

SUMMARY
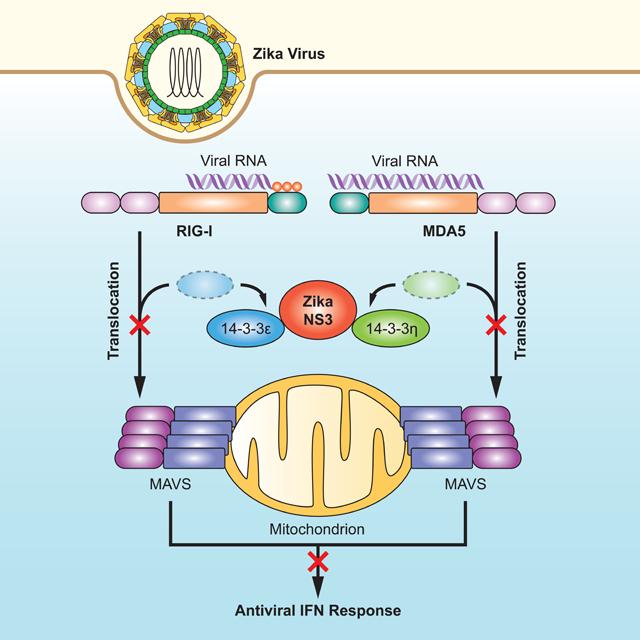
14–3-3 protein family members facilitate the translocation of RIG-I-like receptors (RLRs) to organelles that mediate downstream RLR signaling, leading to interferon production. 14–3-3ε promotes the cytosolic-to-mitochondrial translocation of RIG-I, while 14–3-3η facilitates MDA5 translocation to mitochondria. We show that the NS3 protein of Zika virus (ZIKV) antagonizes antiviral gene induction by RIG-I and MDA5 by binding to and sequestering the scaffold proteins 14–3-3ε and 14–3-3η. 14–3-3-binding is mediated by a negatively-charged RLDP motif in NS3 that is conserved in ZIKV strains of African and Asian lineages and similar to the one found in dengue and West Nile viruses. ZIKV NS3 is sufficient to inhibit the RLR-14–3-3ε/η interaction and to suppress antiviral signaling. Mutational perturbation of 14–3-3ε/η binding in a recombinant ZIKV leads to enhanced innate immune responses and impaired growth kinetics. Our study provides molecular understanding of immune evasion functions of ZIKV, which may guide vaccine and anti-flaviviral therapy development.
Graphical Abstract
eTOC Blurb:
14–3-3 protein family members facilitate the translocation of RIG-I-like receptors to signaling organelles. Riedl et al. show that the NS3 protein of Zika virus physically interacts with the scaffolding proteins 14–3-3ε and 14–3-3η to block cytosol-to-mitochondria translocation of the sensors RIG-I and MDA5 and thereby evade antiviral innate immune responses.
INTRODUCTION
Mosquito-transmitted viral pathogens cause significant morbidity and mortality in humans on a global scale (Ferguson, 2018; Mackenzie et al., 2004). Among these pathogens are members of the Flavivirus genus, such as dengue virus (DENV), West Nile virus (WNV) and the recently emergent Zika virus (ZIKV), which can cause a variety of severe diseases in humans including hemorrhagic shock syndrome (DENV), encephalitis (WNV), and congenital abnormalities in the fetus (ZIKV) (Olagnier et al., 2016; Pierson and Diamond, 2018). Flaviviruses are positive-sense single-stranded RNA viruses that replicate in a variety of cell types, where the virus replication complexes are formed at the endoplasmic reticulum (Hasan et al., 2018; Mukhopadhyay et al., 2005; Neufeldt et al., 2018). For example, ZIKV can replicate in human astrocytes, neuronal cells, and specific cells of the genital tract (both in females and males) in vivo, and in many different cell types in vitro (Miner and Diamond, 2017). The positive-sense RNA genome is translated directly into one large polyprotein which, upon proteolytic cleavage by host and viral enzymes, gives rise to 10 viral proteins [3 structural and 7 nonstructural (NS) proteins]. The NS proteins (NS1–5) have enzymatic activities that are crucial for distinct steps in the virus lifecycle (Mukhopadhyay et al., 2005). In addition, some of the NS proteins of ZIKV (and also other flaviviruses) antagonize the innate immune response (e.g. NS5 antagonizes STAT2), thereby indirectly promoting virus replication (Grant et al., 2016; Wu et al., 2017). No licensed antivirals or vaccines are currently available for ZIKV or most other flaviviruses. As such, understanding the molecular details of ZIKV replication and pathogenesis and viral interactions with the human host is crucial for developing effective countermeasures against ZIKV-induced diseases.
