

Abstract
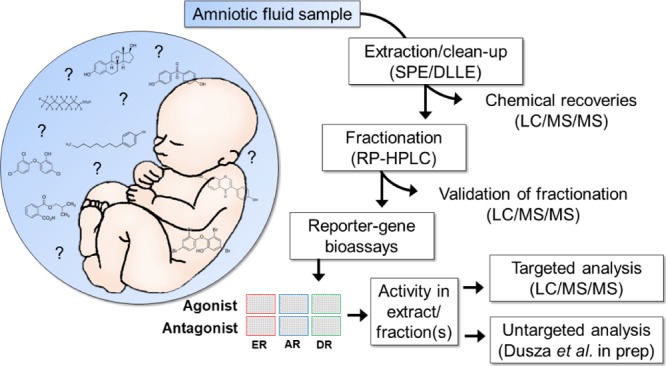
The developing fetus represents a highly sensitive period of exposure to endocrine disrupting compounds (EDCs). However, risk assessment of EDCs is hampered by the lack of data on direct in utero exposure. In this study, we developed a robust analytical methodology for the identification of a wide range of known and unknown EDCs in full-term amniotic fluid (AF). First, a method for extraction and fractionation of a broad range of polar and nonpolar EDCs was developed and validated. Maximal recoveries of reference compounds and minimal interference from the matrix were achieved with a combination of solid phase extraction and dispersive liquid/liquid extraction. Bioassay analysis using cell-based reporter gene assays revealed estrogenic, androgenic, and dioxin-like activity in AF extract corresponding to 1.4 nmol EEQ/L, 76.6 pmol DHT-EQ/L, and 10.1 pmol TEQ/L, respectively. Targeted analysis revealed 13 xenobiotics, phytoestrogens, and endogenous hormones in the AF extract that partly contributed to the bioassay activity. Separation of the complex mixture of chemicals in the AF extract with reversed-phase chromatographic fractionation and subsequent bioassay analysis revealed activity in fractions over a wide range of polarity, indicating diverse (unidentified) substances with potential ED activity. The method developed here represents the first methodological step in an effect-directed analysis approach to identify unknown biologically active compounds in the fetal environment.
Introduction
The developing fetus is highly susceptible to the exposure to endocrine disrupting compounds (EDCs),1−3 a diverse group of exogenous chemicals that can interfere with the endocrine system and hormone action.4 A growing body of literature indicates associations between prenatal exposure to EDCs and developmental, reproductive, and neurobehavioral disorders; endocrine-related cancers; and metabolic diseases.5−10 Despite the increasing concern for developmental origins of adult-onset diseases, the extent of exposure to both known and unknown EDCs in utero is still largely unknown. This significant data gap hampers comprehensive risk assessment of developmental exposures to EDCs.
Maternal matrices, such as blood, urine, and breast milk, are the most commonly used surrogate matrices for prenatal exposure assessment, although it is usually not known to what extent chemicals present in these matrices can reach the fetus.11 Some compounds (e.g. organochlorines) may pass from mother to fetus by active transplacental transfer rather than passive diffusion and may accumulate selectively in the fetal compartment.12,13 Moreover, both placenta and fetal liver express a wide variety of cytochrome P450 enzymes and are metabolically active, resulting in an in utero specific chemical environment.5,14−16 Considering the thousands of chemicals in commercial use that pregnant women might be exposed to and the complex maternal/fetal toxicokinetics, characterization of the whole spectrum of EDCs and their metabolites present in utero is still a major challenge.17 Consequently, new methods to measure fetal “exposome”, including any potential EDCs, are needed.
Amniotic fluid (AF), a product of fetal urination, tracheal secretions, and intra-/transmembranous pathways, accumulates a wide range of environmental xenobiotics throughout pregnancy.18 This fetal repository matrix is unique as it can be collected noninvasively during delivery, it includes maternal/fetal toxicokinetics and provides information on the relevant in utero exposures occurring prenatally.19,20 Although the lipid content of full-term AF is low (approx. 0.15 g/L),21,22 a wide spectrum of hydrophilic and lipophilic xenobiotics has been detected in AF.18,23−28
Effect-directed analysis (EDA) is an effect-based approach used to isolate and identify known and unknown biologically active compounds from a complex mixture.29,30 During EDA, the complexity of the sample is reduced by extraction and fractionation and active fractions are identified with relevant bioassays. Cell-based reporter gene bioassays have been successfully used in EDA studies for the detection of a wide range of EDCs including new and emerging hormone-like contaminants in environmental samples.31−33 However, so far this approach has rarely been applied to human samples and, to date, no EDA methodology has been reported for identifying endocrine disrupting activity in AF.
The application of EDA to AF presents a novel opportunity; however, this approach is challenging considering the diversity of EDCs and the complexity of the matrix, that is, presence of natural hormones, proteins, and lipids that can interfere with bioassay analysis and chemical identification. Therefore, as an important first step in an EDA approach, the goal of this study was to develop a nondiscriminating method to extract and fractionate a wide range of lipophilic and hydrophilic EDCs from AF with low interference from the matrix. Xenobiotics and natural hormones were separated through chromatographic separation, and the activity profile of collected fractions was measured using a battery of cell-based reporter gene bioassays that measured reporter gene expression under the control of important steroid hormone receptors and transcription factors, including the estrogen (ER), androgen (AR), and aryl hydrocarbon (AhR) receptors. Targeted liquid chromatography/mass spectrometry (LC/MS) analysis was used to measure a range of known xenobiotic and natural EDCs that could contribute to the bioassay activity.
Material and Methods
All chemicals and reagents were purchased at the highest commercially available purity and are given in Supporting Information S1.
AF Samples
Samples of AF were kindly provided by Prof. Andrés López Bernal (University of Bristol). The samples were obtained from healthy pregnancies only, with ethical approval and informed consent (U.K. National Health Service, reference E5431, 2008). Approximately, 50 mL of AF was collected during amniotomy at full-term vaginal delivery from each of four anonymous donors and immediately frozen at −80 °C. The samples were pooled, homogenized, aliquoted in volumes of 5 mL in 15 mL polypropylene falcon tubes, and stored at −80 °C.
Method Development
Sample Extraction
A schematic representation of the experimental design for the extraction, fractionation, and the final analysis of AF samples is presented in Figure 1. Two extraction techniques, that is, solid phase extraction (SPE) and dispersive liquid/liquid extractions (DLLE), were compared and tested under different conditions in order to determine the optimal sample extraction method capable of extracting EDCs with a wide range of chemical and physical properties. For this purpose, a mix of reference compounds containing 18 EDCs from 10 different chemical classes, reported previously in AF and covering a wide range of hydrophobicity, was used (Tables 1 and S1). These included natural hormones, phytoestrogens, exogenous polar and nonpolar EDCs, and their hydroxyl metabolites. Two different SPE chemistries, Oasis MCX (mixed-mode, reversed-phase, and cation exchange sorbent) and Oasis HLB (multipurpose reversed-phase sorbent), and two different DLLE extraction solvent combination, dichloromethane (DCM)/acetone and chloroform (CHL)/acetone, were tested. Detailed descriptions of the extraction procedures are given in Supporting Information S2. Different extraction methods were performed with spiked (100 μL of reference mix, Tables 1 and S1) LC/MS grade ultra-pure water (W) or/and spiked AF aliquots. The spiked samples were evaporated and reconstituted in 10% MeOH, and the analyte recoveries were determined by LC–electrospray ionization–tandem MS (LC–ESI–MS/MS). The most effective extraction procedure, that is, a combination of DLLE and SPE, was used during final AF extraction, to maximize analyte recovery.
Figure 1.

Schematic representation of the experimental design for the extraction, fractionation, and the final EDA analysis of AF samples. The extraction efficiency was compared between two SPE sorbents (MCX and HLB), two combinations of DLLE solvents (DCM/acetone and CHL/acetone), and a combination of SPE and DLLE. The nonspiked and spiked final AF extract was fractionated with RP high-performance LC (RP-HPLC). Chemical recoveries in the nonspiked AF extract and in the spiked fractions were analyzed with LC–ESI–MS/MS, whereas nonspiked fractions were tested for their estrogenic (ER-Luc), androgenic (AR-Luc), and dioxin-like (DR-GFP) activity in reporter gene bioassays.
