

Keywords: ferroptosis, ferrostatin-1, glutamate, glutathione peroxidase 4, HT-22 cell, oxidative toxicity, prostaglandin peroxidase synthase 2, reactive oxygen species
Abstract
Ferroptosis is a type of programmed cell death dependent on iron. It is different from other forms of cell death such as apoptosis, classic necrosis and autophagy. Ferroptosis is involved in many neurodegenerative diseases. The role of ferroptosis in glutamate-induced neuronal toxicity is not fully understood. To test its toxicity, glutamate (1.25–20 mM) was applied to HT-22 cells for 12 to 48 hours. The optimal experimental conditions occurred at 12 hours after incubation with 5 mM glutamate. Cells were cultured with 3–12 μM ferrostatin-1, an inhibitor of ferroptosis, for 12 hours before exposure to glutamate. The cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Autophagy was determined by monodansylcadaverine staining and apoptosis by caspase 3 activity. Damage to cell structures was observed under light and by transmission electron microscopy. The release of lactate dehydrogenase was detected by the commercial kit. Reactive oxygen species were measured by flow cytometry. Glutathione peroxidase activity, superoxide dismutase activity and malondialdehyde level were detected by the appropriate commercial kit. Prostaglandin peroxidase synthase 2 and glutathione peroxidase 4 gene expression was detected by real-time quantitative polymerase chain reaction. Glutathione peroxidase 4 and nuclear factor erythroid-derived-like 2 protein expression was detected by western blot analysis. Results showed that ferrostatin-1 can significantly counter the effects of glutamate on HT-22 cells, improving the survival rate, reducing the release of lactate dehydrogenase and reducing the damage to mitochondrial ultrastructure. However, it did not affect the caspase-3 expression and monodansylcadaverine-positive staining in glutamate-injured HT-22 cells. Ferrostatin-1 reduced the levels of reactive oxygen species and malondialdehyde and enhanced superoxide dismutase activity. It decreased gene expression of prostaglandin peroxidase synthase 2 and increased gene expression of glutathione peroxidase 4 and protein expressions of glutathione peroxidase 4 and nuclear factor (erythroid-derived)-like 2 in glutamate-injured HT-22 cells. Treatment of cultured cells with the apoptosis inhibitor Z-Val-Ala-Asp (OMe)-fluoromethyl ketone (2–8 μM), autophagy inhibitor 3-methyladenine (100–400 μM) or necrosis inhibitor necrostatin-1 (10–40 μM) had no effect on glutamate induced cell damage. However, the iron chelator deferoxamine mesylate salt inhibited glutamate induced cell death. Thus, the results suggested that ferroptosis is caused by glutamate-induced toxicity and that ferrostatin-1 protects HT-22 cells from glutamate-induced oxidative toxicity by inhibiting the oxidative stress.
Chinese Library Classification No. R453; R741; Q505
Introduction
Neurodegenerative diseases, including Parkinson’s and Alzheimer’s diseases have complex neurological outcomes with the loss of a large number of neurons as their core feature (Coyle and Puttfarcken, 1993). Further functional analysis has shown that neuronal loss was induced by excessive release of the excitatory transmitter glutamate (Maragakis and Rothstein, 2001); however, the mechanism underlying glutamate induced neurotoxicity is not fully understood.
Oxidative stress is related to many neurological diseases, such as Parkinson’s and Alzheimer’s diseases, amyotrophic lateral sclerosis and ischemic stroke (Kang et al., 2014; Shefa et al., 2019). The occurrence of oxidative stress is marked with the raised output of oxidizing species or a notable decline of antioxidant defenses, such as glutathione (GSH) and superoxide dismutase (SOD) (Schafer and Buettner, 2001). These events lead to the further production of reactive oxygen species (ROS) that eventually reduce the survival of cells. Current research work shows that the biological effects of oxidative stress are dose related and that moderate oxidative stress causes ferroptosis or apoptosis (Dypbukt et al., 1994; Toyokuni, 1999). These results show that ferroptosis and oxidative stress toxicity may act through similar mechanisms (Kang et al., 2014).
Ferroptosis is a type of programmed cell death dependent on iron; it is uniquely different from other forms of cell death such as apoptosis, classic necrosis or autophagy (Dixon et al., 2012; Yao et al., 2019). Ferroptosis has been implicated in multiple pathological diseases, including cancers (Gout et al., 2001; Yu et al., 2015; Hasegawa et al., 2016), heart ischemia/reperfusion injury (Gascón et al., 2016; Baba et al., 2018), acute renal failure (Conrad et al., 2016) and neurodegenerative diseases (Bogdan et al., 2016; Do Van et al., 2016). Some chemicals, such as erastin, and anti-cancer drugs, such as sorafenib, can induce ferroptosis (Lachaier et al., 2014). Recent data indicate that the inhibition of ferroptosis prevents glutamate induced neurotoxicity (Xie et al., 2016). Known inhibitors of ferroptosis include ferrostatin-1 and the iron chelator deferoxamine. Ferrostatin-1 is an aromatic amine that specifically binds with lipid ROS and protects cells against lipid peroxidation (Cao and Dixon, 2016). Our survey of the literature indicates that the role of ferroptosis on glutamate-induced neuronal toxicity is not fully understood.