Among the many different factors that contribute to ZIKV-induced pathogenesis are viral interactions with the mammalian type I interferon (IFN) response, which is essential for the restriction of ZIKV infection both in vitro and in vivo (Lazear et al., 2016; Yockey et al., 2018). Several innate immune sensors and key transcription factors of IFN induction are crucial for ZIKV restriction (Aliota et al., 2016; Lazear et al., 2016; Tripathi et al., 2017). Among those detecting ZIKV infection are the cytoplasmic RNA sensors RIG-I and MDA5 of the RIG-I-like receptor (RLR) family, which initiate an IFN response via TBK1, IRF3, and IRF7, ultimately leading to the transcriptional upregulation of a large suite of IFN-stimulated genes (ISGs) (Bowen et al., 2017; Chazal et al., 2018; Hertzog et al., 2018). Recently, 14–3-3 protein family members have been implicated in innate immunity, where they function to ‘translocate’ RLR sensors to signaling-permitting organelles. Specifically, 14–3-3ε promotes the cytosolic-to-mitochondrial translocation of RIG-I, whereas 14–3-3η facilitates MDA5 translocation to mitochondria, thereby promoting antiviral IFN induction (Lin et al., 2019; Liu et al., 2012).
Given the crucial role of the type I IFN response in restricting ZIKV infection, it is not surprising that ZIKV has evolved to suppress or delay IFN production and IFN-induced signaling, analogous to other flaviviruses. However, the molecular mechanisms and physiological relevance of IFN antagonism by ZIKV have just begun to be elucidated. Here, we show that the NS3 protein of ZIKV antagonizes RIG-I- and MDA5-mediated signaling via molecular mimicry of a cellular 14–3-3-binding motif. A recombinant ZIKV encoding a mutant NS3 protein in which 14–3-3ε/η binding was ablated showed attenuated replication capacity and stimulated elevated innate immune responses.
RESULTS
14–3-3ε and 14–3-3η are critical for the IFN-mediated antiviral response against ZIKV
Members of the 14–3-3 protein family regulate numerous intracellular processes, such as cell cycling, transcription, apoptosis and immunity (Tzivion and Avruch, 2002). 14–3-3ε is an essential component of the TRIM25- and RIG-I-mediated innate immune response to infection with several RNA viruses such as Sendai (SeV) and hepatitis C (HCV) viruses (Liu et al., 2012). Furthermore, recent research showed that 14–3-3η promoted antiviral signaling by MDA5 in the context of HCV infection (Lin et al., 2019). As such, we tested whether 14–3-3ε and 14–3-3η can restrict ZIKV infection in immortalized human fetal astrocytes (SVGA), which are permissive to ZIKV replication and express several key innate immune sensors important for ZIKV detection (Figures S1A and S1B). Ectopic expression of 14–3-3ε or 14–3-3η significantly restricted ZIKV infection in SVGA cells, whereas 14–3-3σ did not (Figure 1A). Ectopically expressed 14–3-3ε or 14–3-3η suppressed ZIKV replication to similar levels as ectopically expressed RIG-I or MDA5 (Figure 1B), suggesting that the anti-ZIKV restriction activity seen for 14–3-3ε and 14–3-3η might be due to their roles in RLR signaling. To test this directly, we determined the effect of 14–3-3ε or 14–3-3η gene silencing on the abundance of ZIKV-induced cytokine, ISG and chemokine transcripts in infected SVGA cells. In parallel, RIG-I and MDA5 also were silenced (Figures 1C and D). Knockdown of endogenous 14–3-3σ, which is not involved in RLR signaling (Lin et al., 2019; Liu et al., 2012), served as an additional control. In agreement with previous work (Esser-Nobis et al., 2019; Hertzog et al., 2018; Ma et al., 2018), RIG-I or MDA5 depletion strongly diminished IFNB1, CCL5 and ISG (IFIT2 and RSAD2) transcript induction upon ZIKV infection in SVGA cells. Similarly, 14–3-3ε and 14–3-3η silencing also markedly reduced virus-induced antiviral gene induction, but 14–3-3σ knockdown did not (Figures 1C and D). Together, these results indicate that 14–3-3ε and 14–3-3η are crucial for cytokine and ISG induction during ZIKV infection, which ultimately inhibits ZIKV replication.
