

Abstract

Catalytic hydrodeoxygenation (HDO) is an effective technology for upgrading pyrolysis bio-oils. Although, in the past years, this process has been extensively studied, the relevance of the cross-reactivity between the numerous chemical components of bio-oil has been scarcely explored. However, molecular coupling can be beneficial for improving the bio-oil characteristics. With the aim of gaining a better understanding of these interactions, this work investigates the catalytic hydrodeoxygenation of mixtures of two typical components of pyrolysis bio-oils: guaiacol and acetic acid. The catalytic tests were carried out employing a bifunctional catalyst based on nickel phosphide (Ni2P) deposited over a commercial nanocrystalline ZSM-5 zeolite. The influence of both hydrogen availability and temperature on the activity and product distribution, was evaluated by carrying out reactions under different H2 pressures (40–10 bar) and temperatures (between 260 and 300 °C). Using blends of both substrates, a partial inhibition of guaiacol HDO occurred because of the competence of acetic acid for the catalytic active sites. Nevertheless, positive interactions were also observed, mainly esterification and acylation reactions, which could enhance the bio-oil stability by reducing acidity, lowering the oxygen content, and increasing the chain length of the components. In this respect, formation of acetophenones, which can be further hydrogenated to yield ethyl phenols, is of particular interest for biorefinery applications. Increasing the temperature results in an increment of conversion but a decrease in the yield of fully deoxygenated molecules due to the production of higher proportion of catechol and related products. Additional experiments performed in the absence of hydrogen revealed that esterification reactions are homogeneously self-catalyzed by acetic acid, while acylation processes are mainly catalyzed by the acidic sites of the zeolitic support.
1. Introduction
Biomass is the most readily available source of renewable carbon to supply the high demand of chemical fuels of the transport sector, among other reasons, due to its good compatibility with the existing infrastructure.1 In particular, the utilization of lignocellulosic biomass as feedstock for biofuels production has attracted a great deal of attention not only because of its abundance and low cost but also due to the fact that this resource does not compete with food production.2 Lignocellulose fast pyrolysis is a relative effective method to produce liquid bio-oils with yields ranging from 40 to 70%.3 However, it is well known that, as a consequence of their poor physicochemical properties (high oxygen content, acidity, corrosivity, and low stability), pyrolysis bio-oils should be further upgraded before they can be used in conventional combustion engines. Hydrodeoxygenation (HDO) process has been proven to be a very promising route for the production of satisfactory liquid biofuels from lignocellulosic biomass4 since it allows biofuel upgrading by oxygen removal in the form of water, enhancing the fuel properties of pyrolysis bio-oils.5
In this context, numerous efforts have been devoted during the past years to the development of efficient and selective hydrodeoxygenation (HDO) catalysts. Typically, HDO catalysts can be classified in several groups regarding the chemical nature of the hydrogenating component: Mo-based sulfides, noble metals, transition metals and metal carbides, nitrides, and phosphides.6−11 This last kind of catalysts, particularly those based on nickel phosphide supported on different solids, has provided encouraging results in the past few years.12 In this sense, the use of acid solids as supports (i.e., ZSM-5 and Al-SBA-15) is of particular interest since they may promote synergic interactions between the metal active phase and their intrinsic acid sites, leading to sequential hydrogenation-dehydration-hydrogenation reactions, which improves the overall HDO efficiency.1,9,13−15
In spite of the extensive research carried out in this field, most of the studies reported in the literature deals with the hydrodeoxygenation of representative single molecules of lignocellulosic bio-oils such as phenol, anisole, guaiacol, or hydroxymethylfurfural (HMF), among others.16−18 However, pyrolysis bio-oils consist of a complex mixture of oxygenated organic compounds, which can include more than 300 different molecules. Thus, this simplified composition overlooks molecular interactions occurring during the upgrading process between the different organic functionalities.19 Conversely, although detailed studies of the HDO of real bio-oils are available in the literature, owing to the analytic difficulties and the intractable complexity of the network of chemical reactions involved, the knowledge of the mechanistic aspects of this upgrading process is rather incomplete.20
The presence of pyrolysis bio-oils of carboxylic acids, mainly acetic and formic, could have a significant impact on the hydroprocessing of other bio-oil components since they can promote acid-catalyzed reactions (dehydration, alkylation, cracking, coke formation, etc.) and react with other functionalized molecules. Likewise, phenolic derivatives, which are relatively abundant in pyrolysis bio-oils (up to 30 wt %), are considered the most refractory compounds to be deoxygenated under hydrogenation conditions. Therefore, the study of the interaction between both types of organic compounds could provide relevant information about the most significant cross-reactivity pathways contributing to the bio-oil upgrading. In this context, Wan et al.21 screened the effect of acetic acid on p-cresol hydrodeoxygenation over different catalysts based on supported noble metals (Ru/C, Pt/C, Ru/Al2O3, Pt/Al2O3, etc.). They found that acetic acid presence improves p-cresol hydrodeoxygenation, in aqueous solutions, by enhancing the selectivity toward methylcyclohexane due to the promotion of dehydration reactions over Ru/C.21 Interesting results have been also reported about the hydrodeoxygenation of guaiacol and propionic acid blends over supported Ni catalysts. In this work, in addition to the components coming from the direct hydrodeoxygenation of individual substrates, mainly cyclohexane and propane, alkylated and esterificated compounds were also detected, which led to an increase in the hydrocarbon chain length of the reaction products, decreasing carbon loss in the gas phase.22 In the same way, HDO of aqueous solutions of guaiacol and acetic acid has been investigated using Ni on red mud at rather harsh operation conditions, achieving a notable yield of C8 hydrocarbons.23 More recently, hydrotreating of methanol solutions containing furfural, hydroxyacetone, guaiacol, and acetic acid over Cu/SBA-15 has shown to lead to the formation of diverse saturated esters and alcohols.24
These reports emphasize the potential interest of coupling reactions for the upgrading of bio-oils. Nevertheless, a deeper understanding of the interactions and cross-reactivity between these components during bio-oil catalytic hydrodeoxygenation is still necessary to gain a better control of the selectivity. Furthermore, there is also a growing interest on using light carboxylic acids such formic acid as a source of hydrogen in catalytic transfer hydrogenation.25 Considering the abundance of these chemicals in bio-oils, it is conceivable to take advantage of them for reducing the hydrogen demand during HDO, with the subsequent advantages for the economy and sustainability of the process.
On this background, this work investigates the catalytic hydrodeoxygenation of mixtures of two typical components of pyrolysis bio-oils: (i) guaiacol, a representative molecule of phenolic monomers of lignin, which possesses a single molecule, both hydroxyl and methoxy functionalities, and (ii) acetic acid, the most abundant carboxylic acid in pyrolysis bio-oils with a concentration typically ranging between 5 and 17 wt %.26 Guaiacol has a tendency to form coke through polymerization and presents high resistance to hydrodeoxygenation,4,27,28 while acetic acid also promotes repolymerization reactions and is the main responsible of bio-oil aging and corrosion during storage.29 Besides, HDO of this carboxylic acid generate volatile products of limited value, and accordingly, several routes for promoting C–C formation with acetic acid has been proposed,30−33 whereas its use as a solvent for the hydrotreating of methoxyphenols has also been considered.34
The study of the cross-reactivity between guaiacol and acetic acid has been carried out, varying the reaction temperature and the hydrogen availability in the reaction media in order to investigate the eventual contribution of catalytic transfer hydrogenation. Likewise, some catalytic tests were performed under an inert atmosphere (N2) to explore the role of nonhydrogenating catalytic centers in the reaction network. A bifunctional system based on nickel phosphide supported on a commercial nanocrystalline ZSM-5 zeolite has been selected as a catalyst for these experiments owing to its remarkable results in a previous investigation with similar substrates, which have shown the excellent activity of this catalyst, comparable to those based on platinum metal group elements at an affordable cost.14
2. Results and Discussion
2.1. Ni2P/ZSM-5 Physicochemical Properties
The crystallinity of both support and bifunctional catalyst as well as the presence of nickel phosphide phases in the loaded material were studied by X-ray diffraction (XRD) (Figure 1). Diffraction patterns show the presence of the characteristic reflections of MFI topology at 2θ ranges of 8–10° and 20–25°, which remain unmodified after nickel phosphide loading. In the pattern corresponding to the loaded catalyst following reduction in H2, the presence of the diffraction peaks at 2θ values of 40.8°, 44.6°, and 47.3° reveal the Ni2P phase formation, which is reported to be the most active nickel phosphide for hydrotreatment processes.35,36 The absence of diffraction signals corresponding to metallic Ni or other nickel phosphide phases denotes the complete conversion of the metal precursors into Ni2P.
