

Keywords: 3×Tg-AD, Aβ3–10-KLH vaccine, Alzheimer's disease, amyloid precursor protein, amyloid-beta, cognitive decline, tau phosphorylation, transgenic mouse
Abstract
Active and passive anti-Aβ immunotherapies have successfully been used for the prevention and treatment of Alzheimer’s disease animal models. However, clinical use of these immunotherapies is not effective, because the vaccination is administered too late. At 1 month of age, 100 μL of Aβ3–10-KLH peptide (vaccine, 2 μg/μL) was subcutaneously injected into the neck of an amyloid precursor protein/presenilin-1/tau transgenic (3×Tg-AD) mouse model. Aβ3–10-KLH peptide was re-injected at 1.5, 2.5, 3.5, 4.5, 5.5, and 6.5 months of age. Serum levels of Aβ antibody were detected by enzyme-linked immunosorbent assay, while spatial learning and memory ability were evaluated by Morris water maze. Immunohistochemistry was used to detect total tau with HT7 and phosphorylated tau with AT8 (phosphorylation sites Ser202 and Thr205) and AT180 (phosphorylation site Thr231) antibodies in the hippocampus. In addition, western blot analysis was used to quantify AT8 and AT180 expression in the hippocampus. The results showed that after vaccine injection, mice produced high levels of Aβ antibody, cognitive function was significantly improved, and total tau and phosphorylated tau levels were significantly reduced. These findings suggest that early active immunization with Aβ3–10-KLH vaccine can greatly reduce tau phosphorylation, thereby mitigating the cognitive decline of 3×Tg-AD mice. This study was approved by the Animal Ethics Committee of China Medical University, China (approval No. 103-316) on April 2, 2016.
Chinese Library Classification No. R456; R392; R741
Introduction
The amyloid-beta (Aβ) cascade hypothesis is the most influential theory used to explain the pathogenesis of Alzheimer’s disease (AD) (Barage and Sonawane, 2015). Genetic, pathological, and biochemical evidence suggest that Aβ deposition acts as an important factor that triggers a series of events (Selkoe and Hardy, 2016; Zhang et al., 2019), which lead to the formation of neurofibrillary tangles (NFTs), neuronal dysfunction, and, finally, dementia (Kozlov et al., 2017; Wijesinghe et al., 2017; Lin et al., 2018). AD has been associated with altered activity of glycogen synthase kinase 3 isozymes, which are shown to contribute to both neurofibrillary tangle and amyloid plaque formation (Hernández et al., 2010). Hyperphosphorylated tau self-assembles to form paired helical filaments and NFTs. Aβ plaques and NFTs disrupt synaptic signaling between neurons and affect normal neuronal function, ultimately leading to cognitive decline (Spires-Jones and Hyman, 2014; Su et al., 2017).
Currently, only symptomatic therapies for AD are available in the clinic. With the progression of AD, the efficacy of symptomatic therapies tends to decrease, and AD progression cannot be fundamentally prevented. Immunotherapies have become one of the most promising methods to reverse or slow AD progression. This treatment strategy uses synthetic peptides or monoclonal antibodies to reduce Aβ load in the brain, thereby slowing disease progression (Ding et al., 2016; Martinez and Peplow, 2019). Active immunotherapy works together with the human immune system to neutralize the toxicity induced by Aβ oligomers (Barrera-Ocampo and Lopera, 2016). Active anti-Aβ immunotherapies effective reduces Aβ load and, thus, exhibit encouraging application prospects.
Since the first report of active immunization targeting Aβ, which terminated the pathological progression of AD in transgenic mice (Schenk et al., 1999), many promising animal experiment results have been tested in pre-clinical trials. However, the first trial involving multiple injections of AN1792 vaccine (aggregated Aβ42) with QS-21 as an adjuvant in AD patients was terminated because of the occurrence of meningoencephalitis in 6% of the study population (Orgogozo et al., 2003). This complication likely resulted from a specific Th1 cell-mediated immune response to QS-21, a potent activator of Th1 cells, and the use of an Aβ42 peptide carrying full-length epitopes activated by T cells. As such, the development of immunotherapies is limited by the risk for developing autoimmune diseases, remarkable side effects, and uncertain therapeutic efficacy.
Although the clinical trial for AN1792 vaccine failed, long-term follow-up of subjects has demonstrated that active immunization with AN1792 vaccine reduced Aβ load (Nicoll et al., 2003) and attenuated neurological deficits (Bayer et al., 2005; Holmes et al., 2008; Vellas et al., 2009). These findings support the potential benefits of immunotherapies, providing Aβ-specific T cell responses are avoided. Second-generation vaccines target the B cell epitope of Aβ. However, the antibodies produced by these vac-cines bind to Aβ monomers, oligomers, fibrils, and even amyloid precursor protein (APP) (Petrushina et al., 2007). Moreover, active immunization with these vaccines was shown to lead to cerebral edema and microbleeds in the brains of AD patients (Lambracht-Washington and Rosenberg, 2013). Therefore, second-generation vaccines do not exhibit obvious therapeutic efficacy. Although active immunotherapy has some proven benefits for AD patients (Lambracht-Washington and Rosenberg, 2013), the main challenges are safety and adverse reactions. Despite a number of shortcomings, immunization with a vaccine remains the best way to mitigate cognitive decline after AD.
