

Necroptosis has been reported in the last years as a type of regulated cell death that can be ignited by a number of specific death receptors, such as FAS, tumor necrosis factor receptor 1 (TNFR1), or pathogen recognition receptors. This cell death subroutine is involved in the response to stress and in homeostatic functions, such as the maintenance of adult T-cell balance (Galluzzi et al., 2018).
Dead receptor activation leads to the recruitment of protein complexes that act as nodes of signaling that can elicit different pathways driving to death or survival, depending on the cell conditions. The TNFR1-dependent pathway (Figure 1) is best known, where binding to TNF-alpha prompts the TNFR1 cytosolic tail to recruit the adaptor proteins, TNFR1-associated death domain, and TNFR-associated factor 2, the receptor-interacting serine/threonine-protein kinase 1 (RIPK1), as well as ubiquitin ligases belonging to cellular inhibitors of apoptosis (cIAP1 and cIAP2), overall forming the protein complex I (Galluzzi et al., 2018). A key molecule in this communication node is RIPK1. RIPK1 not only has the ability to promote caspase-8 activation and apoptosis, but it can also interact with RIPK3 and activate necroptosis. Alternatively, RIPK1 is able to activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in a kinase-independent manner, which prevents apoptosis induced by death receptors. This response involves the expression of the anti-apoptotic protein cellular FAS-associated death domain (FADD)- like interleukin-1beta-converting enzyme-like (FLICE) inhibitory protein (cFLIP), a catalytic inactive homolog of caspase-8. cFLIP is a key molecule in the switch between life and death since it is involved in the regulation of survival and death pathways, as well as in the switch between apoptosis and necroptosis. The main cFLIP variants are the short (cFLIPS) and the long (cFLIPL) isoforms. Both isoforms bind to caspase-8, but cFLIPS blocks caspase-8 activity, while cFLIPL allows some proteolytic activity (Galluzzi et al., 2018; Green, 2019).
Figure 1.
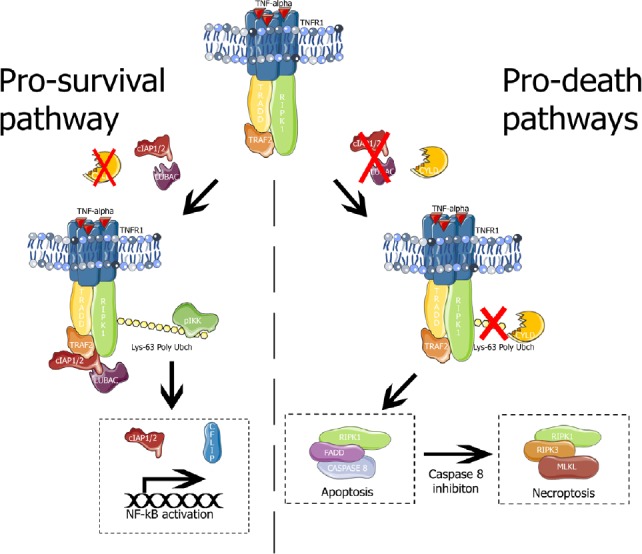
TNFR1 signaling.
Left panel, pro-survival pathway elicited by TNFR1 complex I (TNFR1, TRADD, RIPK1, TRAF2, cIAP1/2 and LUBAC). The E3 ubiquitin ligases cIAP1/2 and LUBAC cause Lys63 polyubiquitination of RIPK1. Polyubiquitinated-RIPK1 serves as scaffold for recruitment and activation of IKK which activates NF-κB promoting the transcription of pro-survival genes (such as cIAP1/2 and cFLIP). Right panel, pro-death pathways. In absence of E3 ubiquitin ligases, or in the presence of the deubiquitinase CYLD, RIPK1 forms the complex II (RIPK1, FADD and caspase 8) which leads to extrinsic apoptosis. If caspase 8 is inhibited, complex II is dissembled and RIPK1 forms necrosome complex (RIPK1, RIPK3 and MLKL) that phosphorylates MLKL, the necroptosis executioner. cFLIP: Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein; cIAP1/2: cellular inhibitors of apoptosis 1/2; CYLD: ubiquitin carboxyl-terminal hydrolase; FADD: Fas-associated death domain; IKK: IκB kinase; LUBAC: linear ubiquitin chain assembly complex; MLKL: mixed lineage domain-like protein; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; RIPK1: receptor-interacting serine/threonine-protein kinase 1; TNFR1: tumor necrosis factor receptor 1; TRADD: receptor-interacting serine/threonine-protein; TRAF2: TNFR-associated factor 2.