Table 1. Reference Compounds Used During Method Development, Including Their Chemical Characteristics, Endocrine Disrupting Mechanism of Action (MoA), EC50 or IC50, Concentrations in Full-Term AF Reported in the Literature and Measured in This Study38,40−54a.
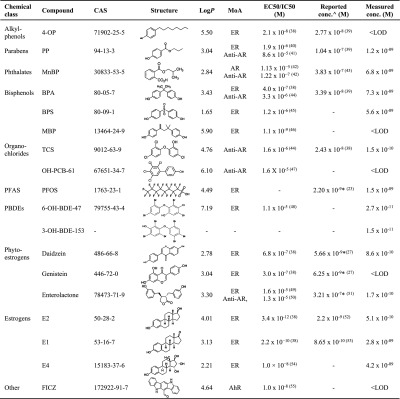
Abbrev: AR, androgenic; ER, estrogenic; AhR, dioxin-like mode of action; PFASs, perfluorinated compounds; PBDEs, polybrominated diphenyl ethers; EC50, half maximal effective concentration; IC50, half maximal inhibitory concentrations; 4-OP, 4-octylphenol; PP, propylparaben; MnBP, mono-n-butyl phthalate; BPA, bisphenol A; BPS, bisphenol S; TCS, triclosan; MBP, 4-methyl-2,4-bis-(p-hydroxyphenyl)pent-1-ene; OH-PCB-61, 2,3,4,5-tetrachloro-4′-biphenylol; PFOS, perfluorooctanesulfonic acid; 6-OH-BDE-47, 6-hydroxy-2,2′,4,4′-tetrabromodiphenyl ether; 3-OH-BDE-153, 3-hydroxy-2,2′,4,4′,5,5′-hexabromodiphenyl ether; E2, 17β-estradiol; E1, estrone; E4, estetrol; FICZ, 6-formylindolo[3,2-b]carbazole; EC50 and IC50, values based on in vitro reporter gene assays; log P, partition coefficient based on octanol–water partitioning, values reported from PubChem or Chemspider database; symbols: () references from the literature; ^ mean concentrations of unconjugated compounds measured at full-term; * concentrations reported in midgestation; - not reported.
HPLC Fractionation
To elucidate the elution profile of the reference compounds, spiked AF extract was fractionated on a reversed phase (RP) Phenomenex Kinetex C18 (100 mm × 2.1 mm, 5 μm pore size) column using an Agilent 1260 HPLC system equipped with a binary pump, fraction collector, and diode array detector. Data analysis and processing was performed with Agilent OpenLAB 1.1 Software. Spiked AF extract was injected (50 μL) at a flow rate of 500 μL/min in 95% HPLC grade water and 5% MeOH. The solvent gradient was held as such for 2 min and then increased linearly reaching 100% MeOH at 48 min. It was held at 100% MeOH for 2 min and then returned to starting conditions over a period of 10 min to equilibrate the column before the next injection. The column temperature was set at 35 °C. Two minute fractions were collected throughout the analytical period, excluding the column equilibrium period, resulting in 25 fractions. Each fraction was evaporated to dryness with a gentle stream of nitrogen (at max 40 °C), the remaining residues were reconstituted in 300 μL of 5% MeOH and analyzed by LC–ESI–MS/MS to determine the analyte recoveries.
Final Sample Preparation
SPE columns (Oasis HLB, 6 cm3, 150 mg, 30 μm particle size, Waters Corp., Milford, MA) were conditioned with 3 mL of MeOH and 4 mL of deionized water. Aliquotes of AF (5 mL) were diluted with 3 mL deionized water. The nonspiked samples were loaded on the cartridges at a rate of 1 mL/min. The aqueous eluent was collected for further extraction with DLLE. The cartridges were rinsed with 3 mL of 5% MeOH and the analytes were eluted with 4 × 2 mL of MeOH. The extraction solvent was evaporated (TurboVap, Zymark Corp.) at 40 °C and under a gentle stream of nitrogen to a final volume of 1 mL. During DLLE, 0.1 M sodium acetate/acetic acid buffer was added to the aqueous eluent to lower the pH (∼4.5). The DLLE was performed twice by the addition of 2 mL of acetone (dispersive solvent) followed by the addition of 500 μL of DCM (extraction solvent). Samples were vortexed, then centrifuged at 4081g for 15 min at 25 °C, organic phases combined, and evaporated to dryness under a gentle stream of nitrogen. The 1 mL of extract remaining after SPE was then transferred to the residue in the DLLE tube and vortexed. The extracts were combined and further concentrated to 1 mL, transferred to an Eppendorf tube, and centrifuged at 20 800g to separate the precipitate formed after sample concentration. To avoid column overload, 50 μL of the final concentrated extract, representing 100 mL AF, was injected three times on a RP-HPLC column, fractionated as above, fractions combined with their complementary fractions, evaporated to dryness, and reconstituted in 300 μL of MeOH. Each fraction (200 μL) was transferred, evaporated to dryness, and reconstituted in 100 μL of dimethyl sulfoxide (DMSO) for bioassay analysis. The remaining 100 μL in MeOH was stored for further chemical analysis. All samples were capped and stored at −20 °C in amber glass vials.
LC–ESI–MS/MS Analysis
Chemical analysis was carried out by LC–ESI–MS/MS using an Agilent 1100 Series LC system coupled with an AB SCIEX API 5000 triple quadrupole MS operating in the MRM mode. The extract (20 μL) was injected on a RP Phenomenex Kinetex C18 column (100 mm × 2.1 mm, 5 μm pore size). The chromatographic parameters (composition and flow rate of the mobile phase) were optimized. Mobile phases included HPLC grade water and MeOH. The ionization was supported via adjustment of pH to 10, through addition of 0.1% ammonia to both mobile phases. The gradient elution program consisted of 10% MeOH (0–2 min), 70% MeOH (2.5–6.5 min), 100% MeOH (7–9 min), 10% MeOH (9.5–11.5 min), and a flow rate of 0.5 mL/min. After elution, the analytes were ionized using a negative ion ESI mode (ESI, TurboIonSpray probe). The optimum operating parameters for ESI were as follows: capillary voltage −4500 V, source temperature 500 °C, curtain gas 15, CAD gas 4, GS1 40, and GS2 30. The column temperature was set at 25 °C. The collision cell parameters and precursor/product ion transitions (one for quantification and one for confirmation) were optimized for each analyte (Table S2).
Reporter Gene Bioassays
The estrogen receptor-mediated luciferase reporter gene (ER-Luc) human breast carcinoma (VM7Luc4E2) cells,34 the AR/glucocorticoid receptor-mediated luciferase reporter gene (AR-Luc) human breast carcinoma (MDA-kb2) cells,35 and the AhR receptor enhanced green fluorescent protein reporter gene (DR-GFP) mouse hepatoma (H1G1.1c3) cells36 were maintained and used to test endocrine activity of extracts and fractions as described in Supporting Information S3. The AF extract, 25 fractions, solvent (DMSO) control, extraction blank, and positive control concentration–response curves were tested in three independent experiments (n=3). 17β-estradiol (E2), dihydrotestosterone (DHT), and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) were used as positive controls for ER and AR-Luc and DR-GFP, respectively. In the anti-ER/AR/DR assays, cells were coexposed to the fraction and positive controls at 4, 150, and 30 pM of E2, DHT, and TCDD, respectively. Cell viability was determined with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Supporting Information S3).
