

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common form of dementia worldwide. As age is the main risk factor, > 97% of all AD cases are of sporadic origin, potentiated by various risk factors associated with life style and starting at an age > 60 years. Only < 3% of AD cases are of genetic origin caused by mutations in the amyloid precursor protein or Presenilins 1 or 2, and symptoms already start at an age < 30 years. In order to study progression of AD, as well as therapeutic strategies, mouse models are state-of-the-art. So far many transgenic mouse models have been developed and used, with mutations in the APP or presenilin or combinations (3×Tg, 5×Tg). However, such transgenic mouse models more likely mimic the genetic form of AD and no information can be given how sporadic forms develop. Several risk genes, such as Apolipoprotein E4 and TREM-2 enhance the risk of sporadic AD, but also many risk factors associated with life style (e.g., diabetes, hypercholesterolemia, stress) may play a role. In this review we discuss the current situation regarding AD mouse models, and the problems to develop a sporadic mouse model of AD.
Keywords: Alzheimer's disease, beta-amyloid, cerebral amyloid angiopathy, cognitive impairment, sporadic and genetic mouse models, tau, vascular risk factors
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative brain disease and characterized by massive loss of memory and learning. The major hallmarks (Figure 1) include the extracellular deposition of beta-amyloid (Aβ) plaques, the intraneuronal accumulation of hyperphosphorylated tau with formation of neurofibrillary tangles (NFT), loss of neurons releasing the neurotransmitter acetylcholine, vascular impairment, glial dysfunctions and inflammation. In about 70% of all cases also a deposition of Aβ within vessel walls has been shown, known as cerebral amyloid angiopathy (CAA) (Selkoe, 2001; Rannikmäe et al., 2013; Masters et al., 2015). The majority of all cases are sporadic, whereas only < 3% are caused by genetic mutations (Selkoe, 2001). The underlying mechanisms leading to sporadic AD are still not fully clear. However, it is assumed, that various (vascular) risk factors associated with life style, such as diabetes, hypercholesterolemia, stress, blood pressure, smoking or heavy metals may play a major role (Andel et al., 2012; Reitz and Mayeux, 2015), but also risk genes can increase the progression of AD.
Figure 1.
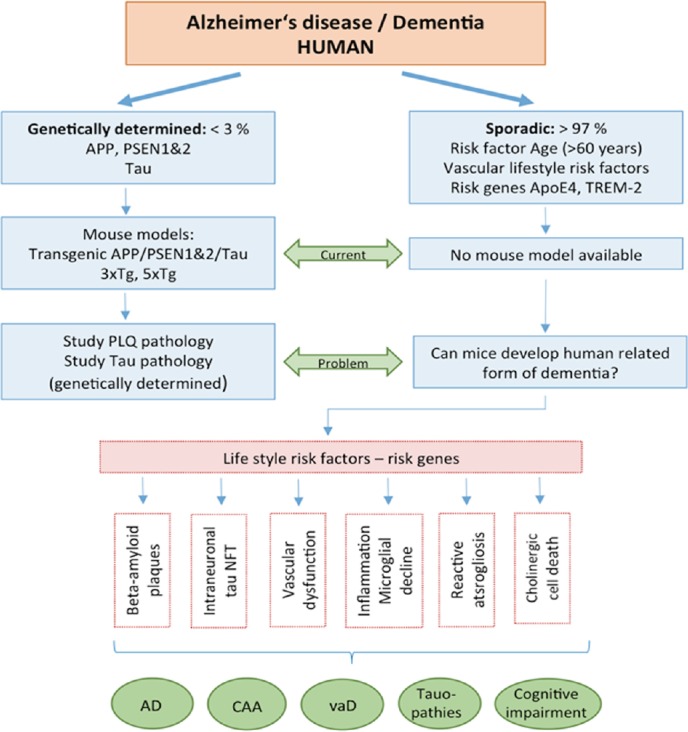
Alzheimer’s disease (AD) is a severe neurodegenerative disorder, characterized by extracellular beta-amyloid plaques (PLQ), intraneuronal tau pathology with formation of neurofibrillary tangles (NFT), vascular dysfunction, inflammation and microglial dysfunction, reactive astrogliosis and cell death of cholinergic neurons.
