

Abstract

A novel macrocyclic construct consisting of a pyrazolopyrimidine scaffold concatenated to a benzene ring through two triazoles has been developed to investigate uncharted chemical space with bioactive potential. The 18-atom macrocycle was assembled via a double copper-catalyzed alkyne–azide cycloaddition (CuAAC) reaction between 1,3-bis(azidomethyl)benzene and a bis-propargylated pyrazolo[3,4-d]pyrimidine core. The resulting macrocycle was functionalized further into a multicyclic analog that displays selective inhibitory activity against the receptor tyrosine kinase AXL.
Introduction
Molecules featuring a ring of 12 or more atoms are formally known as macrocycles. Enabled by their unique structural characteristics, different macrocyclic compounds, originating from natural sources, semisynthesis, or purely synthetic methods, have been shown to display remarkable medicinal properties.1−3 For instance, due to the restriction of their conformational freedom, certain macrocyclic structures possess an enhanced binding selectivity to specific biological targets.4 Rapamycin and its derivatives, so-called rapalogs, represent a classical example of highly selective macrocyclic inhibitors. They have the capacity to selectively bind and block the kinase activity of the multiprotein complex mTORC1 without affecting the parented mTORC2.5 Additionally, macrocyclization has become an essential method to reduce polarity and enhance the proteolytic stability of biologically active peptides.1 Attracted by these benefits, medicinal chemists have recently incorporated the design of macrocyclic rings from nitrogen-rich heterocyclic cores into their toolbox of lead optimization approaches.1−4,6,7 As a result, several macrocyclization strategies reported in the literature have resulted in potent and selective kinase inhibitors,8−11 including the recently approved antineoplastic drug lorlatinib.12
Encouraged by the bioactive properties of the pyrazolopyrimidine derivative eSM119,13 a compound that displays inhibitory activity against three oncogenic receptor tyrosine kinases (AXL, FLT3, and RET, with IC50 values of 0.40, 0.53, and 0.60 μM, respectively), the development of an 18-atom macrocyclic derivative of this compound was investigated (see Figure 1).
Figure 1.

Structure and synthesis/retrosynthetic strategy of eSM119 (left) and macrocycle 1 (right). The novel compound 1 was designed by the incorporation of a second triazole group to enclose the cyclophene ring.
Results and Discussion
A nonsymmetrical dimer-macrocyclization strategy based on a double-copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) reaction was applied for the construction of macrocycle 1 from commercially available 6-chloro-1H-pyrazolo[3,4-d]pyrimidine, 2. The full synthetic route is described in Scheme 1 and the Experimental Section. Briefly, iodination at the C3 position was carried out with N-iodosuccinimide (76%), followed by alkylation with propargyl bromide (46%) to incorporate the first alkyne group. Subsequently, the chloro atom at the C6 position of 4 was substituted via a SNAr reaction14 with propargylamine in tetrahydrofuran (THF) to introduce a second alkyne handle on the pyrazolo[3,4-d]pyrimidine core, giving rise to compound 5 in good yield (80%). The assembly of dialkyne 5 with 1,3-bis(azidomethyl)benzene 6 was achieved using copper iodide, sodium ascorbate, and triethylamine15 to give rise to macrocycle 7 in 23% yield. Since the modest reaction yield obtained was sufficient to progress to the next step of the route, reaction conditions were not optimized further. The synthesis of the macrocyclic analogue of eSM119, that is, compound 1, was completed by the palladium-catalyzed cross-coupling of 7 with 6-(4-Boc-piperazinyl)pyridyl-3-boronic acid pinacol ester, 8, followed by Boc deprotection under acidic conditions (51% yield for two steps).
Scheme 1. Synthesis of the 18-Atom Macrocycle 1 from 6-Chloro-1H-pyrazolo[3,4-d]pyrimidine, 2.

NMR studies of compounds 7, 9, and 1 provided insights into the conformational dynamics of this novel macrocyclic construct. An extremely broad doublet was observed from 4 to 5.2 ppm in the spectra of the three compounds (Figure S1 of the Supporting Information, highlighted by a frame). This flat-looking signal, which was identified as the methylene group bonded to the amino group at the C6 position, is indicative of numerous conformers coexisting at the ambient temperature.16 These conformers can present the methylene protons oriented toward slightly different chemical surroundings, thus inducing different shieldings and consequently different chemical shifts. The presence of multiple conformers is in agreement with in silico calculations obtained with MacroModel,17 which yielded 155 conformers that can be classified into four main groups (see Figure 2 and Supporting. Information).
