyl complex C is formed preferentially from allene hydrometalation.
Figure 4.

Hypothesis for enantiodivergence in aldehyde allylations mediated by gaseous allene vs allyl acetate.
Figure 5.

Computed energy profiles of the kinetic pathways leading to diastereomers D and C and, therefrom, (S)-and (R)-product enantiomers, respectively.
The calculated transition states for allylation of aldehyde 2a by the diastereomeric complexes C and D provide further insight into the origins of enantiodivergence. Complexes C and D are supported by the same antipode of the (S)-tol-BINAP ligand, but their stereogenicity at iridium is opposite. Therefore, examination of the transition states for carbonyl addition will reveal whether enantioselectivity is influenced more by the chirality of the metal center16 or the bisphosphine ligand. The aldehyde addition with C and D occurs by way of Zimmerman-Traxler-type transition states that place the Ar group of the aldehyde in a pseudo-equatorial position (Figure 6).17 In the reaction with D, addition to the (Si)-face of the aldehyde (TS2D-S) to form (S)-3a is 2.2 kcal/mol more favorable than the (Re)-face addition (TS2D-R) to form (R)-3a. TS2D-R is destabilized by the 1,3-diaxial interactions between the aldehyde hydrogen and the benzene ring of the benzoate, which is co-planar with the aldehyde. In TS2D-S, the axial aldehyde hydrogen and the benzoate are on opposite faces of the chair, which relieves steric repulsion. In the allylation transition states with complex C (Figure 6B), because the stereogenicity at iridium is inverted, the (S)-selective transition state (TS2C-S) is now destabilized by the 1,3-diaxial interactions between the aldehyde hydrogen and the benzoate. As such, the most favorable transition state from C is TS2C-R, which eventually leads to the (R)-enantiomer of the homoallylic alcohol product 3a.
Figure 6.

Enantioselectivity-determining aldehyde allylation transition states with π-allyliridium complexes D and C. Activation free energies are with respect to D and C, respectively. The (S)-tol-BINAP ligands are omitted for clarity
In summary, we report the first catalytic enantioselective aldehyde allylations mediated by gaseous allene. These processes exploit a feedstock pronucleophile (allene) in combination with a feedstock reductant (2-propanol) under non-cryogenic conditions with acetone as the sole stoichiometric byproduct. Remarkably, use of allene vs allyl acetate as pronucleophile results in an inversion of enantioselectivity using the same antipode of chiral ligand, (S)-tol-BINAP. The collective experimental and computational data corroborate intervention of diastereomeric π-allyliridium-C,O-benzoate complexes, which arise via allene hydrometalation (from a pentacoordinate iridium hydride) vs allyl acetate ionization (from a square planar iridium species). These data should facilitate the design of related chiral-at-metal complexes for enantioselective catalysis by providing insight into the structural and interactions features of the catalyst that influence enantioselectivity. More broadly, these studies and other work from our laboratory demonstrate how reactions that traditionally have employed organometallic reagents may now be conducted catalytically in the absence of premetalated reagents using abundant feedstocks.1f,18
Supplementary Material
Acknowledgments.
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445, R35-GM128779). Ms. Leyah Schwartz is acknowledged for key preliminary experiments. Mr. Brian Lanigan is thanked for skillful technical assistance. P.L. acknowledges supercomputer resources provided by the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. Single crystal X-ray diffraction data for (S)-Ir-V derived from allyl acetate. This material is available free of charge via the internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews on carbonyl reductive coupling of π-unsaturated feedstocks via hydrogenation, transfer hydrogenation and hydrogen auto-transfer, see:; (a) Ngai M-Y; Kong J-R; Krische MJ Hydrogen-Mediated C-C Bond Formation – A Broad New Concept in Catalytic C-C Coupling. J. Org. Chem 2007, 72, 1063–1072. [DOI] [PubMed] [Google Scholar]; (b) Iida H; Krische MJ Catalytic Reductive Coupling of Alkenes and Alkynes to Carbonyl Compounds and Imines Mediated by Hydrogen. Top. Curr. Chem 2007, 279, 77–104. [Google Scholar]; (c) Hassan A; Krische MJ Unlocking Hydrogenation for C-C Bond Formation: A Brief Overview of Enantioselective Methods. Org. Proc. Res. Devel 2011, 15, 1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed 2019, 58, 10.1002/anie.201905532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For general reviews on metal-catalyzed carbonyl reductive coupling, see:; (a) Metal Catalyzed Reductive C-C Bond Formation; Krische M, Eds.; Topics in Current