Data Analysis
Six concentrations of reference compounds encompassing the entire linear range of the calibration curve were used during LC–ESI–MS/MS analysis. For the quantitative analysis of the reference compounds in the AF extract, LC/MS grade water was injected as calibration point 0. The remaining calibration points were prepared in the matrix to compensate for any matrix induced ion supression during analysis, and the regression lines of each reference compounds were forced through zero. The detection limits (LODs) of measured compounds were calculated as three times the standard deviation (SD) of the extraction blanks that is, a full extraction procedure performed with a nonspiked HPLC grade water (W). Agilent Analyst 1.6.2 software was used for data acquisition and analysis. Analyte relative recovery (%) was calculated in AF spiked with reference compounds (Table 2) as measured concentration/spiked concentration × 100. Matrix factor (MF) was used as a quantitative measurement of matrix effects (ME) and was defined by the compound recoveries in the presence of matrix ions (AF)/compound recoveries in water only (W), where MF ≥ 1 indicates no ME, MF < 1 indicates interference from the matrix.37
Table 2. Analyte Recoveries (%) of the Reference Compounds Extracted with DLLE Using Two Different Extraction Solvent Combinations (DCM/Acetone and CHL/Acetone) and SPE Using Two Different SPE Sorbents (HLB and MCX); Chemical Recoveries (%) ± Relative Standard Deviation (RSD) and MF Measured in the Spiked LC/MS Grade Water (W) vs Spiked AF Extracted with DCM/A, HLB, and a Combination of the Two Extraction Techniques (Final Extraction) as Described in the Method Sectiona.
| DLLE |
SPE |
DLLE—DCM/A |
SPE—HLB |
final extraction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| recovery
(%) (n = 1) |
recovery
(%) (n = 1) |
recovery ± RSD (%) (n = 3) |
recovery ± RSD (%) (n = 3) |
recovery ± RSD (%) (n = 3) | ||||||||
| spike conc. (ng/mL) | DCM/A | CHL/A | HLB | MCX | W | AF | MF | W | AF | MF | AF | |
| 4-OP | 27.6 | 56 | 9 | 6 | 9 | 45 ± 3 | 66 ± 27 | 1.5 | 26 ± 5 | 16 ± 5 | 0.6 | 64 ± 8 |
| PP | 109.2 | 87 | 109 | 81 | 59 | 102 ± 9 | 106 ± 18 | 1.0 | 80 ± 3 | 83 ± 3 | 1.0 | 84 ± 15 |
| MnBP | 107.4 | 48 | 74 | 78 | 62 | 71 ± 4 | 75 ± 13 | 1.1 | 109 ± 14 | 98 ± 12 | 0.9 | 86 ± 15 |
| BPA | 50.7 | 60 | 54 | 70 | 51 | 69 ± 9 | 86 ± 17 | 1.2 | 64 ± 7 | 58 ± 20 | 0.9 | 81 ± 30 |
| BPS | 116.0 | 47 | 35 | 105 | 99 | 54 ± 3 | 44 ± 6 | 0.8 | 77 ± 4 | 50 ± 5 | 0.7 | 115 ± 13 |
| MBP | 102.1 | 65 | 54 | 62 | 76 | 73 ± 4 | 74 ± 27 | 1.0 | 61 ± 13 | 46 ± 8 | 0.8 | 79 ± 32 |
| triclosan | 58.0 | 84 | 26 | 60 | 32 | 72 ± 22 | 59 ± 14 | 0.8 | 40 ± 5 | 33 ± 2 | 0.8 | 104 ± 9 |
| OH-PCB-61 | 100.0 | 62 | 14 | 72 | 55 | 71 ± 32 | 32 ± 8 | 0.4 | 51 ± 4 | 28 ± 7 | 0.5 | 109 ± 3 |
| PFOS | 62.4 | 109 | 85 | 73 | 17 | 78 ± 10 | 90 ± 27 | 1.1 | 65 ± 3 | 52 ± 9 | 0.8 | 98 ± 10 |
| 6-OH-BDE-47 | 49.5 | 81 | 25 | 65 | 28 | 68 ± 34 | 14 ± 6 | 0.2 | 51 ± 7 | 22 ± 6 | 0.6 | 103 ± 10 |
| 3-OH-BDE-157 | 117.4 | 82 | 29 | 47 | 8 | 38 ± 18 | 6 ± 2 | 0.2 | 42 ± 1 | 21 ± 3 | 0.5 | 86 ± 13 |
| daidzein | 104.2 | 82 | 93 | 88 | 55 | 107 ± 7 | 108 ± 11 | 1.0 | 95 ± 3 | 103 ± 6 | 1.1 | 102 ± 5 |
| genistein | 103.8 | 6 | 2 | 65 | 64 | 7 ± 3 | 4 ± 2 | 0.5 | 113 ± 7 | 97 ± 8 | 0.9 | 54 ± 10 |
| enterolactone | 38.7 | 102 | 97 | 83 | 76 | 81 ± 5 | 103 ± 19 | 1.3 | 94 ± 3 | 72 ± 7 | 0.8 | 98 ± 11 |
| E2 | 43.0 | 67 | 63 | 59 | 19 | 67 ± 10 | 87 ± 17 | 1.3 | 80 ± 4 | 70 ± 9 | 0.9 | 87 ± 15 |
| E1 | 55.7 | 68 | 59 | 66 | 15 | 67 ± 5 | 102 ± 29 | 1.5 | 76 ± 2 | 77 ± 8 | 1.0 | 95 ± 9 |
| E4 | 84.0 | 19 | 16 | 98 | 76 | 21 ± 2 | 23 ± 7 | 1.1 | 103 ± 5 | 90 ± 19 | 0.9 | 121 ± 7 |
| FICZ | 45.4 | 32 | 20 | 43 | 43 | 44 ± 25 | 6 ± 9 | 0.1 | 1 ± 1 | 0 ± 0 | – | 6 ± 2 |
| average | 76 | 64 | 48 | 68 | 47 | 63±11 | 60±14 | 0.9 | 68±5 | 57±8 | 0.8 | 87±12 |
n = number of extractions.
Reporter gene activity was corrected for background luminescence/fluorescence activity and expressed either as % relative luminescence units (% RLUs) for the ER and AR-Luc or % relative fluorescence units (% RFUs) for the DR-GFP. For DR-GFP only, fluorescence in the AF extract was additionally corrected for the background fluorescence in the extraction blank. In each assay, the activity in the extracts/fractions was expressed as relative % of luminescence/fluorescence of the highest activity observed in the concentration–response curve of the positive control. In each co-exposure assay, the activity in the positive control (n = 4 per plate) was set at 100% and the anti-ER/AR/DR activity in the samples (i.e., decrease in luminescence/fluorescence) was expressed relative to the positive control. ER/AR/DR activity in the AF extract was transformed into estrogen equivalent units (EEQ), DHT equivalent units (DHT-EQ) and dioxin equivalent units (TEQs) by interpolating the luminescence/fluorescence response elicited by diluted extract from the linear region of the four-parameter sigmoidal Hill concentration–response curve prepared with E2, DHT, and TCDD, respectively. All equivalent units were expressed in picomole or nanomoles per litre of AF (pmol/L or nmol/L). Concentration-response curves and statistical significance of the linear correlation between compound retention time and log P were analyzed/fitted with (GraphPad Software Inc., San Diego, CA).
Results and Discussion
Extraction of EDCs in AF: Method Optimization
EDCs belong to an extremely diverse group of compounds. They include pesticides, phthalates, alkylphenols, flame retardants, polychlorinated biphenyls (PCBs), dioxins, parabens, bisphenols, and synthetic hormones.56 An appropriate extraction technique, capable of extracting chemically and structurally diverse chemicals, is essential for the comprehensive characterization of a broad spectrum of known and novel EDCs that might be present in AF. In this study, a reference mixture consisting of 18 EDCs from 10 different chemical classes was used during the method development to account for this chemical diversity (Tables 1 and S1). Although AF is less complex in its composition than other fetal matrices such as umbilical cord blood or meconium, it contains a mixture of biological components and developmental products, such as cells, nutrients, growth factors, lipids, and proteins, that may interfere with chemical and bioassay analysis. Two extraction methods, SPE and DLLE were tested for their extraction efficiency using a mixture of reference compounds (Table 1) and combination of different SPE sorbents and DLLE solvents. External calibration standards were used for the quantification of compound recoveries with correlation coefficient exceeding 0.99. For DLLE, compound recoveries with DCM and acetone as the extraction solvent and dispersive solvent, respectively, were higher than with chloroform and acetone (Table 2) and higher than with DCM alone (data not shown). The mixture of DCM/acetone as a stronger nonpolar solvent performed better in extracting more hydrophobic compounds [e.g., triclosan, hydroxy-PCB, and polybrominated diphenyl ethers (PBDEs)] than the mixture of chloroform/acetone. However, in both cases, recoveries below 40% were found for E4, genistein, and FICZ. FICZ is stable as a solid but decomposes quickly in solution and in the presence of light and air (half-life of ∼3 h).55 It is thus likely that FICZ was lost due to light and air-induced decomposition or hydrolysis during solvation, extraction, and/or evaporation. In general, weak acids with a high number of hydroxyl groups, such as genistein and E4, have low affinity to partition to nonpolar organic solvents with low dielectric constants such as DCM or chloroform. To aid the partition of polar compounds into the organic phase, DLLE was additionally tested with salting-out. However, addition of sodium chloride resulted in poor organic/aqueous phase separation and the presence of salt residues in the final sample extracts (data not shown).