Differences in the response are mediated, to a large extent, by the ubiquitination process. RIPK1 can be polyubiquitinylated at Lys63 (a non-degradative pathway) by ubiquitin ligases cIAP1 and cIAP2 and the linear ubiquitin chain assembly complex; thus, allowing RIPK1 to activate the prosurvival NF-κB pathway in a kinase-independent manner. A deficiency in ubiquitination or deubiquitination (by ubiquitin carboxyl-terminal hydrolase) releases RIPK1 from complex I and permits its association with FADD and caspase-8 in the cytosol, forming protein complex II, which drives to extrinsic apoptosis (Galluzzi et al., 2018). In conditions where the caspase pathway is inhibited, RIPK1 autophosphorylates and undergoes a conformational change, leading to the oligomerization of RIPK1, which recruits and activates RIPK3. Necroptosis is regulated by RIPK3, which phosphorylates and activates the mixed lineage kinase domain-like protein (MLKL), a pore-forming protein that induces phospholipid scrambling (exposing phosphatidylserine) and disrupts the plasma membrane, resulting in necrotic cell death (Galluzzi et al., 2018; Vandenabeele et al., 2010). MLKL is mainly antagonized by the endosomal sorting complex III, which repairs the membrane at the site of damage in a process that seems to be dependent upon an influx of Ca2+ (Gong et al., 2017).
Inhibition of RIPK1 activity, or its proteasomal degradation, through the alternative K48-linked ubiquitination, blocks death-receptor-induced necroptosis. Also, the necroptosis process can be disrupted by the presence of a dimer complex formed by caspase-8 and cFLIPL. The caspase-8-cFLIPL dimer cleaves RIPK1 and RIPK3 and prevents necroptosis; therefore, the inhibition of caspase-8-c-FLIPL proteolytic activity allows necroptosis to proceed (Galluzzi et al., 2018; Green, 2019).
It must be highlighted that some toll-like receptors and the cytosolic nucleic acid sensor Z-DNA Binding Protein 1 also promote necroptosis; although in this case, RIPK3 is not activated by RIPK1. On the contrary, oligomerized RIPK3 (activated by TRIF) binds RIPK1 and recruits the caspase-8-cFLIPL dimer, which in turn destroys the RIPK3 oligomer. If the caspase-8-c-FLIPL dimer is inhibited or disrupted, necroptosis can proceed. In these conditions, RIPK1 inhibits necroptosis but not the activation of RIPK3 (Green, 2019).
Currently, the Nomenclature Committee of Cell Death indicates that necroptosis critically depends on MLKL, RIPK3, and, at least in some settings, the kinase activity of RIPK1 (Galluzzi et al., 2018); thus, the measure of phosphorylation of MLKL and RIPK3 markers seems to be the most reliable factor to characterize necroptosis. In this regard, in a model of global cerebral ischemia after 72 hours of reperfusion, the presence of necroptosis in the cerebral cortex, has recently been described based on increased mRNA as well as in the increase in the phosphorylation of MLKL (Font-Belmonte et al., 2019). The authors analyzed the main molecules involved in the activation of the necroptosis pathway elicited by the TNFR1 complex in both the cerebral cortex and CA1 hippocampal areas, as these regions have been widely reported to present different vulnerability to ischemia. The onset of necroptosis in the cerebral cortex seems to appear before 48 hours of reperfusion based on the progressive increase in transcription and phosphorylation of MLKL from 48 to 72 hours after reperfusion. In this model, there are no reports of detectable neuronal loss at 48 hours of ischemia, but it is significant after 7 days of ischemia (Anuncibay-Soto et al., 2016). These data support that necroptosis is a subroutine involved in delayed cell death in the cerebral cortex.
The use of salubrinal, an agent that promotes the unfolded protein response (UPR) and alleviates endoplasmic reticulum (ER) stress, has been reported to maintain a neuroprotective effect after 7 days of ischemia (Anuncibay-Soto et al., 2016). Moreover, data extracted from Font-Belmonte et al. (2019), showing a decrease in phosphorylation of MLKL and MLKL transcription, support that this effect involves a decrease in necroptosis activity. However, these authors indicate that some caution has to be taken in the interpretation of the role of ER stress on necroptosis. In this regard, salubrinal has been reported to decrease inflammation in different models of brain damage (Logsdon et al., 2016), including a global cerebral ischemia model (Anuncibay-Soto et al., 2016). Thus, the decrease observed in necroptosis following salubrinal treatment could be a consequence of the UPR-dependent reduction in inflammation or because UPR decreases ER stress, resulting in the alleviation of the necroptosis pathway (Font-Belmonte et al., 2019). In the latter case, ER-stress dependent necroptosis should be independent of proinflammatory ligands.
The analysis of the necroptosis markers provides some interesting hints on the time course of the necroptotic pathway. Thus, RIPK3 phosphorylation, which represents a crucial stage in necroptosome formation and MLKL activation (Galluzzi et al., 2018), decreases from 48 to 72 hours after ischemia and supports the idea of a relatively long temporal sequence in the activation of MLKL rather than a simultaneous response to the activation of the necroptosome. This time course of RIPK3 and MLKL is also supported by treatment with salubrinal. Additionally, the progressive decrease of RIPK3 phosphorylation supports the onset of necroptosis before 48 hours of reperfusion in this model (Font-Belmonte et al., 2019).