Experimental data have shown that the occurrence of ferroptosis is mainly due to the suppression of the cystine/glutamate antiporter system (system Xc –), the restraint of glutathione peroxidase 4 (Gpx4) activity, the accumulation of ROS and the reduction of GSH level (Xie et al., 2016). Gpx4, a unique intracellular antioxidant enzyme, has the ability to directly reduce peroxidized phospholipids in the cell membrane (Dixon et al., 2012; Hasegawa et al., 2016). It has been well documented that Gpx4 plays an vital role in the signal transduction of ferroptosis, decrease of Gpx4 activity significantly reduces the clearance of the lipid peroxides (Conrad and Friedmann Angeli, 2015). Studies have shown that knockdown or overexpression of Gpx4 gene can significantly alter the sensitivity of cells to ferroptosis-inducing agents (Yang et al., 2014). The occurrence and development of ferroptosis by inducers, RSL3 and erastin, resulted in the decline of Gpx4 activity through direct binding of the inducers with Gpx4 and indirectly of glutathione deprivation (Dixon et al., 2012; Yang et al., 2014; Imai et al., 2017). Therefore Gpx4 activity can act as a molecular marker of ferroptosis. The status of nuclear factor erythroid-derived-like 2 (Nrf2) is also a critical element that determines the curative reaction to ferroptosis-targeted therapies in tumor cells (Sun et al., 2016). Nrf2 is known to upregulate oxidative stress-related genes (Brigelius-Flohe et al., 2012). Knockdown of Nrf2 and these Nrf2-targeted genes speeds up erastin-induced ferroptosis in the hepatocellular carcinoma cells (Sun et al., 2016).
We hypothesize that 1) ferroptosis has a crucial effect on glutamate-induced oxidative toxicity, and 2) the inhibitor of ferroptosis, ferrostatin-1, blocks the oxidative toxicity by inhibiting Gpx4. We utilized a glutamate-induced HT-22 cell glutamate toxicity model, with or without the ferroptosis inhibitor ferrostatin-1, to test whether the ferroptosis is involved in glutamate-induced cell toxicity. We examined the levels of oxidative markers such as cytosolic and lipid ROS, malondialdehyde (MDA), SOD, GSH, Gpx4 and Nrf2 to investigate the mechanism of any cytoprotective effect by ferrostatin-1 on cell toxicity. 3) Ferroptosis is a different from other processes of cell death. Therefore we tested the effects of their inhibitors, Z-Val-Ala-Asp (OMe)-fluoromethyl ketone (ZVAD-fmk), 3-methyladenine (3-MA) and necrostatin-1 (Nec-1).
Materials and Methods
Cell culture and drug treatment
HT-22 cells (a mouse hippocampal neurons cell) which were from the Chinese Academy of Sciences (Shanghai, China) were maintained in DMEM medium (Hyclone, South Logan, UT, USA) supplemented with 10% fetal bovine serum, 100 U/mL of penicillin and 100 μg/mL of streptomycin at 37°C in a humidified atmosphere of 5% CO2 (Thermo Fisher Scientific, Waltham, MA, USA).
HT-22 cells were seeded in 6-well culture plate (2 × 105 cells/well). Glutamate (1.25, 2.5, 5, 10, 20 mM) was applied to HT-22 cells for 12, 24, 36, and 48 hours. HT-22 cells were cultured with different concentrations of ferrostatin-1 (3, 6 and 12 μM; Selleck, Shanghai, China) for 16 hours prior to exposure to 5 mM glutamate. After 24 hours, the subsequent analyses were conducted.
Other controls for neuronal protection were the iron chelator deferoxamine mesylate salt (DFO) (25–200 μM; Sigma, St. Louis, MO, USA), OMe-fluoromethyl ketone (ZVAD-fsk) (2–8 μM; Sigma), 3-MA (100–400 μM; Sigma) and Nec-1 (10–40 μM; Sigma). Each was co-treated with 5 mM glutamate for 24 hours at 37°C and 5% CO2 at 1 hour before the 5 mM glutamate treatment.
Cell viability detection
The cell viability was performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Jiang et al., 2014b). The results were determined using the microplate reader (Spectra Max M2e, Sunnyvale, CA, USA) at 490 nm.
Ultrastructure morphological changes of cells
The ultrastructure morphological changes of cells were measured as previously reported (Mei et al., 2014). HT-22 cells were seeded in 6-well culture plate (2 × 105 cells/well) and pretreated with ferostatin-1 (25 μM) for 16 hours and then 5 mM glutamate was added, after which they were incubated for 24 hours. Cells were digested and collected in phosphate buffer saline, immobilized with 2.5% glutaraldehyde in 0.1% sodium chloride buffer, and then 0.1% sodium chloride buffer was fixed with 1% osmium tetroxide. The preparation method comprised of the following steps: trimming, preparing a semi-thin section, positioning, preparing an ultra-thin section, dyeing with the lead acid, observing and taking the photo. Concisely, the cells were pretreated and subjected to transmission electron microscopy (JEM1230, Akishima, Japan) at 20,000× magnification, 80.0 kV accelerating voltage.
Lactate dehydrogenase release assay
Lactate dehydrogenase (LDH) release assay was measured as formerly reported (Wang et al., 2011). HT-22 cells in the logarithmic growth period were digested, collected and diluted according to the description of the LDH assay kit (Beyotime, Nanjing, Jiangsu Province, China). HT-22 cells were cultured in 96-well culture plate (2 × 104 cells/well) at 37°C and incubated with 5% CO2 for 24 hours. Each group had six multiple wells with each well having a volume of 100 μL. Fresh HT-22 cells in DMEM solution containing different concentrations of ferostatin-1 (1, 5, 25 μM) was exposed to 5 mM glutamate for 24 hours. The glutamate group was added with DMEM medium containing 5 mM glutamate. In the treatment group, ferostatin-1 (3, 6, 12 μM) was added 16 hours before the 5 mM glutamate group. After culture at 37°C and 5% CO2 concentration for 24 hours, LDH release was assessed using a LDH assay kit (Beyotime) following the manufacturer’s instruction.