Next to DLLE, two different SPE columns, MCX and HLB, were tested. Both sorbents performed better in extracting hydrophilic compounds than the nonpolar compounds (Table 2). Nevertheless, extraction with HLB resulted in overall higher recoveries, with recoveries above 40% for all compounds except for 4-OP (6%). The two most promising extraction conditions, DLLE (DCM/acetone) and SPE (HLB) were evaluated further for their ME. Lipids, specifically phospholipids, contribute significantly to the ME. They interfere with reversed-phase chromatographic methods, as they are strongly retained on hydrophobic columns and can cause significant ionization suppression during mass spectrometric detection.57 The total amount of lipids in the full-term AF are generally low (∼0.15 g/L),21,22 nevertheless significant preconcentration of the AF during EDA may cause accumulation of lipids and other small organic constituents that may interfere with the bio-/chemical analysis. Considerable interference from the matrix was observed for the strongly hydrophilic hydroxy-PCB (MF 0.4) and hydroxy-PBDEs (MF 0.2) after DLLE extraction (Table 2). MEs for these compounds after HLB-SPE extraction were much less pronounced (MF 0.5–0.6). Considering that lipids (phospholipids) are highly soluble in methanol,58 SPE clean-up with 5% MeOH might have removed part of the lipids adsorbed on the SPE cartridge, resulting in less interference from the matrix. In general, the overall average recoveries and MF for the two extraction methods were comparable; however, SPE-HLB showed lower variability in the recoveries than DLLE (Table 2). Because HLB-SPE recovered more polar compounds with higher efficiency than DLLE, whereas DLLE (DCM/acetone) recovered mid-polar to non-polar compounds with higher efficiency than HLB-SPE, the two methods were combined to maximize the extraction efficiency.
During the final sample preparation, dilution of AF with HPLC-grade water decreased the sample viscosity and improved extraction efficiency (data not shown). Disruption of protein binding was omitted in the pretreatment stage as the amount of total protein in AF is low (∼0.4%) and lower than in maternal blood (∼7%) or umbilical cord blood (∼5%).59−61 The nonpolar compounds that did not adsorb to SPE during loading were recovered with DLLE (DCM/acetone). Here, acidification of the sample (to pH = 4.5) increased the extraction efficiency of the weak acids (data not shown). Prior extraction with SPE resulted in cleaner aqueous eluent for DLLE and better recoveries of nonpolar compounds. During the final extraction, the combination of the off-line SPE and DLLE was successful in recovering reference compounds with high efficiency (Table 2). The relative recoveries ranged from ∼54 to 121%, except for the chemically unstable FICZ (<10%). In general, the average relative recovery in AF was higher for the combined method (87 ± 12%) than for SPE (57 ± 12%) or DLLE (60 ± 14%) alone (Table 2).
HPLC Fractionation
AF extracted with SPE and DLLE was spiked and fractionated on an RP-HPLC column, and each fraction analyzed with LC–ESI–MS/MS. Considering the overall low concentrations of EDCs in AF reported in the literature, lower resolution fractionation (25 fractions) was applied to avoid loss in sensitivity during bioassay analysis. The concentration of the reference compounds in the fractions was quantified with an external calibration curve. Increased column temperature (35 °C) during chromatographic separation resulted in better peak separation (data not shown). In general, good separation of all reference compounds was achieved (Figure 2). A statistically significant correlation was observed between retention time and the compounds log P (r = 0.644, p < 0.001, Figure S2). Natural hormones, E2 (log P 4.01) and E1 (log P 3.69), and the synthetic estrogen BPA (logP 3.43), eluted in fractions 13–15 at mid-retention times (26–30 min) (Figure 2). More polar E4 (log P 2.21), a weaker steroid hormone produced exclusively by the fetal liver during gestation, eluted in the earlier fractions (8–9). As expected, the nonpolar reference compounds (Figure 2 and Table 1), eluted in the late fractions (17–20) with more than 75% of organic solvent in the chromatographic gradient. Although MBP extraction efficiency was high (79%), it was the only compound not detected in any of the fractions. Just like FICZ, MBP can decompose when dissolved in aqueous solution and exposed to sunlight.62 The decomposition during fractionation and evaporation process might, at least partially, contribute to its absence in the fractions. For photosensitive compounds, sample preparation under low UV exposure should be therefore considered.
Figure 2.

Elution profile of reference compounds during RP-HPLC fractionation of spiked AF extract. The total concentration of the reference compound eluted during the whole chromatographic run was set at 100% and the recovery in each individual fraction was reported as relative to it.
Bioassay Analysis
Nonspiked AF was fractionated on an RP-HPLC column (Figures S3 and 1). Serial dilutions of the AF extract and the 25 fractions were tested on six different endpoints from three different reporter gene assays, that is, the AR-Luc, ER-Luc, and DR-GFP in agonistic (Figures 3B and 4A) and antagonistic modes (Figures 3C and 4B). The reporter gene bioassays used are considered highly sensitive with responses to the respective positive controls, that is, E2, DHT and TCDD, comparable to the responses reported in the literature, with EC50 values of 3, 52, and 11 × 10–12 M, respectively (Figure 3A). The diluted AF extract was active in all three bioassays tested (Figure 3B). Estrogenic potency was high with estrogenic activity observed even in the extract diluted 500 times, corresponding to 1.4 nmol (374.5 ng) EEQ/L. The AR and DR activity of the AF extract was found at lower dilutions, corresponding to 76.6 pmol (22.2 ng) DHT-EQ/L and 10.1 pmol (3.3 ng) TEQ/L, respectively (Figure 3B, n = 3). The co-exposure experiments showed no significant antagonistic effect of the AF extract in any of the assays tested (Figure 3C).
Figure 3.
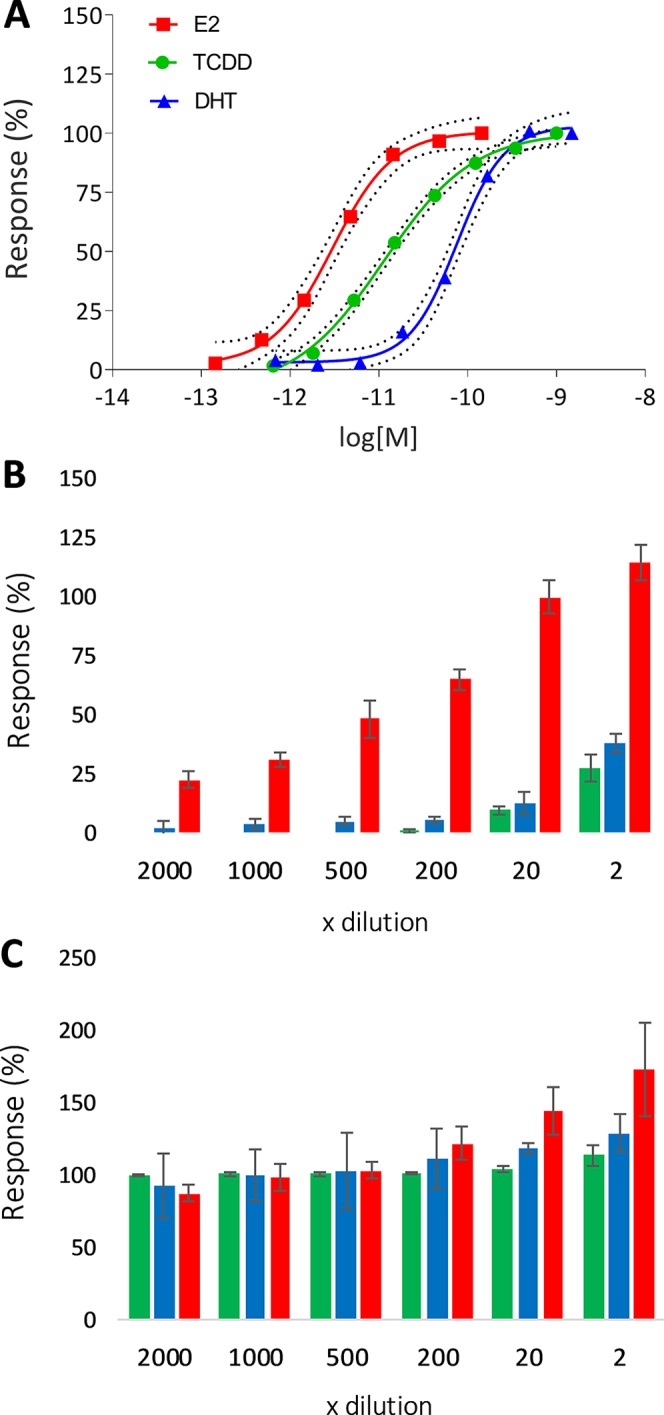
Concentration–response curves (A) with 95% confidence intervals of ER, AR, and DR responsive cell lines exposed to E2 (red), DHT (blue), and TCDD (green), respectively (n = 3). Agonistic (B) and antagonistic (C) activity of the AF extract represented as luminescence response relative to the highest response of the respective positive control (average % ± SD, n = 3). For antagonism experiments (C), extract dilutions were co-exposed with 4, 150, and 30 pM of E2, DHT, and TCDD, respectively. “x dilution” represents the final dilution factor of the AF sample.