Chemistry 279; Springer-Verlag Berlin Heidelberg: Germany, 2007. [Google Scholar]; (b) Moragas T; Correa A; Martin R Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. - Eur. J 2014, 20, 8242–8258. [DOI] [PubMed] [Google Scholar]; (c) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Skucas E; Bower JF; Krische MJ Carbonyl Allylation in the Absence of Preformed Allyl Metal Reagents: Reverse Prenylation via Iridium-Catalyzed Hydrogenative Coupling of Dimethylallene. J. Am. Chem. Soc 2007, 129, 12678–12679. [DOI] [PubMed] [Google Scholar]; (b) Bower JF; Skucas E; Patman RL; Krische MJ Catalytic C-C Coupling via Transfer Hydrogenation: Reverse Prenylation, Crotylation and Allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc 2007, 129, 15134–15135. [DOI] [PubMed] [Google Scholar]
- (4) (a).Han SB; Kim IS; Han H; Krische MJ Enantioselective Carbonyl Reverse Prenylation from the Alcohol or Aldehyde Oxidation Level Employing 1,1-Dimethylallene as the Prenyl Donor. J. Am. Chem. Soc 2009, 131, 6916–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MJ Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C-H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schwartz LA; Holmes M; Brito GA; Gonçalves TP; Richardson J; Ruble JC; Huang K-W; Krische MJ Cyclometallated Iridium-PhanePhos Complexes Are Active Catalysts in Enantioselective Allene-Fluoral Reductive Coupling and Related Alcohol-Mediated Carbonyl Additions that Form Acyclic Quaternary Carbon Stereocenters. J. Am. Chem. Soc 2019, 141, 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For metal-catalyzed allene-carbonyl reductive couplings to form racemic homoallylic alcohols beyond those cited in reference 3, see:; (a) Ngai M-Y; Skucas E; Krische MJ Ruthenium Catalyzed C-C Bond Formation via Transfer Hydrogenation: Branch-Selective Reductive Coupling of Allenes to Paraformaldehyde and Higher Aldehydes. Org. Lett 2008, 10, 2705–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skucas E; Zbieg JR; Krische MJ anti-Aminoallylation of Aldehydes via Ruthenium Catalyzed Transfer Hydrogenative Coupling of Sulfonamido-Allenes: 1,2-Aminoalcohols. J. Am. Chem. Soc 2009, 131, 5054–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR; McInturff EL; Krische MJ Allenamide Hydro-Hydroxyalkylation: 1,2-Aminoalcohols via Ruthenium Catalyzed Carbonyl anti-Aminoallylation. Org. Lett 2010, 12, 2514–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zbieg JR; McInturff EL; Leung JC; Krische MJ Amplification of anti-Diastereoselectivity via Curtin-Hammett Effects in Ruthenium Catalyzed Hydrohydroxyalkylation of 1,1-Disubstituted Allenes: Diastereoselective Formation of All-Carbon Quaternary Centers. J. Am. Chem. Soc 2011, 133, 1141–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sam B; Montgomery TP; Krische MJ Ruthenium Catalyzed Reductive Coupling of Paraformaldehyde to Trifluoromethyl Allenes: CF3-Bearing All-Carbon Quaternary Centers. Org. Lett 2013, 15, 3790–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Oda S; Sam B; Krische MJ Hydroaminomethylation Beyond Carbonylation: Allene-Imine Reductive Coupling by Ruthenium-Catalyzed Transfer Hydrogenation. Angew. Chem. Int. Ed 2015, 54, 8525–8528. [DOI] [PubMed] [Google Scholar]
- (6) (a).Liu RY; Zhou Y; Yang Y; Buchwald SL Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct. J. Am. Chem. Soc 2019, 141, 2251–2256. Also see, [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsai EY; Liu RY; Yang Y; Buchwald SL A Regio- and Enantioselective CuH-Catalyzed Ketone Allylation with Terminal Allenes. J. Am. Chem. Soc 2018, 140, 2007–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For selected reviews on enantioselective carbonyl allylation, see:; (a) Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2794. [DOI] [PubMed] [Google Scholar]; (b) Marek I; Sklute G Creation of Quaternary Stereocenters in Carbonyl Allylation Reactions. Chem. Commun 2007, 1683–1691. [DOI] [PubMed] [Google Scholar]; (c) Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]; (d) Spielmann K; Niel G; de Figueiredoa RM; Campagne JM Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- (8).For selected reviews on enantioselective catalysis via chiral-at-metal complexes, see:; (a) Knight PD; Scott P Predetermination of chirality at octahedral centres with tetradentate ligands: prospects for enantioselective catalysis. Coord. Chem. Rev 2003, 242, 125–143. [Google Scholar]; (b) Bauer EB Chiral-at-Metal Complexes and Their Catalytic Applications in Organic Synthesis. Chem. Soc. Rev 2012, 41, 3153–3167. [DOI] [PubMed] [Google Scholar]; (c) Gong L; Chen L-A; Meggers E Asymmetric Catalysis Mediated by the Ligand Sphere of Octahedral Chiral-at-Metal Complexes. Angew. Chem., Int. Ed 2014, 53, 10868–10874. [DOI] [PubMed] [Google Scholar]