Figure 1.
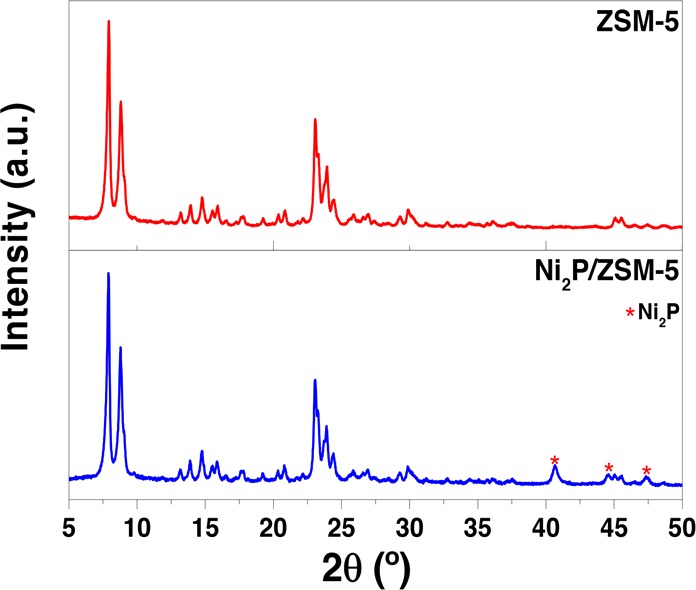
XRD patterns corresponding to raw ZSM-5 and Ni2P/ZSM-5 catalyst.
The main physicochemical properties of both samples are summarized in Table 2. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) analyses indicate that the nickel content in the loaded material is close to the target value (10 wt % Ni). The Ni/P molar ratio estimated was of 1.49, higher than the molar proportion used during the support impregnation ([Ni/P]MOL = 1) due to the partial volatilization of phosphorous during reduction. However, this value is lower than the stoichiometric ratio of the metal-rich Ni2P active phase. Similar results have been frequently found for Ni2P catalysts, indicating that unreacted phosphorus-containing species are also present over the catalysts surface, most likely in the form of P–OH groups, which can act as weak Brönsted acid sites.37
Table 2. Physicochemical Properties of ZSM-5 and Ni2P/ZSM-5 Samples.
| sample | SBETa (m2 g–1) | SMICb (m2 g–1) | SEXT+MSb (m2 g–1) | VPOREc (cm3 g–1) | VMICb (cm3 g–1) | Ni (wt %)d | (Ni/P)MOL | (Si/Al)MOL | Ni2P particle size (nm)e (D50) |
|---|---|---|---|---|---|---|---|---|---|
| ZSM-5 | 452 | 329 | 123 | 0.48 | 0.20 | 42 | |||
| Ni2P/ZSM-5 | 395 | 287 | 108 | 0.55 | 0.17 | 9.6 | 1.49 | 42 | 7.0 |
Calculated by BET method.
Estimated by applying the NL-DFT method.
Total pore volume measured at a P/P0 of ∼0.97.
Determined by ICP-OES.
Measured by TEM image analysis.
Textural properties, estimated from Ar adsorption–desorption isotherms, corresponding to the zeolitic support and impregnated catalyst are also summarized in Table 2. The commercial ZSM-5 support exhibits SBET and SMS+EXT values of 452 and 123 m2/g, respectively. These values are somehow higher than those typically reported for MFI zeolites, which is ascribed to the nanocrystalline nature of this sample. In this sense, transmission electron microscopy (TEM) images, taken over the parent support, show zeolitic nanocrystals with sizes comprised between 30 and 60 nm (Figure 2A). The loading of the active phase causes a decrease in specific surface area of 57 m2/g (approximately a reduction by 12%) due to partial pore blockage caused by metal phosphide nanoparticles, affecting both the microporous network and external surface.14 TEM images of the reduced catalyst reveal that these Ni2P nanoparticles possess a heterogeneous size distribution in the range of 2.5–51 nm and a D50 of 7 nm (Figure 2C and Table 2).
Figure 2.

TEM images acquired over (A) raw ZSM-5 and (B) Ni2P/ZSM-5 catalyst. (C) Ni2P particle size distribution.
The density and strength of acidic sites were evaluated by means of NH3-TPD experiments (Figure S1). The raw zeolitic material exhibits two desorption signals centered around 175 and 360 °C, which involves the existence of acid sites with different strengths. Thus, the peak at low temperatures is assigned to NH3 interacting with the weak acid sites, while the signal registered at higher temperatures should correspond to NH3 bound on stronger acidic sites. After Ni2P incorporation, a notable reduction in the overall acidity value is observed (from 0.39 to 0.27 mmol NH3/g), which is mainly due to removal of the high temperature feature, generally associated with stronger acidic sites. This variation of the TPD profile is attributed to the partial coverage of the zeolitic acid sites by the deposition of metallic nanoparticles and the nucleation of Ni species on the acidic hydroxyl groups.38 Although the particle size distribution is broad, this interaction could favor the formation of some highly dispersed Ni2P nanoclusters during the calcination and reduction process, which can improve the activity of the final bifunctional catalyst. In addition, for the loaded material, the maximum NH3 desorption peak appears centered at 156 °C, which is assigned to the presence of a new acidity, most likely of moderate strength, generated by the nickel phosphide deposition, in particular Niα+ Lewis acid sites and P–OH Brønsted acid sites.17
2.2. Catalytic Hydrodeoxygenation of Guaiacol, Acetic Acid, and Their Blends
Previous reports on the HDO of different phenol-related chemicals over catalysts based on Ni2P revealed that the main products are cyclohexane derivatives, which are obtained with very high yields (>80%).12,14 Studies regarding the hydrodeoxygenation of acetic acid are more scarce, but a recent investigation reported that ethyl acetate and light hydrocarbons (ethylene and ethane) are formed over a Mo2C catalyst at a low hydrogen pressure.39 In the present work, in order to set a reference for comparison purposes, two HDO reactions were carried out using, as feeds, pure guaiacol (R1 in Table 1) and acetic acid (R2 in Table 1) solved in decalin. The catalytic results, expressed in terms of substrate conversion and product selectivities, are presented in Figure 3.
Table 1. Feed Composition and Reaction Conditions Used in the Catalytic Tests Performed.
| reaction | gas | P (bar) | T (°C) | guaiacol (wt %) | acetic acid (wt %) | guaiacol acetate (wt %) | apocynin (wt %) | catalyst |
|---|---|---|---|---|---|---|---|---|
| R1 | H2 | 40 | 260 | 3.3 | Ni2P/ZSM-5 | |||
| R2 | H2 | 40 | 260 | 8 | Ni2P/ZSM-5 | |||
| R3 | H2 | 40 | 260 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R4 | H2 | 30 | 260 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R5 | H2 | 20 | 260 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R6 | H2 | 10 | 260 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R7 | N2 | 2 | 260 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R8 | N2 | 2 | 260 | 3.3 | 8 | ZSM-5 | ||
| R9 | H2 | 40 | 280 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R10 | H2 | 40 | 300 | 3.3 | 8 | Ni2P/ZSM-5 | ||
| R11 | H2 | 40 | 260 | 3.3 | Ni2P/ZSM-5 | |||
| R12 | H2 | 40 | 260 | 3.3 | Ni2P/ZSM-5 | |||
| Blank 1 | N2 | 2 | 260 | 3.3 | 8 | none | ||
| Blank 2 | H2 | 10 | 260 | 3.3 | 8 | none |
Figure 3.

HDO reactions of pure guaiacol (R1) and acetic acid (R2) at 260 °C and 40 bar of H2.