Figure 4.

Agonistic (A) and antagonistic (B) activity of AF fractions in ER (200× diluted, red), AR (2× diluted, blue), and DR (2× diluted, green) responsive cell lines. Antagonistic activity is measured as a decrease in luminescence after co-exposure with 4, 150, and 30 pM of E2, DHT, and TCDD, respectively. Results presented as average % of RLUs of positive control ± SD between experiments (n = 3).
In order to determine if known hormones and xenobiotics contributed to the biological activity measured in vitro, the 18 reference compounds in the nonspiked AF extract were analyzed. From 18 reference compounds, 13 were detected above LODs (Tables 1 and S2). The concentration of natural estrogens, E2 (0.5 nM), E1 (2.8 nM), and E4 (4.2 nM), were detected at levels comparable to the concentrations reported in the literature (Table 1). Most xenobiotics and phytoestrogens were detected at low nM range. The two hydroxy-PBDEs were detected close to LODs (pM range). Considering that the EC50 values for most of the exogenous reference compounds fall within the high nM to μM range (Table 1), their contribution to the observed activity is minor.
To our knowledge, estrogenic, androgenic, and dioxin-like activity have only been reported once before in a recent study by Long et al. (2019) in samples from second trimester AF. The median concentration of the total estrogenic activity was determined at 545.8 ng EEQ/L (range 149.0–1308, n = 75).28 The authors reported a high variation in the levels of E2, ranging from 55 to 1005 E2 ng/L. After E2 subtraction, estrogenic activity induced only by xenoestrogens was estimated at 214.4 ng EEQ/L (n = 68).28 In this study, the combined levels of E2, E1, and E4 measured in AF (Table 1) were determined at 0.56 nmol (152 ng) EEQ/L and contributed to 48% of the activity observed in the ER-Luc bioassay. A combination of xenoestrogens, phytoestrogens, and other minor and weaker steroid estrogens (e.g., estriol (E3),17α-estradiol, hydroxylated estrogen metabolites) are the most likely contributors to the remaining activity.
The combined effect on AR of endogenous androgens and xenoandrogens reported by Long et al. (2019) was in a range of 16–290 ng DHT-EQ/L, and the combined effect on DR of dioxin-like compounds was in a range of 0–2.10 ng TEQ/L. The values obtained in this study fall in the range obtained by Long et al. (2019), except for dioxin-like activity. Here, observed activity was approximately one and a half times higher than the highest value reported by Long et al. (2019). However, direct comparison is somewhat limited. Considering that both hormone levels and the volume of AF changes during pregnancy, full-term AF may significantly differ from second trimester AF in chemical composition and concentration.63 Moreover, the more lipophilic dioxin-like compounds may gradually accumulate in AF throughout pregnancy, resulting in the higher activity observed in this study, that is, samples collected at term compared to samples from mid-gestation.
To further characterize the nature of the chemicals contributing to the biological activity, the AF extract was fractionated and the ER, AR, and DR activity in each fraction was determined. Considering that the ER activity of the 2× diluted extract was above the maximum response of the positive control (∼120% activity, Figure 3B), the fractions tested for estrogenic activity were diluted 200× in order for their response in the bioassay to be interpolated in the linear range of the concentration–response curve of the positive control. In the ER-Luc assay, elevated activity was observed mainly in the early polar fractions (Figure 4A, F2 and F5) and in fractions of intermediate polarity (Figure 4A, F10–11, F14–15). Fraction 10 showed the highest estrogenic activity; however, none of the potent natural estrogens (E1, E2 or E4) eluted in this fraction (Figure 2). The reference compound PFOS, a weak estrogen receptor ligand,64 eluted in fraction 10 and was detected in AF at 1.5 nM (Table 1). However, at 200× dilution, the concentration would be too low to significantly contribute to the observed activity. Nevertheless, it is possible that similar perfluorinated compounds and weak ER ligands [e.g. perfluorooctanoic, perfluorononanoic, and perfluorodecanoic acids]64,65 could elute in this fraction and contribute to the observed activity. Moreover, these compounds have been previously detected in maternal blood and cord blood.66 Estrogenic activity in fractions 2 and 5 could not be explained by any of the reference compounds measured in this study. The retention time at which other agonistic ER activity was observed corresponded well with the retention times of reference estrogenic compounds, that is, natural hormones (E2 and E1, F14–15) and phytoestrogens (enterolactone, F11) (Figures 2 and 4A). Although, E4 was detected in AF (4.25 nM, Table 1) and was shown to elute in fractions 8 and 9 (Figure 2), we did not observe activity in these fractions at 200× dilution (Figure 4A). However, these two fractions showed weak ER activity when tested at lower dilutions (Figure S4). The antiestrogenic activity of environmental chemicals is much less commonly reported than the estrogenic activity. This corroborated our findings, that is, in the antiestrogenic assay, only few fractions showed a small decrease in the luminescence induced by the positive control E2 (Figure 4B).
In this study, three mid-polar (F11, F13, F16) and two non-polar fractions (F21 and F23) showed elevated androgenic activity (Figure 4A). Although we did not analyze the levels of DHT (log P 3.55) and testosterone (log P 3.32), these hormones have similar log P values to BPA and enterolactone (log P 3.3) which eluted in fractions 11 and 13. Both, testosterone and DHT are commonly detected in AF.28,67 It is thus possible that the activity in these fractions are, at least to some extent, caused by endogenous AR ligands. The androgenic activity found in fractions 20 and 21 is noteworthy and could be of xenobiotic origin, as natural androgens are not expected to elute in these highly nonpolar fractions. Antiandrogenic activity, commonly reported in the literature, was observed for multiple fractions (F5, F10, and F16–F20, Figure 4B). MTT assay showed no decrease in cell viability in any of the fractions tested. Interestingly, antiandrogenic activity was not observed in the unfractionated extract, which may be explained by the co-elution of androgenic compounds that, when mixed, could mask the antiandrogenic effects.68 A similar result was observed by Indeveri et al. (2014), who reported lower total antiandrogenic activity in a placental extract than in the summed activities in the fractions.69 The antiandrogenic reference compounds propyl paraben and triclosan eluted in the fractions that showed an antiandrogenic effect, that is, F5 and F17/18, respectively (Figure 4B) and were detected in the nonspiked AF extract (Table 1). Moreover, both compounds were previously reported in maternal blood and cord blood.39 However, the IC50 for their antiandrogenic effects are reported at levels 3–4 orders of magnitude higher than the measured concentrations (Table 1). Therefore, their contribution to the observed activity is likely minimal.
Most dioxin-like compounds are highly lipophilic and include polychlorinated dibenzo-p-dioxins, polychlorinated/polybrominated biphenyls, polychlorinated dibenzofurans, or polycyclic aromatic hydrocarbons.70,71 Reports on quantification of these compounds in utero are surprisingly scarce, with only few publications reporting trace levels of dioxin or dioxin-like compounds in AF.27,72 Despite their lipophilic nature, the high activity observed in this study shows that the extraction method presented here could be successfully used for the detection of dioxin-like activity in AF extract. Consistent with the general chemical characteristics of these compounds, most dioxin-like activity was found in the nonpolar fraction (F19 to F23) (Figure 4A). The observed activity in these late nonpolar fractions indicates that the extraction method presented here extracted compounds with higher log P values than the log P value reported for the most nonpolar reference compound used in this study (6-OH-BDE-47, log P 7.19). Interestingly, weaker dioxin-like activity was also found in earlier fractions (F13–F17), possibly indicating the presence of dioxin-like compounds with intermediate polarity or presence of hydroxylated metabolites. Similar to antiandrogenic activity, the summed dioxin-like activity in the fractions was significantly higher than the activity observed in the unfractionated extract. None of the fractions showed anti-DR activity in the cells co-exposed to TCDD (Figure 4B). This is consistent with the literature, where the majority of reported AhR ligands are agonists.
