

Abstract
The development of new antitubercular agents for the treatment of infections caused by multidrug-resistant (MDR) Mycobacterium tuberculosis is an urgent priority. Pyrrolobenzodiazepines (PBDs) are a promising class of antibacterial agents that were initially discovered and isolated from a range of Streptomyces species. Recently, C8-linked PBD monomers have been shown to work by inhibiting DNA gyrase and have demonstrated activity against M. tuberculosis. However, both PBD monomers and dimers are toxic to eukaryotic cells, limiting their development as antibacterial agents. To eliminate the toxicity associated with PBDs and explore the effect of C8-modification with a known antibacterial agent with the same mechanism of action (i.e., ciprofloxacin, a gyrase inhibitor), we synthesized a C8-linked PBD–ciprofloxacin (PBD–CIP, 3) hybrid. The hybrid compound displayed minimum inhibitory concentration values of 0.4 or 2.1 μg/mL against drug-sensitive and drug-resistant M. tuberculosis strains, respectively. A molecular modeling study showed good interaction of compound 3 with wild-type M. tuberculosis DNA gyrase, suggesting gyrase inhibition as a possible mechanism of action. Compound 3 is a nontoxic combination hybrid that can be utilized as a new scaffold and further optimized to develop new antitubercular agents.
Introduction
Tuberculosis (TB) is one of the most serious bacterial infections, with around 10 million new cases and an average of almost 1.3 million deaths in 2017 alone.1 Novel strategies are required to treat TB and specifically target drug-resistant strains of Mycobacterium tuberculosis, which are increasing worldwide. A lengthy multidrug regimen is needed to eradicate the infection, which complicates the treatment of TB and threatens our attempts to control this pandemic. Therefore, there is an urgent need for new, improved antibiotics that will shorten treatment times.2 Only a few new TB drugs are in clinical trials, and since the 1960s, there have only been two new antibiotics, bedaquiline and delaminid, developed for treatment; these drugs are only given to patients with extensively drug-resistant (XDR)-TB or pre-XDR-TB. The WHO End-TB strategy aims to reduce TB deaths by 95% and to cut new cases by 90% between 2015 and 2035 with the ultimate aim to end the global TB epidemic.3 This goal will only be achieved through improved TB control measures and innovative tools for the discovery of new interventions. M. tuberculosis has evolved many mechanisms for avoiding the effects of antibiotics. Its waxy, impermeable coat does not allow for the easy penetration of antibiotics, and, in addition, the organism possesses efflux systems that allow for the rapid expulsion of antibiotics.4 One approach for TB drug development is the potentiation of existing antibiotics, and this could accelerate drug discovery efforts.
Recently, the PBD class of sequence-specific DNA minor groove binding agents has been recognized as potential chemotherapeutic and antibacterial agents.5−8 They have a kinetic preference for 5′-Py–G–Py-3′ sequences,9 while 5′-AGA-3′ has been established as the thermodynamically preferred sequence.10 PBDs are able to act as covalent binding agents by positioning themselves securely within the DNA minor groove and selecting for GC-rich sequences. This is made possible by the particular three-dimensional (3D) shape afforded to them by a chiral center at their C11a(S)-position. PBDs can also form an aminal linkage between their C11-position and the C2-NH2 group of a guanine base once securely within the minor groove, due to the presence of a “soft” electrophilic imine moiety at their N10–C11-position.11,12 Naturally occurring PBD monomers such as anthramycin, tomaymycin, neothramycin, and sibiromycin form adducts that span three base pairs, with guanine in the central position,13,14 while the larger synthetic C8-linked PBD monomers and PBD dimers can span up to eight base pairs.15−18 The side chains of PBD monomers play a key role in DNA sequence recognition and fit of the PBDs within the DNA minor groove, which has been linked to their cytotoxicity.7 C8-substituents with pyrrole and imidazole heterocycles,15 biaryls (e.g., 4-(1-methyl-1H-pyrrol-3-yl)-benzenamine (MPB)),16 and benzofused rings (e.g., benzofuran and benzothiophene)17,18 have been shown to facilitate binding and stabilization of DNA, while large bulky groups like porphyrins reduce the ability of PBDs to bind to duplex DNA due to steric factors.19 C8-linked PBD monomers have shown notable antibacterial activity against MDR Gram-positive bacteria6,8 and M. tuberculosis.20 More recently, DNA gyrase inhibition has been reported as an additional mechanism of action of antibacterial PBDs.6 A study by Brucoli et al. evaluated 12 C8-linked PBD-heterocyclic polyamide conjugates for antitubercular activity and DNA sequence selectivity.20 These PBD conjugates exhibited significant growth inhibition of M. tuberculosis H37Rv, with minimum inhibitory concentrations (MICs) ranging from 0.08 to 5.19 μg/mL. As is the case for other PBD compounds, these compounds demonstrated cytotoxicity toward mammalian cells due to DNA binding. To eliminate the cytotoxicity associated with PBDs,16,18 we aimed to develop an antitubercular C8-PBD conjugate that could not be accommodated easily within the DNA minor groove, due to the presence of a large C8-substituent while retaining antimycobacterial activity.