Figure 2.
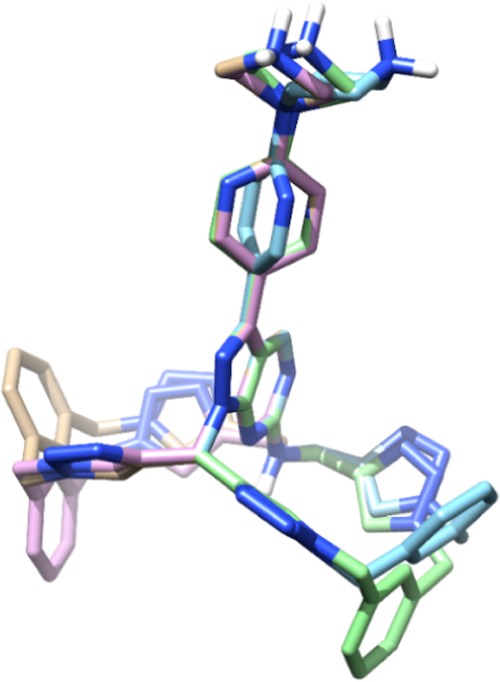
Representative three-dimensional (3D) structure models (superimposed through the pyrazolopyrimidine ring) of four of the most significant types of conformers found for 1 after macrocyclic sampling with MacroModel.
Next, 1H NMR analysis of 1 was performed over a range of temperatures (27, 40, 60, and 80 °C), aiming to overcome the rotational energy barrier and increase the rate of exchange between the conformers. As shown in Figure 3A, the NMR spectra show the coalescence of the signals as the temperature increases, eventually merging into a single averaged peak at 4.64 ppm that integrates for two protons. Given the moderate temperature required for conformer interconversions (single peak starts to form at 40 °C), it is expected that under physiological conditions, compound 1 could exchange between conformers to potentially find energetically favorable conformations that increase cell permeability and/or binding to target proteins in an effective manner.
Figure 3.

(A) 1H NMR spectra of 1 at different temperatures. (B, C) two-dimensional (2D) HSQC analysis of 1 at 27 and 80 °C.
The methylene peak annotation was corroborated by a 2D heteronuclear single-quantum correlation (HSQC) analysis (Figure 3B,C). Although no carbon–proton coupling was seen at 27 °C, a strong correlation between the carbon peak at 36.7 ppm of the 13C NMR spectrum and the proton signal at 4.64 ppm was clearly observed when the experiment was performed at 80 °C.
Given the structural and conformational differences between macrocycle 1 and its open analogue eSM119 (see Figure 1), the biological properties of 1 were expected to differ from those of eSM119. To evaluate this, biochemical assays were performed to test the capacity of 1 to inhibit the enzymatic activity of a selection of closely related protein kinases: TAM family members TYRO3, AXL, and MER and receptor tyrosine kinases RET and FLT3. This focused panel of kinases was selected based on the reported selectivity profile of eSM119.13 Enzymatic studies (based on 33P incorporation) were performed by Reaction Biology Corp. using a 10-point semi-log dilution series.18 The calculated IC50 values are compiled in Table 1.
Table 1. IC50 Values (in μM) for 1 and eSM119 against a Selection of Recombinant Tyrosine kinases.
Member of the TAM subfamily.
The study revealed that compound 1 moderately inhibits AXL kinase with an IC50 value of 13 μM. Notably, it did not elicit the inhibition of TYRO3, MER, and RET (estimated IC50 > 1 mM) and exhibited an IC50 value of 53.5 μM for FLT3, a potency over 4-fold lower than that against AXL. The inhibitory properties of macrocycle 1 clearly contrast with the similar levels of activity displayed by eSM119 for AXL and kinases FLT3 and RET (1.3- and 1.5-fold differences, respectively). Thus, the large difference in activity displayed by macrocycle 1 against AXL and RET, with IC50 values differing by >75-fold, is particularly relevant. The improved selectivity profile of 1 is important because, despite many efforts, the development of selective AXL inhibitors has proved to be extremely challenging, with no selective inhibitors of AXL kinase activity reported to date.19 This includes bemcentinib, an orally available kinase inhibitor in clinical development that is marketed as an AXL-selective inhibitor,19 although it has been shown by several groups to equally inhibit FLT3 and other kinases.13,20,21 To the best of our knowledge, compound 1 (eOC148) is the first one to display distinctly superior activity for AXL compared to its most closely related kinases MER, RET, and FLT3. However, it is also important to note that the herein reported macrocyclization strategy negatively impacted the potency of the analog to inhibit AXL relative to the open conformation (32.5-fold reduction in potency), indicating that there is still room for improvement.