The B cell epitope is located in the Aβ1–15 region, however, the Aβ3–10 fragment preserves the immuno-genicity of the fragment to elicit a sufficient immune response (Gilman et al., 2005) while avoiding Th1 immune inflammation. Therefore, the Aβ3–10 fragment was used as the major component of the vaccine peptide. Sha et al. (2012) reported that Aβ3–10 can induce high levels of anti-Aβ antibody while maintaining a low level of Th1 immune response.
In this study, we combined Aβ3–10 peptide with keyhole limpet hemocyanin (KLH) to induce high levels of T cell-mediated Th2 immune response and anti-Aβ antibody KLH is a well-known carrier protein with high immunogenicity (Swerdlow et al., 1996). Frenkel et al. (2001) immunized AD mouse models with Aβ37–42 coupled to KLH and obtained high levels of anti-Aβ antibody. In this study, we used potent Freund’s adjuvant as an Aβ3–10-KLH adjuvant to induce a strong Th2 immune response in animals (Walsh et al., 1999). There is evidence that Aβ3–10-KLH can induce a strong Th2-polarized anti-Aβ antibody response and inhibit Aβ deposition in APP/presenilin-1 (PS1) mice (Ding et al., 2016).
APP/PS1/tau transgenic mouse models (3×Tg-AD) harboring mutant forms of APP, PS1M146V, and tau P301L can mimic AD pathology in humans. Indeed, levels of various indicators in 3×Tg-AD mice are closer to those of humans compared with double-transgenic AD mouse models used in a previous study (Oddo et al., 2006a). In 3×Tg-AD mice, deficits in cognitive function occur as early as 4 months, which parallels the time of formation of extracellular Aβ deposits in the hippocampus (Clinton et al., 2007; Mastrangelo and Bowers, 2008). After 8–12 months, large amounts of both Aβ and phosphorylated tau (p-tau) deposits are present in the cerebral cortex, leading to cognitive decline (Billings et al., 2005; Giménez-Llort et al., 2007, 2010; García-Mesa et al., 2011). 3×Tg-AD mice are one of the few AD mouse models involving both Aβ and p-tau pathology, which allows multiple treatments to regulate protein formation throughout the life cycle. Thus, 3×Tg-AD mice represent an ideal model for an overall assessment of AD. The role of Aβ in tau pathology has also been shown in 3×Tg-AD mice, and damage to cerebral neurons gradually leads to cognitive dysfunction (Van der Jeugd et al., 2018). However, the molecular mechanism of Aβ in hyperphosphorylation of tau protein remains poorly understood. In this study, we actively immunized 3×Tg-AD mice with Aβ3–10-KLH vaccine at 1 month of age, a time at which there were no Aβ plaques (Winton et al., 2011), to determine the efficacy of active immunization with Aβ3–10-KLH vaccine and investigate whether early active immunization with Aβ3–10-KLH vaccine can slow the progression of AD.
The objectives of this study were to investigate the pathological mechanism of AD by quantitative comparison of anti-Aβ antibody in vivo in 3×Tg-AD mice, and assess whether the increase in anti-Aβ oligomer antibody concentration by early active immunization with Aβ3–10-KLH vaccine affects the learning and memory abilities of 3×Tg-AD mice.
Materials and Methods
Aβ3–10-KLH vaccine synthesis
A cysteine (Cys) was added to the C-terminus of the Aβ3–10 amino acid sequence H-Glu-Phe-Arg-His-Asp-Ser-Gly-Tyr-COOH to couple KLH. From the C-terminus to the N-terminus, amino acids were sequentially linked to form a polypeptide chain (EFRHDSGYC) using a solid-phase peptide synthesis technique. The synthesized polypeptide was purified to > 90% purity by high performance liquid chromatography. Its molecular weight was determined to be 1113.17 by mass spectrometry. KLH was coupled to Cys to form Aβ3–10-KLH vaccine. The above procedure was performed by GenScript Co., Ltd. (Nanjing, Jiangsu Province, China).
Animals and immunization methods
Twenty-four male or female 3×Tg-AD mice (Oddo et al., 2003) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Twelve strain-matched C57BL/6/129S nTg mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. [Beijing, China; License No. SYXK (Jing) 2017-0033] and used as controls. All mice were bred and maintained at the Laboratory Animal Center of China Medical University (China).
Experiments were performed in accordance with National Institutes of Health (NIH; Bethesda, MD, USA) guidelines on use of laboratory animals and approved by the Animal Ethics Committee of China Medical University (approval No. 103-316) on April 2, 2016. Mice used in this study were housed individually with 12-hour light/dark cycles and provided ad libitum access to food and water.