We selected ciprofloxacin, a known gyrase inhibitor, as the C8-substituent to develop a PBD–ciprofloxacin hybrid molecule as ciprofloxacin possesses the same mechanism of action as PBDs.6 Ciprofloxacin and other fluoroquinolones are used in the management of MDR-TB (Figure 1), but the emergence of fluoroquinolone-resistant M. tuberculosis is a cause for significant concern. Alternative approaches such as the development of hybrid molecules can overcome resistance, extend the life span of current treatments, and expand the pool of first-line drugs that are available for TB treatment. Ciprofloxacin hybrids with both small molecules21 and macromolecules22,23 have been used as a strategy to improve activity and overcome resistance in both Gram-positive and Gram-negative bacteria. We conjugated a C8-linker-containing PBD to ciprofloxacin through its piperazine moiety. This conjugation allowed the carboxylic acid group of ciprofloxacin to remain unmodified, which is crucial for its activity and provided a C8-PBD hybrid with a large bulky substituent, which would influence the DNA binding and hence the toxicity of the PBDs. The hybrid demonstrated no DNA binding and was found to be nontoxic against eukaryotic cell lines. It was anticipated that the synthesized PBD–CIP hybrid would maintain its ability to interact with DNA gyrase due to its structural similarity with C8-PBD monomers, and an in silico study demonstrated good interaction with M. tuberculosis DNA gyrase. The hybrid compound displayed MIC values of 0.4 or 2.1 μg/mL against drug-sensitive and drug-resistant M. tuberculosis strains, respectively, which is comparable to the MIC values reported for C8-linked PBDs against M. tuberculosis.20 This is the first report of a nontoxic C8-linked PBD hybrid with activity against M. tuberculosis and is a starting point for the development of nontoxic PBD molecules that do not bind DNA as new antibiotics for TB treatment.
Figure 1.

Structure of selected fluoroquinolones and pyrrolobenzodiazepine with reported activity against M. tuberculosis.
Results and Discussion
Design, In Silico Modeling, and Synthesis of Fluoroquinolone–Pyrrolobenzodiazepine Hybrid
The fluoroquinolone–pyrrolobenzodiazepine hybrid (compound 3, Figure 2) was designed by considering the structure–activity relationship data available for fluoroquinolone and pyrrolobenzodiazepine classes of molecules. The substituted 4-quinolone ring and the tricyclic pyrrolobenzodiazepine system were connected through a C4 chemical spacer. The C8-position of the PBD A-ring and C7-position of ciprofloxacin were selected for chemical modification as the SAR data for these chemical classes indicated that structural changes are allowed in these positions. The OH group of the C8-position of the PBD was linked to the C7-piperazine group of ciprofloxacin via a 4C linker with a terminal carboxylic acid group.
Figure 2.

Design of the fluoroquinolone–pyrrolobenzodiazepine hybrid.
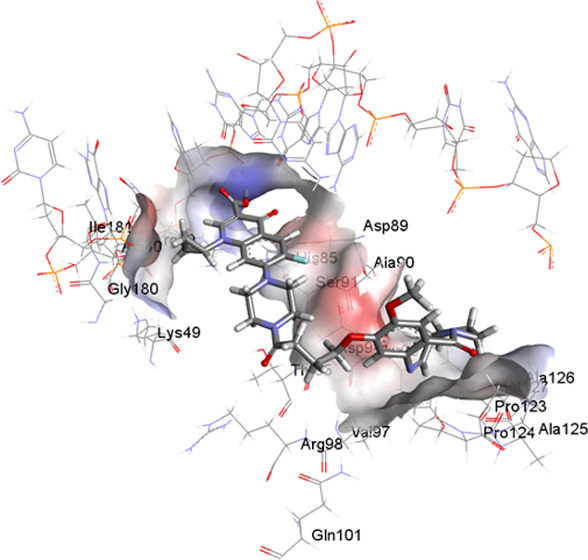
To explore the ability of compound 3 to interact with DNA gyrase enzyme, it was docked with both wild-type and S95T mutant gyrase (present within M. tuberculosis strains of East Asian, Central Asian, and EuroAmerican lineages) from M. tuberculosis. Compound 3 showed a −35.84 kcal/mol free energy of binding and a ChemScore of 30.67 compared to a −27.45 kcal/mol free energy of binding and a ChemScore of 24.89 observed for ciprofloxacin. The free energy of binding and ChemScore for compound 3 against the S95T mutant were comparable to those observed for ciprofloxacin (−28.1 vs −27.5 kcal/mol and 24.45 vs 25.46, respectively). Compound 3 showed greater interaction with both wild-type and S95T mutant gyrase enzyme gyrA (Figures 3 and S1, Tables S1–S4) compared to ciprofloxacin. The ciprofloxacin component of the compound showed hydrogen bond interactions with arginine 39 and alanine 986 residues, while the PBD component showed hydrophobic interactions with proline 124 and tyrosine 93 residues. The interaction of compound 3 with GyrA S95T was comparable to that observed for ciprofloxacin (Figure S2). This modeling data suggested that compound 3 could interact with GyrA, occupying a binding pocket close to the ciprofloxacin binding pocket. Therefore, it was decided to synthesize the hybrid compound and evaluate its activity against M. tuberculosis strains.
Figure 3.

Two-dimensional (2D) and 3D structures of ciprofloxacin (A, B) and compound 3 (C, D) within the binding site of the wild type of DNA gyrase A.
Compound 3 was synthesized according to the synthetic scheme reported in Scheme 1. The N10 alloc–tetrahydropyranyl (THP) protected pyrrolobenzodiazepine core 1 was synthesized according to the nine-step literature-reported procedure16. Commercially available ciprofloxacin ethyl ester 1 was coupled to the protected pyrrolobenzodiazepine core structure 2 to form the amide bond connection required to link the two pharmacophores. N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDCI) and 4-dimethylaminopyridine (DMAP) were selected as the coupling system for the formation of the amide bond. This was followed by hydrolysis of the ciprofloxacin ethyl ester to generate the active carboxylic acid. Finally, the N10 alloc–THP group was deprotected in the presence of palladium to generate the reactive imine group between N10 and C11 of the pyrrolobenzodiazepine structure. The final compound (3, PBD–CIP) was purified using liquid column chromatography with a dichloromethane (DCM)/methanol gradient. The compound was fully characterized by NMR and MS spectrometric techniques prior to biological evaluation.