AD can be distinguished into a genetic (< 3% of patients affected) and sporadic (majority of > 97% of all cases) form of the disease. In genetic AD the major risk genes amyloid precursor protein (APP) and presenilin 1&2 (PSEN1&2) are mutated. Transgenic mouse models with mutations in these genes have been developed and allow to study the progression of beta-amyloid and tau pathologies. Age (> 60 years) is the main risk factor to develop sporadic AD, and two risk factors (Apoliprotein E4 and TREM-2) increase the risk, and many risk factors associated with life style play a role in progression. However, so far, the initial causes for sporadic AD are not known and no mouse model for sporadic AD is available. In addition, also other forms of dementia with cognitive impairment have been reported, showing partial overlap with AD, such as cerebral amyloid angiopathy (CAA), vascular dementia (vaD) or pure tauopathies (such as frontotemporal lobe dementia).
The literature search was performed by an extensive PubMed research including the keywords of “Alzheimer’s disease, sporadic, mouse models, cerebral amyloid angiopathy, vascular risk factors”. The time frame was set to data from 1990 to 2019.
In the last decades, various mouse models of AD have been generated to mimic the major neuropathological hallmarks in AD. These models are one of the most important research tools in AD as they provide marked advantage toward the understanding of the AD pathogenesis. Such models allow to study the progression of Aβ and tau pathologies, but also to explore therapeutic strategies. The major problem currently is, that these mice mimic some, but not all of the key pathologies of human AD (Hall and Roberson, 2012). In this review we will discuss (a) the current status of AD mouse models, (b) the problems associated with these models and (c) if mice can develop a human related disease and (d) how we can progress to develop a mouse model of sporadic AD (Figure 1).
Genetic Mouse Models of Alzheimer’s Disease
AD-associated genes can be divided into those in which mutations cause autosomal dominant AD and those in which polymorphisms serve as risk factors for AD. Mutations in three major genes are known causing autosomal dominant AD: an overexpression of the amyloid precursor protein (APP) gene leads to an increased Aβ production (mainly toxic Aβ42) and mutations in the Presenilin genes 1 and 2 (PSEN1, PSEN2) cause activation of the gamma-secretase complex and subsequent cleavage of APP with increased Aβ production (Hall and Roberson, 2012). In 1995, a breakthrough reported the first transgenic mouse model using platelet derived growth factor-β encoding human APP with typical Aβ plaques and cognitive impairment (Games et al., 1995). Many more mouse models have been created meanwhile, such as the Tg2576 mice (Hsiao et al., 1996; Elder et al., 2010) or the APP_Swedish Dutch Iowa mice (APP_SweDI) (Davis et al., 2004) or the APP23 (Sturchler-Pierrat et al., 1997), J20 (Mucke et al., 2000) or TgCRND8 (Chishti et al., 2001) mice. Novel transgenic mice have been generated by crossing PS1/PS2 with APP, which potentiated the formation of plaques (Holcomb et al., 1998; Elder et al., 2010). Finally, more complex models have been created, combining three APP and two PS1 mutations (5×FAD), resulting in a rapid onset of Aβ deposition, impaired memory and also a loss of neurons (Oakley et al., 2006; Elder et al., 2010).
The central question in the role of tau in the Aβ cascade is, whether if Aβ drives the tau pathology or vice versa or if both pathologies have synergistic effects. It is well known that tau is a microtubule-associated protein and physiologically stabilizes and regulates axonal transport. However, the interaction between Aβ and tau seems to be more complex and by far not fully explored. Thus, this problem forced the generation of tau specific transgenic mice. However, transgenic mice overexpressing human tau mainly cause a “frontotemporal like-dementia”, but do not lead to a typical AD pathology (Hall and Roberson, 2012). These results forced researchers to generate transgenic mouse models combining human tau and APP. One of the first such models is the TAPP model, generated by crossing Tg2576 and JNPL3 mice, expressing P301L human tau (Lewis et al., 2001). Another transgenic model combines the APP, PS1 and tau mutations (3×Tg mice) and these mice develop first Aβ plaques and then NFT (Hall and Roberson, 2012; Oddo et al., 2003). In order to overcome the intrinsic drawbacks due to the APP over-expression, e.g., a destruction of endogenous gene loci, second-generation mouse models were generated. Due to the change of three amino acids, differing between mouse and human APP, it was possible to create a human APP knock-in model, harbouring Swedish and Beyreuther/Iberian mutations with/without arctic mutations within the APP gene. This model generates the Aβ pathology as well as neuroinflammation and memory deficits depending on the age (Saito et al., 2014). Limitations of the APP over-expression are described extensively in a review (Sasaguri et al., 2017).