Detection of oxidative stress markers
HT-22 cells were seeded in 6-well culture plate (2 × 105 cells/well) and pretreated with ferostatin-1 (25 μM) for 16 hours and then 5 mM glutamate was added, after which they were incubated for 24 hours. HT-22 cells were added to the lysis solution for 5 minutes, centrifuged and removed, and the precipitate was discarded. Gpx4 activity was tested as previously described (Pan et al., 2008). SOD activity was tested by nitro Blue Tetrazolium staining method (Flohe and Gunzler, 1984; Guo et al., 2012). MDA level was tested by the thiobarbituric acid method (Heath and Packer, 1968).
Caspase 3 activity assay
Caspase 3 activity assay was performed as formerly reported (Su et al., 2014). Briefly, the manufacturer’s instructions (Beyotime) were followed for all reagents and procedures. The yellow fluorescence was measured after 1 hour incubation at 37°C in the dark.
4′,6-Diamidino-2-phenylindole staining
4′,6-Diamidino-2-phenylindole (DAPI) fluorescent staining was tested as previously reported (Szczurek et al., 2014). Concisely, the cells were incubated with the DAPI dye at room temperature in the dark for 10 minutes and the cells were examined under a fluorescence microscope (BX60; Olympus, Tokyo, Japan) with a Leica DC 500 camera (Wetzlar, Germany).
Flow cytometry
The content of cytoplasmic ROS containing C11-BODIPY (581/591) and lipid ROS was detected with fluorescent H2DCF-DA as a probe. HT-22 cells were resuspended in H2DCF-DA and C11-BODIPY (581/591) serum-free medium with a final concentration of 10 μM. Cells were cultured for 30 minutes and shaken for 3–5 minutes C11-BODIPY (581/591) to ensure full contact between the fluorescent probe and the cells. Afterward, the cells were washed three times with fresh medium to fully remove the H2DCF-DA and C11-BODIPY (581/591). ROS and lipid ROS were levels measured by flow cytometry (Beckman Coulter, Brea, CA, USA). After preparation of the HT-22 cells, described as above, the cells were analyzed with a flow cytometer (Beckman Coulter). ROS and lipid ROS levels were measured as previously described (Su et al., 2014). The monodansylcadaverine (MDC) level test was carried out as previously reported (Mei et al., 2014) and the mean fluorescence intensity for 10,000 cells/sample was determined.
Western blot analysis
Western blot analysis was used as formerly reported (Jiang et al., 2014a). Briefly, aliquots of protein were transferred onto nitrocellulose filter membranes (Bio-Rad, Hercules, CA, USA) after SDS-PAGE electrophoresis. After blocking with nonfat milk for 1 hour, nitrocellulose filter membranes were incubated with primary antibodies (rabbit anti-mouse Gpx4 antibody, 1:1000, ab125066, Abcam, Shanghai, China; rabbit anti-Nrf2 antibody, 1:1000, ab137550, Abcam) overnight at 4°C. Peroxidase-conjugated goat anti-rabbit IgG (ZSGB-BIO, Beijing, China) was diluted 1:500 and incubated at 25°C for 1.5 hours. Protein bands were analyzed by Image lab 5.2.1 (Bio-Rad).
Real-time quantitative polymerase chain reaction
Real-time quantitative polymerase chain reaction analysis was carried out as formerly reported (Zhu et al., 2012). Total RNA was isolated with TRIzol Reagent (Gibco/Brl, Grand Island, NY, USA) and the real-time quantitative polymerase chain reaction was carried out with the QuantiNova SYBR Green PCR Kit (Bimake, Houston, TX, USA) following the manufacturer’s instructions. The results were acquired after 40 cycles of amplification. Quantification data represented the mean of three experiments. The threshold cycles (Ct) were conclusive for each gene and gene expression levels were evaluated using 2–ΔΔCt. The primers used in the experiments are listed in Table 1.
Table 1.
The primer sequences of the target genes in this study
| Gene | Forward | Reverse | Product size (bp) |
|---|---|---|---|
| Ptgs2 | 5′-AAG ACT TGC CAG GCT GAA CT-3′ | 5′-CTT CTG CAG TCC AGG TTC AA-3′ | 485 |
| Gpx4 | 5′-GCA GGA GCC AGG AAG TAA TC-3′ | 5′-GGC TGG ACT TTC ATC CAT TT-3′ | 1342 |
| β-Actin | 5′-AGT GTG ACG TTG ACA TCC GT-3′ | 5′-TGC TAG GAG CCA GAG CAG TA-3′ | 120 |
Statistical analysis
All data discussed in this study were presented as the mean ± standard deviation (SD). The data were analyzed using one-way analysis of variance by GraphPad Prism 5.0 for Windows (GraphPad Software, San Diego, CA, USA). Significance of difference between experimental groups was analyzed by the Tukey’s post-hoc test, with P < 0.05 considered to be statistically significant.
Results
Ferroptosis is involved in glutamate-induced toxicity in HT-22 cells
Dose-response tests evaluated the effect of treatment of HT-22 cells using different concentrations glutamate and showed decreased survival of neurons over time (MTT assay). Cultures with glutamate significantly decreased cell viability, P < 0.01, (Figure 1A). The cell survival rate was near its lowest in HT-22 cells treated with 5 mM glutamate for 12 hours. These conditions were selected for the model.