Under these reaction conditions (260 °C and 40 of H2), guaiacol shows a very high conversion value, being close to 89%, and a selectivity toward cyclohexane of 85%. These results confirm the high activity for HDO of phenolic derivatives of the Ni2P/ZSM-5 catalytic system, as it was previously reported in the literature.14 In addition to cyclohexane, methylcyclopentane (∼7.5%) and hexane (∼3%) are also detected but in a much lower proportion. The presence of this last compound denotes the occurrence of ring-opening reaction followed by its contraction, which are catalyzed by the acid sites of the zeolitic support. Minor amounts of other compounds such as benzene, anisole, veratrole, or phenol are also identified in the reaction media. The presence of these components indicates the existence of other minority transformation routes (e.g., demethylation), as previously reported.22 The concentration of gas products, mainly CH4 and CO generated by guaiacol demethylation and decarbonylation, was very low: <0.25 wt % referred to the initial guaiacol amount (see Figure S2).
Acetic acid exhibits a conversion close to 31% (R2), being almost three times lower than for guaiacol, probably due to the higher substrate concentration present in the reaction mixture (3.3 vs 8 wt %) and the more severe temperature needed for the efficient hydrodeoxygenation of acetic acid.30,40 In this case, ethyl acetate is the only compound detected in the liquid phase, which is formed by the esterification between acetic acid and ethanol generated from acetic acid hydrogenation. The absence of ethanol in the liquid mixture suggests that, under these conditions, it easily reacts with the unconverted acetic acid as soon as it is formed, in accordance with previous reports.39,41 Furthermore, acetone is not detected in this assay, suggesting that ketonization reactions are not significant over Ni2P/ZSM-5 catalysts under these conditions. The contribution of gaseous fraction is low but appreciably higher than for guaiacol HDO, ∼2.5 wt % referred to the acetic acid fed into the reaction media, being mostly composed of CH4, CO, and a much lower concentration of ethane (see Figure S2). Formation of the majority of gases can be ascribed to the thermal decomposition of acetic acid in the reducing environment. On the other hand, ethane is produced from the complete hydrodeoxygenation of acetic acid, as it has been found previously using different catalysts.42,43
Previous catalytic tests with single-component solutions provide substantial information about the reactivity of guaiacol and acetic acid under hydrotreating conditions. However, pyrolysis bio-oil upgrading actually involves a convoluted reaction network resulting from the interactions between molecules with different functional groups, either existing initially or being created in this complex liquid matrix. Thus, in order to ascertain the intermolecular reactivity between two of the most abundant families in pyrolysis bio-oils (carboxylic acids and methoxyphenols), hydrodeoxygenation experiments have been carried out using acetic acid and guaiacol blends. The first catalytic assay (R3) was carried out under the same reaction conditions as those employed in the previous experiments with single components (260 °C, 40 bar of H2, and 3.3% guaiacol and 8% of acetic acid dissolved in decalin). The time evolution of both the conversion and the product distribution obtained in this catalytic test is depicted in Figure 4. For the sake of comparison with the previous experiments performed using pure model compounds as feeds, both conversion and product selectivity were estimated referred to each substrate. Although this description of the data leads to certain duplicity, as products of coupling appear in both guaiacol and acetic acid graphs (e.g., guaiacol acetate is considered in both the selectivity of guaiacol and acetic acid), it facilitates the discussion of the transformation routes.
Figure 4.

Variation of the conversion and the selectivity with respect to (A) guaiacol and (B) acetic acid as a function of the reaction time obtained in the catalytic hydrotreating reaction (R3) of the blend at 260 °C and 40 bar of H2.
As it can be appreciated in Figure 4, some conversion (ca. 10% for guaiacol and 12.5% for acetic acid) takes place during heating up of the reactor (the onset time of reaction is taken when the reactor reaches the selected temperature) mainly due to production of guaiacol acetate and in lower extent ethylguaiacol, following reaction paths discussed in detail below. On the other hand, the degree of transformation attained for guaiacol and acetic acid in the blend after 120 min are lower than those obtained when using single components. This reduction in conversion is especially pronounced for guaiacol, which drops from 89 to 35%, while for acetic acid, the conversion decreases slightly from 31 to 26%. This behavior is in contrast with the reports on the HDO of oxygenated aromatics in aqueous solutions with no acidic catalyst such as Ru/C, where acetic acid has a promoting effect by facilitating dehydration of intermediate cyclic alcohols.23 In the present case, these data evidence a competition for the catalytic active centers between both substrates, presumably explained by the acetic acid preferential adsorption on the active sites of the catalyst because of its smaller molecular size and its tendency to form carboxylates, which inhibits the guaiacol hydrodexygenation.44,45 In this respect, the lower electronic density of Ni in the phosphide may favor a strong adsorption of acetic acids and, subsequently, the partial deactivation of Ni2P/ZSM-5,17 as compared with Ni/ZSM-5, which does not show any significant loss of activity when treating guaiacol and propionic acid blends.22
Temporal evolution of the guaiacol-derived products shows a strong decay of the guaiacol acetate concentration with reaction time, while the proportion of other molecules such catechol, cyclohexane, anisole, and catechol acetate increases initially but tends to level off at longer reaction times. In contrast, the concentration of ethyl catechol rises appreciably after 120 min of reaction, while ethyl guaiacol selectivity is high initially but grows only slightly with the progress of the reaction. Considering the final selectivity toward cyclohexane, the value is much lower than that attained for the pure feed (∼21 vs 85%), denoting a reduction in the HDO efficiency. As a consequence, intermediate products of HDO such as anisole (∼7.5%), phenol (∼7.2%), and especially catechol (∼9%) appear in larger proportion in the reaction mixture than in the case of the tests performed with pure guaiacol. In addition, esterification products, such as guaiacol acetate (∼15%), catechol acetate (∼2.6%), and diacetoxybenzene (∼2.4%), generated by the reaction between the phenolic substrate and some of its HDO intermediates with acetic acid, are also detected. Thus, ester selectivity (ca. 15%) is almost as much as that of cyclohexane, which is the product of complete HDO. That product distribution is in sharp contrast with that obtained from HDO of pure guaiacol, which led to selectivity toward alkanes of more than 95%. Remarkably, such large variations in product distribution were not reported for Ni/ZSM-5 when dealing with propionic acid and guaiacol mixtures, also suggesting that the partial deactivation of Ni2P by acetic acid modulates the product distribution of the blends with guaiacol. Anyhow, through esterification and ethylation reactions, acetic acid is consumed, forming molecules with longer chain length. Simultaneously, both oxygen (removed as H2O in esterification) content and acidity are decreased, without requiring any additives such alcohols and with lower hydrogen consumption.24,44
Under these reaction conditions, esterification products, particularly guaiacol acetate, could be further hydrodeoxygenated. Therefore, in order to ascertain the behavior of guaiacol acetate under HDO conditions, an additional catalytic test was carried out using guaiacol acetate dissolved in decalin (3.3 wt %) as a feed (Figure S3). In this case, almost total conversion is achieved (∼99%), with guaiacol as the major reaction product with a selectivity of 48%. Partial guaiacol acetate decomposition into guaiacol and acetic acid reflects the existence of reversible equilibrium and the low reactivity of this aromatic ester for hydrogenation. This fact can be detrimental for the deep deoxygenation of blends containing this chemical. As a result of the subsequent guaiacol hydrodeoxygenation, anisole (∼14%), phenol (∼2%), and some amount of cyclohexane (∼5%) were also detected in the catalytic tests with guaiacol acetate. In addition, a small proportion of ethyl guaiacol appears in the reaction mixture. As it is discussed in detail below, ethyl guaiacol (mainly 4-ethyl, but other isomers are also detected) is a product of the partial hydrogenation of apocynin, formed previously as a result of the C-acylation of guaiacol with acetic acid. However, alternatively, apocynin could be also generated by means of the Fries rearrangement of guaiacol acetate via an intermolecular pathway, which is catalyzed by acid sites.32,46
Interestingly, ethylated products, mainly ethylguaiacol (∼16%) and, in lower proportion, ethyl phenol (∼4%) and ethyl catechol (2.5%), also appear in the reaction of guaiacol and acetic acid blends, as it can be appreciated in Figure 4. Previous literature results have shown that further deoxygenation to yield ethyl benzene is possible using aqueous blends of guaiacol and acetic acid under higher temperature and pressure with Ni-based catalysts.23 In the present case, these ethylated products could be formed by the sequential hydrodeoxygenation of acetophenones (mainly apocynin, but other isomers are also detected) formed as a result of the direct C-acylation of guaiacol with acetic acid. To confirm this statement, HDO reaction of apocynin was performed under the same conditions, 260 °C and 40 bar of H2 (Figure S4), using 3.3 wt % of substrate. HDO of apocynin mostly leads to ethyl guaiacol (∼69%, mainly 4-ethyl-guaiacol) and ethyl cyclohexane (∼21%) with a conversion close to 88%. The low proportion of ethyl phenol and the absence of ethyl catechol suggest their rapid conversion toward the final deoxygenated product, ethyl cyclohexane, favored by the absence of acetic acid in the reaction media. This result confirms that apocynin, previously generated by acylation of guaiacol with acetic acid and, possibly, by Fries rearrangement of guaiacol acetate, experiences the following cascade of reactions: apocynin → ethyl guaiacol → ethyl catechol → ethyl phenol → ethyl cyclohexane. A similar reaction scheme has been proposed for the HDO of acetophenone over Pt/Al2O3, which shows the easy formation of ethyl benzene.47 This is a remarkable result because, through this transformation, the complete deoxygenation of the acetyl group is achieved without carbon loss, producing appreciable yield of ethyl cyclohexane, which is a hydrocarbon in the gasoline range. Moreover, it is important to highlight that acetophenones are themselves highly valuable fine chemicals with applications in cosmetic and pharmaceutical industries. In particular, apocynin has been investigated as an anti-inflammatory drug.48 Accordingly, it can be also of interest to stop the progress of the hydrotreating reaction in order to increase the selectivity to acetophenones.