Although such mice partly develop both important AD pathologies, they still do not reflect the complete complex “sporadic nature of human AD”. A list of all current available mouse models can be found at https://www.alzforum.org/ research-models.
Sporadic Mouse Models of Alzheimer’s Disease and Related Dementia
As mentioned above, more than 97% of all AD cases are of sporadic origin and age is the main risk factor. The causes for initiation and progression of sporadic AD are unknown, but it seems to become clear that sporadic AD already starts 20–30 years before first symptoms are seen. It seems likely, that the progression is influenced by two main parameters: genetic risk factors and risk factors associated with life style, possibly associated with vascular risk factors (Figure 1).
Genetic risk factors in sporadic Alzheimer’s disease
The strongest genetic risk factor gene for late onset AD is apolipoprotein E4 (ApoE4) (Figure 1). ApoE consists of three alleles (E2, E3, E4), but in AD the ApoE4 allele seems to be the most prominent risk factor. Interestingly, the E2 allele reduces the risk to develop AD and the allele E3 does not influence the risk. Although this gene is only present in about 15% of the population, the E4 allele occurs in 40-65% of all AD cases (Hall and Roberson, 2012; Rincon and Wright, 2013). ApoE knock-out mice (apoE-/-) have been created to demonstrate the role of ApoE in the brain, especially to address the role of ApoE in glial or neuronal function. Transgenic mice have been generated by combinations of ApoE and PDAPP, Tg2576, J9 or 5xFAD (Tai et al., 2011). A limitation of the models is that the murine and human forms of ApoE are only 70% homologous (Tai et al., 2011)
The second recently discovered risk factor for late-onset sporadic AD is the “triggering receptor expressed on myeloid cells 2” (TREM-2). Individuals expressing this gene suffer from a 4x higher risk to develop AD (King, 2018). This gene encodes a receptor expressed in myeloid cells, mediating inflammatory responses. Specifically, the very rare variant p.Arg47His increases the risk for AD. The disease triggering mechanism of TREM-2 is mainly linked to microglial activation. It is well known that in AD microglia become activated, migrate to plaques to eliminate plaques; however, over decades the microglial function is diminished. Interestingly, in patients with TREM2 risk variants a stronger plaque-associated neuritic dystrophy together with a lower coverage of microglia is present. The effects on the microglial clustering due to the p.Arg47His variant of TREM2 have been tested in a mouse model of amyloidosis (Carmona et al., 2018; Song et al., 2018), which may reflect a form of late onset sporadic AD.
(Vascular) risk factors associated with life style in sporadic Alzheimer’s disease
Beside the genetic risk factors, also life style age-related risk factors, possibly vascular risk factors, play a major role (Figure 1). This suggestion is based on the overlap of AD with vascular dementia and the hypothesis that AD is a vascular disease (de la Torre and Stefano, 2000; de la Torre, 2004; Humpel, 2011). The suggested risk factors associated with lifestyle and subsequently with sporadic AD are: hypertension, high blood pressure, diabetes type II, inflammation, hypoxia and stroke, stress, homocysteine, alcohol, smoking and the exposure to heavy metals or other not yet discovered factors.
Hypertension: High blood pressure is highly linked to a damage of the blood-brain barrier (BBB) and an increase of the receptor for advanced glycated end products expression, which causes a dysfunction of the Aβ clearing process (Carnevale et al., 2016). Mouse models with hypertension increase the risk to develop sporadic AD. In an APP_PS1 mouse model the infusion of hypertensive doses of angiotensin II caused cerebrovascular alterations and an increase in vascular Aβ depositions (Broquères-You et al., 2014).
Diabetes and hypercholesterolemia: Type 2 diabetes is the most common form of diabetes exhibiting the major hallmarks of insulin resistance and hyperinsulinemia. Moreover, also hypercholesterolemia, obesity and hypertension are major co-factors, all together affecting Aβ clearing through the BBB and hyperphosphorylation of tau (Sims-Robinson et al., 2010). In mouse models, either the injection of streptozotozin and/or a high fat diet causes diabetes-like symptoms with an increase of γ-secretase activity (Kimura, 2016).