Figure 1. 14–3-3ε and 14–3-3η inhibit ZIKV replication by mediating antiviral gene expression.

(A) Viral titers in the supernatant of SVGA cells that were transfected with empty vector or the indicated FLAG-tagged 14–3-3 proteins and then infected with ZIKV (BRA/2015; MOI 0.1) for the indicated times, determined by plaque assay and presented as PFU/mL.
(B) Frequency of infected A549 cells that were transfected with either empty vector or plasmids encoding the indicated FLAG-tagged proteins for 42 h and then infected with ZIKV (BRA/2015; MOI 0.2) for 48 h, assessed by flow cytometry using an antibody recognizing the ZIKV E protein.
(C) qRT-PCR analysis of the indicated transcripts in SVGA cells that were transfected with the indicated siRNAs for 24 h and then infected with ZIKV (BRA/2015; MOI 1) for 24 or 48 h.
(D) Silencing efficiency of indicated genes for the experiment shown in (C), determined by qRT-PCR and normalized to cellular GAPDH.
Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 [Student’s t-test in (A)]; *p < 0.01, **p < 0.001, ***p < 0.0001 [ANOVA in (B, C)]. Data are representative of two (A, C, D) or three (B) independent experiments. See also Figure S1.
ZIKV NS3 interacts with 14–3-3ε using a phosphomimetic RLDP motif and competes with RIG-I for 14–3-3ε binding
Key molecules in innate immune signaling are targets of viral antagonism (Beachboard and Horner, 2016; Chan and Gack, 2016b). As such, one might expect that ZIKV antagonizes RLR signaling by either targeting the sensors or the regulatory proteins critical for the RLR response. Indeed, the NS3 protein of DENV evades RIG-I signaling in a proteolysis-independent manner by binding to 14–3-3ε using a negatively-charged 64-RxEP-67 motif that mimics a phosphorylatable motif found in cellular 14–3-3 interactors (Chan and Gack, 2016a). We evaluated whether ZIKV, which is closely related to DENV, contains a similar phosphomimetic motif in its NS3 protein. Sequence alignment of the NS3 proteins from different ZIKV strains, including the prototypical African strain isolated in Uganda in 1947 (MR 766) and the more recent epidemic French Polynesian (H/PF/2013) and Brazilian (Brazil Paraiba 2015; hereafter referred to as BRA/2015) strains, revealed that the NS3 proteins of all strains analyzed (total of 673) encode a potential phosphomimetic 14–3-3-binding motif, 64-RLDP-67, containing a central negatively-charged Asp residue (D66) (Figures 2A and S2A). Notably, the motif found in ZIKV NS3 is identical to that found in WNV NS3 at the corresponding position (RLDP), and similar to the one in DENV NS3, which harbors a phosphomimetic Glu (E66) residue instead of D66 (motif RIEP or RLEP) (Chan and Gack, 2016a). In contrast, none of the analyzed NS3 proteins from yellow fever (YFV), Japanese encephalitis (JEV), tick-borne encephalitis (TBEV) viruses, or the distantly related HCV encoded a phosphomimetic RxE/DP motif at the corresponding location (Figures 2A and S2A).
Figure 2. ZIKV NS3 binds to human 14–3-3ε and competes with RIG-I for the 14–3-3ε interaction.

(A) Amino acid sequence of the NS3 region containing the phosphomimetic 14–3-3-interaction motif Rx(E/D)P (residues 64–67; red) in ZIKV, DENV and WNV, as well as the corresponding NS3 regions in the related Flaviviridae family members JEV, YFV, TBEV, and HCV (blue). Representative virus strains are shown (see Figure S2A for a summary of the complete sequence analysis). Of note, cladogram shown is not to scale.
(B) Upper panel: Coomassie-stained complexes of affinity-purified FLAG-tagged ZIKV NS3 (H/PF/2013) from transiently transfected HEK 293T cells using anti-FLAG agarose. Purified FLAG-tagged ZIKV NS1 (H/PF/2013) as well as cells transfected with empty vector served as controls. Asterisks denote the FLAG-NS1 and FLAG-NS3 proteins. Arrow indicates the band that identified 14–3-3ε and 14–3-3η by MS analysis. Lc, antibody light chain. Hc, antibody heavy chain.