Another interesting aspect of the study is the increase of TNFR1 and IκB transcripts after the onset of necroptosis, which suggests that this cell death subroutine induces an inflammatory effect in the cerebral cortex that could activate NF-κB in a possible attempt to promote cell survival. The treatment with salubrinal reduces IκB and TNFR1 transcript levels, which seems to support the hypothesis of the necroptosis-induced inflammatory effect and the subsequent prosurvival response (Font-Belmonte et al., 2019). These authors also observed increases in the expression of the anti-apoptotic protein cFLIPL, which, together with the increases in IκB, is hypothesized to be involved in the attempt to promote a prosurvival response and block necroptosis and apoptosis in the cerebral cortex. This prosurvival response is not observed in CA1 and could also be involved in the differential vulnerability to ischemia between the cerebral cortex and CA1. Treatment with salubrinal results in decreased necroptosis, which blocks cFLIPL levels in the cerebral cortex but not in CA1, providing additional support to the hypothesis that the cerebral cortex can elicit a survival response induced by necroptosis that cannot be elicited by CA1.
Based on the lack of changes in the TNFR1 protein levels, it has been suggested that the onset of necroptosis in the global cerebral ischemia model could be TNFR1 independent, and the activation of TNFR1 would be elicited by the inflammation caused by necroptosis (Font-Belmonte et al., 2019). This hypothesis could be supported by the effects of ER stress on necroptosis that they observed in the cerebral cortex. In this regard, TNFR1-independent pathways have been related to ER stress in other organs, such as the lungs (Kim et al., 2018) and the heart (Zhu et al., 2018).
Finally, the report of Font-Belmonte highlights the differences in the necroptotic response in cerebral cortex and CA1, where data on current necroptosis markers are less consistent in the hippocampus. Necroptosis in the hippocampus has been reported in the same cerebral ischemia model but based on total RIPK3 protein levels (Vieira et al., 2014). However, some controversial data makes it difficult to assure the extension of this subroutine in CA1. Although some markers, such as MLKL transcription, provide support to the presence of necroptosis in CA1, stronger markers, such as phosphorylation of MLKL are not detected (Font-Belmonte et al., 2019). Thus, evidence of necroptosis in CA1 is weaker compared to that observed in the cerebral cortex, supporting the idea of different subroutine pathways in different brain structures.
In summary, global cerebral ischemia results in the activation of the necroptotic pathway in the cerebral cortex after 72 hours of reperfusion, which seems to be responsible for the neuronal demise observed after 7 days of reperfusion. The alleviation of ER stress by salubrinal treatment decreases, but does not eliminate, this cell death subroutine.
This work was supported by MINECO and FEDER funds (RTC-2015-4094-1); by Junta de Castilla y León (LE025P17); and by Neural therapies SL (NT-Dev-01). Paloma González-Rodríguez is granted from Junta de Castilla y León (EDU/529/2017). Enrique Font-Belmonte is supported by a grant from the University of León.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Alavala Matta Reddy, Adikavi Nannaya University, India.
P-Reviewer: Matta Reddy A; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Anuncibay-Soto B, Pérez-Rodríguez D, Santos-GAldiano M, Font E, Regueiro-Purriños M, Fernández-López A. Post-ischemic salubrinal treatment results in a neuroprotective role in global cerebral ischemia. J Neurochem. 2016;138:295–306. doi: 10.1111/jnc.13651. [DOI] [PubMed] [Google Scholar]
- 2.Font-Belmonte E, Ugidos IF, Santos-Galdiano M, González-Rodríguez P, Anuncibay-Soto B, Pérez-Rodríguez D, Gonzalo-Orden M, Fernández-López A. Post-ischemic salubrinal administration reduces necroptosis in a rat model of global cerebral ischemia. J Neurochem. 2019 doi: 10.1111/jnc.14789. doi: 10.1111/jnc.14789. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR. ESCRT-III Acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300.e16. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green DR. The coming decade of cell death research: Five riddles. Cell. 2019;177:1094–1107. doi: 10.1016/j.cell.2019.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H, Zamel R, Bai XH, Lu C, Keshavjee S, Keshavjee S, Liu M. Ischemia reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol. 2018;315:L595–608. doi: 10.1152/ajplung.00069.2018. [DOI] [PubMed] [Google Scholar]
- 7.Logsdon AF, Lucke-Wold BP, Nguyen L, Matsumoto RR, Turner RC, Rosen CL, Huber JD. Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res. 2016;1643:140–151. doi: 10.1016/j.brainres.2016.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2018;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 9.Vieira M, Fernandes J, Carreto L, Anuncibay-Soto B, Santos M, Han J, Fernández-López A, Duarte CB, Carvalho AL, Santos AE. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis. 2018;68:26–36. doi: 10.1016/j.nbd.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/MPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]