Twenty-four 1-month-old 3×Tg-AD mice were randomly divided into an Aβ3–10-KLH group and phos-phate-buffered saline (PBS) group (n = 12/group). A total of 1 mg Aβ3–10-KLH peptide was dissolved in PBS until the inoculation concentration reached 2 μg/μL. Dissolved peptides were emulsified with Freund’s Complete Adjuvant (Sigma, St. Louis, MO, USA) at 1:1 (v/v) for the first immunization and with Freund’s Incomplete Adjuvant (Sigma) at 1:1 (v/v) for the following immunizations. In the Aβ3–10-KLH group, mice were actively immunized by subcutaneous injection of 100 μL of the above prepared mixtures in the neck at 1, 1.5, 2.5, 3.5, 4.5, 5.5, and 6.5 months of age (Ding et al., 2016). In the PBS group, 100 μL of PBS was identically injected at each time point. Twelve 1-month-old C57BL/6/129S wild-type (WT) mice used as controls (WT group) were raised identically to Aβ3–10-KLH and PBS groups, but without other treatments.
Six mice from each group were used for serum detection of anti-Aβ antibody and evaluation of learning and memory abilities. The remaining six mice in each group were used for immunohistochemistry and western blot assay.
Detection of serum levels of anti-Aβ antibody
Prior to the first immunization and 10 days after each immunization with Aβ3–10-KLH vaccine, blood samples were collected from the internal iliac vein for detection of anti-Aβ antibody in the serum. The anti-Aβ antibody was separated from endogenous Aβ in serum using a low-pH dissociation method. The serum was diluted at 1:100 with dissociation solution (PBS + 1.5% bovine serum albumin + 0.2 M glycine-HCl, pH 3.5) and incubated at room temperature for 20 minutes. Next, the diluted serum was centrifuged at 14,000 × g for 20 minutes at room temperature in a centrifuge tube (Millipore, Billerica, MA, USA; 50,000 Dalton molecular weight cut-off). The sample reservoir was inverted in another tube. Serum samples were centrifuged at 1000 × g for 2 minutes. Next, the isolated endogenous anti-Aβ antibody solution was collected and its pH value was adjusted with 1 M Tris buffer solution (pH 9.0) to 7.0. Serum level of anti-Aβ antibody was de-tected by enzyme-linked immunosorbent assay (ELISA). An Aβ3–10-coated 96-well ELISA plate (Corning Inc., Corning, NY, USA) was incubated with pre-dissociated serum, post-dissociated serum, and serial dilu-tions of a standard BAM-10 antibody (Signet, Dedham, MA, USA). After the addition of secondary antibody goat anti-mouse IgG conjugated with horseradish peroxidase (1:5000), serum samples were incubated at room temperature for 1 hour and developed with 1-step TMB. Optical densities at 450 nm were measured using a microplate reader (BioTek Instruments, Winooski, VT, USA). Serum level of anti-Aβ antibody was quantified using a calibration curve generated by serial dilution of BAM-10.
Morris water maze test
Morris water maze testing was performed in mice at 7 months of age to evaluate their spatial learning and memory abilities. The Morris water maze (Huaibei Zhenghua Biologic Apparatus Facilities Co., Ltd., Huaibei, Anhui Province, China) used in this study consisted of a circular stainless steel pool (diameter of about 100 cm) filled with water to a depth of 30 cm. The water was made opaque with dry milk. The pool was divided into the same four quadrants namely northwest, northeast, southwest, and southeast, which were labeled north, south, east, and west, respectively. The camera above the pool was connected to a computer via a Noldus video tracking system (Ethovision; Noldus Information Technology, Beijing, China) to record the swimming trajectory of mice during the experiment.
Place navigation test
The hidden platform was submerged 2 cm below the water surface for 5 days of place navigation testing. Mice were placed facing the wall of the pool in each quadrant. The time required for the mouse to find the hidden platform, i.e. latency, was recorded. If a mouse did not locate the platform within 60 seconds, it would be manually guided to the platform, and the latency would be recorded as 60 seconds. According to the latency and swimming trajectory to find the platform, the mouse’s ability and strategy to search for the hidden platform were analyzed. The latency period to find the underwater platform and swimming distance of each rat were recorded. A shorter delay time and trajectory length indicated stronger learning ability. The swimming trajectory of rats was recorded with a camera and analyzed using behavioral record software, EthoVision XT 8 (Noldus Information Technology).
Spatial exploration test
On day 6 of the experiment, the hidden platform was removed. Mice were placed facing the wall of the pool in the quadrant not adjacent to the one where the platform had been located during the test described above. The time required for a mouse to first arrive at the original platform location, number of crossings of the original platform location, time spent in the quadrant where the platform was located, and swimming trajectory were recorded to assess the spatial memory ability of each mouse.
Tissue specimens
After water maze tests, mice were deeply anesthetized by intraperitoneal injection of 10% chloral hydrate (Sigma). Thoracic surgery was performed and the left ventricle was rapidly perfused with normal saline. Next, brains were harvested and halved using a median-sagittal cut. Left hemispheres were embedded and fixed for immunohistochemistry, while right hemispheres were preserved in a liquid nitrogen tank for subsequent western blot assays.