Alteration of glucose mechanisms: It is well known that an impaired glucose mechanism and neuronal damage clearly correlate in AD. N-acetylglucosamine is an end-product of the glucose mechanism and activates the hexosamine pathway. An interesting polymer (connected to N-acetylglucosamine) is Chitin, which accumulates in plaques of sporadic AD (Turano et al., 2015).
Inflammation: Many studies show that neuroinflammation is not a factor on its own to enhance the risk of AD, however, it is assumed, that the combination of several risk factors lead to chronic inflammation, which deteriorates AD pathology (Kinney et al., 2018). Lipopolysaccharide is a well-known drug to induce inflammatory responses in mice (Foidl and Humpel, 2019).
Hypoxia and stroke: Stroke interrupts the oxygen and energy supply to the brain, leading to cerebrovascular damage and dysfunctions in the brain, which is associated with the onset of vascular dementia and/or AD. Moreover, hypoxia is strongly linked to other risk factors for AD and vascular dementia, such as hypertension, diabetes, hyperhomocysteinemia or smoking (Vijayan and Reddy, 2016).
Stress: Chronic stress in midlife but also post-traumatic stress disorders are in a strong relationship of developing dementia and AD in later life stages. A mouse model shows this relationship including increased levels of soluble Aβ in the cerebrospinal fluid (Justice et al., 2015; Justice, 2018).
Hyperhomocysteinemia: An overactivation of homocysteine is widely known to worsen the disease process, while vitamins B12 or folate can counteract symptoms (Farina et al., 2017). We have shown in an animal model that hyperhomocysteinemia caused severe memory impairment and a decline of cholinergic neurons including vascular microbleedings (Pirchl et al., 2010).
Alcohol: There are conflicting results regarding the role of alcohol and its effect on dementia. On the one hand there is the assumption, that a low to moderate use of alcohol (especially of red wine) has protective effects. However, many studies insist on the opposite effect (Huang et al., 2016). In a previous study, we showed that ethanol together with CaCl2 caused dysfunction of platelet APP processing both in rodent and human platelets (Ehrlich and Humpel, 2014).
Smoking: Smoking is a critical factor especially in vascular diseases, but also in the pathogenesis of AD. Transgenic APPswe/PS1 mice exposed to cigarette smoke showed an increased formation of Aβ plaques, inflammatory responses, alterations in tau phosphorylation around the plaques, but no neuronal decline (Moreno-Gonzalez et al., 2013).
Heavy metals: It is well known that an overload or accumulation of metals in the brain leads to toxic reactions like oxidative stress or disrupted mitochondrial functions (Chen et al., 2018). Copper intoxication both in wildtype and in transgenic mice caused an increased production of Aβ, together with vascular impairment (Singh et al., 2013). Similarly, iron overload leads to an increased plaque formation and behavioral impairment, as shown in APP/PS1 mice (Becerril-Ortega et al., 2014).
Taken together, many mouse models have been described where a single stimulus caused some typical AD hallmarks, mainly cognitive impairment, but do not fully reflect the disease. It seems likely, that many risk factors are linked to AD progression, such as diabetes and high-fat diet, cholesterol levels and inflammation. Thus it is hypothesized that the combination of many of such (vascular) risk factors associated with life style may initiate and/or potentiate the progression of sporadic AD.
Mouse model of cerebral amyloid angiopathy
A typical hallmark in AD is the deposition of Aβ in the vessel walls known as CAA (Figure 1). However, while in AD plaques mainly the toxic Aβ42 form aggregates, in vessels mainly the Aβ40 form is deposited (DeSimone et al., 2017). Further, CAA occurs also alone without any AD pathology. Mouse models of CAA though, are also quite rare and also here, the sporadic manner of the disease formation is of utmost importance. In our recent study, we (Foidl and Humpel, 2019) exposed wildtype C57BL6 mice for up to 56 weeks with different subchronic mild vascular risk factors. We showed for the first time, that the treatment with various vascular risk factors leads to a marked cognitive impairment. This was accompanied by a strong damage and decline of vessels, BBB disruption, cerebral vascular bleedings and inflammatory responses. However, we could not see typical Aβ plaques or tau NFT, and thus we suggest the establishment of a novel mouse model for CAA. Our data highly suggest that the combination of several mild lifestyle risk factors over months does not lead to a sporadic mouse model of AD.