Scheme 1. Synthetic Scheme for PBD–CIP (3).

Evaluation of DNA Binding Ability of Compound 3
PBDs are known to interact covalently with the DNA minor groove, which is the key source of their eukaryotic toxicity. The ability of compound 3 to bind and stabilize DNA was evaluated using a Förster resonance energy transfer (FRET)-based DNA melting assay24 using two fluorophore-labeled oligonucleotide sequences (sequence F1: 5′-FAM-TAT-ATA-TAG-ATA-TTT-TTT-TAT-CTA-TAT-ATA-3′-TAMRA and sequence F2: 5′-FAM-TAT-AGA-TAT-AGA-TAT-TTT-ATA-TCT-ATA-TCT-ATA-3′-TAMRA) to understand the effect of the relatively bulky ciprofloxacin building block. Netropsin, a known DNA minor groove binder, and a previously reported C8-linked PBD with antibacterial activity, PBD-Py-Py (GWL-78, Figure 1), were used as positive controls. Netropsin showed a 16 °C stabilization of sequence F1 and a 13.5 °C stabilization of sequence F2 at 1 μM; GWL-78 showed an 18 °C stabilization of sequence F1 and a 16.5 °C stabilization of sequence F2. However, compound 3 showed weak stabilization of both DNA sequences, with ΔTm values between 1 and 1.5 °C at a 1 μM concentration (drug/DNA ratio 5:1) (Table 1). The result suggests that the bulky ciprofloxacin substitution at the C8-position of the PBD reduces the ability of compound 3 to interact and stabilize the DNA sequences. Most PBD monomers stabilize duplex DNA sequences in the range of 8–18 °C and show low micromolar to nanomolar IC50s against eukaryotic cell lines.5 As DNA binding and stabilization are linked to the eukaryotic toxicity of pyrrolobenzodiazepines,25 this lack of DNA binding observed for compound 3 was expected to reduce the toxicity of the newly synthesized hybrid.
Table 1. DNA Duplex Stabilization for Compound 3 and Control Compoundsa.
| ΔTm at 1 μM |
||
|---|---|---|
| compounds | sequence F1 | sequence F2 |
| 3 | 1.1 ± 0.2 | 1.5 ± 0.5 |
| GWL-78 | 18.0 ± 0.2 | 16.5 ± 0.5 |
| netropsin | 16.0 ± 0.3 | 13.5 ± 0.2 |
| ciprofloxacin | 0.4 ± 0.2 | 0.2 ± 0.3 |
ΔTm values are reported in the table at °C in comparison to the oligo only. Assay conducted in triplicate.
Evaluation of Eukaryotic Toxicity
The aim of the study is to develop a compound with antitubercular activity, as well as selective toxicity toward prokaryotes. However, DNA stabilization has been associated with cytotoxicity for PBD-type covalent minor groove binders.25 Therefore, the eukaryotic cytotoxicity of the synthesized compounds was tested in the cervical cancer cell line HeLa and the nontumor lung fibroblast WI-38 using an MTT assay with GWL-78 as the control.26 Compound 3 did not show any notable toxicity (>90% survival at 24 h) at the highest concentration (20 μM) tested, while 24% HeLa cells and 41% WI-38 cells survived after 24 h incubation with 20 μM GWL-78. These results are encouraging, especially as standard C8-linked PBD monomers tend to show high levels of cytotoxicity.
Microbiological Evaluation of Compound 3
The activity of compound 3 and ciprofloxacin was determined using M. tuberculosis H37Rv and two clinical M. tuberculosis isolates of Beijing/W lineage, 1192/015 and 08/00483E (multidrug-resistant, Table S5). Both clinical isolates harbor mutations within the DNA gyrase gene (gyrA) (Table 2); however, these mutations do not confer resistance to fluoroquinolones including ciprofloxacin.
Table 2. Mutations Present in M. tuberculosis Strains 1192/015 and 08/00483E gyrA But Absent in M. tuberculosis H37Rv.
| position | mutation | annotation | gene | description |
|---|---|---|---|---|
| 7362 | G → C | E21Q (GAG → CAG) | gyrA | DNA gyrase A |
| 7585 | G → C | S95T (AGC → ACC) | gyrA | DNA gyrase A |
| 9304 | G → A | G668D (GGC → GAC) | gyrA | DNA gyrase A |
A modified resazurin microtiter assay in combination with a modified Gompertz function27 was used to generate MIC values for compound 3 and ciprofloxacin. A dose-dependent response of bacterial growth inhibition was observed for all strains exposed to compound 3 or ciprofloxacin, thereby confirming antimicrobial activity for both compounds (Figures 4 and 5).
Figure 4.

Effect of compound 3 and GWL-78 against HeLa and WI-38 cell line. The lack of cytotoxicity observed for compound 3 was statistically significant compared to that for GWL-78.
Figure 5.

Growth inhibition of M. tuberculosis strains by compound 3 or ciprofloxacin. Dose-dependent growth inhibition of M. tuberculosis strains H37Rv (black), M. tuberculosis 1192/015 (red), and M. tuberculosis 08/00483E (blue) by ciprofloxacin (0.094–6.036 μM) (A) and compound 3 (0.048–3.1 μM) (B). Twofold serial dilutions of each antibiotic were evaluated. The antibiotic activity was ascertained by the addition of resazurin, and the optical density (OD570nm) was measured. The OD570nm measurements were converted to a percentage of the OD570nm measurements for the control that contained no antibiotic. The values plotted are a percentage of the growth without antibiotic and are the mean values of three independent experiments ± standard deviation and analyzed using Student’s t test. Vertical lines indicate modified Gompertz-determined MIC values.