Figure 1.

Effect of ferrostatin-1 on glutamate-induced toxicity in HT-22 cells.
(A) The decreased cell viability, measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, was induced by increasing doses of glutamate treatment. (B) The effect of ferrostatin-1 on the viability of HT-22 cells incubated with 5 mM glutamate, as measured by MTT assay. (C) The effect of ferrostatin-1 on the HT-22 cells. Viability assayed with MTT. (D) The effect of ferrostatin-1 on the lactate dehydrogenase (LDH) level in the HT-22 cells, with and without the glutamate treatment. Data are expressed as the mean ± SD. #P < 0.05, ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. glutamate group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represented the mean of three experiments in duplicates. HT-22 cell: A mouse hippocampal neurons cell.
Ferrostatin-1 was pre-administered to determine if ferroptosis is involved in glutamate-induced toxicity. The results indicated that ferrostatin-1 dose-dependently reduced cell death, with a maximum effect at a dose of 12 μM (Figure 1B), but had no significant effect on the survival of HT-22 cells deprived of glutamate (Figure 1C). However, ferrostatin-1 did not rescue apoptosis induced by 1-methyl-4-phenylpyridinium in PC12 cells (data not shown). The LDH assay showed that after glutamate treatment the release of LDH significantly increased, which was inhibited by ferrostatin-1 treatment (P < 0.01; Figure 1D).
DAPI staining was utilized to assess the apoptosis of cells in our study. The number of cells decreased after glutamate treatment (Figure 2B). However, co-treatment with ferrostatin-1 and glutamate significantly increased cell growth (Figure 2C).
Figure 2.

Effect of ferrostatin-1 on the morphological features of HT-22 cells.
(A–C) 4′,6-Diamidino-2-phenylindole (DAPI) staining of HT-22 cells in (A) control, (B) Glu (5 mM), (C) Glu (5 mM) + Fer-1 (12 μM) groups. The white arrows indicate cell nucleus. (D–F) Transmission electron microscopy was used to observe the mitochondria. The white arrow indicates mitochondria. The mitochondria membrane density increased after the Glu treatment (E) compared with the control group (D) but the changes were smaller in the Glu + Fer-1 group (F). Scale bars: 100 μm in A–C, 200 μm in D–F. Fer-1: Ferrostatin-1; Glu: glutamate; HT-22 cell: a mouse hippocampal neurons cell.
The morphological features of mitochondria were observed under transmission electron microscopy. The results showed that mitochondria of the 5 mM glutamate treatment group (Figure 2E) appeared smaller than controls (Figure 2D), and the membrane density increased compared with the control group. Glutamate (5 mM) plus ferrostatin-1 (12 μM) reduced the morphologic changes induced by glutamate (Figure 2F).
Other types of cell death inhibitor fail to influence cell survival in HT-22 cells treated with glutamate
Different types of cell death inhibitors including apoptosis inhibitor ZVAD-fmk, autophagy inhibitor 3-MA, necrosis inhibitor Nec-1, and iron chelator DFO were co-treated with glutamate to determine whether these inhibitors affected the survival rate of HT-22 cells treated with glutamate. ZVAD-fmk (2–8 μM), 3-MA (100–400 μM) and Nec-1 (10–40 μM) were unsuccessful defenses against glutamate-induced cell death (Figure 3A–C). However, the iron chelator DFO significantly prevented HT-22 cell death in the glutamate toxicity model (P < 0.01) (Figure 3D). This suggests that ferroptosis plays an important role in glutamate-induced cell toxicity.
Figure 3.
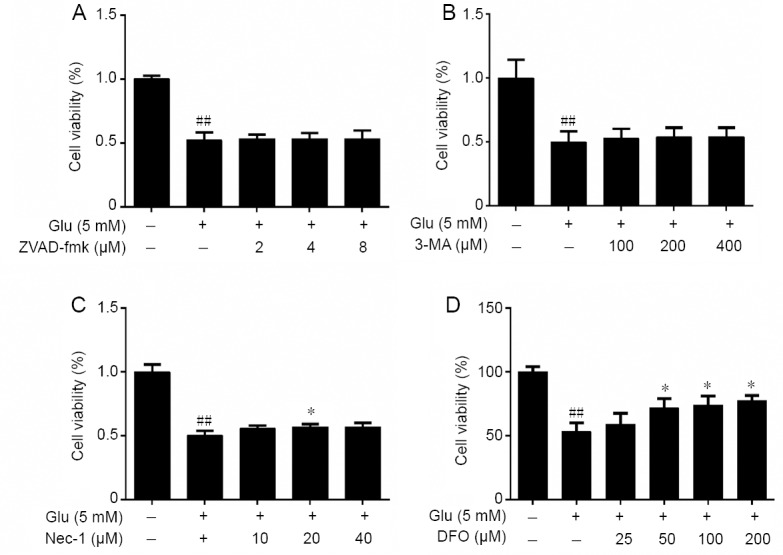
Effects of other types of cell death inhibitor including ZVAD-fmk (A), 3-MA (B), Nec-1 (C) and DFO (D) on the cell viability of HT-22 cells treated with Glu as detected by MTT.
Cell viability are expressed the optical density ratio to control group. Data are expressed as the mean ± SD. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. Glu group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represented the mean of three experiments in duplicates. 3-MA: 3-Methyladenine; DFO: deferoxamine mesylate salt; Glu: glutamate; HT-22 cell: a mouse hippocampal neurons cell; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Nec-1: necrostatin-1; ZVAD-fmk: Z-Val-Ala-Asp (OMe)-fluoromethyl ketone.