This study shows for the first time that a combination of off-line SPE and DLLE can be used to extract a broad-spectrum of EDCs from AF with low interference from the matrix. Although it is generally assumed that the chemical concentrations in AF are lower than those detected in maternal urine, serum, or blood, we show here that the sample preconcentration, fractionation, and the use of cell-based reporter gene assays provide a sensitive method for successful detection of a range of endocrine disrupting activities in AF extract. The biological activity found in fractions of differing polarity gives a first indication of the diversity of endogenous and exogenous ER, AR, and AhR ligands present in AF. The method presented here sets the foundation for further qualitative and quantitative identification of known and unknown EDCs with biological activity, a necessary step forward in hazard characterization and risk assessment of EDC exposure in utero.
Acknowledgments
We thank Prof. Andrés López Bernal (University of Bristol) for providing samples of AF, Dr. Lillemor Asplund (Stockholm University) for supplying hydroxylated-PBDE standards and Dr. Eelco Ruijter (Vrije Universtiteit Amsterdam) for his help with MBP synthesis. We thank Prof. M. Lamoree (Vrije Universiteit Amsterdam) for her critical review of the manuscript. We acknowledge the financial contribution of Brunel University London and Utrecht University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.9b04255.
Additional information on MBP synthesis, extraction procedures, cell culture, parameters used during LC–ESI–MS/MS analysis, and RP-HPLC fractionation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Perera F.; Herbstman J. Prenatal Environmental Exposures, Epigenetics, and Disease. Reprod. Toxicol. 2011, 31, 363–373. 10.1016/j.reprotox.2010.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallozzi M.; Bordi G.; Garo C.; Caserta D. The Effect of Maternal Exposure to Endocrine Disrupting Chemicals on Fetal and Neonatal Development: A Review on the Major Concerns. Birth Defects Res., Part C. 2016, 108, 224–242. 10.1002/bdrc.21137. [DOI] [PubMed] [Google Scholar]
- Beszterda M.; Frański R. Endocrine Disruptor Compounds in Environment: As a Danger for Children Health. Pediatr. Endocrinol. Diabetes Metab. 2018, 24, 88–95. 10.18544/pedm-24.02.0107. [DOI] [PubMed] [Google Scholar]
- Zoeller R. T.; Brown T. R.; Doan L. L.; Gore A. C.; Skakkebaek N. E.; Soto A. M.; Woodruff T. J.; Vom Saal F. S. Endocrine-Disrupting Chemicals and Public Health Protection: A Statement of Principles from the Endocrine Society. Endocrinology 2012, 153, 4097–4110. 10.1210/en.2012-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker J. D. Exposure to Environmental Endocrine Disruptors and Child Development. Arch. Pediatr. Adolesc. Med. 2012, 166, 952–958. 10.1001/archpediatrics.2012.241. [DOI] [PubMed] [Google Scholar]
- Heindel J. J. Endocrine Disruptors and the Obesity Epidemic. Toxicol. Sci. 2003, 76, 247–249. 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- Vaiserman A. Early-life Exposure to Endocrine Disrupting Chemicals and Later-life Health Outcomes: An Epigenetic Bridge?. Aging Dis. 2014, 5, 419–29. 10.14336/AD.2014.0500419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macon M. B.; Fenton S. E. Endocrine Disruptors and the Breast: Early Life Effects and Later Life Disease. J. Mammary Gland Biol. Neoplasia 2013, 18, 43–61. 10.1007/s10911-013-9275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler J.; Hamers T.; van Eck van der Sluijs-van de Bor M.; Schoeters G.; van der Ven L.; Eggesbo M.; Koppe J.; Feinberg M.; Trnovec T. The OBELIX Project: Early Life Exposure to Endocrine Disruptors and Obesity. Am. J. Clin. Nutr. 2011, 94, 1933S–1938S. 10.3945/ajcn.110.001669. [DOI] [PubMed] [Google Scholar]
- Martin-Gronert M. S.; Ozanne S. E. Mechanisms Underlying the Developmental Origins of Disease. Rev. Endocr. Metab. Disord. 2012, 13, 85–92. 10.1007/s11154-012-9210-z. [DOI] [PubMed] [Google Scholar]
- Whyatt R. M.; Barr D. B. Measurement of Organophosphate Metabolites in Postpartum Meconium as a Potential Biomarker of Prenatal Exposure: A Validation Study. Environ. Health Perspect. 2001, 109, 417–420. 10.1289/ehp.01109417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M. H. B.; Karimi M.; Lyche J. L.; Lie E.; Polder A.; Mdegela R. H.; Skaare J. U.; Manyilizu W. B.; Solhaug A.; Mokiti F.; et al. Prenatal Exposure to Persistent Organic Pollutants in Northern Tanzania and Their Distribution between Breast Milk, Maternal Blood, Placenta and Cord Blood. Environ. Res. 2019, 170, 433–442. 10.1016/j.envres.2018.12.026. [DOI] [PubMed] [Google Scholar]
- Vizcaino E.; Grimalt J. O.; Fernández-Somoano A.; Tardon A. Transport of Persistent Organic Pollutants across the Human Placenta. Environ. Int. 2014, 65, 107–115. 10.1016/j.envint.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Prouillac C.; Lecoeur S. The Role of the Placenta in Fetal Exposure to Xenobiotics: Importance of Membrane Transporters and Human Models for Transfer Studies. Drug Metab. Dispos. 2010, 38, 1623–1635. 10.1124/dmd.110.033571. [DOI] [PubMed] [Google Scholar]
- Fisher M.; Arbuckle T. E.; Mallick R.; LeBlanc A.; Hauser R.; Feeley M.; Koniecki D.; Ramsay T.; Provencher G.; Bérubé R.; et al. Bisphenol A and Phthalate Metabolite Urinary Concentrations: Daily and across Pregnancy Variability. J. Expo. Sci. Environ. Epidemiol. 2015, 25, 231–239. 10.1038/jes.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkola J.; Pelkonen O.; Pasanen M.; Raunio H. Xenobiotic-Metabolizing Cytochrome P450 Enzymes in the Human Feto-Placental Unit: Role in Intrauterine Toxicity. Crit. Rev. Toxicol. 1998, 28 (1), 35–72. 10.1080/10408449891344173. [DOI] [PubMed] [Google Scholar]
- Bergman Å.; Heindel J.; Jobling S.; Kidd K.; Zoeller R. T. State-of-the-science of endocrine disrupting chemicals, 2012. Toxicol. Lett. 2012, 211, S3. 10.1016/j.toxlet.2012.03.020. [DOI] [Google Scholar]
- Geer L. A.; Pycke B. F. G.; Sherer D. M.; Abulafia O.; Halden R. U. Use of Amniotic Fluid for Determining Pregnancies at Risk of Preterm Birth and for Studying Diseases of Potential Environmental Etiology. Environ. Res. 2015, 136, 470–481. 10.1016/j.envres.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood M. A.; Gilbert W. M.; Sherman M. P. Amniotic Fluid: Not Just Fetal Urine Anymore. J. Perinatol. 2005, 25, 341–348. 10.1038/sj.jp.7211290. [DOI] [PubMed] [Google Scholar]
- Lozano J.; García-Algar O.; Vall O.; de la Torre R.; Scaravelli G.; Pichini S. Biological Matrices for the Evaluation of In Utero Exposure to Drugs of Abuse. Ther. Drug Monit. 2007, 29, 711–734. 10.1097/ftd.0b013e31815c14ce. [DOI] [PubMed] [Google Scholar]
- Singh E. J.; Zuspan F. P. Amniotic Fluid Lipids in Normal Human Pregnancy. Am. J. Obstet. Gynecol. 1973, 117, 919–925. 10.1016/0002-9378(73)90062-8. [DOI] [PubMed] [Google Scholar]