MIC values were calculated for three M. tuberculosis strains (H37Rv, Beijing susceptible strain 1192/015, and Beijing drug-resistant strain 08/00483E) that were exposed to either ciprofloxacin or compound 3 at concentration ranges of 0.094–6.036 and 0.048–3.1 μM, respectively. Three independent replicate experiments were performed for each strain and each antibiotic. An MIC was determined for each replicate experiment using a modified Gompertz function. The three independent MIC values for each strain were used to calculate the mean MIC and standard deviation from the mean.
The MICs of ciprofloxacin for both clinical isolates were not significantly different from the MIC calculated for M. tuberculosis H37Rv (Figure 4A and Table 3). In contrast, the exposure of M. tuberculosis strains to compound 3 resulted in markedly different dose-dependent responses, and the MIC values recorded for M. tuberculosis Beijing/W strains were significantly greater than the MIC recorded for M. tuberculosis H37Rv.
Table 3. MIC Values ± Standard Deviation, As Determined Using the Modified Gompertz Function for M. tuberculosis H37Rv, M. tuberculosis 1192/015, and M. tuberculosis 08/00483E, Which Were Exposed to Ciprofloxacin and Compound 3.
| MIC [μM] |
||
|---|---|---|
| strain reference | ciprofloxacin | compound 3 |
| H37Rv | 0.453 ± 0.088 | 0.413 ± 0.0004 |
| 1192/015 | 0.386 ± 0.078 | 1.903 ± 0.031 |
| 08/00483E | 0.415 ± 0.101 | 2.153 ± 0.136 |
The MIC value for compound 3 against H37Rv (0.413 ± 0.0004 μg/mL) is comparable to that reported for C8-linked PBD monomers with traditional heteroaromatic building blocks (MIC range 5.19–0.08 μg/mL) by Brucoli et al.20 However, C8-PBD monomers with heteroaromatic building blocks are cytotoxic15 and not considered desirable candidates for development as antimicrobial agents. Therefore, the activity observed for the noncytotoxic compound 3 is encouraging and provides an opportunity to further improve the activity of this chemical series through medicinal chemistry optimization. Compound 3 had comparable activity to ciprofloxacin in inhibiting the growth of M. tuberculosis H37Rv, suggesting that the increased molecular level interaction of compound 3 with DNA gyrase, as indicated by molecular modeling data, did not result in a more active compound against M. tuberculosis compared to ciprofloxacin.
Although inhibitory to the growth of clinical M. tuberculosis strains (1192/015 and 08/00483E) at low micromolar concentrations, compound 3 is approximately fivefold less active than ciprofloxacin against these strains. The molecular modeling study suggested greater interaction with S95T mutant gyrase A, which is present in these strains, compared to that of ciprofloxacin. Therefore, this reduction in activity observed for compound 3 was surprising. In the absence of a biochemical assay to directly measure the inhibition of the mutant S95T enzyme, it is not possible to fully ascertain the reason behind the reduction in activity. The relatively higher molecular weight of compound 3 might play a role in its ability to cross the mycobacterial membrane, particularly if cell wall alteration contributes to the resistance of these clinical strains against antitubercular agents. These results suggest that the addition of pyrrolobenzodiazepine to ciprofloxacin has reduced its activity against mutant strains through an unknown mechanism. Potential explanations for this observation include greater affinity of efflux pumps for compound 3 or reduced permeability of compound 3 through the mycobacterial cell wall. This is supported by the observation that Beijing/W M. tuberculosis strains (including M. tuberculosis 1192/015 and 08/00483E) harbor a deletion of the RD105 region of the genome (Rv0071–Rv0074) that results in the thickening of the cell wall and reduced intracellular concentrations of antibiotics.28 The deletion of the RD105 region results in a gene fusion of Rv0071 and Rv0074 generating a gene product with carbopeptidase activity that can cleave peptidoglycan, potentially enhancing the cross-linking of peptidoglycan to increase the thickness of the cell wall. Further experimentation would be required to elucidate these hypotheses. Calculation of c log P by ChemBioOffice 16.0 showed that compound 3 was more hydrophobic (c log P 1.85) compared to ciprofloxacin (c log P −0.72). This increased hydrophobicity may reduce uptake and therefore the intracellular concentration of compound 3 in the Beijing/W strains due to the hydrophilic nature of peptidoglycan. This can also affect the efflux potential of the compound as molecules with more hydrophobic side chains are considered as preferred substrates of efflux pumps.29
The mutations noted in Table 2 may further reduce the ability of compound 3 to bind effectively with the mutated DNA gyrase A, although the molecular modeling study suggested comparable binding affinities for both wild-type and S95T mutant enzymes. Despite compound 3 being less active than ciprofloxacin in some strains, the identification of a new hybrid chemical class with activity against M. tuberculosis provides the drug development pipeline with new chemical scaffolds to work on. Both the pharmacophores, pyrrolobenzodiazepine and fluoroquinolones, which are present in the hybrid compound 3 have shown clinical utility in cancer and infection, respectively.
Compound 3 displayed a dose-dependent inhibition of growth of both the clinical and laboratory, M. tuberculosis strains, at concentrations in the low micromolar range. Although the activity of compound 3 was reduced in the clinical strains compared to that of H37Rv, this was not unexpected as reduced activity has been observed previously in these clinical strains (unpublished data). It is very promising that inhibition was observed in these clinical strains. An increase in the MIC observed in the Beijing strains was likely to be due to phenotypic resistance mechanisms, such as reduced permeability, which will be investigated further. These findings highlight the need for the evaluation of novel compounds against different lineages and clinically adapted strains. Novel drugs are needed for TB treatment, and compound 3 is a starting point for further development, particularly as the toxic effects of DNA binding have been removed. This noncytotoxic and novel chemical scaffold can be used to develop new antitubercular agents or as a chemical biology tool to study antimicrobial resistance in M. tuberculosis.
