

Intracellular signal transduction controlling neuronal development and survival is conveyed by second messengers that are often differentially regulated over space and time. The highly polarized morphology of neurons is conferred by a network of regulatory signaling pathways that determine axon guidance and dendrite formation. Among these, cyclic adenosine monophosphate (cAMP) is a second messenger that is critical for numerous neuronal functions and known to activate and integrate a variety of downstream pathways. In the central nervous system (CNS), cAMP-dependent signaling is involved in growth cone motility, neuronal metabolism, axon extension in vitro, neuroprotection, and survival in vivo. The complexity of cAMP-dependent neuronal physiology and function raises fundamental questions about the mechanisms determining the specificity by which cAMP can regulate these processes. A growing body of evidence suggests that fidelity in cAMP signaling is often achieved by the temporal and spatial restriction of cAMP fluxes to discrete cellular compartments where cAMP diffusion is limited by phosphodiesterase-catalyzed degradation. Despite evidence showing the importance of cAMP for neuronal survival and axon pathfinding (Goldberg et al., 2002), much remains to be learned regarding the architecture supporting cAMP compartmentation in neurons.
For many years, the only known source of cAMP in neurons were transmembrane, G-protein dependent adenylyl cyclases (ACs, tmACs), which generate cAMP fluxes in response to neurohumoral stimulation. These enzymes are located mainly on the plasma membrane, but can also be present on internalized vesicles, at the nuclear envelope, and on other organelles. Besides tmACs, there is also a calcium- and bicarbonate-sensitive soluble AC (sAC). Unlike tmACs, sAC is distributed throughout the cytoplasm, at centrioles and within nucleus and mitochondria. Notably, electrical activity and bicarbonate, that both elevate sAC activity, have been shown to increase retinal ganglion cell (RGC) growth and survival, while siRNA-mediated sAC inhibition resulted in decreased axon elongation in vitro and survival in vivo (Corredor et al., 2012). In contrast, AC1/AC8 double knockout or pharmacological inhibition did not affect RGC survival and morphology. In addition, overexpression of sAC was sufficient to overcome myelin-associated inhibitory signals in vitro and stimulate axon regeneration in vivo (Martinez et al., 2014). As neurons employ multiple cAMP synthesizing enzymes, it is unclear whether sAC alone drives cAMP-dependent survival and growth signaling in neurons, although sAC does appear to play a critical role in RGC and photoreceptor differentiation (Shaw et al., 2016). Downstream of adenylyl cyclase, cAMP signaling complexity is further diversified by multiple cAMP effector proteins including protein kinase A (PKA) and exchange protein activated by cAMP (Epac), both crucial for developing neurons. Notably, it has been shown that cAMP/PKA activity in axons can be elevated by brain derived neurotrophic factor priming membrane insertion of TrkB receptor (Cheng et al., 2011). This allows for prolongation of PKA signaling and creates a positive feedback response in the developing axon. Similarly, activated Epac enhances neurite outgrowth and regeneration of dorsal root ganglion neurons. However, these two cAMP effectors may also play opposite roles. How then is spatial control of these cAMP-mediated opposite processes achieved?
According to a now well-established model, spatial-temporal control of cAMP signaling in neurons is ensured through compartmentalization and local regulation of cAMP fluxes. In particular, PKA is compartmentalized into separately regulated pools of protein kinase by binding to a heterogeneous family of multivalent scaffolds named A-kinase anchoring proteins (AKAPs) (Kapiloff et al., 2014). Compartmentation is conferred by AKAPs that bind preferentially either RII or RI PKA subunits, with RII being the more abundant subunit in neurons, with each AKAP containing its own unique localization domain and distinct intracellular location. In addition to differential localization, AKAPs typically bind other signaling enzymes, as well as upstream activators and downstream signaling pathway effectors, such that almost all PKA-dependent signaling is believed to take place within a scaffold-organized “signalosome”. In this context, AKAP anchoring can shape dynamics of both PKA and cAMP signaling by bringing together regulatory proteins such as phosphodiesterases (PDEs) and specific isoforms of ACs. Scaffolding at AKAP microdomains permits crosstalk between cAMP and extracellular signal-regulated kinases and with calcium-dependent phosphatases such as calcineurin that in turn regulate transcription factors such as nuclear factor of activated T-cells and myocyte enhancer factor 2 (Murphy et al., 2014). These functional signalosomes serve as critical nodes for signal integration and are central to directing the course of axon growth in developing hippocampal neurons.