To shed light on the preferential inhibition of AXL over RET, docking studies were conducted using the available crystal structures of the proteins (PDB entries: 5U6B(22) and 2IVV,23 respectively). Computational strategy and analyses are described in full in the Supporting Information. The 155 conformational structures of 1 yielded by MacroModel sampling were docked at the active site of the AXL kinase domain using Autodock 4.2.6.24 As shown in Figure 4A, the predicted binding pose of compound 1 displays the piperazino-pyridinyl branch oriented toward a negatively charged region (in red) corresponding to the glycine-rich G-loop of the N-lobe (see Supporting Information). The positively charged piperazine (protonated at physiological pH) forms a H-bond with Gly545 in one of the most flexible parts of the N-lobe, responsible for positioning the γ-phosphate of ATP for catalysis (Supporting Information, Figure S4). Moreover, its newly incorporated triazole ring faces the Asp690 residue of the DFG motif and the catalytically essential Lys567, with which it forms two H-bonds. Together with the H-bond formed by the piperazine ring with the backbone chain of the G-loop, these interactions support the capacity of compound 1 to inhibit AXL kinase activity.
Figure 4.

(A) Columbic surface representation of the AXL active site with the predicted binding pose of 1 (light yellow). Red and blue areas represent negatively and positively polarized areas, respectively. (B) Antiproliferative activity of compound 1 (30 μM) against a panel of cancer cell lines. The cell viability was analyzed with PrestoBlue at day 5. Error bars: ± standard deviation (SD) from n = 2.
Analogous docking studies were also performed with the crystal structure of RET kinase (PDB entry: 2IVV).23 The predicted binding pose of 1 is orientated in a fashion similar to that in AXL; however, the formation of H-bonds with Lys567 are precluded due to a more tightened disposition of the ATP-binding cleft that forces the ligand to move slightly away from the Lys567 residue. This pose indicates a lower binding affinity, thus providing the rationale for the preferential binding of compound 1 to AXL kinase over RET.
Finally, the antiproliferative properties of 1 were tested across a panel of eight cell lines: breast cancer MDA-MB-231, ZR-75-1, and MCF7, glioma U87, colorectal cancer HCT116, lung adenocarcinoma A549, and leukemia MOLM-13 and MV4–11 cells. Cells were treated with compound 1 at 30 μM for 5 days, and the cell viability was determined using the PrestoBlue reagent. As shown in Figure 4B, compound 1 displayed antiproliferative activity against MV4–11 cells, but not against the other cancer cell lines. This is important because MV4–11 is an acute myeloid leukemia cell line that has shown to be dependent on AXL signaling for survival.25 To confirm this, MV4–11 cells were treated with macrocycle 1 over a range of concentrations (0.3–30 μM). The resulting dose–response curve (see Supporting Information, Figure S7) provided an half- maximal effective concentration (EC50) value of 16.6 μM. The fact that compound 1 displays anticancer activity against a cell line that is reliant on AXL activity for proliferation but not in non-AXL-dependent cancer cell lines further highlights the selectivity profile of this novel compound. The similar potency of 1 against the recombinant AXL protein (IC50 = 13 μM) and in MV4–11 cells (EC50 = 16.6 μM) indicates that this macrocyclic construct possesses a high capacity to cross the cell membrane.
Conclusions
In conclusion, the synthesis and characterization of a novel 18-atom macrocyclic structure comprising four concatenated rings is herein reported. The construct was readily assembled using a nonsymmetrical dimer macrocyclization strategy based on a double CuAAC reaction. The macrocycle, which has been built from a pyrazolopyrimidine scaffold, was designed to create a conformationally constrained analog of the multikinase inhibitor eSM119 and displayed moderate while selective inhibition of AXL kinase, offering a new optimization pathway to block the activity of this cancer-associated protein kinase with superior selectivity. Importantly, the reported synthetic method provides a practical route for the combinatorial generation of libraries of 18-atom macrocycles by varying the benzene ring, the 6:5 fused heterocyclic core, and their substituents, to explore additional chemical space with pharmacological potential. The docking studies also suggest that the on-target potency could be improved by locking the structure at the right conformation. Future efforts will be focused on optimizing the structure substituents and the macrocyclic ring size and conformational freedom, for example, by switching the benzene ring to 5-membered heterocycles or making homologs7 by eliminating one or more of the macrocyclic CH2 groups.