Lower panel: Anti-FLAG immunoblot (IB) analysis of affinity-purified FLAG-tagged NS3 and NS1 from the complexes shown above. Asterisks denote the FLAG-NS1 and FLAG-NS3 proteins.
(C) Binding of GST-NS3 from ZIKV (H/PF/2013) and DENV (NGC; positive control) or GST (negative control) to HA-tagged 14–3-3ɛ in HEK 293T cells transiently transfected for 42 h to express those proteins, determined by GST-pulldown (PD) and IB using anti-HA and anti-GST.
(D) Interaction of NS3 with endogenous 14–3-3ε in SVGA cells infected with ZIKV (BRA/2015; MOI 1) for 48 h, assessed by immunoprecipitation (IP) with anti-14–3-3ε, followed by IB with anti-NS3 and anti-14–3-3ε. Whole cell lysates (WCLs) were probed with anti-NS3, anti-14–3-3ε and anti-Actin (loading control).
(E) Binding of GST-NS3 from the indicated ZIKV strains to endogenous 14–3-3ε in HEK 293T cells transfected for 42 h to express those proteins, assessed by GST-PD and IB using anti-14–3-3ε and anti-GST. Cells transfected with GST-NS3 from YFV (strain 17D) served as negative control.
(F) Binding of GST-fused ZIKV NS3 (BRA/2015) WT and KIKP to endogenous 14–3-3ε in transiently transfected HEK 293T cells, determined as in (E).
(G) Binding of endogenous 14–3-3ε to FLAG-RIG-I in HEK 293T cells that were co-transfected with either vector or increasing amounts of GST-NS3 from ZIKV (BRA/2015) for 42 h and then infected with SeV (50 HAU/mL) for 22 h, determined by FLAG-PD and IB with anti-14–3-3ε and anti-FLAG. WCLs were immunoblotted with anti-GST, anti-14–3-3ε and anti-Actin (loading control).
Data are representative of one (B), two (C), four (D–F), or three (G) independent experiments. See also Figure S2.
The presence of a potential 14–3-3-interaction motif in ZIKV NS3 led us to hypothesize that ZIKV NS3 might interact with 14–3-3ε and thereby antagonize the RIG-I signaling pathway. Indeed, mass spectrometry (MS) analysis of affinity-purified FLAG-NS3 from ZIKV (strain H/PF/2013) identified 14–3-3ε as interaction partner, visible as a ~30 kDa band present only in the ZIKV NS3 sample but not vector or ZIKV NS1 controls (Figures 2B and S2B). Notably, MS analysis of this band also identified 14–3-3η, although with lower peptide frequency than for 14–3-3ε (Figure S2B). Co-immunoprecipitation (co-IP) confirmed that GST-fused NS3 (ZIKV, H/PF/2013) efficiently bound to HA-tagged 14–3-3ε, as did GST-NS3 from DENV (Figure 2C). In support of these findings, the NS3 protein from ZIKV (BRA/2015 strain) also readily bound to endogenous 14–3-3ε during infection of SVGA astrocytes, where their binding increased with augmented expression of NS3 during the course of infection (Figures 2D and S2C). As the prototypical African lineage strain MR 766 differs in pathogenesis from more recent, epidemic ZIKV isolates (of the Asian lineage) (Pierson and Diamond, 2018), we assessed whether the NS3 proteins from different ZIKV strains varied in their 14–3-3ε-binding capacity. In contrast to YFV NS3, which does not encode a 14–3-3-binding motif and thus served as a negative control, the NS3 proteins of all three ZIKV strains tested (MR 766, H/PF/2013 and BRA/2015) bound to endogenous 14–3-3ε (Figure 2E), suggesting that 14–3-3ε binding is a conserved feature of multiple ZIKV strains. To test whether the ZIKV NS3–14-3–3ε interaction requires the phosphomimetic motif, we mutated 64-RLDP-67 to 64-KIKP-67 in ZIKV NS3 (termed hereafter NS3 KIKP), analogous to our 14–3-3ε-binding-deficient DENV NS3 KIKP mutant (Chan and Gack, 2016a), by substituting R64 and the negatively-charged D66 residue for positively-charged lysine (K) residues, and in addition, mutating L65 to I65. In contrast to the wild-type (WT) ZIKV NS3 protein, which efficiently bound 14–3-3ε, NS3 KIKP showed reduced 14–3-3ε-binding capacity (Figure 2F), indicating that the RLDP motif in ZIKV NS3 is important for the interaction with 14–3-3ε. Finally, since phosphomimetic motifs have been shown to outcompete cellular (usually phosphorylated) 14–3-3-interacting partners (Chan and Gack, 2016a), we tested whether ZIKV NS3 competes with RIG-I for 14–3-3ε interaction. A competitive binding assay showed that RIG-I and 14–3-3ε interacted upon infection with SeV, a virus that stimulates RIG-I signaling, as previously shown (Liu et al., 2012). However, in the presence of ZIKV NS3, the binding between RIG-I and 14–3-3ε was inhibited in a dose-dependent manner (Figure 2G), suggesting that ZIKV NS3 disrupted the physical interaction of the two host proteins. Collectively, these data indicate that ZIKV NS3 encodes a phosphomimetic RLDP motif that allows it to compete with RIG-I for 14–3-3ε binding.