Experimental Section
Chemistry
Solvents and reagents have been obtained from several commercial sources, including Sigma-Aldrich, Fluorochem, Alfa Aesar, and Fisher Scientific. Thin-layer chromatography analyses were performed on plates coated with silica gel (Merck, F254 plates, silica gel 60). The final plates were visualized under ultraviolet radiation (UV) at either 254 or 365 nm. Manual flash chromatography utilized in the purification of the crude of the reaction was performed using silica gel as a stationary phase (silica gel from Merck, 230–400 mesh). The solvents were routinely evaporated under vacuum using an RC-600 evaporating system (from KNF), equipped with an SC-920 G vacuum pump (KNF). The final compounds, when thermally stabile, were dried using a VacuumTherm (Thermo Scientific) vacuum oven overnight. Glassware used was dried in a UN55 oven (Memmert) at 200 °C.
1H and 13C nuclei nuclear magnetic (NMR) analyses were performed on a Spectrospin 400 MHz spectrometer (Bruker) equipped with a SampleXpress (from Bruker) autosampler system, using deuterated solvents for the preparation of the samples. The obtained spectra were analyzed using Topspin 7.1 software (Bruker). The chemical shifts were reported relative to trimethylsilane (TMS), used as a standard (0.00 ppm). Signals were identified and described as singlet (s), doublet (d), triplet (t), quartet (q), or multiplets (m). Coupling constants were reported in Hertz (Hz). LC–MS analyses were performed on a Waters Alliance 2695 system (Waters), with elution in gradient. High-performance liquid chromatography (HPLC)-grade solvents were used as a mobile phase, while a Monolithic C18 50 × 4.60 mm2 column (Phenomenex) was used as a stationary phase. The UV detection was performed using a Waters 2996 photoarray detector (Waters). Mass spectra were collected in both ESI+ and ESI– modes by a Waters QZ instrument (Waters) coupled to the HPLC system.
Synthesis of (S)-1-Cyclopropyl-6-fluoro-7-(4-(4-((7-methoxy-5-oxo-2,3,5,11a-tetrahydro-1H-benzo[e]pyrrolo[1,2-a][1,4]diazepin-8-yl)oxy)butanoyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3)
Alloc, THP protected pyrrolobenzodiazepine core 2 (200 mg, 0.38 mmol, 1 equiv), and ciprofloxacin ethyl ester 1 (1.1 equiv) were dissolved in dimethylformamide (10 mL). EDCI (2 equiv) and 4-dimethylaminopyridine (1.75 equiv) were added to the solution, which was left under a magnetic stirrer overnight at room temperature under a nitrogen atmosphere. The reaction went to completion after 15 hours and was monitored by LC–MS. Ethyl acetate (30 mL) was added to the solution, and the organic phase was washed with a NaHCO3 aqueous saturated solution (2 × 10 mL), citric acid aqueous saturated solution (2 × 10 mL), and brine (2 × 10 mL). The organic phase was collected, dried over MgSO4, and evaporated under vacuum to give the crude of reaction, which was purified by column chromatography on silica gel (mobile phase: 95/5, DCM/MeOH, v/v), obtaining 150 mg of the protected PBD–ciprofloxacin ethyl ester conjugate (3a). The obtained product 3a was fully characterized using NMR and MS spectroscopy (ESI). The protected compound 3a was dissolved in a 1:1 mixture of dioxane/MeOH (6 mL) and underwent basic hydrolysis, being treated with an aqueous solution of NaOH 0.1 M (excess). The reaction went to completion. The reaction mixture was evaporated under vacuum, H20 (20 mL) was added to the crude, and subsequently acidified until pH 2, treating with HCl of a 0.1 M aqueous solution. The aqueous phase was extracted with ethyl acetate (3 × 10 mL) that was subsequently collected, dried over MgSO4, and evaporated under vacuum to give the corresponding protected PBD–ciprofloxacin derivative (120 mg). The obtained compound (120 mg) was dissolved in DCM (3 mL) and treated with tetrakisPd(0) triphenylphosphine (0.05 equiv) and pyrrolidine (1.2 equiv). The reaction went to completion in 20 minutes, and the crude of reaction was evaporated under vacuum and purified by column chromatography on silica gel (mobile phase: DCM/MeOH, 93/7, v/v) to give the final PBD–ciprofloxacin compound 3 (PBD–CIP) as a white solid (62 mg, yield calculated on three steps: 37%).
1H NMR (400 MHz, CHLOROFORM-d) δ: 8.78 (s, 1H), 8.05 (d, J = 12.84 Hz, 1H), 7.66 (d, J = 4.28 Hz, 1H), 7.52 (s, 1H), 7.34 (d, J = 7.05 Hz, 1H), 6.84 (s, 1H), 4.12–4.22 (m, 2H), 3.94 (s, 3H), 3.68–3.85 (m, 6H), 3.50–3.62 (m, 2H), 3.31 (br. s., 5H), 2.58–2.68 (m, 2H), 2.28–2.35 (m, 2H), 2.22–2.28 (m, 2H), 2.03–2.10 (m, 2H), 1.39 (q, J = 5.96 Hz, 2H), 1.10–1.24 (m, 3H).