However, also transgenic mice can develop a CAA pathology. The transgenic APPDutch mouse model mimics the genetic variant of hereditary cerebral haemorrhage with amyloidosis – Dutch type (HCHWA-D), harbouring the human APP51 with E693Q mutation under the murine Thy1 promoter (Herzig et al., 2004). These mice are impaired in cognition, but the onset of CAA is very late (22–25 months) which makes this model technically problematic (Herzig et al., 2004). Another transgenic mouse model (the APP_SweDI) also develops vessel occlusions but also at later stages (Davis et al., 2004).
Platelets as a risk factor for sporadic Alzheimer’s disease?
It is very interesting to note that platelets (thrombocytes) contain high amounts of APP and are able to produce and release Aβ40 into the blood, which may play a role in blood clotting. We hypothesize that platelets may play a crucial role in initiation of AD, because platelets are dysfunctional in AD (Plagg and Humpel, 2015). It seems likely, that lesions in brain vessels over a long time period together with BBB disruption and bleedings may cause an overactivation and dysfunction of platelets. Platelets can contain APP and can produce and release Aβ and may also produce toxic Aβ42 under certain conditions (Humpel, 2017).
Problems arising in mouse models of Alzheimer’s disease–missing translational links?
The major problem in the current mouse models arises, that the available transgenic models only mimic the rare genetic form of AD with an early onset and no established mouse models for sporadic AD are available. These transgenic mice either develop plaques alone or NFT alone, or in combination both pathologies. But these mice do not exhibit all AD pathologies (CAA, inflammation, cholinergic cell death or glial activation) and most importantly, we do not get any information how sporadic AD is initiated and progresses. Thus, it is hypothesized, that AD is the result of a combination of genetic, life style and environmental factors. Therefore, the generation of only a single pathology cannot reflect the complexity of a late onset form of sporadic AD.
Mice do not reflect the most important risk factor in humans, which is an old age. While the average life time of mice is between 22–28 months, sporadic AD develops in humans at an age of 60 years. Thus, one needs to ask, if the old aged mouse can really mimic a human individual of an age of 60 years. And more seriously asked, can a mouse develop a sporadic disease with a life time of only 22–28 months? This represents a major conflict, as the immune system of young animals is notably different compared to aged individuals.
There is also a difference in the murine and human overexpression of APP. Plaques in humans pretend to have a lower core density with an amorphous structure and are more difficult to dissolve than murine plaques (Balducci and Forloni, 2011). Moreover, in transgenic mice plaques were found to exhibit a very large size compared to the human form, which makes it difficult to compare (Saito, 2018).
Another major concern is the structural difference between human and murine Aβ. So far it is well established that the human Aβ42 peptide can aggregate and form the toxic plaques, thus in all mouse models human Aβ was overexpressed or in cell culture models mainly Aβ with a human sequence is used. Mouse Aβ42 shows a different amino acid sequence and it is not fully clear if it can aggregate under physiological conditions. In fact, mouse Aβ is less susceptible in forming β-sheet structures and aggregated fibrils and murine Aβ is less neurotoxic as it generates less reactive oxygen species compared to human Aβ (Lv et al., 2013). However, we showed that endogenous rat Aβ can form Aβ-like depositions when exposed to low pH and ApoE4 in brain slices (Marksteiner and Humpel, 2008). Thus, the central question is: what are the exact molecular and cellular mechanisms that induce aggregation of murine Aβ?
Based on the common amyloid hypothesis it is suggested that Aβ plaques cause the activation of hyper-phosphorylated tau and subsequent formation of NFT. While mouse models may mimic such a situation, so far in APP transgenic models, Aβ did not clearly initiate the tau pathology. We recently showed that okadaic acid caused hyperphosphorylation of specific tau epitopes, especially of p-tau396, but no formation of NFTs in organotypic brain slices (Foidl and Humpel, 2018). Okadaic acid injection into mice is useful to get a better insight to the mechanisms of hyperphosphorylated tau also in combination with the complex connection of Aβ and tau.
To date, the amyloid hypothesis is discussed critically, as the correlation between amyloid plaques and AD is partly doubted (Morris et al., 2014). So far, the hypothesis has failed in clinical trials, as the current available drugs are not counteracting the amyloid pathology. Though, as mentioned above, there is also a strong focus on anti-amyloid treatments targeting Aβ by active and passive immunization. But it has to be discussed as well, that Aβ deposition occurs also in cognitive normal subjects (as imaged with Pittsburgh PET compound), which leads to a challenge in the diagnosis and the question when the Aβ plaques correlate with cognitive decline and AD progression (Morris et al., 2014). A possible explanation is based on suggestions, that soluble oligomers of Aβ are the actual toxic factor and not the fibrillary Aβ42 within plaques. These soluble Aβ oligomers are supposed to play a role in very early stages of AD, due to the processing of APP even before plaques are formed (Morris et al., 2014; Mroczko et al., 2018).