Experimental Section
General Information
Chemicals were purchased from Sigma-Aldrich or Fisher. NMR spectra were recorded at room temperature (rt) on a 500 MHz Bruker Avance III spectrometer, unless otherwise stated. Chemical shifts are reported in ppm relative to the solvent peak. MS were obtained in a Bruker MicroTOF II. Analytical thin-layer chromatography (TLC) was performed using TLC Silica gel 60 F254 plates and visualized under a UV lamp. Purifications were carried out by flash chromatography using a silica gel 220–440 mesh (Sigma-Aldrich). Compounds used in the biological experiments were >95% pure, as measured by high-performance liquid chromatography (HPLC) using an UV–vis 254 nm detector. Method: eluent A: water and trifluoroacetic acid (0.4%); eluent B: acetonitrile; A/B = 95:5 to 20:80 in 6 min, isocratic 1 min, 20:80 to 95:5 in 0.1 min, and isocratic 2 min. Stock solutions (100 mM) were prepared in dimethylsulphoxide (DMSO).
Synthesis of 6-Chloro-3-iodo-1-propargyl-1H-pyrazolo[3,4-d]pyrimidine (4)
Compound 4 was synthesized by the iodination of compound 2 into 6-chloro-3-iodo-1H-pyrazolo[3,4-d]pyrimidine (3), as previously described.11 Subsequently, to a suspension of 3 (560 mg, 2 mmol) in anhydrous dimethylformamide (DMF) (10 mL) at 0 °C under an argon atmosphere was added sodium hydride 60% in mineral oil in small amounts (120 mg, 3 mmol), followed by the dropwise addition of a solution of propargyl bromide in toluene (80% w/w in toluene, 0.32 mL, 3 mmol). The mixture was heated under microwave irradiation at 150 °C for 1.5 h. The solvents were then evaporated, and the residue was redissolved with a mixture of EtOAc (30 mL) and water (15 mL). The aqueous layer was extracted twice with EtOAc (15 mL), and the combined organic layers were dried with anhydrous Na2SO4, filtered, and evaporated. The crude extract was purified by flash chromatography using DCM as the eluent to afford compound 4(13) as a white solid (293 mg, 46%). 1H NMR (500 MHz, CDCl3) δ 8.80 (s, 1H), 5.26 (d, J = 2.5, 2H), 2.44 (t, J = 2.5, 1H).
Synthesis of 3-Iodo-N,1-dipropargyl-1H-pyrazolo[3,4-d]pyrimidine-6-amine (5)
To a solution of 4 (800 mg, 2.5 mmol) in THF (15 mL) was added propargylamine (0.72 mL, 11.32 mmol) dropwise. The mixture was heated at 120 °C under microwave irradiation for 1 h. The solvent was evaporated, and the crude extract was purified by recrystallization from MeOH to afford compound 5 as a beige solid (600 mg, 80%). 1H NMR (500 MHz, DMSO-d6) δ 8.56 (m, 1H), 8.16 (bs, 1H), 5.06 (s, 2H), 4.17 (dd, J = 6.0, 2.5, 2H), 3.38 (t, J = 2.5, 1H), 3.06 (s, 1H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 161.6, 155.1, 154.8, 112.8, 94.1, 81.9, 78.7, 76.0, 73.1, 35.9, 30.8. high-resolution mass spectrometry (HRMS) [electrospray ionization-time of flight (ESI-TOF)] m/z: [M + H]+ Calcd for C11H9IN5 337.9897; Found 337.9890.
Synthesis of 1,3-Bis(azidomethyl)benzene (6)
Compound 6 was synthesized as previously described.26
Synthesis of 13-Iodo-11H,31H,71H-9-aza-1(1,6)-pyrazolo[3,4-d]pyrimidine-3(4,1),7(1,4)-ditriazole-5(1,3)-benzenecyclononaphane (7)
To a solution of 5 (337 mg, 1 mmol) in dioxane/H2O (20/3 mL) was added a solution of 6 (188 mg, 1 mmol), CuI (66 mg, 0.4 mmol), sodium ascorbate (40 mg, 0. 2 mmol), and triethylamine (TEA) (0.11 mL, 0.8 mmol) in dioxane (1 mL). The mixture was stirred at room temperature for 36 h. The solvent was evaporated, and the crude extract was purified by flash chromatography using DCM/MeOH 10/0.4 as the eluent to afford a white solid (120 mg, 23%). 1H NMR (500 MHz, DMSO-d6) δ 8.52 (s, 1H), 8.34 (t, J = 6.5, 1H), 7.71 (s, 1H), 7.55 (s, 1H, H), 7.41–7.33 (m, 3H), 6.95 (s, 1H), 5.58–5.33 (m, 6H), 5.12–4.04 (bd, 2H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 161.6, 155.0, 154.2, 146.5, 143.9, 137.0, 136.8, 129.0, 128.2, 128.1, 126.7, 122.0, 121.4, 112.4, 92.9, 52.6, 52.5, 41.9, 36.3. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H17IN11 526.0708; Found 526.0701.