Molecular Docking of the Compounds to Wild and Mutant Gyrase A
M. tuberculosis gyrase A structure was obtained by homology modeling using the Swiss Model webserver. The sequence identity (SID) and similarity (SIM) of the target with the template crystal structure of S. aureus gyrase A (PDB ID code 2XCS) were 55.83% and 0.45, respectively. The structure was amended using Accelrys discovery studio and minimized using the AMBER 16.0 package program. AutoDock SMINA was used for blind molecular docking of ciprofloxacin and compound 3 to the homology modeled minimized structure of gyrase A from M. tuberculosis to determine the best binding pocket. Blind molecular docking was followed by GOLD to perform flexible molecular docking and determining more precise and evaluated energies and scores by docking the compounds into the SMINA identified binding pocket. The GOLD program calculates a fitness score, which takes into account the ligand binding position and the ability of the ligand to stabilize the complex. The fitness score, ChemScore, was used to determine the best-docked pose for each compound. For molecule docking of the mutant form of gyrase A, Ser84 was altered to Leu using PyMOL software, and the new structure of gyraseA_S84L was minimized using Sybyl program. GOLD molecular docking was performed on the minimized structure of the mutant enzyme, and the ligand conformational flexibility along with the partial flexibility of the protein was thoroughly examined by a genetic algorithm (GA). The default parameters were selected (100 population sizes, 5 for the number of islands, 100 000 number of operations, and 2 for the niche size) for each run, and the maximum number of runs was set to 20 for each compound. The cutoff values of 4.0 Å for van der Waals distance and 2.5 Å (dH–X) for hydrogen bonds were employed.
Microbiological Evaluation
Bacterial Strains and Reagents
Mycobacterium tuberculosis strains, H37Rv, 1192/015, and 08/00483E (multidrug-resistant (MDR)), were used in the modified resazurin microtiter assay (REMA) plate method. M. tuberculosis Beijing strains 1192/015 and 08/00483E were obtained from the PHE National Mycobacterial Reference Laboratory.
Reagents used were Middlebrook 7H9 medium (BD Difco), OADC supplemented with 0.5% glycerol (24388.295, VWR Chemicals) and 0.2% Tween 80 (P1754, Sigma), CAMR Mycobacterium medium MOD2 (CMM MOD2),30 ciprofloxacin (Sigma-Aldrich, cat no. 33434), and 0.02% resazurin solution (10 mg resazurin sodium salt (R7017, Sigma-Aldrich) in 50 mL phosphate-buffered saline, pH 7.4 (Severn Biotech) containing 5% Tween 80).31 All of the antibiotics were dissolved in dimethyl sulphoxide (DMSO) (Sigma-Aldrich D2650) to a stock concentration of 10 mg/mL.
Preparation of Bacterial Inocula
Bacterial strains were grown in 50 mL of OADC-supplemented, Middlebrook 7H9 medium with 0.5% glycerol and 0.2% Tween 80 in vented Erlenmeyer flasks, shaken at 37 °C for 5 days. Inoculum for the REMA method was prepared to an OD540nm of 0.5, from Middlebrook 7H9 cultures by diluting cells in CMM MOD2 medium.
Modified Resazurin Microtiter Assay (REMA) Plate Method
A modification of the REMA plate method previously described by Nateche et al. in 2006 was used.32,33 A 10 mg/mL stock solution of each antibiotic was prepared in DMSO. A twofold serial dilution of each antibiotic stock solution was performed in CMM MOD2 medium. The two antibiotics evaluated were ciprofloxacin (0.094–6.036 μM) and compound 3 (0.048–3.1 μM). Each antibiotic concentration of 100 μL was added to six replicate wells in a 96-well plate (734-1554, VWR). Three of the wells were “drug-only”, and another three wells were “test wells” used to assess the activity of the antibiotic. Bacterial culture (10 μL) was added to the test wells to give a final OD540nm of 0.05. Three further wells contained 100 μL of medium and 10 μL of bacteria as “drug-free” controls to confirm the growth of the bacteria in the absence of an antibiotic. The drug-only controls allowed for confirmation that there was an absence of colorimetric interference from the antibiotics. Bacteria were diluted to an optical density (OD540nm) of 0.5, and 10 μL of the inoculum was added to each of the test wells and the drug-free controls. Medium (10 μL) was added to the drug-only wells. The plates were incubated for 7 days at 37 °C while shaking at 200 rpm. Drug activity was assessed by adding 11 μL resazurin to each well of the 96-well plate and then incubating the plate at room temperature for 6 hours. The optical density (OD570nm) was measured in each well. The OD570nm measurements were converted to a percentage of the drug-free control; data were expressed as a percentage of the growth without an antibiotic.
Statistical Analyses
The average OD570nm for each drug concentration was fitted to a sigmoidal curve using the modified Gompertz function (y = A + Ce-e(B(x – M))), and the minimum inhibitory concentration (MIC) was identified from the point of inflexion of the lower asymptote.27 This was applied to the mean of the OD570nm of four biological repeats to produce a graphical representation for each condition. An unpaired, two-tailed, Student’s t test was used to determine significance between biological repeats.
Acknowledgments
The authors thank King’s College London Health School for a Ph.D. studentship to P.P. (award no. JGA3804), Medical Research Council (award no. MC_PC_13065), and Public Health England and Department of Health for supporting this work. We acknowledge the PHE National Mycobacterium Reference Laboratory (NMRL) for providing M. tuberculosis Beijing strains 1192/015 and 08/00483E. The views expressed in this publication are those of the authors and not necessarily those of Public Health England or the Department of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b00834.
Atomic-level interactions of compound 3 in binding sites; mutations conferring antibiotic resistance; synthesis and characterization of PBD core 2; molecular models of ciprofloxacin–gyrase interaction; and mutant data (PDF)
Author Present Address
§ Chemistry Department, Oncology Business Unit, Nerviano Medical Sciences S.r.l., 20014 Nerviano, Milan, Italy
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report 2018; World Health Organization, 2018.