The functional significance of anchored signaling has been studied mainly in term of postsynaptic transmission. One of the best described neuronal AKAP compartments is organized by AKAP79/AKAP150, which is anchored to the plasma membrane by an N-terminal domain binding phosphatidylinositol-4,5-bisphosphate under regulation by calmodulin and protein kinase C (PKC) (Wild and Dell’Acqua, 2018). By scaffolding different combinations of protein phosphatases and kinases, AKAP79/150 participates in a response to fluctuations in intracellular second messengers such as cAMP, phospholipids and calcium. By coupling L-type voltage-dependent calcium channels with calcium/calmodulin-dependent phosphatase calcineurin and other downstream effectors, AKAP79/150 can integrate neuronal electrical activity with the activation of calcium sensitive enzymes, including ACs and PDEs (Murphy et al., 2014). Several other AKAPs are expressed in CNS neurons likely contributing to the regulation of neuronal growth, survival and response to different stimuli. For example, mitochondrial AKAP1 functions to maintain respiratory chain activity, control superoxide production and participate in mitochondrial fission. Its deletion impairs calcium homeostasis in neurons exposed to glutamate by loss of PKA phosphorylation of the mitochondrial fission enzyme dynamin-related protein 1 (Wild and Dell’Acqua, 2018). In proliferating cells, AKAP9 localizes to the centrosome and by bringing together PKA and PDE4D, modulates compartmentalized dynamics of cAMP over the cell cycle. PDE4D scaffolding is crucial for cell cycle progression, as disruption of centrosomal cAMP microdomain by PDE4D3 displacement resulted in accumulation of cells in prophase (Terrin et al., 2012).
Recently we have characterized the role of the perinuclear scaffold muscle A-kinase anchoring protein α (mAKAPα/AKAP6) in neuroprotection of retinal ganglion cells. In the brain and RGCs, mAKAP is expressed as the alternatively spliced α-isoform, which differs from the mAKAPβ isoform found in myocytes by inclusion of an extended N-terminal domain. mAKAP was originally identified as a PKA scaffold and was later found to bind cAMP-specific PDE4D3, AC2 and AC5, thereby tethering all components necessary for local regulation of cAMP fluxes within a perinuclear compartment (Figure 1). The activity of PDEs has been continuously linked to survival and neuronal outgrowth. For example, the PDE4-specific inhibitor rolipram promotes axon regeneration, reduces formation of a glial scar, and improves functional recovery after spinal cord lesion (Nikulina et al., 2004), although the mechanism of its action is not fully understood, and there have been some controversy regarding whether cAMP is always beneficial. Further research identified more than 20 mAKAPα-binding partners forming multimolecular signalosomes that transduce cAMP, calcium, phospholipid, mitogen-activated protein kinase and hypoxic signaling. Activation of a specific subset of AC is necessary to elevate cAMP in a compartment and can be influenced by variety of intrinsic and extrinsic factors. Similarly, the regulation of cyclic nucleotide turnover by PDEs cannot be understood without broader insight as their concerted action is required to buffer local cAMP elevations and to prevent the non-specific activation of downstream targets. But how does the balance between ACs and PDEs in a mAKAPα compartment mediates survival and axon growth?
Figure 1.