The temporal evolution of acetic acid conversion and product distribution in the R3 test is displayed in Figure 4B, which shows that, although the total conversion increases only slightly with reaction time, the selectivity varies remarkably. In particular, the concentration of ethyl acetate increases initially and then reaches a plateau, while, as mentioned above, the proportion of guaiacol acetate progressively diminishes. The proportion of methyl acetate is high and gradually rises with reaction time, while the ethanol concentration is only significant after 100 min. Interestingly, some acetone is produced during heating up possibly due to ketonization in zeolite centers, as previously reported,30 but its concentration swiftly drops. Following 120 min of reaction, the major reaction products are ethyl acetate and methyl acetate with selectivity values of ∼39 and ∼30%, respectively. As it has been mentioned before, ethyl acetate is generated by the reaction of acetic acid with ethanol formed during its HDO process. In this case, a small amount of ethanol (ca. 4%) has been also observed in the reaction mixture. Formation of methyl acetate can proceed from acetic acid esterification with the methanol produced by demethoxylation of guaiacol derivatives, as it has been reported elsewhere.34 The selectivity toward the reaction products coming from the cross-reactivity of guaiacol and acetic acid, denoted as phenolic derivatives, are relatively low (<25%), and this can be attributed to the high proportion of acetic acid employed in the reaction feed.
With the information extracted from the catalytic tests discussed above, a reaction pathway for the HDO of acetic acid and guaiacol blends is proposed in Figure 5, which, for the guaiacol transformations, implies three main routes: (i) esterification to yield guaiacol acetate, which is reversible and hardly produce any other chemical; (ii) demethylation and subsequent esterification with acetic acid to eventually generate catechol acetate and then diacetoxybenzene; and (iii) acylation and successive hydrodeoxygenation, first leading to ethylguaiacol and, eventually, to ethyl cyclohexane as an end product, which is a molecule of interest in fuel processing. In addition, the direct transformations of each molecule take place simultaneously following the scheme described elsewhere, yielding, as final products of the HDO of guaiacol and acetic acid, cyclohexane and ethyl acetate, respectively.40,49
Figure 5.

Proposed cross-reaction mechanism of HDO of guaiacol and acetic acid blends. Please note that the positional isomers represented are those detected in higher concentrations but mixtures of regioisomers are generally obtained.
2.2.1. Influence of the Hydrogen Pressure on the Catalytic Hydrodeoxygenation of Guaiacol/Acetic Acid Blends
Results of the previous section have proven the existence of synergetic effects in the hydroprocessing of acetic acid and guaiacol blends, leading to the generation of esterification products and ethylated derivatives with longer hydrocarbon length than the initial substrates. The product distribution in these catalytic processes can be influenced by the hydrogen pressure in the reaction media. In this regard, an excess of hydrogen could favor the formation of alkanes, while partial deoxygenation would be predominant at lower H2 pressures. Interestingly, the possibility of using carboxylic acids as a source of hydrogen, for the deoxygenation of guaiacol, is a potentially attractive route for the upgrading of bio-oils. Thus, to get further information about the role of hydrogen availability, in addition to the assays carried out under 40 bar, a series of hydrodeoxygenation tests were performed using pressures of 30 (R4), 20 (R5), and 10 bar (R6) of H2. The conversion and product distribution corresponding to these experiments are depicted in Figure 6.
Figure 6.

Conversion and products selectivity respect to (A) guaiacol and (B) acetic acid obtained in catalytic HDO reactions of the blend at 260 °C as a function of H2 pressure: Catalytic tests were performed at 40 bar (R3), 30 bar (R4), 20 bar (R5), and 10 bar (R6).
As it can be appreciated, the decrease in the hydrogen pressure from 40 to 30 bar, representing around 108 and 81% of the stoichiometric amount of H2 required for the oxygen removal without C–C bond saturation, respectively, do not produce significant variations in the catalytic results. Thus, considering the results attained at 120 min, similar conversion values for both substrates (35 and 23% for guaiacol and acetic acid, respectively) are obtained, and only a small reduction in the selectivity toward HDO products is observed. In the case of guaiacol, the cyclohexane selectivity decreases from 20 to 18%, while the proportion of catechol slightly increases from 9 to 11%. For acetic acid, the selectivity toward ethyl acetate lowers from 40 to 34%, whereas that for methyl acetate increases from 30 to 35%. These results indicate that the amount of hydrogen available in the reaction media is still enough to produce HDO reactions. However, when lower hydrogen pressures are employed (10 and 20 bar, equivalent to 54 and 27% of the required H2 for full deoxygenation, respectively), the conversion values of both model compounds are significantly diminished. This reduction is more marked for guaiacol (40 vs 28–31%) than for acetic acid (25 vs 19–21%), most likely owed to the preferential adsorption of acetic acid on the catalyst active sites, which can be detrimental for the conversion of the phenol derivative. As consequence, for guaiacol, the selectivity values toward the HDO products such as anisole, phenol, and, especially, cyclohexane are lowered. In the case of acetic acid, the product distribution remains basically unchanged under 20 bar of H2 (54% of the H2 required for full oxygen removal), but a significant reduction in ethyl acetate is observed at 10 bar of H2 (34 vs 16%). These results denote that, under 10 bar of H2 (27% of the H2 required for complete oxygen removal), there is a relevant restriction in hydrogen availability for both substrates. Likewise, at a H2 pressure of 10 bar, the proportion of guaiacol acetate is higher than in previous reactions (∼22%). This is an expected result since, when the hydrogen pressure is decreased, the HDO route is appreciably inhibited, favoring the esterification reaction. On the other hand, the proportion of acetophenones and their ethylated derivatives are similar for these experiments, suggesting that hydrogen does not play a key role on the initial acylation reactions. Furthermore, gas phase analyses (see Figure S2) also reflect a sharp reduction in the formation of ethane as the hydrogen pressure decreases, while CO and CH4 production is only slightly diminished.