- Mdluli K.; Kaneko T.; Upton A. The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harbor Perspect. Med. 2015, 5, a021154 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviglione M.; Sulis G. Tuberculosis 2015: burden, challenges and strategy for control and elimination. Infect. Dis. Rep. 2016, 8, 6570. 10.4081/idr.2016.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L.; Parish T.; Balganesh M.; Ainsa J. A. Antituberculosis drugs: reducing efflux= increasing activity. Drug Discovery Today 2017, 22, 592–599. 10.1016/j.drudis.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Mantaj J.; Jackson P. J. M.; Rahman K. M.; Thurston D. E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem., Int. Ed. 2017, 56, 462–488. 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriollo P.; Hind C. K.; Picconi P.; Nahar K. S.; Jamshidi S.; Varsha A.; Clifford M.; Sutton J. M.; Rahman K. M. C8-linked pyrrolobenzodiazepine monomers with inverted building blocks show selective activity against multidrug resistant Gram-positive bacteria. ACS Infect. Dis. 2018, 4, 158–174. 10.1021/acsinfecdis.7b00130. [DOI] [PubMed] [Google Scholar]
- Rahman K. M.; Jackson P. J.; James C. H.; Basu B. P.; Hartley J. A.; de la Fuente M.; Schatzlein A.; Robson M.; Pedley R. B.; Pepper C. GC-targeted C8-linked pyrrolobenzodiazepine–biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. 10.1021/jm301882a. [DOI] [PubMed] [Google Scholar]
- Rahman K. M.; Rosado H.; Moreira J. B.; Feuerbaum E.-A.; Fox K. R.; Stecher E.; Howard P. W.; Gregson S. J.; James C. H.; de la Fuente M.; Waldron D. E.; Thurston D. E.; Taylor P. W. Antistaphylococcal activity of DNA-interactive pyrrolobenzodiazepine (PBD) dimers and PBD-biaryl conjugates. J. Antimicrob. Chemother. 2012, 67, 1683–1696. 10.1093/jac/dks127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K. M.; Vassoler H.; James C. H.; Thurston D. E. DNA Sequence Preference and Adduct Orientation of Pyrrolo 2,1-c 1,4 benzodiazepine Antitumor Agents. ACS Med. Chem. Lett. 2010, 1, 427–432. 10.1021/ml100131b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvvada M. S.; Forrow S. A.; Hartley J. A.; Stephenson P.; Gibson I.; Jenkins T. C.; Thurston D. E. Inhibition of bacteriophage T7 RNA polymerase in vitro transcription by DNA-binding pyrrolo 2,1-c 1,4 benzodiazepines. Biochemistry 1997, 36, 2478–2484. 10.1021/bi952490r. [DOI] [PubMed] [Google Scholar]
- Rahman K. M.; James C. H.; Bui T. T.; Drake A. F.; Thurston D. E. Observation of a single-stranded DNA/pyrrolobenzodiazepine adduct. J. Am. Chem. Soc. 2011, 133, 19376–19385. 10.1021/ja205395r. [DOI] [PubMed] [Google Scholar]
- Thurston D. E.; Bose D. S.; Thompson A. S.; Howard P. W.; Leoni A.; Croker S. J.; Jenkins T. C.; Neidle S.; Hartley J. A.; Hurley L. H. Synthesis of sequence-selective C8-linked pyrrolo [2, 1-c][1, 4] benzodiazepine DNA interstrand cross-linking agents. J. Org. Chem. 1996, 61, 8141–8147. 10.1021/jo951631s. [DOI] [PubMed] [Google Scholar]
- Hertzberg R. P.; Hecht S. M.; Reynolds V. L.; Molineux I. J.; Hurley L. H. DNA-sequence specificity of the pyrrolo 1,4 benzodiazepine antitumor antibiotics - Methidiumpropyl-EDTA-iron(ii) footprinting analysis of DNA-binding sites for anthramycin and related drugs. Biochemistry 1986, 25, 1249–1258. 10.1021/bi00354a009. [DOI] [PubMed] [Google Scholar]
- Hurley L. H.; Reck T.; Thurston D. E.; Langley D. R.; Holden K. G.; Hertzberg R. P.; Hoover J. R. E.; Gallagher G.; Faucette L. F. Pyrrolo 1,4 benzodiazepine antitumor antibiotics - relationship of DNA alkylation and sequence specificity to the biological-activity of natural and synthetic compounds. Chem. Res. Toxicol. 1988, 1, 258–268. 10.1021/tx00005a002. [DOI] [PubMed] [Google Scholar]
- Wells G.; Martin C. R. H.; Howard P. W.; Sands Z. A.; Laughton C. A.; Tiberghien A.; Woo C. K.; Masterson L. A.; Stephenson M. J.; Hartley J. A.; Jenkins T. C.; Shnyder S. D.; Loadman P. M.; Waring M. J.; Thurston D. E. Design, Synthesis, and Biophysical and Biological Evaluation of a Series of Pyrrolobenzodiazepine–Poly(N-methylpyrrole) Conjugates. J. Med. Chem. 2006, 49, 5442–5461. 10.1021/jm051199z. [DOI] [PubMed] [Google Scholar]
- Rahman K. M.; Jackson P. J. M.; James C. H.; Basu B. P.; Hartley J. A.; de la Fuente M.; Schatzlein A.; Robson M.; Pedley R. B.; Pepper C.; Fox K. R.; Howard P. W.; Thurston D. E. GC-Targeted C8-Linked Pyrrolobenzodiazepine-Biaryl Conjugates with Femtomolar in Vitro Cytotoxicity and in Vivo Antitumor Activity in Mouse Models. J. Med. Chem. 2013, 56, 2911–2935. 10.1021/jm301882a. [DOI] [PubMed] [Google Scholar]