Immunohistochemistry
Mouse brains were paraffin-embedded and sliced. Six hippocampal sections from each 3×Tg-AD mouse were selected for immunohistochemical staining of total tau with HT7 antibody, and p-tau with AT8 (phosphorylation sites Ser202 and Thr205) and AT180 (phosphorylation site Thr231) antibodies.
Dried sections were dewaxed and dehydrated using a series of alcohol gradients, treated with 3% hydrogen peroxide for 15 minutes at room temperature to inactivate endogenous peroxidase, and washed three times with 0.01 M PBS for 5 minutes each. Subsequently, sections were treated with Lab’s antigen retrieval reagent for 20 minutes and washed three times with 0.01 M PBS for 5 minutes each. Sections were then incubated with goat serum blocking solution at room temperature for 30 minutes, followed by mouse anti-HT7 (1:1000; Invitrogen, Carlsbad, CA, USA), mouse anti-phospho-tau phospho-dependent AT8 (1:1000; Ser202/Thr205; Invitrogen), and AT180 (1:1000; Thr231; Invitrogen) monoclonal antibodies (Invitrogen) at 4°C overnight. The next day, sections were washed three times with PBS for 5 minutes each, incubated with secondary antibody biotin-labeled goat anti-mouse IgG (1:200; Abcam, Cambridge, MA, USA) at room temperature for 1 hour, and washed three times with PBS for 5 minutes each. Thereafter, sections were incubated with streptomycin-avidin-biotin-peroxidase complex (SABC; Wuhan Boster Biological Technology, Ltd., Wuhan, Hunan Province, China) at room temperature for 30 minutes, washed three times with PBS for 5 minutes each, developed with 3,3′-diaminobenzidine (Wuhan Boster Biological Technology), and counter-stained with hematoxylin. Under an optical microscope, images were taken at 10× and 20× magnifications. Image-Pro Plus 6.0 image analysis software (Media Cybernetics, Inc., Silver Spring, MD, USA) was used for semi-quantitative analysis of images.
Western blot analysis
Mouse hippocampal tissue was homogenized and centrifuged. Supernatant was collected for protein separation using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Beyotime Institute of Biotechnology, Beijing, China). Separated protein was then transferred onto a polyvinylidene fluoride (PVDF) membrane, and 10% skim milk was added to cover the membrane for 2 hours while shaking to prevent nonspecific binding of antibodies. After washing with PBS containing 0.1% Tween-20, proteins on the PVDF membrane were separately labeled using primary rabbit anti-mouse AT8 (Ser202/Thr205; 1:500; Invitrogen) and rabbit anti-mouse AT180 (Thr231; 1:1000; Invitrogen) at 4°C overnight. After washing with PBS containing 0.1% Tween-20, proteins were labeled with goat anti-mouse horseradish peroxidase-conjugated antibody (1:5000; Invitrogen) at room temperature for 2 hours. Western blot signals were detected by enhanced chemiluminescence (Millipore). Relative optical density of protein blots was quantified using ImageJ software (NIH). GAPDH was used as a loading control.
Statistical analysis
All data are expressed as the mean ± SD, and were statistically analyzed with SPSS 16.0 software (SPSS, Chicago, IL, USA). One-way analysis of variance and least significant difference tests were used for com-parisons between groups. A level of P < 0.05 was considered statistically significant.
Results
Aβ3–10-KLH vaccine immunizes 3×Tg-AD mice to produce high levels of anti-Aβ antibody
Serum anti-Aβ antibody was not detected by ELISA until the second immunization. Levels were significantly increased after the fourth immunization and reached 46.808 ± 10.121 μg/mL by the last immunization (P < 0.01, vs. PBS group; Figure 1).
Figure 1.
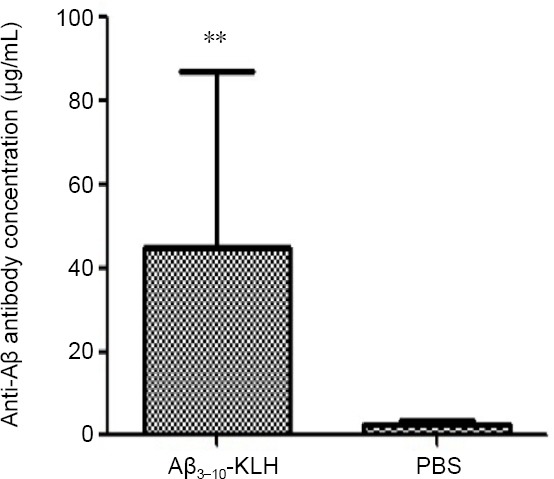
Serum level of anti-Aβ antibody in 3×Tg-AD mice after the last immunization with Aβ3–10-KLH vaccine.