Moreover, also the extensive neuronal loss in patients with AD is not reflected in its entirety in mice (Franco and Cedazo-Minguez, 2014). Several studies examined a hippocampal loss of neurons in APPxPS1 mice or even in a more drastic form in APP_Swedish-London transgenic mice crossed with PS1M233T/L235L mutations or in 5xFAD mice featuring 3xAPP and 2xPS1 mutations (Balducci and Forloni, 2011). We also reported previously that in transgenic APP_SweDI mice a retrograde-induced cell death of cholinergic neurons occurs when plaques are present, including a cognitive decline (Foidl et al., 2016).
The missing translational link is also seen in the fact, that therapeutic strategies clearly were successful in mouse models, but did not show positive results in humans. A typical example is vaccination, which was successful to clear Aβ plaques in mouse brains but not in humans (King, 2018). Promising results have been shown recently in a study, where 5xFAD mice were treated with 3K3A-activated protein C, which enhanced the BBB-integrity, improved behavioral skills and most importantly, it reduced the appearance of Aβ (Lazic et al., 2019).
Future Perspectives and Outlook
Genetic mouse models of AD are still the most common strategy to work on experimental AD research. Novel possibilities as the CRISPR-Cas9 gene editing seems to provide interesting options both in treatment and production of animal models (Rohn et al., 2018). Though, the major problem in the current mouse model arises, that on the one hand there is a general lack of sporadic mouse models of AD and on the other hand, that research understands sporadic mouse models mainly in regard to ApoE4 investigation. Therefore, it is still of utmost importance to focus more on several risk-factors, also linked to vascular disturbances. Also, the generation of mouse models for other forms of dementia, such as frontotemporal lobe dementia, may add more information. In this context, our novel CAA mouse model (Foidl and Humpel, 2019) adds additional information about factors which may or may not induce sporadic AD. The following major question arises: why does the treatment with (vascular) risk factors not lead to a sporadic form of AD in mice in our sporadic CAA model? A possible explanation may point again to age as the major risk factor in human AD, since the short lifespan of mice may prevent the progression of AD and may not mimic all aspects of the human disease. In future, many additional risk factors (associated with life style) must be tested to induce the sporadic form of AD. This may become a pivotal step to close the translational gap between animal models and the human form of the disease.
Additional file: Open peer review reports 1 (112.4KB, pdf) –3 (112.7KB, pdf) .
Footnotes
Conflicts of interest: None declared.
Financial support: This work was supported by the Austrian Science Funds (P24734-B24).
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Willian Orlando Castillo, University of São Paulo, Brazil; Zhefan Stephen Chen, The Chinese University of Hong Kong, China; Alain Buisson, Université Grenoble Alpes, France.
Funding: This work was supported by the Austrian Science Funds (P24734-B24).