We also measured the classical markers of apoptosis and autophagy; the activation of caspase-3 and the level of MDC (Xie et al., 2016) to test if apoptosis and autophagy occurred in glutamate-induced cell toxicity and whether they are involved in the protective effect of ferrostatin-1 on HT-22 cells. As shown in Figure 4A, there was a small increase in caspase-3 activity in the glutamate treated group but addition of ferrostatin-1 had no further effect. The flow cytometry was utilized to monitor the intracellular MDC level. Our results showed that the fluorescence intensity of glutamate-treated group was markedly raised in comparison with the control group (P < 0.01), but there was no significant difference in MDC level between the glutamate-treated group and ferrostatin-1 group (Figure 4B).
Figure 4.
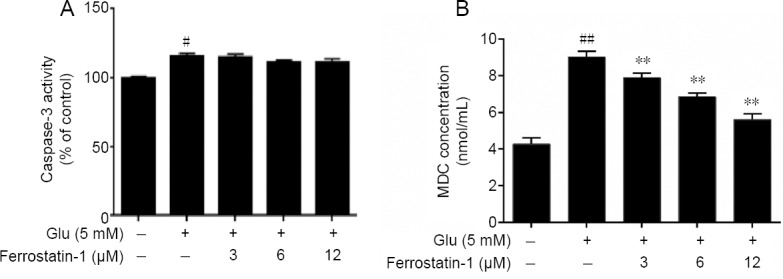
Effects of glutamate and ferrostatin-1 on apoptosis and autophagy measured as the activity of caspase-3 (A) and MDC (B) in HT-22 cells.
Data are expressed as the mean ± SD. #P < 0.05, ##P < 0.01, vs. control group; **P < 0.01, vs. Glu group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represented the mean of three experiments in duplicates. Glu: Glutamate; HT-22 cell: a mouse hippocampal neurons cell; MDC: monodansylcadaverine.
Ferrostatin-1 reduces the expression of ROS and lipid peroxidation in glutamate-treated HT-22 cells
Ferroptosis is characterized by lipid peroxidation, raising the possibility that the protective effect of ferrostatin-1 on glutamate-induced cell toxicity is by preventing lipid peroxidation (Latunde-Dada, 2017). Thus we measured the cytosolic and lipid ROS, SOD and MDA, which are factors associated with lipid peroxidation. Treatment with glutamate resulted in a time-dependent increase in cytosolic and lipid ROS measured by flow cytometry using DCFH-DA and C11-BODIPY as probes, indicating that ferrostatin-1 could significantly reduce the cytosolic and lipid ROS (P < 0.01, vs. glutamate group; Figure 5A & B).
Figure 5.
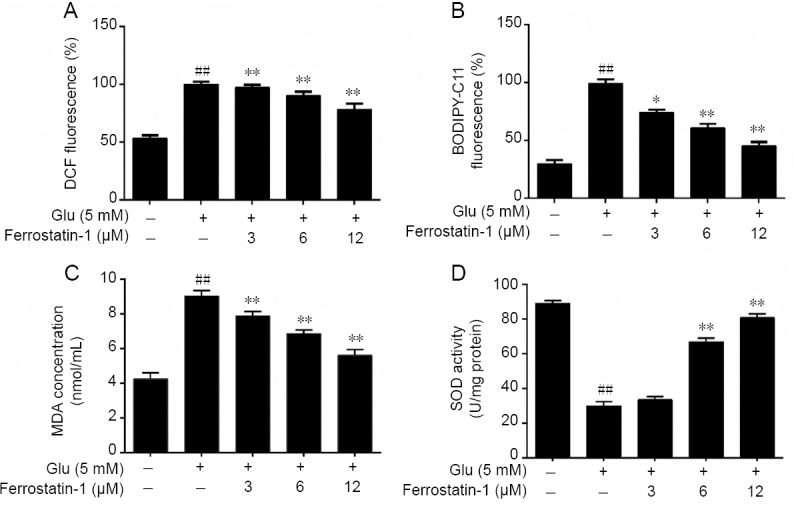
Effects of ferrostatin-1 on glutamate-induced ROS and lipid peroxidation level in Glu-treated HT-22 cells.
(A, B) The effect of ferrostatin-1 on cytosolic (A) and lipid (B) ROS level in Glu-treated HT-22 cells by flow cytometry. (C, D) The effect of ferrostatin-1 on MDA (C) and SOD (D) level in Glu-treated HT-22 cells. Data are expressed as the mean ± SD. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. Glu group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represent the mean of three experiments in duplicates. Glu: Glutamate; HT-22 cell: a mouse hippocampal neurons cell; MDA: malondialdehyde; ROS: reactive oxygen species; SOD: superoxide dismutase.
We measured the expression of MDA and SOD to further examine whether glutamate could induce lipid peroxidation in HT-22 cells. As shown in Figure 5C, after pretreatment with glutamate, the intracellular MDA concentration of HT-22 cells markedly increased, which was prevented by ferrostatin-1 treatment. The results also showed that after glutamate treatment the SOD activity markedly decreased in contrast to the control group. However, pretreatment with ferrostatin-1 significantly increased SOD activity compared with the glutamate group (P < 0.01; Figure 5D).
Effect of ferrostatin-1 on the expression of prostaglandin peroxidase synthase 2, Nrf2 and Gpx4 in glutamate treated HT-22 cells
To investigate the mechanism of the cytoprotective effect of ferrostatin-1 in glutamate-treated cells, we examined the expression of the mRNAs, prostaglandin peroxidase synthase 2 (PTGS2) and Gpx4. A total of 5 mM glutamate increased the expression of PTGS2 in HT-22 cells, whereas ferrostatin-1 suppressed the PTGS2 up-regulation by glutamate measured by real-time quantitative polymerase chain reaction (P < 0.01, vs. control group; Figure 6A).