Active immunizations with Aβ3–10-KLH vaccine produced a high level of anti-Aβ antibody. Data are expressed as the mean ± SD (n = 12). **P < 0.01, vs. PBS group (one-way analysis of variance followed by least significant difference test). The experiment was performed in triplicate. 3×Tg-AD: Amyloid precursor protein/presenilin-1/tau transgenic mouse models; Aβ: amyloid-beta; KLH: keyhole limpet hemocyanin; PBS: phosphate-buffered saline.
Active immunization with Aβ3–10-KLH vaccine improves behavioral performance of 3×Tg-AD mice
After seven immunizations, the cognitive function of 3×Tg-AD mice (7 months of age) was evaluated using the Morris water maze test. As shown in Figure 2, in the place navigation test, latency to find the hidden platform in the Aβ3–10-KLH group was significantly reduced compared with the PBS group (P < 0.01). In the spatial exploration test, the time required for mice to arrive at the original platform location was significantly reduced, the time spent in the quadrant where the platform was located was significantly increased, and the number of crossings of the original platform location was significantly increased in the Aβ3–10-KLH group compared with the PBS group (P < 0.05 or P < 0.01). Thus, Morris water maze test results suggest that active immunization with Aβ3–10-KLH vaccine cam greatly improve the cognitive function of 3×Tg-AD mice.
Figure 2.

Active immunization with Aβ3–10-KLH vaccine greatly improves the cognitive function of 3×Tg-AD mice as shown by water maze tests.
(A) In the place navigation test, the time required for a mouse to find the hidden platform in the Aβ3–10-KLH group was significantly shortened compared with the PBS group (**P < 0.01). (B) Spatial exploration test. (a) Time required for a mouse to first arrive at the original platform location; (b) time spent in the quadrant where the platform was located; (c) number of crossings of the original platform location. Compared with the PBS group, the time required for a mouse to first arrive at the original platform location was significantly shortened, time spent in the quadrant where the platform was located was prolonged, and number of crossings of the original platform location was increased (*P < 0.05, **P < 0.01). (C) Swimming trajectory of mice in each group in behavioral tests. (a–c) Place navigation test, (d–f) spatial exploration test. Data are expressed as the mean ± SD of n = 6 mice in each group, and analyzed by one-way analysis of variance followed by least significant difference test. The experiment was performed in triplicate. 3×Tg-AD: Amyloid precursor protein/presenilin-1/tau transgenic mouse models; Aβ: amyloid-beta; KLH: keyhole limpet hemocyanin; PBS: phosphate-buffered saline; WT: wild-type.
Active immunization with Aβ3–10-KLH vaccine reduces AT8- and AT180-immunoactive p-tau, and HT7-immunoreactive total tau in 3×Tg-AD mice
Immunohistochemical staining results revealed weak positivity for AT8- and AT180-immunoreactive p-tau and HT7-immunoreactive total tau in the Aβ3–10-KLH group, but strong positivity in the PBS group. However, no AT8- or AT180-immunoreactive p-tau or HT7-immunoreactive total tau were detected in the WT group. Semi-quantitative analysis of immunohistochemical results indicated that AT8, AT180, and HT7 immunore-activities in the Aβ3–10-KLH group were significantly reduced compared with the PBS group (P < 0.05; Figure 3).
Figure 3.

Active immunization with Aβ3–10-KLH vaccine reduced AT8 (A), AT180-immunoreactive phosphorylated (p) tau protein (B), and HT7-immunoreactive total tau (C) in the hippocampus of 3×Tg-AD mice at 7 months of age.
Representative micrographs of AT8, AT180, and HT7 immunohistochemical staining in the hippocampus of mice (left, original magnification,10×; right, original magnification, 20×). AT8, AT180, and HT7 immunoreactivity in the hippocampus of mice in the Aβ3–10-KLH group was weak, but was strong in the PBS group. No AT8, AT180, or HT7 immunoreactivity was detected in the wild-type (WT) group. AT8, AT180, and HT7 immunoreactive cells are stained brown. Semi-quantitative analysis of AT8, AT180, and HT7 immunoreactivities. Data are expressed as the mean ± SD of n = 6 mice in each group. *P < 0.05, vs. PBS group (one-way analysis of variance followed by the least significant difference test). 3×Tg-AD: Amyloid precursor protein/presenilin-1/tau transgenic mouse models; Aβ: amyloid-beta; KLH: keyhole limpet hemocyanin; PBS: phosphate-buffered saline; WT: wide-type.
Active immunization with Aβ3–10-KLH vaccine reduces protein expression of p-tau isoforms (AT8 and AT180) in 3×Tg-AD mice
Western blot assay results indicated that the expression of AT8- and AT180-immunoreactive p-tau in the hippocampus of 7-month-old 3×Tg-AD mice was significantly lower in the Aβ3–10-KLH group compared with the PBS group (P < 0.05; Figure 4).
Figure 4.

Active immunizations with Aβ3–10-KLH vaccine reduced protein expression of p-tau (AT8 and AT180).