- Biezenski J. J.; Pomerance W.; Goodman J. Studies on the origin of amniotic fluid lipids. Am. J. Obstet. Gynecol. 1968, 102, 853–861. 10.1016/0002-9378(68)90514-0. [DOI] [PubMed] [Google Scholar]
- Jensen M. S.; Nørgaard-Pedersen B.; Toft G.; Hougaard D. M.; Bonde J. P.; Cohen A.; Thulstrup A. M.; Ivell R.; Anand-Ivell R.; Lindh C. H.; et al. Phthalates and Perfluorooctanesulfonic Acid in Human Amniotic Fluid: Temporal Trends and Timing of Amniocentesis in Pregnancy. Environ. Health Perspect. 2012, 120, 897–903. 10.1289/ehp.1104522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradman A.; Barr D. B.; Claus Henn B. G.; Drumheller T.; Curry C.; Eskenazi B. Measurement of Pesticides and Other Toxicants in Amniotic Fluid as a Potential Biomarker of Prenatal Exposure: A Validation Study. Environ. Health Perspect. 2003, 111, 1779–1782. 10.1289/ehp.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippat C.; Wolff M. S.; Calafat A. M.; Ye X.; Bausell R.; Meadows M.; Stone J.; Slama R.; Engel S. M. Prenatal Exposure to Environmental Phenols: Concentrations in Amniotic Fluid and Variability in Urinary Concentrations during Pregnancy. Environ. Health Perspect. 2013, 121, 1225–1231. 10.1289/ehp.1206335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. F.; Chernyak S. M.; Domino S. E.; Batterman S. A.; Loch-Caruso R. Concentrations and Speciation of Polybrominated Diphenyl Ethers in Human Amniotic Fluid. Sci. Total Environ. 2012, 417–418, 294–298. 10.1016/j.scitotenv.2011.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster W.; Chan S.; Platt L.; Hughes C. Detection of Endocrine-Disrupting Chemicals in Samples of Second Trimester Human Amniotic Fluid. J. Clin. Endocrinol. Metab. 2000, 85, 2954–2957. 10.1210/jcem.85.8.6850. [DOI] [PubMed] [Google Scholar]
- Long M.; Ghisari M.; Kjeldsen L.; Wielsøe M.; Nørgaard-Pedersen B.; Mortensen E. L.; Abdallah M. W.; Bonefeld-Jørgensen E. C. Autism Spectrum Disorders, Endocrine Disrupting Compounds, and Heavy Metals in Amniotic Fluid: A Case-Control Study. Mol. Autism. 2019, 10, 1. 10.1186/s13229-018-0253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Giesy J. P.; Lee J.-S.; Lee J.-H.; Khim J. S. Effect-Directed Analysis: Current Status and Future Challenges. Ocean Sci. J. 2016, 51, 413–433. 10.1007/s12601-016-0038-4. [DOI] [Google Scholar]
- Brack W. Effect-Directed Analysis: A Promising Tool for the Identification of Organic Toxicants in Complex Mixtures?. Anal. Bioanal. Chem. 2003, 377, 397–407. 10.1007/s00216-003-2139-z. [DOI] [PubMed] [Google Scholar]
- Zwart N.; Nio S. L.; Houtman C. J.; De Boer J.; Kool J.; Hamers T.; Lamoree M. H. High-Throughput Effect-Directed Analysis Using Downscaled in Vitro Reporter Gene Assays to Identify Endocrine Disruptors in Surface Water. Environ. Sci. Technol. 2018, 52, 4367–4377. 10.1021/acs.est.7b06604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon E.; van Velzen M.; Brandsma S. H.; Lie E.; Løken K.; de Boer J.; Bytingsvik J.; Jenssen B. M.; Aars J.; Hamers T.; et al. Effect-Directed Analysis To Explore the Polar Bear Exposome: Identification of Thyroid Hormone Disrupting Compounds in Plasma. Environ. Sci. Technol. 2013, 47, 8902–8912. 10.1021/es401696u. [DOI] [PubMed] [Google Scholar]
- Houtman C. J.; Van Oostveen A. M.; Brouwer A.; Lamoree M. H.; Legler J. Identification of Estrogenic Compounds in Fish Bile Using Bioassay-Directed Fractionation. Environ. Sci. Technol. 2004, 38, 6415–6423. 10.1021/es049750p. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Denison M. S. Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals. In Vitr. Mol. Toxicol. 2000, 13, 67–82. [PubMed] [Google Scholar]
- Wilson V. S.; Bobseine K.; Lambright C. R.; Gray L. E. A Novel Cell Line, MDA-Kb2, That Stably Expresses an Androgen- and Glucocorticoid-Responsive Reporter for the Detection of Hormone Receptor Agonists and Antagonists. Toxicol. Sci. 2002, 66, 69–81. 10.1093/toxsci/66.1.69. [DOI] [PubMed] [Google Scholar]
- Nagy S. R.; Sanborn J. R.; Hammock B. D.; Denison M. S. Development of a Green Fluorescent Protein-Based Cell Bioassay for the Rapid and Inexpensive Detection and Characterization of Ah Receptor Agonists. Toxicol. Sci. 2002, 65, 200–210. 10.1093/toxsci/65.2.200. [DOI] [PubMed] [Google Scholar]
- Silvestro L.; Tarcomnicu I.; Rizea S.. Matrix Effects in Mass Spectrometry Combined with Separation Methods—Comparison HPLC, GC and Discussion on Methods to Control These Effects. Tandem Mass Spectrometry—Molecular Characterization; InTech, 2013. [Google Scholar]
- Stoner M. A.; Yang C. Z.; Bittner G. D. A Robotic BG1Luc Reporter Assay to Detect Estrogen Receptor Agonists. Toxicol. In Vitro 2014, 28, 916–925. 10.1016/j.tiv.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar S.; Sood S.; Showkat S.; Lite C.; Chandrasekhar A.; Vairamani M.; Barathi S.; Santosh W. Detection of Phenolic Endocrine Disrupting Chemicals (EDCs) from Maternal Blood Plasma and Amniotic Fluid in Indian Population. Gen. Comp. Endocrinol. 2017, 241, 100–107. 10.1016/j.ygcen.2016.05.025. [DOI] [PubMed] [Google Scholar]
- Okubo T.; Yokoyama Y.; Kano K.; Kano I. ER-dependent estrogenic activity of parabens assessed by proliferation of human breast cancer MCF-7 cells and expression of ERα and PR. Food Chem. Toxicol. 2001, 39, 1225–1232. 10.1016/s0278-6915(01)00073-4. [DOI] [PubMed] [Google Scholar]
- Satoh K.; Nonaka R.; Ohyama K.-i.; Nagai F. Androgenic and Antiandrogenic Effects of Alkylphenols and Parabens Assessed Using the Reporter Gene Assay with Stably Transfected CHO-K1 Cells (AR-EcoScreen System). J. Health Sci. 2005, 51, 557–568. 10.1248/jhs.51.557. [DOI] [Google Scholar]
- Shen O.; Du G.; Sun H.; Wu W.; Jiang Y.; Song L.; Wang X. Comparison of in Vitro Hormone Activities of Selected Phthalates Using Reporter Gene Assays. Toxicology Letters 2009, 191, 9–14. 10.1016/j.toxlet.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Huang P.; Kuo P.; Chou Y.; Lin S.; Lee C. Association between prenatal exposure to phthalates and the health of newborns☆. Environ. Int. 2009, 35, 14–20. 10.1016/j.envint.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Di Paolo C.; Kirchner K.; Balk F. G. P.; Muschket M.; Brack W.; Hollert H.; Seiler T.-B. Downscaling Procedures Reduce Chemical Use in Androgen Receptor Reporter Gene Assay. Sci. Total Environ. 2016, 571, 826–833. 10.1016/j.scitotenv.2016.07.059. [DOI] [PubMed] [Google Scholar]
- Rosenmai A. K.; Dybdahl M.; Pedersen M.; Alice van Vugt-Lussenburg B. M.; Wedebye E. B.; Taxvig C.; Vinggaard A. M. Are Structural Analogues to Bisphenol A Safe Alternatives?. Toxicol. Sci. 2014, 139, 35–47. 10.1093/toxsci/kfu030. [DOI] [PubMed] [Google Scholar]
- Okuda K.; Fukuuchi T.; Takiguchi M.; Yoshihara S. i.; Hu J.; Liu J. Novel Pathway of Metabolic Activation of Bisphenol A-Related Compounds for Estrogenic Activity. Drug Metab. Dispos. 2011, 39, 1696–1703. 10.1124/dmd.111.040121. [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information . PubChem Database. Source=ChEMBL, AID=255211. https://pubchem.ncbi.nlm.nih.gov/bioassay/255211.