P-Reviewers: Castillo WO, Chen ZS, Buisson A; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Andel R, Crowe M, Hahn EA, Mortimer JA, Pedersen NL, Fratiglioni L, Johansson B, Gatz M. Work-related stress may increase the risk of vascular dementia. J Am Geriatr Soc. 2012;60:60–67. doi: 10.1111/j.1532-5415.2011.03777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balducci C, Forloni G. APP transgenic mice: their use and limitations. Neuromolecular Med. 2011;13:117–137. doi: 10.1007/s12017-010-8141-7. [DOI] [PubMed] [Google Scholar]
- 3.Becerril-Ortega J, Bordji K, Fréret T, Rush T, Buisson A. Iron overload accelerates neuronal amyloid-β production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol Aging. 2014;35:2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 4.Broquères-You D, Dere E, Benessiano J, Lévy BI, Poittevin M, Mariani J, Cifuentes D, Kubis N, Merkulova-Rainon T, Bonnin P, Pocard M. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension. 2014;65:218–224. doi: 10.1161/HYPERTENSIONAHA.114.04139. [DOI] [PubMed] [Google Scholar]
- 5.Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018;17:721–730. doi: 10.1016/S1474-4422(18)30232-1. [DOI] [PubMed] [Google Scholar]
- 6.Carnevale D, Perrotta M, Lembo G, Trimarco B. Pathophysiological links among hypertension and Alzheimer’s disease. High Blood Press Cardiovasc Prev. 2016;23:3–7. doi: 10.1007/s40292-015-0108-1. [DOI] [PubMed] [Google Scholar]
- 7.Chen P, Miah MR, Aschner M, Perry G. Metals and Neurodegeneration F1000Research. F1000 Faculty Rev-366. 2018;5:1–13. [Google Scholar]
- 8.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 9.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 10.de la Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 11.de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: The role of constitutive nitric oxide. Brain Res Rev. 2000;34:119–136. doi: 10.1016/s0165-0173(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 12.DeSimone CV, Graff-Radford J, El-Harasis MA, Rabinstein AA, Asirvatham SJ, Holmes DR. Cerebral amyloid angiopathy: diagnosis, clinical implications, and management strategies in atrial fibrillation. J Am Coll Cardiol. 2017;70:1173–1182. doi: 10.1016/j.jacc.2017.07.724. [DOI] [PubMed] [Google Scholar]
- 13.Ehrlich D, Humpel C. Effects of ethanol on aggregation, serotonin release, and amyloid precursor protein processing in rat and human platelets. Platelets. 2014;25:16–22. doi: 10.3109/09537104.2013.764979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elder GA, Gama Sosa MA, De Gasperi R. Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med. 2010;77:69–81. doi: 10.1002/msj.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farina N, Jernerén F, Turner C, Hart K, Tabet N. Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Exp Gerontol. 2017;99:146–150. doi: 10.1016/j.exger.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Foidl BM, Do-Dinh P, Hutter-Schmid B, Bliem HR, Humpel C. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol Learn Mem. 2016;136:86–96. doi: 10.1016/j.nlm.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foidl BM, Humpel C. Differential hyperphosphorylation of Tau-S199, -T231 and -S396 in organotypic brain slices of Alzheimer Mice. A model to study early Tau hyperphosphorylation using okadaic acid. Front Aging Neurosci. 2018;10:1–15. doi: 10.3389/fnagi.2018.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foidl BM, Humpel C. Chronic treatment with five vascular risk factors causes cerebral amyloid angiopathy but no Alzheimer pathology in C57BL6 mice. Brain Behav Immun. 2019;78:52–64. doi: 10.1016/j.bbi.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Franco R, Cedazo-Minguez A. Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front Pharmacol. 2014;5:146. doi: 10.3389/fphar.2014.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Games D, Adams D, Alessandrini R, Barbour R, Borthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F ß-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 21.Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88:3–12. doi: 10.1016/j.brainresbull.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Stürchler-Pierrat C, Bürki K, Van Duinen SG, Maat-Schieman MLC, Staufenbiel M, Mathews PM, Jucker M. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 23.Holcomb L, Gordon MN, Mcgowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 24.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 25.Huang WJ, Zhang X, Chen WW. Association between alcohol and Alzheimer’s disease (Review) Exp Ther Med. 2016;12:1247–1250. doi: 10.3892/etm.2016.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol. 2011;46:225–232. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humpel C. Platelets: their potential contribution to the generation of beta-amyloid plaques in Alzheimer’s disease. Curr Neurovasc Res. 2017;14:290–298. doi: 10.2174/1567202614666170705150535. [DOI] [PubMed] [Google Scholar]