(A, B) Immunoblotting results of AT8 (phosphorylation sites Ser202 and Thr205) and AT180 (phosphorylation site Thr231). Protein expression levels of AT8 and AT180 were significantly lower in the Aβ3–10-KLH group compared with the PBS group (*P < 0.05). Data are expressed as mean ± SD (n = 6), and analyzed by one-way analysis of variance followed by the least significant difference test. All experiments were conducted at least in triplicate. 3×Tg-AD: Amyloid precursor protein/presenilin-1/tau transgenic mouse models; Aβ: amyloid-beta; KLH: keyhole limpet hemocyanin; PBS: phosphate-buffered saline; WT: wild-type.
Discussion
In this study, we used Aβ3–10-KLH vaccine to immunize 3×Tg-AD mice at 1 month of age, a time at which Aβ protein deposits do not form plaques. The Aβ3–10 fragment, which is a major component of the Aβ3–10-KLH vaccine used in this study, does not contain a T cell epitope that can cause an inflammatory response; instead, it contains a B cell epitope that retains an immune response. Morris water maze test results indicated that seven active immunizations with Aβ3–10-KLH vaccine induced transgenic mice to produce high levels of anti-Aβ antibody, which prevented and eliminated the production of Aβ protein without triggering the amyloid cascade, reduced production of tau protein (mainly p-tau) and, thereby, mitigated cognitive decline of 3×Tg-AD mice.
Our previous studies showed that Aβ3–10 peptide binding to KLH adjuvant causes few inflammatory re-sponses (Sha et al., 2012; Sengupta et al., 2016). In addition, this combination exhibits stronger ability to remove Aβ protein deposition and inhibit neuroinflammatory reactions, and better alleviates cognitive decline. Moreover, this immune effect persisted 4 months after immunizations. Results from this study suggest that early active immunization with Aβ3–10-KLH vaccine can elicit production of high levels of anti-Aβ antibody in 3×Tg-AD mice, thus enhancing their learning and memory abilities. In addition to avoiding inflammation, Aβ3–10-KLH vaccine reduced the production of p-tau, which is a key factor for improving cognitive function. The major kinases involved in p-tau pathology phosphorylate multiple sites, including AT8 and AT180; therefore, AT8 and AT180 reflect p-tau levels.
This study is the first to report active immunization with Aβ3–10-KLH vaccine in 3×Tg-AD mice (Rasool et al., 2013). Immunohistochemistry of total tau and p-tau was performed in the hippocampus, which is a major brain region responsible for learning and memory abilities. In this study, we evaluated whether an increase in anti-Aβ antibody levels induced by active immunization with Aβ3–10-KLH vaccine could reduce total tau and p-tau in the hippocampus of 3×Tg-AD mice. In the early phase, but not late phase, p-tau protein was removed from the brain by injection of an anti-Aβ antibody. Importantly, these cleared Aβ deposits led to the subsequent clearance of early tau pathology, which was greatly dependent on its phosphorylation status. In the late phase, hyperphosphorylated tau protein aggregates appeared to be unaffected by anti-Aβ antibody therapy. Early brain injury in AD is likely reversible, and early treatment of brain injury may reverse the pathological changes of AD (Winton et al., 2011). Therefore, in this study, active immunization with Aβ3–10-KLH vaccine was performed before the pathological deposition of Aβ in animal models. This induced high levels of anti-Aβ antibody, prevented the production of Aβ, effectively reduced the production of p-tau, and blocked Aβ cascade reaction. Notably, injection of the anti-amyloid oligomer antibody A11 resulted in pathological decreases in p-tau and Aβ in 3×Tg-AD mice, which supports the association of Aβ oligomers with Aβ and tau pathology (Oddo et al., 2006b; Seino et al., 2010). Previous studies reported that passive immunization reduced the level of Aβ oligomers and directly induced GSK-3β activation and tau protein phosphorylation (Ma et al., 2006; Oddo et al., 2006a). Notably, although Aβ has been shown to play a central role in the pathogenesis of AD, including memory impairment, synaptic loss, and neuronal cell death (Cleary et al., 2005), the mechanism underlying tau protein phosphorylation in AD remains poorly understood.
Aβ exerts neurotoxicity through the presence of tau protein, which synergizes with Aβ to aggravate neuronal cell dysfunction (Denver and McClean, 2018). In the absence of hippocampal neurons expressing tau protein, Aβ deposition does not cause degenerative changes in nerve cells. Results from another study suggest that the reduction of Aβ alone cannot mitigate cognitive decline in AD mice. Indeed, the learning and memory abilities of AD mice were only greatly enhanced when Aβ and tau protein were simultaneously reduced (Oddo et al., 2006a). In this study, we actively immunized 3×Tg-AD mice with Aβ3–10-KLH vaccine to produce high levels of anti-Aβ antibody, which significantly reduced p-tau protein and insoluble tau protein while preventing Aβ oligomer production, thereby enhancing the cognitive function of AD mice. Results from this study showed that active immunization with Aβ3–10-KLH vaccine could not only prevent Aβ deposition, but also reduce tau protein phosphorylation without triggering the Aβ cascade reaction. Therefore, active immunization with Aβ3–10-KLH vaccine can simultaneously act on Aβ and tau protein, thus producing encouraging therapeutic effects.