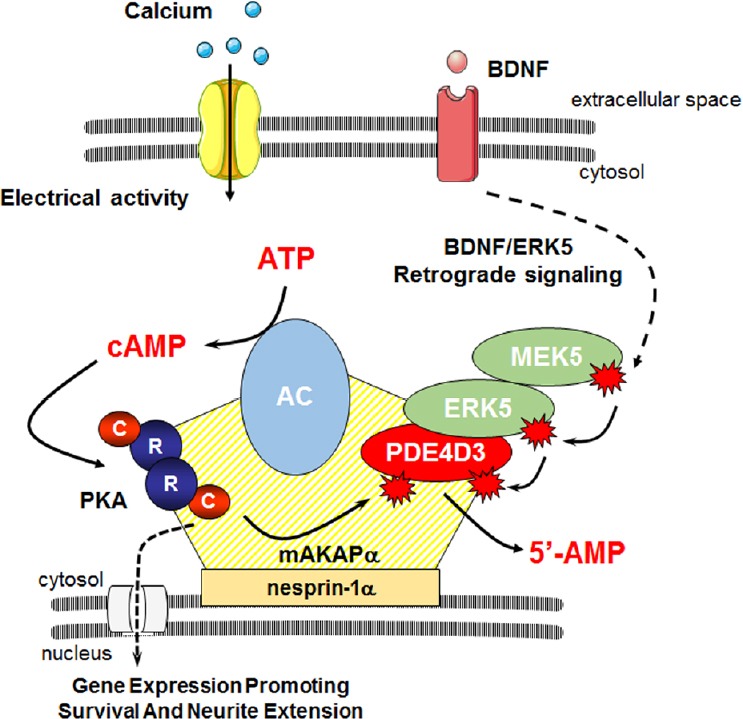
Model of cAMP signaling regulation in the perinuclear compartment.
Neuronal membrane depolarization promotes calcium entry and activation of calcium-sensitive adenylyl cyclases (ACs) leading to increased intracellular cAMP concentration. cAMP binds tetrameric protein kinase A (PKA) and results in dissociation of regulatory (R) and catalytic (C) subunits. At mAKAPα signalosomes, PKA may then phosphorylate phosphodiesterase 4D3 increasing its cAMP hydrolytic activity, thus providing a negative feedback loop by which PKA controls its own activity. mAKAPα also may provide a platform for integration of signals induced by neurotrophins such as brain-derived neurotrophic factor (BDNF). In response to BDNF, mitogen-activated protein kinase kinase 5 (MEK5) activates extracellular signal-regulated kinase 5 (ERK5), which phosphorylates PDE4D3 inhibiting cAMP hydrolysis. This neurotrophin-dependent mechanism regulating PDE4D3 activity may allow for increased local cAMP and induction of genes involved in neuronal growth and survival. cAMP: Cyclic adenosine monophosphate.
It is well established that cAMP/PKA signaling is required for activity dependent neurite outgrowth (Goldberg et al., 2002). We have shown that mAKAPα is nonessential during RGC development, but is indispensable for cAMP and neurotrophin-enhanced neuronal survival following optic nerve injury (Wang et al., 2015). Using novel molecular tools to locally modulate cAMP level at mAKAPα, we recently demonstrated that cAMP-dependent events in the perinuclear compartment are necessary for the extension of hippocampal neurons in vitro (Boczek et al., 2019). mAKAP is localized to the nuclear envelope of myocytes and neurons by binding the integral membrane protein nesprin-1α. Increasing local cAMP levels with a constitutively active adenylyl cyclase catalytic domain – nesprin-1α fusion protein was sufficient to promote neurite outgrowth in a manner similar to depolarization by KCl. cAMP fluxes in the mAKAPα compartment are controlled, at least in part, by PDE4D3, and, conversely, suppression of cAMP signaling at mAKAPα signalosomes by expression of a constitutively-active PDE-nesprin-1α fusion protein attenuated axon elongation and blocked any rescue effects of KCl. Although we cannot exclude that membrane depolarization may activate some mAKAPα-independent signaling, it is apparent that the perinuclear compartment defined by mAKAPα is required for electrical activity-dependent neuronal outgrowth. Lastly, we demonstrated that PDE4D3 anchoring disruption using a peptide that displaced PDE4D3 from mAKAPα signalosomes could both enhance hippocampal and RGC neurite extension in vitro, as well as confer neuroprotection after RGC injury in vivo, providing a potential strategy for the therapeutic translation of our findings (Boczek et al., 2019).