- 28.Justice NJ, Huang L, Tian JB, Cole A, Pruski M, Hunt AJ, Flores R, Zhu MX, Arenkiel BR, Zheng H. Posttraumatic stress disorder-like induction elevates β-amyloid levels, which directly activates corticotropin-releasing factor neurons to exacerbate stress responses. J Neurosci. 2015;35:2612–2623. doi: 10.1523/JNEUROSCI.3333-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Justice NJ. The relationship between stress and Alzheimer’s disease. Neurobiol Stress. 2018;8:127–133. doi: 10.1016/j.ynstr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura N. Diabetes mellitus induces Alzheimer’s disease pathology: Histopathological evidence from animal models. Int J Mol Sci. 2016;17:503. doi: 10.3390/ijms17040503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King A. The search for better animal models of Alzheimer’s disease. Nature. 2018;559:13–15. doi: 10.1038/d41586-018-05722-9. [DOI] [PubMed] [Google Scholar]
- 32.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv. 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazic D, Sagare AP, Nikolakopoulou AM, Griffin JH, Vassar R, Zlokovic BV. 3K3A-activated protein C blocks amyloidogenic BACE1 pathway and improves functional outcome in mice. J Exp Med. 2019;216:279–293. doi: 10.1084/jem.20181035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 35.Lv X, Li W, Luo Y, Wang D, Zhu C, Huang ZX, Tan X. Exploring the differences between mouse mAβ 1-42 and human hAβ 1-42 for Alzheimer’s disease related properties and neuronal cytotoxicity. Chem Commun. 2013;49:5865–5867. doi: 10.1039/c3cc40779a. [DOI] [PubMed] [Google Scholar]
- 36.Marksteiner J, Humpel C. Beta-amyloid expression, release and extracellular deposition in aged rat brain slices. Mol Psychiatry. 2008;13:939–952. doi: 10.1038/sj.mp.4002072. [DOI] [PubMed] [Google Scholar]
- 37.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Prim. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 38.Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 2013;4:1495. doi: 10.1038/ncomms2494. [DOI] [PubMed] [Google Scholar]
- 39.Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:1–21. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, Lewczuk P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J Neural Transm (Vienna) 2018;125:177–191. doi: 10.1007/s00702-017-1820-x. [DOI] [PubMed] [Google Scholar]
- 41.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aß 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Eldik L Van, Berry R, Vassar R. Intraneuronal ß-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 44.Pirchl M, Ullrich C, Humpel C. Differential effects of short- and long-term hyperhomocysteinaemia on cholinergic neurons, spatial memory and microbleedings in vivo in rats. Eur J Neurosci. 2010;32:1516–1527. doi: 10.1111/j.1460-9568.2010.07434.x. [DOI] [PubMed] [Google Scholar]
- 45.Plagg B, Humpel C. Platelets in Alzheimer's Disease. The Non-Thrombotic Role of Platelets in Health and Disease. IntechOpen 2015. 2015:191–217. [Google Scholar]
- 46.Rannikmäe K, Samarasekera N, Martînez-Gonzâlez NA, Salman RAS, Sudlow CLM. Genetics of cerebral amyloid angiopathy: Systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2013;84:901–908. doi: 10.1136/jnnp-2012-303898. [DOI] [PubMed] [Google Scholar]
- 47.Reitz C, Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol. 2015;88:640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rincon F, Wright CB. Vascular cognitive impairment. Curr Opin Neurol. 2013;26:29–36. doi: 10.1097/WCO.0b013e32835c4f04. [DOI] [PubMed] [Google Scholar]
- 49.Rohn TT, Kim N, Isho NF, Mack JM. The potential of CRISPR/Cas9 gene editing as a treatment strategy for Alzheimer’s disease. J Alzheimers Dis Parkinsonism. 2018 doi: 10.4172/2161-0460.1000439. doi: 10.4172/2161-0460.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saito T, Saido TC. Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol. 2018;9:211–218. doi: 10.1111/cen3.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 52.Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017;36:2473–2487. doi: 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selkoe DJ. Alzheimer’s disease: genes, proteins, therapy. Physiol Rev. 2001;81:742–760. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 54.Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6:551–559. doi: 10.1038/nrneurol.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh I, Sagare AP, Coma M, Perlmutter D, Gelein R, Bell RD, Deane RJ, Zhong E, Parisi M, Ciszewski J, Kasper RT, Deane R. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc Natl Acad Sci U S A. 2013;110:14771–14776. doi: 10.1073/pnas.1302212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018;215:745–760. doi: 10.1084/jem.20171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold K-H, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tai LM, Youmans KL, Jungbauer L, Yu C, Ladu MJ. Introducing human APOE into Aβ transgenic mouse models. Int J Alzheimers Dis. 2011;2011:810981. doi: 10.4061/2011/810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turano E, Busetto G, Marconi S, Guzzo F, Farinazzo A, Commisso M, Bistaffa E, Angiari S, Musumeci S, Sotgiu S, Bonetti B. Neurotoxicity and synaptic plasticity impairment of N-acetylglucosamine polymers: Implications for Alzheimer’s disease. Neurobiol Aging. 2015;36:1780–1791. doi: 10.1016/j.neurobiolaging.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 60.Vijayan M, Reddy PH. Stroke, vascular dementia, and Alzheimer’s disease: molecular links. J Alzheimers Dis. 2016;54:427–443. doi: 10.3233/JAD-160527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.