- Basher M. A.; Rahman K. M.; Jackson P. J.; Thurston D. E.; Fox K. R. Sequence-selective binding of C8-conjugated pyrrolobenzodiazepines (PBDs) to DNA. Biophys. Chem. 2017, 230, 53–61. 10.1016/j.bpc.2017.08.006. [DOI] [PubMed] [Google Scholar]
- Corcoran D. B.; Lewis T.; Nahar K. S.; Jamshidi S.; Fegan C.; Pepper C.; Thurston D. E.; Rahman K. M. Effects of Systematic Shortening of Noncovalent C8 Side Chain on the Cytotoxicity and NF-κB Inhibitory Capacity of Pyrrolobenzodiazepines (PBDs). J. Med. Chem. 2019, 62, 2127–2139. 10.1021/acs.jmedchem.8b01849. [DOI] [PubMed] [Google Scholar]
- Raju G.; Srinivas R.; Santhosh Reddy V.; Idris M. M.; Kamal A.; Nagesh N. Interaction of Pyrrolobenzodiazepine (PBD) Ligands with Parallel Intermolecular G-Quadruplex Complex Using Spectroscopy and ESI-MS. PLoS ONE 2012, 7, e35920 10.1371/annotation/d51e5efe-c697-4400-bac2-67027fbb88b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucoli F.; Guzman J. D.; Basher M. A.; Evangelopoulos D.; McMahon E.; Munshi T.; McHugh T. D.; Fox K. R.; Bhakta S. DNA sequence-selective C8-linked pyrrolobenzodiazepine heterocyclic polyamide conjugates show anti-tubercular-specific activities. J. Antibiot. 2016, 69, 843–849. 10.1038/ja.2016.43. [DOI] [PubMed] [Google Scholar]
- Domalaon R.; Idowu T.; Zhanel G. G.; Schweizer F. Antibiotic Hybrids: the Next Generation of Agents and Adjuvants against Gram-Negative Pathogens?. Clin. Microbiol. Rev. 2018, 31, e00077 10.1128/CMR.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Chen Y.; Xu Z.; Wang Z.; Zeng Q.; Fan H. Switchable Control of Antibiotic Activity: A Shape-Shifting “Tail” Strategy. Bioconjugate Chem. 2018, 29, 74–82. 10.1021/acs.bioconjchem.7b00599. [DOI] [PubMed] [Google Scholar]
- Schmidt M.; Harmuth S.; Barth E. R.; Wurm E.; Fobbe R.; Sickmann A.; Krumm C.; Tiller J. C. Conjugation of Ciprofloxacin with Poly(2-oxazoline)s and Polyethylene Glycol via End Groups. Bioconjugate Chem. 2015, 26, 1950–1962. 10.1021/acs.bioconjchem.5b00393. [DOI] [PubMed] [Google Scholar]
- Renčiuk D.; Zhou J.; Beaurepaire L.; Guédin A.; Bourdoncle A.; Mergny J.-L. A FRET-based screening assay for nucleic acid ligands. Methods 2012, 57, 122–128. 10.1016/j.ymeth.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Gregson S. J.; Howard P. W.; Hartley J. A.; Brooks N. A.; Adams L. J.; Jenkins T. C.; Kelland L. R.; Thurston D. E. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J. Med. Chem. 2001, 44, 737–748. 10.1021/jm001064n. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Lambert R.; Pearson J. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J. Appl. Microbiol. 2000, 88, 784–790. 10.1046/j.1365-2672.2000.01017.x. [DOI] [PubMed] [Google Scholar]
- Qin L.; Wang J.; Lu J.; Yang H.; Zheng R.; Liu Z.; Huang X.; Feng Y.; Hu Z.; Ge B. A deletion in the RD105 region confers resistance to multiple drugs in Mycobacterium tuberculosis. BMC Biol. 2019, 17, 7 10.1186/s12915-019-0628-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H.; Basina M.; Nguyen V.; Rosenberg E. Y. Multidrug Efflux Pump AcrAB of Salmonella typhimuriumExcretes Only Those β-Lactam Antibiotics Containing Lipophilic Side Chains. J. Bacteriol. 1998, 180, 4686–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James B.; Williams A.; Marsh P. The physiology and pathogenicity of Mycobacterium tuberculosis grown under controlled conditions in a defined medium. J. Appl. Microbiol. 2000, 88, 669–677. 10.1046/j.1365-2672.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Gold B.; Roberts J.; Ling Y.; Quezada L. L.; Glasheen J.; Ballinger E.; Somersan-Karakaya S.; Warrier T.; Nathan C. Visualization of the Charcoal Agar Resazurin Assay for Semi-quantitative, Medium-throughput Enumeration of Mycobacteria. JoVE - J. Visualized Exp. 2016, e54690 10.3791/54690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateche F.; Martin A.; Baraka S.; Palomino J. C.; Khaled S.; Portaels F. Application of the resazurin microtitre assay for detection of multidrug resistance in Mycobacterium tuberculosis in Algiers. J. Med. Microbiol. 2006, 55, 857–860. 10.1099/jmm.0.46513-0. [DOI] [PubMed] [Google Scholar]
- Mosaei H.; Molodtsov V.; Kepplinger B.; Harbottle J.; Moon C. W.; Jeeves R. E.; Ceccaroni L.; Shin Y.; Morton-Laing S.; Marrs E. C. L.; Wills C.; Clegg W.; Yuzenkova Y.; Perry J. D.; Bacon J.; Errington J.; Allenby N. E. E.; Hall M. J.; Murakami K. S.; Zenkin N. Mode of Action of Kanglemycin A, an Ansamycin Natural Product that Is Active against Rifampicin-Resistant Mycobacterium tuberculosis. Mol. Cell 2018, 72, 263–274 e5. 10.1016/j.molcel.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.