With the aim of exploring the hydrogen influence over the formation of acetophenones and ethylated compounds, an additional catalytic test was carried out using the same reaction conditions but under an inert atmosphere (2 bar of N2, R7). Likewise, in order to further ascertain the role of the Ni2P phase and that of acid centers provided by the zeolitic support, two additional reactions were also performed using (i) the raw ZSM-5 zeolite (R8) and (ii) a blank experiment without any catalyst (Blank 1). All these results are depicted in Figure 7. Under an inert atmosphere, the conversion values achieved in these experiments for both substrates are in the same range as those attained using different hydrogen pressures (assays R3–R6). In contrast, the product distribution is entirely different. Thus, over the bifunctional Ni2P/ZSM-5 and over the raw support, guaiacol acetate, formed by the esterification of both substrates, is the main reaction product, with selectivity percentages of 85 and 88%, respectively. The guaiacol acetate yield is even higher in the blank experiment without catalysts. Accordingly, it can be concluded that formation of this molecule is homogeneously self-catalyzed by acetic acid, with a selectivity value higher than 99% (regarding to guaiacol). This observation agrees well with the information reported in the literature for similar phenolic derivatives.32 Furthermore, it is important to stress that, basically, the same results were obtained employing a reduced pressure of hydrogen (10 bar) in a test without a catalyst (Blank 2, Figure S5), confirming that hydrogen does not exert any influence in esterification reactions. Regarding acetic acid, the selectivity toward guaiacol acetate is somewhat lower (ca. 90%) in the blank test due the formation of a small amount of methyl acetate coming from acetic acid esterification with methanol, the latter being produced by demethoxylation of guaiacol. These results clearly indicate that hydrogen transfer reactions from acetic acid are not significant under these mild conditions and, accordingly, hydrogen supply is necessary to achieve a significant degree of deoxygenation. In this respect, it is also worth noting that in the absence of hydrogen, acetic acid decomposition is remarkably reduced, as indicated by the low concentration of CO and, especially, methane, which are only detectable over Ni2P/ZSM-5 (see Figure S2). This fact suggests that active centers of nickel phosphide play a role on this last process.
Figure 7.

Conversion and products selectivity respect to guaiacol (left) and acetic acid (right) obtained in blank and catalytic reactions at 260 °C and using an inert atmosphere (2 bar of N2) over Ni2P/ZSM-5 (R7), ZSM-5 (R8), and without a catalyst (Blank 1).
Using Ni2P/ZSM-5 and ZSM-5 materials, selectivities toward C-acylated compounds (acetophenones) obtained under an inert atmosphere are low but in the same range (≤5%) than those obtained using different H2 pressures, corroborating that its formation is not influenced by the presence of hydrogen. In these latter experiments, the selectivity toward these products is slightly higher over the raw zeolite than over the bifunctional catalytic system (5 vs 3%), suggesting that this reaction is catalyzed by the acid sites of the zeolitic support, which are partially blocked by the Ni2P nanoparticles during their impregnation, as discussed in section 2.1. Also, an increase in methyl acetate production is observed when ZSM-5 is used, denoting that demethoxylation of guaiacol can be promoted by acid sites of the zeolite.
2.2.2. Influence of the Temperature on the Catalytic Hydrodeoxygenation of Guaiacol/Acetic Acid Blends
In order to determine the effect of temperature on the different transformation routes of the guaiacol and acetic acid blends (see scheme of Figure 5), additional catalytic assays were performed over Ni2P/ZSM-5 at 40 bar of H2 during 120 min at temperatures ranging between 260 and 300 °C. The variation of the conversion and the selectivity for both guaiacol and acetic acid as a function of temperature are plotted in Figure 8. This graph show that the conversion of guaiacol increases almost linearly with temperature, reaching 60% at 300 °C, whereas product distribution changes remarkably. Guaiacol acetate contribution drops with increasing temperature to reach only 5% at 300 °C, and at the same time, the selectivity toward cyclohexane progressively decreases, being only 7% at 300 °C as compared to 12% at 240 °C. On the contrary, the contribution of catechol and, in a lesser extent, ethylcatechol rises with increasing the operation temperature, while ethylguaiacol concentration is only slightly affected by temperature. Although, the increment of HDO activity at higher temperatures could be expected, similarly to the present case, an increase in demethoxylation and a decrease in full deoxygenation of anisole with rising the operation temperature have been reported over Ni2P/SiO2.50
Figure 8.

Variation of the conversion and the selectivity with respect to guaiacol (top) and acetic acid (bottom) as a function of the operation temperature obtained in the catalytic hydrotreating reaction of the guaiacol and acetic acid blend at 40 bar of H2 for 120 min: 260 °C (R3), 280 °C (R9), and 300 °C (R10).
In contrast with the behavior of guaiacol, conversion of acetic acid varies slightly with temperature. However, the product distribution presents a clear evolution with a gradual increment of methyl acetate and an almost parallel diminution of ethyl acetate with rising the temperature. Ethanol production increases moderately with temperature, while the proportion of phenolic derivatives slightly increases. This is due to the fact that the lower production of guaiacol acetate is compensated at a high temperature by the formation of ethylated aromatics, as it is observed in the corresponding product distribution of guaiacol.
In summary, differentiated reaction processes occur in parallel during the hydrotreating of guaiacol and acetic acid blends using a Ni2P/ZSM-5 bifunctional catalytic system, giving rise to a complex reaction network, as it is unraveled in this work. Along with the direct HDO of guaiacol and acetic acid, esterification and acylation reactions take place simultaneously, leading to the formation of molecules with larger chain length and lower oxygen content. Although esterification has a limited interest for the production of valuable molecules, the C–C bond formation between both substrates is particularly relevant because it allows an efficient deoxygenation without significant carbon losses, stabilizing the reaction media by means of acetic acid neutralization. In contrast, deep deoxygenation of these binary mixtures to render pure alkanes is more challenging, and lower HDO yields are obtained with these binary blends than in the case of the tests performed with single components. As expected, decreasing the H2 pressure is detrimental for the formation of alkanes, but remarkably, partial deoxygenation to produce oxygenated aromatics such as anisole or ethyl guaiacol is still important. These general trends observed in the treatment of guaiacol and acetic acid blends over Ni2P/ZSM-5 catalysts are accentuated with increasing temperature because these more severe operation conditions are detrimental for the total deoxygenation, as higher yields of chemicals such ethyl catechol and ethylguaiacol are obtained at 300 °C. This route of acylation and selective hydrogenation is highly promising to avoid carbon losses in the gas phase, and accordingly, developing specific catalysts for promoting those routes of transformation can have a positive impact for bio-oil upgrading.
3. Conclusions
This work explores the molecular interactions produced during the hydrodeoxygenation of mixtures of two typical components of pyrolysis bio-oil: guaiacol, a representative monomer of lignin depolymerization, and acetic acid, the most abundant carboxylic acid present in these bio-oils. The catalytic system selected for this study, a bifunctional catalyst based on Ni2P loaded on a commercial nanocrystalline ZSM-5 zeolite, is quite active for the HDO of pure substrates, especially for the conversion of the phenolic derivative. However, when acetic acid and guaiacol blends are processed, the HDO efficiency is reduced, especially for guaiacol conversion because of the competence and preferential adsorption of acetic acid over the catalytically active centers. In exchange, several potentially interesting intermolecular interactions were also observed using the Ni2P/ZSM-5 catalyst during the simultaneous hydroprocessing of both substrates:
-
1.
Esterification reaction between acetic acid and guaiacol produces mainly guaiacol acetate, which is self-catalyzed by acetic acid. Although the formation of this ester can contribute to stabilize the bio-oils, this route is of limited practical interest because of the reversibility of the process and the reduced reactivity in HDO conditions of guaiacol acetate.
-
2.
Direct C-acylations, leading to the formation of acetophenones (mainly apocynin), which are high-value fine chemicals, with potential applications in cosmetic or pharmaceutical industries.
-
3.
Formation of guaiacol ethylated derivatives by the hydrodeoxygenation of the acetophenones previously produced. In particular, ethyl guaiacol is readily formed under mild operation conditions, and its yield increases moderately with temperature.