Synthesis of 13-[6-(4-Boc-piperazin-1-yL)pyridin-3-yl]- 11H,31H,71H-9-aza-1(1,6)-pyrazolo[3,4-d]pyrimidin-3(4,1),7(1,4)-ditriazole-5(1,3)-benzenecyclononaphane (9)
To a suspension of 7 (58.5 mg, 0.11 mmol) in DMF/H2O (5/0.5 mL) was added 6-(4-Boc-piperazin-1-yl)pyridine-3-boronic acid pinacol ester (8, 65 mg, 0.17 mmol), sodium carbonate (24 mg, 0.17 mmol), triphenylphosphine (6 mg, 20% mol), and palladium(II) acetate (2 mg, 5% mol). The mixture was irradiated by microwave at 120 °C for 1 h. The solvent was evaporated and the crude extract was purified by flash chromatography using DCM/MeOH 10/0.6 as the eluent to afford a white solid (40 mg, 55%). 1H NMR (500 MHz, DMSO-d6) δ 9.15 (s, 1H), 8.74 (s, 1H), 8.26–8.05 (m, 2H), 7.72 (s, 1H), 7.53 (s, 1H), 7.44–7.31 (m, 3H), 6.95 (s, 2H), 5.48 (s, 6H), 5.03–3.96 (bd, 2H), 3.58 (s, 4H), 3.45 (s, 4H), 1.43 (s, 9H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 160.8, 158.4, 155.3, 154.4, 154–0, 146.8, 145.7, 144.3, 141.4, 137.0, 136.9, 135.5, 129.0, 128.2, 128.0, 126.7, 121.9, 121.4, 117.8, 107.2, 105.1, 79.0, 52.6, 52.6, 44.2, 41.8, 36.2, 28.1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C33H37N14O2 661.3218; Found 661.3232.
Synthesis of 13-[6-(Piperazin-1-yL)pyridin-3-yl]- 11H,31H,71H-9-aza-1(1,6)-pyrazolo[3,4-d]pyrimidine-3(4,1),7(1,4)-ditriazole-5(1,3)-benzenecyclononaphane (1)
A solution of 9 (19 mg, 0.029 mmol) in trifluoroacetic acid (TFA)/H2O (4/0.5 mL) was stirred at room temperature for 2 h. The solvent was evaporated, and the crude extract was purified by reversed-phase chromatography using H2O/MeOH (6/4) as the eluent to afford a white solid (15 mg, 92%). 1H NMR (27 °C) (500 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.77 (s, 1H), 8.20 (s, 1H), 8.14 (d, J = 8.5, 1H), 7.72 (s, 1H), 7.54 (s, 1H), 7.37 (s, 3H), 7.02 (d, J = 8.5, 1H), 6.95 (s, 1H), 5.18–4.02 (bd, 2H), 5.50 (s, 6H), 3.74 (s, 4H), 3.13 (s, 4H). 1H NMR (80 °C) (500 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.74 (s, 1H), 8.12 (d, J = 8.5, 1H), 7.85 (s, 1H), 7.55 (s, 1H), 7.38 (m, 4H), 6.99 (d, J = 8.5, 1H), 6.89 (s, 1H), 5.49 (s, 6H), 4.64 (s, 2H), 3.76 (s, 4H), 3.15 (s, 4H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 160.8, 158.0, 155.3, 154.4, 146.8, 145.6, 144.2, 141.3, 137.0, 136.9, 135.7, 129.0, 128.2, 128.0, 126.6, 121.9, 121.4, 118.4, 107.4, 105.1, 52.6, 52.5, 43.1, 42.5, 41.8, 36.2. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H29N14 561.2694; Found 561.2659.