Figure 6.

Effect of ferrostatin-1 on the expression of PTGS2 (A) and Gpx4 (B) after Glu treatment in HT-22 cells.
The data present as the mean ± SD. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. Glu group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represent the mean of three experiments in duplicates. Glu: Glutamate; Gpx4: glutathione peroxidase 4; HT-22 cell: a mouse hippocampal neurons cell; PTGS2: prostaglandin peroxidase synthase 2.
As shown in Figure 7A, 5 mM glutamate significantly decreased Nrf2 compared with the control group, measured by western blot assay. Ferrostatin-1 reduced the deficit. The expressions of Gpx4 and Gpx4 decreased after glutamate treatment (P < 0.01) and were significantly antagonized by ferrostatin-1 treatment (Figures 6B and 7B).
Figure 7.

Effect of ferrostatin-1 on the expression of Nrf2 (A) and Gpx4 (B) after Glu treatment in HT-22 cells.
Western blot data represent as the mean ± SD. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. Glu group (one-way analysis of variance followed by Tukey’s post-hoc test). Quantification data represent the mean of three experiments in duplicates. Glu: Glutamate; Gpx4: glutathione peroxidase 4; HT-22 cell: a mouse hippocampal neurons cell; Nrf2: nuclear factor erythroid-derived-like 2.
Discussion
As described above, the mechanism underlying glutamate induced neurotoxicity is not well understood. The HT-22 cells lack functional glutamate receptors and therefore have been widely used as a model for the analysis of the mechanism of glutamate-induced neurotoxicity. The toxicity induced in HT-22 cells by glutamate could be rescued by a ferroptosis inhibitor but not by an apoptosis inhibitor, autophagy inhibitor or necrosis inhibitor. Thus, our present study demonstrated that ferroptosis plays a crucial role in glutamate-induced oxidative toxicity, which is in accordance with our previous study (Wu et al., 2012). The cell activity assays that are classical markers of apoptosis and autophagy, such as caspase-3 and MDA, confirmed that apoptosis and autophagy are not involved in glutamate-induced cell toxicity. On the other hand, our data showed that the cell toxicity was inhibited by the iron chelator DFO, which strongly suggests the cell toxicity induced in HT-22 cells by glutamate is dependent on iron. The morphology experimental results showed that the change of the mitochondrial morphology and the bilayer membrane density and other structural characteristics from the glutamate-induced HT-22 cells was consistent with a recent research report on ferroptosis (Cao and Dixon, 2016). Thus we can confirm that ferroptosis is a major process in the cell toxicity induced by glutamate treatment.
Although it has been reported that glutamate-induced HT-22 cell death was significantly inhibited by the ferroptosis inhibitor ferrostatin-1 (Liu et al., 2015), the mechanism had not been uncovered. In our present study, ferrostatin-1 significantly reduced the up-regulation of cytoplasmic and lipid ROS that are associated with glutamate-induced cell death by ferroptosis. However, other types of cell death inhibitors including ZVAD-fmk, 3-MA, Nec-1 failed to reduce glutamate-induced cell death. To the best of our knowledge, these findings have never been reported before. Our other investigations showed that ferrostatin-1 blocked glutamate-induced downregulation of PTGS2, Nrf2 and Gpx4. PTGS2 is one of the key enzymes in the synthesis of prostaglandins, which increases the activity of peroxidase and the level of ROS and affect the balance of oxidation. It has been previously reported that PTGS2 encoding cyclooxygenase-2 is significantly up-regulated in ferroptosis, indicating the important role of PTGS2 (Xie et al., 2016). In this study, we showed that glutamate upregulated PTGS2 in HT-22 cells, co-treatment of cells with ferrostatin-1 suppressed the PTGS2 induction by glutamate, suggesting that ferrostatin-1 may protect cells from toxicity by inhibiting the expression of PTGS2. Our results suggested that PTGS2 upregulation may be downstream of the lipid peroxidation that occurs during ferroptosis.
Nrf2 initiates an endogenous antioxidant response element and activates the transcription of a large number of antioxidant enzymes downstream, including Gpx4 and heme oxygenase-1. We previously found that Nrf2 had a protective effect on dopaminergic neurons (Jiang et al., 2014a). In this research, we demonstrated that the expression of Nrf2 decreased significantly after glutamate treatment, which was prevented by ferrostatin-1. This confirms that glutamate causes oxidative toxicity affecting cell viability, whereas ferrostatin-1 protects cells by inhibiting oxidative toxicity. Our results are consistent with the previous study demonstrating that Nrf2 protects against ferroptosis in hepatocellular carcinoma cells (Sun et al., 2016) and shows in addition that Nrf2 up-regulation may underlie the cytoprotective mechanism of ferrostatin-1.