Recently, an Aβ monoclonal antibody manufactured by Biogen was shown to reduce brain Aβ levels and mitigate cognitive decline in AD patients (Vaillancourt, 2016). Identification of specific antibodies for Aβ oligomers is important for the development of AD therapy. Our previous studies confirmed that active immunization with Aβ3–10-KLH vaccine prevents Aβ deposition via Aβ oligomers, inhibits neuroinflammatory reaction, and obviously alleviates cognitive deficits; moreover, the elicited immune effects persisted 4 months later (Ding et al., 2016). Compared with the PBS group, seven active immunizations with Aβ3–10-KLH vaccine resulted in significant decreases in AT8- and AT180-immunoreactive p-tau, and HT7-immunoreactive total tau. Notably, the decrease in total tau is greatly attributable to the decrease in p-tau. Our previous study also revealed that in 3×Tg-AD mice, HT7 staining was decreased in part because Aβ reduction lowers non-phosphorylated HT7-immunoreactive tau protein with normal function. As hyperphosphorylation of p-tau is a key step in the formation of NFTs, Aβ cell-mediated tau protein clearance is considered to be dependent on the phosphorylation state of p-tau.
In this study, we also determined the optimal timing for active immunization with vaccines. We actively immunized 3×Tg-AD mice at 1 month of age by subcutaneous injection of Aβ3–10-KLH vaccine. Our results showed that serum level of anti-Aβ antibody was increased, p-tau expression was reduced, the degree of phosphorylation of tau protein was decreased, and cognitive decline of 3×Tg-AD mice was mitigated. These results suggest that early active immunization with Aβ3–10-KLH vaccine prevented and eliminated Aβ protein without triggering the Aβ cascade reaction, reduced tau protein phosphorylation and, thereby, is an encouraging method for effective treatment of AD.
Taken together, early active immunization with Aβ3–10-KLH vaccine not only avoids the inflammatory reaction caused by the traditional Aβ vaccine, but also produces high levels of anti-Aβ antibody to block the Aβ deposition-caused pathological amyloid cascade, and effectively reduces tau protein phosphorylation and the formation of neurofibrillary tangles, thereby mitigating cognitive decline. There are certain limitations to this study. As such, it is necessary to perform in-depth studies involving multiple intervention opportunities and targets, and comprehensively analyze the results of various studies to explore a more reasonable and effective active immunization program.
Additional file: Open peer review reports 1 (110.4KB, pdf) and 2 (122KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that they have no competing interests.
Financial support: This work was supported by the National Natural Science Foundation of China, No. 81371227 (to YPC). The funding body played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication.
Institutional review board statement: This study was approved by the Animal Ethics Committee of China Medical University, China (approval No. 103-316) on April 2, 2016.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Hans-Gert Bernstein, Otto-von-Guericke University Magdeburg, Germany; Alain Buisson, Université Joseph Fourier Grenoble, France; Cristina Agliardi, IRCCS Fondazione Don Carlo Gnocchi, Italy.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81371227 (to YPC).
P-Reviewers: Bernstein HG, Buisson A, Agliardi C; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Van Deusen A, Yu J, Song LP; T-Editor: Jia Y
References
- 1.Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015;52:1–18. doi: 10.1016/j.npep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Barrera-Ocampo A, Lopera F. Amyloid-beta immunotherapy: the hope for Alzheimer disease? Colomb Med (Cali) 2016;47:203–212. [PMC free article] [PubMed] [Google Scholar]
- 3.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 4.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 5.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 6.Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denver P, McClean PL. Distinguishing normal brain aging from the development of Alzheimer’s disease: inflammation, insulin signaling and cognition. Neural Regen Res. 2018;13:1719–1730. doi: 10.4103/1673-5374.238608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding L, Meng Y, Zhang HY, Yin WC, Yan Y, Cao YP. Active immunization with the peptide epitope vaccine Abeta3-10-KLH induces a Th2-polarized anti-Abeta antibody response and decreases amyloid plaques in APP/PS1 transgenic mice. Neurosci Lett. 2016;634:1–6. doi: 10.1016/j.neulet.2016.09.050. [DOI] [PubMed] [Google Scholar]
- 9.Frenkel D, Kariv N, Solomon B. Generation of auto-antibodies towards Alzheimer’s disease vaccination. Vaccine. 2001;19:2615–2619. doi: 10.1016/s0264-410x(00)00501-6. [DOI] [PubMed] [Google Scholar]
- 10.García-Mesa Y, López-Ramos JC, Giménez-Llort L, Revilla S, Guerra R, Gruart A, Laferla FM, Cristòfol R, Delgado-García JM, Sanfeliu C. Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J Alzheimers Dis. 2011;24:421–454. doi: 10.3233/JAD-2011-101635. [DOI] [PubMed] [Google Scholar]