Kinase Screening Assay
The radioisotope-based assay ([γ-33P]-ATP) consisted of measuring 33P incorporation induced by each kinase (AXL, TYRO3, MER, RET, or FLT3) on the substrate poly [Glu, Tyr] 4:1 in the presence of eSM119 and compound 1 relative to DMSO. Experiments were performed by Reaction Biology Corp.. Compounds’ IC50 values were determined from 1:3 dilution curves starting at 10 μM (10 points in total) with 10 μM ATP.
Cell Viability Assays
MDA-MB-231, MCF7, U87, HCT116, and A549 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 2 mM L-glutamine and incubated in a Heracell 240i tissue culture incubator at 37 °C and 5% CO2. ZR-75-1, MOLM-13, and MV4-11 cells were grown under the same conditions with Roswell Park Memorial Institute (RPMI) instead of DMEM. Cells were plated in 96-well plates at 1500 (HCT116 and A549), 2000 (MDA-MB-231, MCF7, and U87), or 4000 cells/well (MOLM-13 and MV4–11 cells) in 95 (suspension cells) or 100 μL (for adherent cells) of supplemented medium. Adherent cells were incubated for 48 h in an incubator at 37 °C and 5% CO2. The media was then aspirated from each well and replaced with 95 μL of fresh medium. Suspension cells were treated on the same date that they were seeded. Then, 5 μL of a DMSO solution of compound 1 at 600 μM was added to each well containing cells to obtain a final concentration of 30 μM. Untreated cells were incubated with DMSO (0.1% v/v). After 5 d, the PrestoBlue cell viability reagent (10 μL) was added to each well, and the plates incubated for 0.5–4 h (MV4–11 required 3 h of incubation). Fluorescence emission was detected using an Envision fluorescence plate reader (excitation 540 nm, emission 590 nm). All conditions were normalized to the untreated cells (100%). For the dose–response study, MV4–11 cells were seeded in 96-well plates as described before. Serial 1:3 dilutions of compound 1 in DMSO were prepared in a separate 96-well intermediate plate. Then, 5 μL from the intermediate plate was added to each well containing cells. The determination of cell viability was performed at day 5 as described before, and the results were normalized to the untreated cells (100%). Curves were fitted using a four-parameter logistic fit with the minimum value constrained to zero using the GraphPad Prism software to calculate EC50 values.
Acknowledgments
O.C.-L. and B.L. thank Spain Ministry of Education, Culture and Sport and the Erasmus + Traineeship programme for funding, respectively. C.T. thanks the CMVM for a Principal’s Scholarship. F.F. acknowledges support from Universidad de Granada. SHM and AU-B are grateful to Scottish Power and CRUK for funding. The authors also thank the Centro de Supercomputación de la Universidad de Granada (CSIRC) for computing resources and Prof Sandro Cosconati (DiSTABiF, Università della Campania Luigi Vanvitelli, Caserta, Italy) for his help with the docking studies.
Glossary
Abbreviations
- CuAAC
copper(I)-catalyzed alkyne–azide cycloaddition
- DMSO
dimethylsulphoxide
- EC50
half-maximal effective concentration
- ppm
parts per million
- rt
room temperature
- SD
standard deviation
- TLC
thin-layer chromatography.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03525.
Macrocycle conformational sampling, docking studies, dose–response studies of 1 in MV4–11 cells, and NMR spectra of 4, 5, 7, 9, and 1 (PDF)
Author Present Address
§ Department of Chemistry – Centre for Imaging Science, University of Loughborough, Epinal Way, Loughborough LE11 3TU, U.K. (B.L.).
Author Present Address
∥ Istituto Italiano di Tecnología, Via Morego 30, Genova 16163, Italy (S.H.M.).