In contrast with the formation of the final products of the direct HDO (cyclohexane and ethane), these cross-reactions are not so sensitive to the hydrogen availability. In fact, esterification is the major process in the absence of hydrogen, and although the formation of ethyl guaiacol from apocynin requires hydrogen, selectivities do not change dramatically when H2 pressure varies in the 10 to 40 bar range. However, it is important to note that the overall conversion decreases when reducing the hydrogen supply, and therefore the yield of these products is lower at reduced H2 pressure. Furthermore, these tests indicate that hydrogen transfer from acetic acid is not significant under the present conditions. On the other hand, the increase in the temperature facilitates demethoxylation reactions, leading to a larger proportion of catechol and related compounds, while the production of full deoxygenated molecules such as cyclohexane decreases appreciably.
These results, obtained with this simple blend, illustrate the magnitude of the challenges faced when dealing with real bio-oils, which contain hundreds of different compounds. Using Ni2P/ZSM-5 as a catalyst may result in a relatively poor HDO activity due to the partial blocking of the active center by acetic acid. Accordingly, if the goal is the production of hydrocarbons, it may be advisable to perform an initial step of ketonization of the bio-oils to remove the carboxylic acids before the HDO step. However, it also worth noting that the occurrence of reactions between phenolic components and carboxylic acids found in the pyrolysis bio-oils during hydroprocessing can be also beneficial if the most promising routes can be promoted. In this respect, increasing the chain length eventually can lead to the formation of hydrocarbons in the gasoline range such as ethyl cyclohexane, although the total yield of hydrocarbons is lower for these binary blends. Overall, the above described molecular interactions can contribute to achieve a higher stability and quality of the upgraded bio-oil, with lower hydrogen consumption than in a pure hydrodeoxygenation process.
4. Experimental Section
4.1. Preparation of Ni2P/ZSM-5 Bifunctional Catalyst
The catalyst was prepared by impregnation process of a commercial ZSM-5 zeolite with a total loading of 10 wt % of nickel using an aqueous solution of Ni(NO3)2·6H2O (Sigma-Aldrich) and (NH4)2HPO4 (Sigma-Aldrich) and a Ni/P molar ratio of 1 in order to favor the formation of the Ni2P metal-rich active phase.51 The impregnated solid was dried overnight at room temperature and then at 120 °C for 24 h and subsequently calcined at 500 °C for 5 h under static air conditions. Afterward, the phosphate catalyst obtained was reduced at 650 °C in a tubular furnace for 3 h in a hydrogen flow (80 mL·min–1) and finally passivated by progressive introduction of an air flow at room temperature.12 The zeolitic support is a commercial nanocrystalline ZSM-5 kindly supplied by Clariant with [Si/Al]MOL = 42 and SBET = 452 m2/g.
4.2. Catalyst Characterization
XRD patterns corresponding to the zeolitic support and the Ni2P loaded catalyst were recorded with a Philips PW 3040/00 X’Pert diffractometer using Cu Kα radiation at 45 kV and 40 mA. Ni, P, and Al contents were determined by means of ICP-OES analyses carried out on a Perkin Elmer Optima 7300 AD instrument after acidic digestion of the catalyst. TEM images of these materials were acquired using a PHILIPS TECNAI 20T microscope working at 200 kV. The Ni2P particle size distribution was estimated with Image J software from TEM images taken over the supported catalytic system. Textural properties of both parent zeolite and bifunctional catalyst were determined from Ar adsorption–desorption isotherms at 87 K measured on a Quantachrome AUTOSORB iQ system. The surface area was calculated using the BET (Brunauer–Emmett–Teller) equation, while surface values corresponding to both the microporous system and external surface were estimated using the nonlocal density functional theory (NL-DFT) method. The total pore volume was determined at a relative pressure of 0.98. The acidity of the samples was evaluated by temperature-programmed desorption of ammonia (NH3-TPD) employing a Micromeritics AUTOCHEM 2910 analyzer. These assays were performed using a quartz microreactor, heating the sample from room temperature up to 600 °C for 1 h with an inert gas. Then, the material was cooled down to 100 °C and saturated with a 10 vol % NH3/He mixture, removing the physisorbed ammonia by an inert gas flow. Finally, the temperature was linearly increased up to 550 °C with a heating ramp of 10 °C/min. The desorbed ammonia was monitored by a thermal conductivity detector (TCD) detector.
4.3. Catalytic Activity Tests
Catalytic assays were performed in a 100 mL stainless steel high-pressure stirred batch reactor using different reaction mixtures. First, hydrotreating tests of the individual compounds were carried out at 260 °C and 40 bar of hydrogen using the following feeds: (i) 3.3 wt % guaiacol in decalin and (ii) 8 wt % of acetic acid in decalin. Then, a mixture of 3.3 wt % guaiacol and 8 wt % of acetic acid in decalin [(guaiacol/acetic acid)MOL of 0.2] was tested at different temperatures (260, 280, and 300 °C) and hydrogen pressures (10, 20, 30, and 40 bar of H2, with ∼37 bar as the stoichiometric amount of H2 required for the oxygen removal without C–C bond saturation). In addition, several catalytic tests were performed under an inert atmosphere (2 bar of N2) at 260 °C. Likewise, hydrodeoxygenation reaction tests using both guaiacol acetate and apocynin as substrates (3.3 wt % in decalin) were also performed as a reference since they were detected as reaction intermediates. Table 1 summarizes all the catalytic experiments performed in this work and the reaction conditions employed.
In a typical experiment, 50 mL of the corresponding feed was loaded into the reactor together with 150 mg of catalyst. After that, the system was purged three times with 3 bar of N2 and then pressurized until the corresponding reaction pressure (H2 o N2) at room temperature. Subsequently, the reactor was heated up to the selected reaction temperature (260–300 °C) and kept at that temperature along the reaction time under vigorous stirring (1000 rpm) for 2 h. In addition, the time evolution of the catalytic activity was studied by carrying out assays at 30, 60, 90, and 120 min at 260 °C and 40 bar of H2. After that, the reactor was cooled down with a water/ice mixture. Reagents and products were analyzed by a precalibrated gas chromatograph (Agilent, 7890A) using an FID (flame ionization detector) detector and an HP-INNOWAX column and by a GC–MS (Bruker) provided with a BP-5 column. Response factors obtained after calibration with commercial standards of the main components are collected in Table S1. Gas products were collected in a sampling bag and analyzed in a dual-channel Agilent CP-4900 Micro Gas Chromatograph equipped with a TCD detector.
The catalytic activity was evaluated in terms of substrate conversion, XSUBS (%), and product selectivity toward i product, Si (%), according to the following equations
where n0 and n are the initial and final moles of substrate in the mixture, respectively, and ni represent the moles of product i in the final mixture. In the reactions performed using guaiacol and acetic acid blends as feeds, the selectivity values were estimated referred to each individual substrate. The carbon balance of the catalytic experiments considering both the liquid and gas fraction ranged in all cases between 95 and 100 mol %.
Acknowledgments
The authors thank the Spanish “Ministry of Economy and Competiveness” for their financial support through the project CATPLASBIO (CTQ2014-60209-R) and the “Regional Government of Madrid” and European Structural Funds for the RESTOENE2 (S2013/MAE-2882) project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03221.
Response factors, NH3-TPD, gas production, guaiacol acetate HDO reaction, apocynin HDO reaction, and Blank 2 reaction (PDF)
Author Present Address
§ Present address: Instituto de Catálisis y Petroleoquímica, CSIC, c/ Marie Curie, 2, Cantoblanco, Madrid, E28049, Spain.
The authors declare no competing financial interest.