Our current study also found that ferrostatin-1 protected HT-22 cells against the down-regulation of Gpx4-induced by glutamate. We demonstrated that the down-regulation of Gpx4-induced by glutamate not only contributed to the death of HT-22 nerve cells but specifically to neuronal ferroptosis. This supports the hypothesis that glutamate induces ferroptosis through inhibition of Gpx4. Since Gpx4 is an antioxidant enzyme that reduces oxidized cholesterol (Conrad and Friedmann Angeli, 2015), it is convincing to conclude that ferrostatin-1 protects HT-22 cells by inhibiting oxidative toxicity. Ablation of Gpx4 induces neuronal loss, therefore systemic ablation of Gpx4 would be fatal to mice (Seiler et al., 2008). Moreover, conditional deletion of Gpx4 triggers rapid degeneration of spinal motor neurons probably through ferroptosis in mice (Chen et al., 2015). RSL3 and Erastin can direct bind to Gpx4 causing the decline of the Gpx4 activity in cancer cell ferroptosis (Dixon et al., 2012; Yang et al., 2014), intimating a special role for Gpx4 in the cancer cell.
In our aim to investigate the underlying principle of the protective effect of ferrostatin-1 on HT-22 cells we examined the relative markers of oxidative toxicity. Our results showed that ferrostatin-1 significantly inhibited the production of cytoplasmic and lipid ROS, and reversed glutamate-induced suppression of GSH and Gpx in HT-22 cells, suggesting that ferrostatin-1 protects HT-22 cells by blocking oxidative toxicity. The mitochondria of glutamate treated HT-22 cells were smaller in size and their membrane density greater than the control group, whereas the changes were less pronounced in the ferrostatin-1 group, suggesting that ferrostatin-1 protects the mitochondria from damages.
Accumulation of ROS is one of the hallmarks of ferroptosis. We found that the ferroptosis inhibitor, ferrostatin-1, can effectively inhibit both the cytosolic and lipid ROS production, these are similar results to those found with HT-1080 cancer cells (Gao et al., 2015; Kwon et al., 2015). Our experiments confirm that ferroptosis is caused by the production of cellular cytoplasmic and lipid ROS. DAPI staining and MDC levels were significantly boosted in glutamate-treated cells, which suggested that apoptosis and autophagy also occur. However, although ferrostatin-1 significantly increased cell viability compared with the glutamate reference group, the fluorescence intensities of DAPI and MDC were not affected. Neither autophagy nor apoptosis in cells would be expected in the timescale of this experiment. Rather, we have shown that ferroptosis is the main form of cell death during glutamate induced complex cell death in the HT-22 cells.
We chose one inhibitor, ferrostatin-1, to test the occurrence of ferroptosis in a glutamate-induced cell death model and decided to pre-treat the cells with ferrostatin-1 to protect them from glutamate induced injury. In future, the effects of more than one inhibitor might acquire more information on the molecular mechanisms. Studies on the protective effects of ferrostatin-1 in animal models in vivo should also be explored. It has been reported that the metabolic glutamate receptor 5 (GLU5) in HT-22 cell membranes reacts with glutamate to promote ERK1/2 and subsequent peroxidation (Kritis et al., 2015; Sato et al., 2016). It would be useful to explore how mGLU5 may regulate the ferroptosis.
Our study indicates that most of glutamate-induced HT-22 cell death occurs by ferroptosis. Ferroptosis is an iron-dependent non-apoptotic cell death induced in the cell by reversing the cystine/glutamate transporter (Xc−) in the cells reducing the antioxidant GSH (Dixon et al., 2012). Overall, glutamate treatment not only causes obvious depletion of intracellular GSH but also inhibits cellular uptake of cystine, which then leads to inactivate Gpx and increases cytosolic and lipid ROS level. These trigger oxidative stress and disorders in the cellular redox state, eventually causing cell ferroptosis. Our research suggests that Nrf2 and Gpx4 up-regulation may be the basis of the cytoprotective mechanism of ferrostatin-1.
Two lines of future research are necessary to understand the process. The first would be to explore ferroptosis in animal experiments in vivo. The second arises from our results that suggest ferroptosis occurred in HT-22 cells damaged by glutamate. However, the glutamic acid-damaged HT-22 cell model may result in other means of cell death, including apoptosis and necrosis. The circumstances and time scales relative to those of ferroptosis need further study. In summary, our results strongly reveal that ferroptosis plays a vital function in glutamate-induced neuronal damage. We found that ferrostatin-1 antagonized neurotoxicity induced by glutamate through inhibiting oxidative toxicity, and shed light on the ferroptosis inhibitors as potential therapeutic agents for the treatment of neurodegenerative diseases.
Additional file: Open peer review report 1 (103.9KB, pdf) .
Acknowledgments
We profoundly appreciate the assistance of the Professor Jing-Gen Liu (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, China) and the Professor Walter H. Hsu (Department of Biomedical Sciences, Iowa State University, USA) for providing nice advice and thoughtful feedback on the manuscript.
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest. All authors had final decision of report content, interpretation of data, and the decision to submit the report for publication.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Chih-Li Lin, Chung Shan Medical University, China.