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Marsault E.; Peterson M. L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Giordanetto F.; Kihlberg J. Macrocyclic Drugs and Clinical Candidates: What Can Medicinal Chemists Learn from Their Properties?. J. Med. Chem. 2014, 57, 278–295. 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- Iyoda M.; Yamakawa J.; Rahman M. J. Conjugated Macrocycles: Concepts and Applications. Angew. Chem., Int. Ed. 2011, 50, 10522–10553. 10.1002/anie.201006198. [DOI] [PubMed] [Google Scholar]
- Villar E. A.; Beglov D.; Chennamadhavuni S.; Porco J. A. Jr.; Kozakov D.; Vajda S.; Whitty A. How Proteins Bind Macrocycles. Nat. Chem. Biol. 2014, 10, 723–731. 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C.; Carragher N. O.; Unciti-Broceta A. eCF309: a Potent, Selective and Cell-Permeable mTOR Inhibitor. Med. Chem. Commun. 2016, 7, 471–477. 10.1039/C5MD00493D. [DOI] [Google Scholar]
- Bogdan A. R.; James K. Synthesis of 5-Iodo-1,2,3-triazole-Containing Macrocycles Using Copper Flow Reactor Technology. Org. Lett. 2011, 13, 4060–4063. 10.1021/ol201567s. [DOI] [PubMed] [Google Scholar]
- Bogdan A. R.; James K. Efficient Access to New Chemical Space Through Flow—Construction of Druglike Macrocycles Through Copper-Surface-Catalyzed Azide–Alkyne Cycloaddition Reactions. Chem. - Eur. J. 2010, 16, 14506–14512. 10.1002/chem.201002215. [DOI] [PubMed] [Google Scholar]
- William A. D.; Lee A. C.-H.; Blanchard S.; Poulsen A.; Teo E. L.; Nagaraj H.; Tan E.; Chen D.; Williams M.; Sun E. T.; Goh K. C.; Ong W. C.; Goh S. K.; Hart S.; Jayaraman R.; Pasha M. K.; Ethirajulu K.; Wood J. M.; Dymock B. W. Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1 (8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma. J. Med. Chem. 2011, 54, 4638–4658. 10.1021/jm200326p. [DOI] [PubMed] [Google Scholar]
- William A. D.; Lee A. C.-H.; Goh K. C.; Blanchard S.; Poulsen A.; Teo E. L.; Nagaraj H.; Lee C. P.; Wang H.; Williams M.; Sun E. T.; Hu C.; Jayaraman R.; Pasha M. K.; Ethirajulu K.; Wood J. M.; Dymock B. W. Discovery of Kinase Spectrum Selective Macrocycle (16E)-14-Methyl-20-oxa-5,7,14,26-tetraazatetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3, 5,8(27),9,11,16,21,23-decaene (SB1317/TG02), a Potent Inhibitor of Cyclin Dependent Kinases (CDKs), Janus Kinase 2 (JAK2), and Fms-like Tyrosine Kinase-3 (FLT3) for the Treatment of Cancer. J. Med. Chem. 2012, 55, 169–196. 10.1021/jm201112g. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu J.; Zhang W.; Stashko M. A.; Nichols J.; Miley M. J.; Norris-Drouin J.; Chen Z.; Machius M.; DeRyckere D.; Wood E.; Graham D. K.; Earp H. S.; Kireev D.; Frye S. V. Design and Synthesis of Novel Macrocyclic Mer Tyrosine Kinase Inhibitors. ACS Med. Chem. Lett. 2016, 7, 1044–1049. 10.1021/acsmedchemlett.6b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver A. L.; Zhang W.; Liu Q.; Jiang X.; Stashko M. A.; Nichols J.; Miley M. J.; Norris-Drouin J.; Machius M.; DeRyckere D.; Wood E.; Graham D. K.; Earp H. S.; Kireev D.; Frye S. V.; Wang X. Discovery of Macrocyclic Pyrimidines as MerTK-Specific Inhibitors. ChemMedChem 2017, 12, 207–213. 10.1002/cmdc.201600589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W.; Richardson P. F.; Bailey S.; Brooun A.; Burke B. J.; Collins M. R.; Cui J. J.; Deal J. G.; Deng Y. L.; Dinh D.; Engstrom L. D.; He M.; Hoffman J.; Hoffman R. L.; Huang Q.; Kania R. S.; Kath J. C.; Lam H.; Lam J. L.; Le P. T.; Lingardo L.; Liu W.; McTigue M.; Palmer C. L.; Sach N. W.; Smeal T.; Smith G. L.; Stewart A. E.; Timofeevski S.; Zhu H.; Zhu J.; Zou H. Y.; Edwards M. P. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J. Med. Chem. 2014, 57, 4720–4744. 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Myers S. H.; Temps C.; Houston D. R.; Brunton V. G.; Unciti-Broceta A. Development of Potent Inhibitors of Receptor Tyrosine Kinases by Ligand-Based Drug Design and Target-Biased Phenotypic Screening. J. Med. Chem. 2018, 61, 2104–2110. 10.1021/acs.jmedchem.7b01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciti-Broceta A.; Pineda-de-las-Infantas M. J.; Díaz-Mochón J. J.; Romagnoli R.; Baraldi P. G.; Gallo M. A.; Espinosa A. Regioselective Synthesis of 9-Alkyl-6-chloropyrido [3,2-e][1,2,4]triazolo[4,3-a]pyrazines. Reactivity of Aliphatic and Aromatic Hydrazides. J. Org. Chem. 2005, 70, 2878–2880. 10.1021/jo0478398. [DOI] [PubMed] [Google Scholar]