Undoubtedly, whether activity-dependent cAMP signals driving neuroprotection and neuroregeneration are orchestrated by mAKAPα deserves further study. As our in vitro experiments were performed on embryonic hippocampal neurons and neonatal RGCs, which are known for their enhanced potential to grow and elongate axons, it will be necessary to verify if similar effects can be detected for mature neurons. A combinatorial reactivation of a developmental growth program and elevation of cAMP in the perinuclear compartment may be required to obtain meaningful levels of either neurite extension in vitro or more importantly for axonal regeneration after in vivo injury for adult neurons. It is also possible that revealing a pro-regenerative potential will require synergistic modulation of cAMP in other cellular compartments by both intrinsic and extrinsic stimuli. In this regard, live-cell imaging using cAMP/PKA FRET sensors targeted to different compartments could provide data relevant to the upstream regulation of cAMP signaling in subcellular microdomains. Currently used sensors based on Epac protein or sensitive to PKA activity are able to track changes in cAMP concentration in submicromolar range, thus providing excellent tools for such experiments. The use of AKAP anchoring disruptors should clarify how interactions with binding partners within different signalosomes affect these processes. Application of these tools should give us a new, profound insight into molecular mechanisms of cAMP signaling compartmentalization and help to identify novel targets for neuroprotection and regeneration in the mature CNS. In the meantime, as expression of the PDE4D3 anchoring disruptor peptide enhanced RGC survival after optic nerve crush, providing proof-of-concept for a novel gene therapy for RGC neuroprotection, whether PDE4D3 anchoring disruption can be effective in diseases such as glaucoma should be explored given the great unmet need for new therapies for this prevalent cause of human blindness.
This work was supported in part by National Institutes of Health Grant EY026766 (to MSK).
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Alavala Matta Reddy, Adikavi Nannaya University, India; Giacinto Bagetta, University of Calabria, Italy.
P-Reviewers: Matta Reddy A, Bagetta G; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Boczek T, Cameron EG, Yu W, Xia X, Shah SH, Castillo Chabeco B, Galvao J, Nahmou M, Li J, Thakur H, Goldberg JL, Kapiloff MS. Regulation of neuronal survival and axon growth by a perinuclear cAMP compartment. J Neurosci. 2019;39:5466–5480. doi: 10.1523/JNEUROSCI.2752-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A. 2011;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corredor RG, Trakhtenberg EF, Pita-Thomas W, Jin X, Hu Y, Goldberg JL. Soluble adenylyl cyclase activity is necessary for retinal ganglion cell survival and axon growth. J Neurosci. 2012;32:7734–7744. doi: 10.1523/JNEUROSCI.5288-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA. Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron. 2002;33:689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- 5.Kapiloff MS, Rigatti M, Dodge-Kafka KL. Architectural and functional roles of A kinase-anchoring proteins in cAMP microdomains. J Gen Physiol. 2014;143:9–15. doi: 10.1085/jgp.201311020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez J, Stessin AM, Campana A, Hou J, Nikulina E, Buck J, Levin LR, Filbin MT. Soluble adenylyl cyclase is necessary and sufficient to overcome the block of axonal growth by myelin-associated factors. J Neurosci. 2014;34:9281–9289. doi: 10.1523/JNEUROSCI.1434-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell’Acqua ML. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. 2014;7:1577–1588. doi: 10.1016/j.celrep.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786–8790. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw PX, Fang J, Sang A, Wang Y, Kapiloff MS, Goldberg JL. Soluble adenylyl cyclase is required for retinal ganglion cell and photoreceptor differentiation. Invest Ophthalmol Vis Sci. 2016;57:5083–5092. doi: 10.1167/iovs.16-19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC, Mongillo M, Sawa A, Jordanides NE, Mountford JC, Zaccolo M. PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J Cell Biol. 2012;198:607–621. doi: 10.1083/jcb.201201059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Cameron EG, Li J, Stiles TL, Kritzer MD, Lodhavia R, Hertz J, Nguyen T, Kapiloff MS, Goldberg JL. Muscle a-kinase anchoring protein-alpha is an injury-specific signaling scaffold required for neurotrophic- and cyclic adenosine monophosphate-mediated survival. EBioMedicine. 2015;2:1880–1887. doi: 10.1016/j.ebiom.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wild AR, Dell’Acqua ML. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol Ther. 2018;185:99–121. doi: 10.1016/j.pharmthera.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]