Supplementary Material
References
- Rogers K. A.; Zheng Y. Selective Deoxygenation of Biomass-Derived Bio-Oils within Hydrogen-Modest Environments: A Review and New Insights. ChemSusChem 2016, 9, 1750–1772. 10.1002/cssc.201600144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K.; Maheria K. C.; Kumar Dalai A. Bio-Oil Valorization: A Review. Renewable Sustainable Energy Rev. 2013, 23, 91–106. 10.1016/j.rser.2013.02.036. [DOI] [Google Scholar]
- Bridgwater A. V. Review of Fast Pyrolysis of Biomass and Product Upgrading. Biomass Bioenergy 2012, 38, 68–94. 10.1016/j.biombioe.2011.01.048. [DOI] [Google Scholar]
- Patel M.; Kumar A. Production of Renewable Diesel through the Hydroprocessing of Lignocellulosic Biomass-Derived Bio-Oil: A Review. Renewable Sustainable Energy Rev. 2016, 58, 1293–1307. 10.1016/j.rser.2015.12.146. [DOI] [Google Scholar]
- Furimsky E. Hydroprocessing Challenges in Biofuels Production. Catal. Today 2013, 217, 13–56. 10.1016/j.cattod.2012.11.008. [DOI] [Google Scholar]
- Nie L.; de Souza P. M.; Noronha F. B.; An W.; Sooknoi T.; Resasco D. E. Selective Conversion of M-Cresol to Toluene over Bimetallic Ni–Fe Catalysts. J. Mol. Catal. A: Chem. 2014, 388-389, 47–55. 10.1016/j.molcata.2013.09.029. [DOI] [Google Scholar]
- Chen C.; Chen G.; Yang F.; Wang H.; Han J.; Ge Q.; Zhu X. Vapor Phase Hydrodeoxygenation and Hydrogenation of M -Cresol on Silica Supported Ni, Pd and Pt Catalysts. Chem. Eng. Sci. 2015, 135, 145–154. 10.1016/j.ces.2015.04.054. [DOI] [Google Scholar]
- Lu Q.; Zhang Y.; Tang Z.; Li W.; Zhu X. Catalytic Upgrading of Biomass Fast Pyrolysis Vapors with Titania and Zirconia/titania Based Catalysts. Fuel 2010, 89, 2096–2103. 10.1016/j.fuel.2010.02.030. [DOI] [Google Scholar]
- Shi H.; Chen J.; Yang Y.; Tian S. Catalytic Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Nickel Phosphide Catalysts: Remarkable Support Effect. Fuel Process. Technol. 2014, 118, 161–170. 10.1016/j.fuproc.2013.08.010. [DOI] [Google Scholar]
- Hwu H. H.; Chen J. G. Surface Chemistry of Transition Metal Carbides. Chem. Rev. 2005, 105, 185–212. 10.1021/cr0204606. [DOI] [PubMed] [Google Scholar]
- Hargreaves J. S. J. Heterogeneous Catalysis with Metal Nitrides. Coord. Chem. Rev. 2013, 257, 2015–2031. 10.1016/j.ccr.2012.10.005. [DOI] [Google Scholar]
- Berenguer A.; Sankaranarayanan T. M.; Gómez G.; Moreno I.; Coronado J. M.; Pizarro P.; Serrano D. P. Evaluation of Transition Metal Phosphides Supported on Ordered Mesoporous Materials as Catalysts for Phenol Hydrodeoxygenation. Green Chem. 2016, 18, 1938–1951. 10.1039/C5GC02188J. [DOI] [Google Scholar]
- Yang Y.; Ochoa-Hernández C.; Pizarro P.; de la Peña O’Shea V. A.; Coronado J. M.; Serrano D. P. Influence of the Ni/P Ratio and Metal Loading on the Performance of NixPy/SBA-15 Catalysts for the Hydrodeoxygenation of Methyl Oleate. Fuel 2015, 144, 60–70. 10.1016/j.fuel.2014.12.008. [DOI] [Google Scholar]
- Berenguer A.; Bennett J. A.; Hunns J.; Moreno I.; Coronado J. M.; Lee A. F.; Pizarro P.; Wilson K.; Serrano D. P. Catalytic Hydrodeoxygenation of M-Cresol over Ni2P/hierarchical ZSM-5. Catal. Today 2018, 304, 72–79. 10.1016/j.cattod.2017.08.032. [DOI] [Google Scholar]
- Berenguer A.; Gutiérrez-Rubio S.; Linares M.; Ochoa-Hernández C.; Moreno I.; García-Fierro J. L.; Coronado J. M.; Serrano D. P.; Pizarro P. On the Feasibility of Using Hierarchical ZSM-5 and Beta Zeolites as Supports of Metal Phosphides for Catalytic Hydrodeoxygenation of Phenol. Energy Technol. 2019, 7, 1900214. 10.1002/ente.201900214. [DOI] [Google Scholar]
- Song H.; Gong J.; Song H.; Li F. A Novel Surface Modification Approach for Synthesizing Supported Nickel Phosphide Catalysts with High Activity for Hydrodeoxygenation of Benzofuran. Appl. Catal., A 2015, 505, 267–275. 10.1016/j.apcata.2015.08.007. [DOI] [Google Scholar]
- Wu S. K.; Lai P. C.; Lin Y. C. Atmospheric Hydrodeoxygenation of Guaiacol over Nickel Phosphide Catalysts: Effect of Phosphorus Composition. Catal. Lett. 2014, 144, 878–889. 10.1007/s10562-014-1231-7. [DOI] [Google Scholar]
- Ted Oyama S.; Onkawa T.; Takagaki A.; Kikuchi R.; Hosokai S.; Suzuki Y.; Bando K. K. Production of Phenol and Cresol from Guaiacol on Nickel Phosphide Catalysts Supported on Acidic Supports. Top. Catal. 2015, 58, 201–210. 10.1007/s11244-015-0361-5. [DOI] [Google Scholar]
- Zhang Q.; Chang J.; Wang T.; Xu Y. Review of Biomass Pyrolysis Oil Properties and Upgrading Research. Energy Convers. Manage. 2007, 48, 87–92. 10.1016/j.enconman.2006.05.010. [DOI] [Google Scholar]
- Auersvald M.; Shumeiko B.; Staš M.; Kubička D.; Chudoba J.; Šimáček P. Quantitative Study of Straw Bio-Oil Hydrodeoxygenation over a Sulfided NiMo Catalyst. ACS Sustainable Chem. Eng. 2019, 7, 7080–7093. 10.1021/acssuschemeng.8b06860. [DOI] [Google Scholar]
- Wan H.; Chaudhari R. V.; Subramaniam B. Aqueous Phase Hydrogenation of Acetic Acid and Its Promotional Effect on P -Cresol Hydrodeoxygenation. Energy Fuels 2013, 27, 487–493. 10.1021/ef301400c. [DOI] [Google Scholar]
- Sankaranarayanan T. M.; Kreider M.; Berenguer A.; Gutiérrez-Rubio S.; Moreno I.; Pizarro P.; Coronado J. M.; Serrano D. P. Cross-Reactivity of Guaiacol and Propionic Acid Blends during Hydrodeoxygenation over Ni-Supported Catalysts. Fuel 2018, 214, 187–195. 10.1016/j.fuel.2017.10.059. [DOI] [Google Scholar]
- Agblevor F. A.; Jahromi H. Aqueous Phase Synthesis of Hydrocarbons from Reactions of Guaiacol and Low Molecular Weight Oxygenates. ChemCatChem 2018, 10, 5201–5214. 10.1002/cctc.201800982. [DOI] [Google Scholar]
- Chen J.; Cai Q.; Lu L.; Leng F.; Wang S. Upgrading of the Acid-Rich Fraction of Bio-Oil by Catalytic Hydrogenation-Esterification. ACS Sustainable Chem. Eng. 2017, 5, 1073–1081. 10.1021/acssuschemeng.6b02366. [DOI] [Google Scholar]
- Gilkey M. J.; Xu B. Heterogeneous Catalytic Transfer Hydrogenation as an Effective Pathway in Biomass Upgrading. ACS Catal. 2016, 6, 1420–1436. 10.1021/acscatal.5b02171. [DOI] [Google Scholar]
- Demirbas A. The Influence of Temperature on the Yields of Compounds Existing in Bio-Oils Obtained from Biomass Samples via Pyrolysis. Fuel Process. Technol. 2007, 88, 591–597. 10.1016/j.fuproc.2007.01.010. [DOI] [Google Scholar]