- Mercado-Feliciano M.; Bigsby R. M. Hydroxylated Metabolites of the Polybrominated Diphenyl Ether Mixture DE-71 Are Weak Estrogen Receptor-α Ligands. Environ. Health Perspect. 2008, 116, 1315–1321. 10.1289/ehp.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information . PubChem Database. Source=824, AID=1259403. https://pubchem.ncbi.nlm.nih.gov/bioassay/1259403.
- National Center for Biotechnology Information . PubChem Database. Source=824, AID=1259247. https://pubchem.ncbi.nlm.nih.gov/bioassay/1259247.
- Engel S. M.; Levy B.; Liu Z.; Kaplan D.; Wolff M. S. Xenobiotic Phenols in Early Pregnancy Amniotic Fluid. Reprod. Toxicol. 2006, 21, 110–112. 10.1016/j.reprotox.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mazor M.; Hershkowitz R.; Ghezzi F.; Cohen J.; Silber A.; Levy J.; Leiberman J. R.; Glezerman M. Maternal plasma and amniotic fluid 17?-estradiol, progesterone and cortisol concentrations in women with successfully and unsuccessfully treated preterm labor. Arch. Gynecol. Obstet. 1996, 258, 89–96. 10.1007/bf00626029. [DOI] [PubMed] [Google Scholar]
- Freeman R.; Lev-Gur M.; Koslowe R.; Schulman H.; Gatz M. Maternal Plasma and Amniotic Fluid Levels of Estradiol, Estrone, Progesterone, and Prolactin in Early Pregnancy. Obstet. Gynecol. 1984, 63, 507–510. [PubMed] [Google Scholar]
- Gérard C.; Mestdagt M.; Tskitishvili E.; Communal L.; Gompel A.; Silva E.; Arnal J.-F.; Lenfant F.; Noel A.; Foidart J.-M.; et al. Combined Estrogenic and Anti-Estrogenic Properties of Estetrol on Breast Cancer May Provide a Safe Therapeutic Window for the Treatment of Menopausal Symptoms. Oncotarget 2015, 6, 17621–17636. 10.18632/oncotarget.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Creech K. L.; Zuercher W. J.; Willson T. M. Gram-Scale Synthesis of FICZ, a Photoreactive Endogenous Ligand of the Aryl Hydrocarbon Receptor. Sci. Rep. 2019, 9, 9982. 10.1038/s41598-019-46374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A. C.; Chappell V. A.; Fenton S. E.; Flaws J. A.; Nadal A.; Prins G. S.; Toppari J.; Zoeller R. T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. 10.1210/er.2015-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmical J.; Brown S. The Impact of Phospholipids and Phospholipid Removal on Bioanalytical Method Performance. Biomed. Chromatogr. 2016, 30, 710–720. 10.1002/bmc.3686. [DOI] [PubMed] [Google Scholar]
- Ghosh C.; Gaur S.; Shinde P.; Chakraborty B. A Systematic Approach to Overcome the Matrix Effect during LC-ESI-MS/MS Analysis by Different Sample Extraction Techniques. J. Bioequivalence Bioavailab. 2011, 03, 122–127. 10.4172/jbb.1000072. [DOI] [Google Scholar]
- Tisi D. K.; Emard J. J.; Koski K. G. Total Protein Concentration in Human Amniotic Fluid Is Negatively Associated with Infant Birth Weight. J. Nutr. 2004, 134, 1754–1758. 10.1093/jn/134.7.1754. [DOI] [PubMed] [Google Scholar]
- Robertson E. G.; Cheyne G. A. Plasma Biochemistry in Relation to Oedema of Pregnancy. J. Obstet. Gynaecol. Br. Commonw. 1972, 79, 769–776. 10.1111/j.1471-0528.1972.tb12918.x. [DOI] [PubMed] [Google Scholar]
- Moniz C. F.; Nicolaides K. H.; Bamforth F. J.; Rodeck C. H. Normal Reference Ranges for Biochemical Substances Relating to Renal, Hepatic, and Bone Function in Fetal and Maternal Plasma throughout Pregnancy. J. Clin. Pathol. 1985, 38, 468–472. 10.1136/jcp.38.4.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao Y.; Shimazu M.; Kohra S.; Nagae M.; Ishibashi Y.; Tominaga N.; Ishibashi H.; Yoshihara S.; Arizono K. Photodecomposition and Bioconcentration of a Bisphenol A Metabolite in Medaka, Oryzias latipes. J. Health Sci. 2004, 50, 576–580. 10.1248/jhs.50.576. [DOI] [Google Scholar]
- Nagamani M.; McDonough P. G.; Ellegood J. O.; Mahesh V. B. Maternal and Amniotic Fluid Steroids throughout Human Pregnancy. Am. J. Obstet. Gynecol. 1979, 134, 674–680. 10.1016/0002-9378(79)90649-5. [DOI] [PubMed] [Google Scholar]
- Benninghoff A. D.; Bisson W. H.; Koch D. C.; Ehresman D. J.; Kolluri S. K.; Williams D. E. Estrogen-like Activity of Perfluoroalkyl Acids in Vivo and Interaction with Human and Rainbow Trout Estrogen Receptors in Vitro. Toxicol. Sci. 2011, 120, 42–58. 10.1093/toxsci/kfq379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen L. S.; Bonefeld-Jørgensen E. C. Perfluorinated Compounds Affect the Function of Sex Hormone Receptors. Environ. Sci. Pollut. Res. 2013, 20, 8031–8044. 10.1007/s11356-013-1753-3. [DOI] [PubMed] [Google Scholar]
- Needham L. L.; Grandjean P.; Heinzow B.; Jørgensen P. J.; Nielsen F.; Patterson D. G.; Sjödin A.; Turner W. E.; Weihe P. Partition of Environmental Chemicals between Maternal and Fetal Blood and Tissues. Environ. Sci. Technol. 2011, 45, 1121–1126. 10.1021/es1019614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood M. Y.; Saxena B. B. Testosterone and Dihydrotestosterone in Maternal and Cord Blood and in Amniotic Fluid. Am. J. Obstet. Gynecol. 1977, 129, 37–42. 10.1016/0002-9378(77)90815-8. [DOI] [PubMed] [Google Scholar]
- Weiss J. M.; Hamers T.; Thomas K. V.; Leonards P. E. G.; Lamoree M. H.; van der Linden S. Masking Effect of Anti-Androgens on Androgenic Activity in European River Sediment Unveiled by Effect-Directed Analysis. Anal. Bioanal. Chem. 2009, 394, 1385–1397. 10.1007/s00216-009-2807-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiveri P.; Horwood J.; Abdul-Sada A.; Arrebola J. P.; Olea N.; Hill E. M. Analytical Methodology for the Profiling and Characterization of Androgen Receptor Active Compounds in Human Placenta. Reprod. Toxicol. 2014, 47, 102–110. 10.1016/j.reprotox.2014.06.004. [DOI] [PubMed] [Google Scholar]
- van den Berg M.; Denison M. S.; Birnbaum L. S.; Devito M. J.; Fiedler H.; Falandysz J.; Rose M.; Schrenk D.; Safe S.; Tohyama C.; et al. Polybrominated Dibenzo-p-Dioxins, Dibenzofurans, and Biphenyls: Inclusion in the Toxicity Equivalency Factor Concept for Dioxin-like Compounds. Toxicol. Sci. 2013, 133, 197–208. 10.1093/toxsci/kft070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machala M.; Vondrácek J.; Bláha L.; Ciganek M.; Neca J. V. Aryl Hydrocarbon Receptor-Mediated Activity of Mutagenic Polycyclic Aromatic Hydrocarbons Determined Using in Vitro Reporter Gene Assay. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2001, 497 (1–2), 49–62. 10.1016/S1383-5718(01)00240-6. [DOI] [PubMed] [Google Scholar]
- Luzardo O. P.; Mahtani V.; Troyano J. M.; Álvarez de la Rosa M.; Padilla-Pérez A. I.; Zumbado M.; Almeida M.; Burillo-Putze G.; Boada C.; Boada L. D.; et al. Determinants of Organochlorine Levels Detectable in the Amniotic Fluid of Women from Tenerife Island (Canary Islands, Spain). Environ. Res. 2009, 109, 607–613. 10.1016/j.envres.2009.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.