- Haldón E.; Nicasio M. C.; Pérez P. J. Copper-Catalysed Azide–Alkyne Cycloadditions (CuAAC): an Update. Org. Biomol. Chem. 2015, 13, 9528–9550. 10.1039/C5OB01457C. [DOI] [PubMed] [Google Scholar]
- Conejo-García A.; Campos J. M.; Entrena A.; Sánchez-Martín R. M.; Gallo M. A.; Espinosa A. Conformational Dynamics of a Bispyridinium Cyclophane. J. Org. Chem. 2003, 68, 8697–8699. 10.1021/jo0301292. [DOI] [PubMed] [Google Scholar]
- Watts K. S.; Dalal P.; Tebben A. J.; Cheney D. L.; Shelley J. C. Macrocycle Conformational Sampling with MacroModel. J. Chem. Inf. Model. 2014, 54, 2680–2696. 10.1021/ci5001696. [DOI] [PubMed] [Google Scholar]
- Fraser C.; Dawson J. C.; Dowling R.; Houston D. R.; Weiss J. T.; Munro A.; Muir M.; Harrington L.; Webster S. P.; Frame M.; Brunton V.; Patton E. E.; Carragher N. O.; Unciti-Broceta A. Rapid Discovery and Structure–Activity Relationships of Pyrazolopyrimidines That Potently Suppress Breast Cancer Cell Growth via SRC Kinase Inhibition with Exceptional Selectivity over ABL Kinase. J. Med. Chem. 2016, 59, 4697–4710. 10.1021/acs.jmedchem.6b00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. H.; Brunton V. G.; Unciti-Broceta A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016, 59, 3593–3608. 10.1021/acs.jmedchem.5b01273. [DOI] [PubMed] [Google Scholar]
- Kimani S. G.; Kumar S.; Bansal N.; Singh K.; Kholodovych V.; Comollo T.; Peng Y.; Kotenko S. V.; Sarafianos S. G.; Bertino J. R.; Welsh W. J.; Birge R. B. Small Molecule Inhibitors Block Gas6-Inducible TAM Activation and Tumorigenicity. Sci. Rep. 2017, 7, 43908 10.1038/srep43908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L.; Zhang Z.; Gao D.; Chan S.; Luo J.; Tu Z. C.; Zhang Z. M.; Ding K.; Ren X.; Lu X. Quinolone Antibiotic Derivatives as New Selective Axl Kinase Inhibitors. Eur. J. Med. Chem. 2019, 166, 318–327. 10.1016/j.ejmech.2019.01.065. [DOI] [PubMed] [Google Scholar]
- Gajiwala K. S.; Grodsky N.; Bolanos B.; Feng J.; Ferre R.; Timofeevski S.; Xu M.; Murray B. W.; Johnson T. W.; Stewart A. The Axl Kinase Domain in Complex with a Macrocyclic Inhibitor Offers First Structural Insights into an Active TAM Receptor Kinase. J. Biol. Chem. 2017, 292, 15705–15716. 10.1074/jbc.M116.771485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles P. P.; Murray-Rust J.; Kjaer S.; Scott R. P.; Hanrahan S.; Santoro M.; Ibanez C. F.; McDonald N. Q. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 16, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Batalla I.; Schultze A.; Wroblewski M.; Erdmann R.; Heuser M.; Waizenegger J. S.; Riecken K.; Binder M.; Schewe D.; Sawall S.; Witzke V.; Cubas-Cordova M.; Janning M.; Wellbrock J.; Fehse B.; Hagel C.; Krauter J.; Ganser A.; Lorens J. B.; Fiedler W.; Carmeliet P.; Pantel K.; Bokemeyer C.; Loges S. Axl, a Prognostic and Therapeutic Target in Acute Myeloid Leukemia Mediates Paracrine Crosstalk of Leukemia Cells with Bone Marrow Stroma. Blood 2013, 122, 2443–2452. 10.1182/blood-2013-03-491431. [DOI] [PubMed] [Google Scholar]
- Ghosh K.; Kar D.; Joardar S.; Sahub D.; Ganguly B. Azaindole-1,2,3-triazole Conjugate as Selective Fluorometric Sensor for Dihydrogenphosphate. RSC Adv. 2013, 3, 16144–16151. 10.1039/c3ra42702a. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.