- (9).For a recent authoratitive review on diastereo- and enantiodivergent catalytic processes, see:; Beletskaya IP; Nájera C; Yus M Stereodivergent Catalysis. Chem. Rev 2018, 118, 5080–5200. [DOI] [PubMed] [Google Scholar]
- (10).Studies on the enantioselective π-allyliridium C,O-benzoate-catalyzed carbonyl allylations mediated by allyl acetate are longstanding and encompass a vast number of examples, including applications in natural product total synthesis. In all cases, the enantiofacial selectivity of aldehyde addition using allyl acetate as a pronucleophile corresponds to that shown in the present study. For key primary literature, see:; (a) Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium Catalyzed Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Using Allyl Acetate as an Allyl Metal Surrogate. J. Am. Chem. Soc 2008, 130, 6340–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium Catalyzed Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level via Transfer Hydrogenative Coupling of Allyl Acetate: Departure from Chirally Modified Allyl Metal Reagents in Carbonyl Addition. J. Am. Chem. Soc 2008, 130, 14891–14899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hassan A; Lu Y; Krische MJ Elongation of 1,3-Polyols via Iterative Catalyst-Directed Carbonyl Allylation from the Alcohol Oxidation Level. Org. Lett 2009, 11, 3112–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Han SB; Hassan A; Kim I-S; Krische MJ Total Synthesis of (+)-Roxaticin: A Departure from Stoichiometric Chiral Reagents, Auxiliaries, and Premetalated Nucleophiles in Polyketide Construction. J. Am. Chem. Soc 2010, 132, 15559–15561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schmitt DC; Dechert-Schmitt A-MR; Krische MJ Iridium-Catalyzed Allylation of Chiral β-Stereogenic Alcohols: Bypassing Discrete Formation of Epimerizable Aldehydes. Org. Lett 2012, 14, 6302–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dechert-Schmitt A-MR; Schmitt DC; Krische MJ Protecting-group-Free Diastereoselective C-C Coupling of 1,3-Glycols and Allyl Acetate through Site-Selective Primary Alcohol Dehydrogenation. Angew. Chem. Int. Ed 2013, 52, 3195–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For related deuterium labelling studies on corresponding allyl acetate mediated carbonyl allylations, see reference 10b.
- (12).Klei SR; Golden JT; Tilley TD; Bergman RG Iridium-Catalyzed H/D Exchange into Organic Compounds in Water. J. Am. Chem. Soc 2002, 124, 2092–2093 and references cited therein. [DOI] [PubMed] [Google Scholar]
- (13).The influence of remote electron donating or withdrawing groups on enantioselectivity can be understood on this basis. More Lewis acidic catalysts accelerate rate-determining carbonyl addition. For the less Lewis acidic catalyst Ir-IV where X = OMe (see Table 1), the kinetic catalyst diastereomer C slowly isomerizes to the thermodynamically preferred catalyst diastereomer D, which delivers the opposite enantiomer, resulting in an erosion in enantioselectivity.
- (14).An inversion of enantioselectivity is also observed in corresponding reactions of dimethyl allene vs allyl acetate:; (a) Han SB; Kim IS; Han H; Krische MJ Enantioselective Carbonyl Reverse Prenylation from the Alcohol or Aldehyde Oxidation Level Employing 1,1-Dimethylallene as the Prenyl Donor. J. Am. Chem. Soc 2009, 131, 6916–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) (Addition/Correction) Han SB; Kim IS; Han H; Krische MJ Enantioselective Carbonyl Reverse Prenylation from the Alcohol or Aldehyde Oxidation Level Employing 1,1-Dimethylallene as the Prenyl Donor. J. Am. Chem. Soc 2010. 132, 12517–12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).DFT calculations were performed at the ωB97X-D/6–311+G(d,p)-SDD/CPCM(PhMe)//B3LYP-D3/6–31G(d)-SDD level of theory. See Supporting Information for computational details.
- (16) (a).Chen S; Huang X; Meggers E; Houk KN Origins of Enantioselectivity in Asymmetric Radical Additions to Octahedral Chiral-at-Rhodium Enolates: A Computational Study. J. Am. Chem. Soc 2017, 139, 17902–17907. [DOI] [PubMed] [Google Scholar]; (b) Tutkowski B; Meggers E; Wiest O Understanding Rate Acceleration and Stereoinduction of an Asymmetric Giese Reaction Mediated by a Chiral Rhodium Catalyst. J. Am. Chem. Soc 2017, 139, 8062–8065. [DOI] [PubMed] [Google Scholar]; (c) Chen S; Zheng Y; Cui T; Meggers E; Houk KN, Arylketone π-Conjugation Controls Enantioselectivity in Asymmetric Alkynylations Catalyzed by Centrochiral Ruthenium Complexes. J. Am. Chem. Soc 2018, 140, 5146–5152. [DOI] [PubMed] [Google Scholar]
- (17).Grayson MN; Krische MJ; Houk KN, Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For a practical perspective on green chemistry, see:; Rossen K Greening Organic Chemistry with Process Chemistry. J. Org. Chem 2019, 84, 4580–4582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.