- 11.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 12.Gimenez-Llort L, Garcia Y, Buccieri K, Revilla S, Sunol C, Cristofol R, Sanfeliu C. Gender-specific neuroimmunoendocrine response to treadmill exercise in 3×Tg-AD mice. Int J Alzheimers Dis. 2010;2010:128354. doi: 10.4061/2010/128354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: A role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31:125–147. doi: 10.1016/j.neubiorev.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Hernández F, Gómez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010;223:322–325. doi: 10.1016/j.expneurol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 16.Kozlov S, Afonin A, Evsyukov I, Bondarenko A. Alzheimer’s disease: as it was in the beginning. Rev Neurosci. 2017;28:825–843. doi: 10.1515/revneuro-2017-0006. [DOI] [PubMed] [Google Scholar]
- 17.Lambracht-Washington D, Rosenberg RN. Advances in the development of vaccines for Alzheimer’s disease. Discov Med. 2013;15:319–326. [PMC free article] [PubMed] [Google Scholar]
- 18.Lin W, Yang LK, Zhu J, Wang YH, Dong JR, Chen T, Wang D, Xu XM, Sun SB, Zhang L. Deep brain stimulation for the treatment of moderate-to-severe Alzheimer’s disease: Study protocol for a prospective self-controlled trial. Clin Trials Degener Dis. 2018;3:66–70. [Google Scholar]
- 19.Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, Frautschy SA, Cole GM. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res. 2006;83:374–384. doi: 10.1002/jnr.20734. [DOI] [PubMed] [Google Scholar]
- 20.Martinez B, Peplow PV. Amelioration of Alzheimer’s disease pathology and cognitive deficits by immunomodulatory agents in animal models of Alzheimer’s disease. Neural Regen Res. 2019;14:1158–1176. doi: 10.4103/1673-5374.251192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastrangelo MA, Bowers WJ. Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer’s disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 2008;9:81. doi: 10.1186/1471-2202-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 23.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006a;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 24.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006b;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 25.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 26.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 27.Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, Patel A, Head E, Cribbs DH, Agadjanyan MG. Alzheimer’s disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Abeta species in amyloid precursor protein transgenic mice. J Neurosci. 2007;27:12721–12731. doi: 10.1523/JNEUROSCI.3201-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasool S, Martinez-Coria H, Wu JW, LaFerla F, Glabe CG. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg-AD mice. J Neurochem. 2013;126:473–482. doi: 10.1111/jnc.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 30.Seino Y, Kawarabayashi T, Wakasaya Y, Watanabe M, Takamura A, Yamamoto-Watanabe Y, Kurata T, Abe K, Ikeda M, Westaway D, Murakami T, Hyslop PS, Matsubara E, Shoji M. Amyloid beta accelerates phosphorylation of tau and neurofibrillary tangle formation in an amyloid precursor protein and tau double-transgenic mouse model. J Neurosci Res. 2010;88:3547–3554. doi: 10.1002/jnr.22516. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sengupta U, Nilson AN, Kayed R. The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42–49. doi: 10.1016/j.ebiom.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sha S, Xing XN, Guo WS, Li Y, Zong LX, Guo R, Cao YP. In vivo electroporation of a new gene vaccine encoding ten repeats of Abeta3-10 prevents brain Abeta deposition and delays cognitive impairment in young Tg-APPswe/PSEN1dE9 mice. Neurochem Res. 2012;37:1534–1544. doi: 10.1007/s11064-012-0748-7. [DOI] [PubMed] [Google Scholar]
- 34.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su F, Yun P, Liu X, Shen X, Li CL, Li RY. Novel bio-mimetic receptors for early detection of Alzheimer’s disease biomarkers. Zhongguo Zuzhi Gongcheng Yanjiu. 2017;21:5552–5557. [Google Scholar]
- 36.Swerdlow RD, Ebert RF, Lee P, Bonaventura C, Miller KI. Keyhole limpet hemocyanin: structural and functional characterization of two different subunits and multimers. Comp Biochem Physiol B Biochem Mol Biol. 1996;113:537–548. doi: 10.1016/0305-0491(95)02091-8. [DOI] [PubMed] [Google Scholar]
- 37.Vaillancourt DE. Aducanumab reduces Aβ plaques in Alzheimer’s disease. Mov Disord. 2016;31:1631. doi: 10.1002/mds.26833. [DOI] [PubMed] [Google Scholar]
- 38.Van der Jeugd A, Parra-Damas A, Baeta-Corral R, Soto-Faguás CM, Ahmed T, LaFerla FM, Giménez-Llort L, D’Hooge R, Saura CA. Reversal of memory and neuropsychiatric symptoms and reduced tau pathology by selenium in 3xTg-AD mice. Sci Rep. 2018;8:6431–6431. doi: 10.1038/s41598-018-24741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M AN1792 (QS-21)-251 Study Team. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6:144–151. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 41.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 42.Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Abeta peptides in 3xTg-AD mice: implications for tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Zhang L, Liu JJ, Zhao Y, Liu Y, Lin JW. N-butylphthalide affects cognitive function of APP/PS1 transgenic mice (Alzheimer’s disease model) Zhongguo Zuzhi Gongcheng Yanjiu. 2019;23:3025–3030. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.