- Mortensen P. M.; de Carvalho H. W. P.; Grunwaldt J.-D.; Jensen P. A.; Jensen A. D. Activity and Stability of Mo2C/ZrO2 as Catalyst for Hydrodeoxygenation of Mixtures of Phenol and 1-Octanol. J. Catal. 2015, 328, 208–215. 10.1016/j.jcat.2015.02.002. [DOI] [Google Scholar]
- Kallury R. K. M. R.; Restivo W. M.; Tidwell T. T.; Boocock D. G. B.; Crimi A.; Douglas J. Hydrodeoxygenation of Hydroxy, Methoxy and Methyl Phenols with Molybdenum Oxide/nickel Oxide/alumina Catalyst. J. Catal. 1985, 96, 535–543. 10.1016/0021-9517(85)90321-5. [DOI] [Google Scholar]
- Oasmaa A.; Solantausta Y.; Arpiainen V.; Kuoppala E.; Sipilä K. Fast Pyrolysis Bio-Oils from Wood and Agricultural Residues. Energy Fuels 2010, 24, 1380–1388. 10.1021/ef901107f. [DOI] [Google Scholar]
- Resasco D. E.; Crossley S. P. Implementation of Concepts Derived from Model Compound Studies in the Separation and Conversion of Bio-Oil to Fuel. Catal. Today 2015, 257, 185–199. 10.1016/j.cattod.2014.06.037. [DOI] [Google Scholar]
- Ciddor L.; Bennett J. A.; Hunns J. A.; Wilson K.; Lee A. F. Catalytic Upgrading of Bio-Oils by Esterification. J. Chem. Technol. Biotechnol. 2015, 90, 780–795. 10.1002/jctb.4662. [DOI] [Google Scholar]
- Duong N. N.; Wang B.; Sooknoi T.; Crossley S. P.; Resasco D. E. Enhancing the Acylation Activity of Acetic Acid by Formation of an Intermediate Aromatic Ester. ChemSusChem 2017, 10, 2823–2832. 10.1002/cssc.201700394. [DOI] [PubMed] [Google Scholar]
- Herron J. A.; Vann T.; Duong N.; Resasco D. E.; Crossley S.; Lobban L. L.; Maravelias C. T. A Systems-Level Roadmap for Biomass Thermal Fractionation and Catalytic Upgrading Strategies. Energy Technol. 2017, 5, 130–150. 10.1002/ente.201600147. [DOI] [Google Scholar]
- Vriamont C. E. J. J.; Chen T.; Romain C.; Corbett P.; Manageracharath P.; Peet J.; Conifer C. M.; Hallett J. P.; Britovsek G. J. P. From Lignin to Chemicals: Hydrogenation of Lignin Models and Mechanistic Insights into Hydrodeoxygenation via Low-Temperature C–O Bond Cleavage. ACS Catal. 2019, 9, 2345–2354. 10.1021/acscatal.8b04714. [DOI] [Google Scholar]
- Koike N.; Hosokai S.; Takagaki A.; Nishimura S.; Kikuchi R.; Ebitani K.; Suzuki Y.; Oyama S. T. Upgrading of Pyrolysis Bio-Oil Using Nickel Phosphide Catalysts. J. Catal. 2016, 333, 115–126. 10.1016/j.jcat.2015.10.022. [DOI] [Google Scholar]
- Serrano D. P.; Aguado J.; Escola J. M.; Rodríguez J. M.; Peral Á. Hierarchical Zeolites with Enhanced Textural and Catalytic Properties Synthesized from Organofunctionalized Seeds. Chem. Mater. 2006, 18, 2462–2464. 10.1021/cm060080r. [DOI] [Google Scholar]
- Lee Y.; Oyama S. Bifunctional Nature of a SiO2-Supported Ni2P Catalyst for Hydrotreating: EXAFS and FTIR Studies. J. Catal. 2006, 239, 376–389. 10.1016/j.jcat.2005.12.029. [DOI] [Google Scholar]
- Zhang L.; Fu W.; Yu Q.; Tang T.; Zhao Y.; Zhao H.; Li Y. Ni2P Clusters on Zeolite Nanosheet Assemblies with High Activity and Good Stability in the Hydrodesulfurization of 4,6-Dimethyldibenzothiophene. J. Catal. 2016, 338, 210–221. 10.1016/j.jcat.2016.02.029. [DOI] [Google Scholar]
- Kumar A.; Phadke S.; Bhan A. Acetic Acid Hydrodeoxygenation on Molybdenum Carbide Catalysts. Catal. Sci. Technol. 2018, 8, 2938–2953. 10.1039/C8CY00358K. [DOI] [Google Scholar]
- Joshi N.; Lawal A. Hydrodeoxygenation of Acetic Acid in a Microreactor. Chem. Eng. Sci. 2012, 84, 761–771. 10.1016/j.ces.2012.09.018. [DOI] [Google Scholar]
- Ying X.; Tiejun W.; Longlong M.; Guanyi C. Upgrading of Fast Pyrolysis Liquid Fuel from Biomass over Ru/γ-Al2O3 Catalyst. Energy Convers. Manage. 2012, 55, 172–177. 10.1016/j.enconman.2011.10.016. [DOI] [Google Scholar]
- Onyestyák G.; Novodárszki G.; Wellisch Á. F.; Kalló D.; Thakur A. J.; Deka D. Co and Ni Catalysts Loaded on Typical Well-Ordered Micro- and Mesoporous Supports for Acetic Acid Reduction. React. Kinet., Mech. Catal. 2017, 121, 109–119. 10.1007/s11144-016-1134-2. [DOI] [Google Scholar]
- Shangguan J.; Olarte M. V.; Chin Y.-H. C. (Cathy). Mechanistic Insights on C O and C C Bond Activation and Hydrogen Insertion during Acetic Acid Hydrogenation Catalyzed by Ruthenium Clusters in Aqueous Medium. J. Catal. 2016, 340, 107–121. 10.1016/j.jcat.2016.04.024. [DOI] [Google Scholar]
- Milina M.; Mitchell S.; Pérez-Ramírez J. Prospectives for Bio-Oil Upgrading via Esterification over Zeolite Catalysts. Catal. Today 2014, 235, 176–183. 10.1016/j.cattod.2014.02.047. [DOI] [Google Scholar]
- Alexander M. R.; Beamson G.; Blomfield C. J.; Leggett G.; Duc T. M. Interaction of Carboxylic Acids with the Oxyhydroxide Surface of Aluminium: Poly(acrylic Acid), Acetic Acid and Propionic Acid on Pseudoboehmite. J. Electron Spectrosc. Relat. Phenom. 2001, 121, 19–32. 10.1016/S0368-2048(01)00324-3. [DOI] [Google Scholar]
- Martin R. Uses of the Fries Rearrangement for the Preparation of Hydroxyarylketones. A Review. Org. Prep. Proced. Int. 1992, 24, 369–435. 10.1080/00304949209356226. [DOI] [Google Scholar]
- González C.; Marín P.; Díez F. V.; Ordóñez S. Hydrodeoxygenation of Acetophenone over Supported Precious Metal Catalysts at Mild Conditions: Process Optimization and Reaction Kinetics. Energy Fuels 2015, 29, 8208–8215. 10.1021/acs.energyfuels.5b02112. [DOI] [Google Scholar]
- Peters E. A.; Hiltermann J. T. N.; Stolk J. Effect of Apocynin on Ozone-Induced Airway Hyperresponsiveness to Methacholine in Asthmatics. Free Radical Biol. Med. 2001, 31, 1442–1447. 10.1016/S0891-5849(01)00725-0. [DOI] [PubMed] [Google Scholar]
- Zhao H. Y.; Li D.; Bui P.; Oyama S. T. Hydrodeoxygenation of Guaiacol as Model Compound for Pyrolysis Oil on Transition Metal Phosphide Hydroprocessing Catalysts. Appl. Catal., A 2011, 391, 305–310. 10.1016/j.apcata.2010.07.039. [DOI] [Google Scholar]
- Li Y.; Fu J.; Chen B. Highly Selective Hydrodeoxygenation of Anisole, Phenol and Guaiacol to Benzene over Nickel Phosphide. RSC Adv. 2017, 7, 15272–15277. 10.1039/C7RA00989E. [DOI] [Google Scholar]
- Oyama S. Effect of Phosphorus Content in Nickel Phosphide Catalysts Studied by XAFS and Other Techniques. J. Catal. 2002, 210, 207–217. 10.1006/jcat.2002.3681. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.