P-Reviewer: Lin CL; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
References
- 1.Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H, Matsui T. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314:H659–H668. doi: 10.1152/ajpheart.00452.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brigelius-Flohe R, Muller M, Lippmann D, Kipp AP. The yin and yang of nrf2-regulated selenoproteins in carcinogenesis. Int J Cell Biol. 2012;2012:486147. doi: 10.1155/2012/486147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–28106. doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol. 2015;2:e995047. doi: 10.4161/23723556.2014.995047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 9.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi: 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Dypbukt JM, Ankarcrona M, Burkitt M, Sjoholm A, Ström K, Orrenius S, Nicotera P. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. J Biol Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 12.Flohe L, Gunzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 13.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gascón S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, Deshpande A, Heinrich C, Karow M, Robertson SP, Schroeder T, Beckers J, Irmler M, Berndt C, Angeli JP, Conrad M, Berninger B, Götz M. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 16.Guo C, He Z, Wen L, Zhu L, Lu Y, Deng S, Yang Y, Wei Q, Yuan H. Cytoprotective effect of trolox against oxidative damage and apoptosis in the NRK-52e cells induced by melamine. Cell Biol Int. 2012;36:183–188. doi: 10.1042/CBI20110036. [DOI] [PubMed] [Google Scholar]
- 17.Hasegawa M, Takahashi H, Rajabi H, Alam M, Suzuki Y, Yin L, Tagde A, Maeda T, Hiraki M, Sukhatme VP, Kufe D. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget. 2016;7:11756–11769. doi: 10.18632/oncotarget.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heath RL, Packer L. Photoperoxidation in isolated chloroplasts. I. Kinetics and stoichiometry of fatty acid peroxidation. Arch Biochem Biophys. 1968;125:189–198. doi: 10.1016/0003-9861(68)90654-1. [DOI] [PubMed] [Google Scholar]
- 19.Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by gpx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170. doi: 10.1007/82_2016_508. [DOI] [PubMed] [Google Scholar]
- 20.Jiang G, Wu H, Hu Y, Li J, Li Q. Gastrodin inhibits glutamate-induced apoptosis of PC12 cells via inhibition of CaMKII/ASK-1/p38 MAPK/p53 signaling cascade. Cell Mol Neurobiol. 2014a;34:591–602. doi: 10.1007/s10571-014-0043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang G, Hu Y, Liu L, Cai J, Peng C, Li Q. Gastrodin protects against MPP(+)-induced oxidative stress by up regulates heme oxygenase-1 expression through p38 MAPK/Nrf2 pathway in human dopaminergic cells. Neurochem Int. 2014b;75:79–88. doi: 10.1016/j.neuint.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Tiziani S, Park G, Kaul M, Paternostro G. Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun. 2014;5:3672. doi: 10.1038/ncomms4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kritis AA, Stamoula EG, Paniskaki KA, Vavilis TD. Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci. 2015;9:91. doi: 10.3389/fncel.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–24403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, Galmiche A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–6422. [PubMed] [Google Scholar]
- 26.Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Wang W, Li Y, Xiao Y, Cheng J, Jia J. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull. 2015;38:1234–1239. doi: 10.1248/bpb.b15-00048. [DOI] [PubMed] [Google Scholar]
- 28.Maragakis NJ, Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol. 2001;58:365–370. doi: 10.1001/archneur.58.3.365. [DOI] [PubMed] [Google Scholar]
- 29.Mei W, Dong C, Hui C, Bin L, Fenggen Y, Jingjing S, Cheng P, Meiling S, Yawen H, Xiaoshan W, Guanghui W, Zhiwu C, Qinglin L. Gambogenic acid kills lung cancer cells through aberrant autophagy. PLoS One. 2014;9:e83604. doi: 10.1371/journal.pone.0083604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Q, Huang K, He K, Lu F. Effect of different selenium sources and levels on porcine circovirus type 2 replication in vitro. J Trace Elem Med Biol. 2008;22:143–148. doi: 10.1016/j.jtemb.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Sato K, Yamanaka Y, Asakura Y, Nedachi T. Glutamate levels control HT22 murine hippocampal cell death by regulating biphasic patterns of Erk1/2 activation: role of metabolic glutamate receptor 5. Biosci Biotechnol Biochem. 2016;80:712–718. doi: 10.1080/09168451.2015.1107466. [DOI] [PubMed] [Google Scholar]
- 32.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 33.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J, Huh Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res. 2019;14:749–756. doi: 10.4103/1673-5374.249218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su J, Cheng H, Zhang D, Wang M, Xie C, Hu Y, Chang HC, Li Q. Synergistic effects of 5-fluorouracil and gambogenic acid on A549 cells: activation of cell death caused by apoptotic and necroptotic mechanisms via the ROS-mitochondria pathway. Biol Pharm Bull. 2014;37:1259–1268. doi: 10.1248/bpb.b13-00972. [DOI] [PubMed] [Google Scholar]
- 36.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szczurek AT, Prakash K, Lee HK, Zurek-Biesiada DJ, Best G, Hagmann M, Dobrucki JW, Cremer C, Birk U. Single molecule localization microscopy of the distribution of chromatin using Hoechst and DAPI fluorescent probes. Nucleus (Calcutta) 2014;5:331–340. doi: 10.4161/nucl.29564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toyokuni S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol Int. 1999;49:91–102. doi: 10.1046/j.1440-1827.1999.00829.x. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Zhu G, Yang S, Wang X, Cheng H, Wang F, Li X, Li Q. Paeonol prevents excitotoxicity in rat pheochromocytoma PC12 cells via downregulation of ERK activation and inhibition of apoptosis. Planta Med. 2011;77:1695–1701. doi: 10.1055/s-0030-1271033. [DOI] [PubMed] [Google Scholar]
- 40.Wu HY, Jiang GL, Li QL. Protection of gastrodin on PC12 cell injury induced by glutamate via inhibition of cell apoptosis. Zhongguo Linchuang Yaoli Xue yu Zhiliao Xue. 2012;17:1361–1367. [Google Scholar]
- 41.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, Fu XH, Hu Y, Liu C, Kong XH, Feng SQ. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. 2019;14:532–541. doi: 10.4103/1673-5374.245480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu G, Wang X, Wu S, Li Q. Involvement of activation of PI3K/Akt pathway in the protective effects of puerarin against MPP+-induced human neuroblastoma SH-SY5Y cell death. Neurochem Int. 2012;60:400–408. doi: 10.1016/j.neuint.2012.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.