ZIKV NS3 inhibits the translocation of RIG-I from the cytosol to mitochondria and antiviral signaling
As 14–3-3ε has been shown to mediate the translocation of RIG-I from the cytosol to mitochondria and mitochondria-associated membranes for promoting antiviral signaling (Liu et al., 2012), we assessed endogenous RIG-I protein abundance in the cytosolic and mitochondrial fractions of ZIKV-infected SVGA cells (Figure 3A). Compared to SeV infection, infection with ZIKV only minimally induced mitochondrial translocation of RIG-I; RIG-I remained primarily in the cytosol during ZIKV infection (Figure 3A), suggesting that RIG-I translocation and thereby its downstream signaling are inhibited by ZIKV. GST-NS3 from ZIKV (strain H/PF/2013) suppressed SeV-induced activation of an IFN-β-promoter-driven luciferase activity in a dose-dependent manner (Figure 3B), indicating that NS3 of ZIKV is sufficient to block RIG-I signaling and further suggesting that this antagonism is independent of the proteolytic activity of NS3, which requires co-expression of the viral NS2B protein (Kang et al., 2017). Similarly, ZIKV NS3 (strain H/PF/2013) suppressed IFN-β-promoter activation following ectopic expression of RIG-I-2CARD, the constitutively active form of RIG-I (Gack et al., 2007) (Figure 3C). Consistent with their ability to efficiently bind 14–3-3ε, GST-NS3 from ZIKV strains MR 766, H/PF/2013 and BRA/2015 also suppressed SeV-mediated IFN-β promoter activation (Figure 3D). Expression of ZIKV NS3 also reduced the protein expression of ISG15, ISG54, and RIG-I (itself an ISG) stimulated by SeV, as did NS3 from DENV (Figure 3E). Moreover, SeV-induced MAVS filament formation was impaired in cells that ectopically expressed ZIKV NS3 (Figure 3F). In comparison, 14–3-3-binding-deficient ZIKV NS3 KIKP minimally blocked endogenous MAVS filament formation (Figure 3F), which supports the idea that RIG-I-signal inhibition by NS3 depends on the RLDP motif. Finally, to corroborate that ZIKV NS3-mediated antagonism primarily occurs at the level of RIG-I, and not downstream of it, we compared the effect of ZIKV NS3 expression on signaling induced by ectopic expression of RIG-I 2CARD or MAVS, which are each sufficient to activate downstream signaling when overexpressed. ZIKV NS3 dampened IFN-β promoter activation induced by RIG-I 2CARD, but had no effect on IFN-β luciferase activity mediated by FLAG-MAVS (Figure 3G). Taken together, these results indicate that the NS3 protein of ZIKV interferes with the cytosol-to-mitochondria translocation step of RIG-I, thereby dampening downstream signaling and antiviral gene expression.
Figure 3. NS3 of ZIKV inhibits the RIG-I signaling pathway.

(A) Upper panels: IB analysis of endogenous RIG-I in the cytosolic and mitochondrial fractions of SVGA cells that were mock-infected, or infected with SeV (50 HAU/mL) or ZIKV (BRA/2015, MOI 1) for 24 h. IB analysis of GAPDH (cytosolic protein) and MAVS (mitochondrial protein) served as controls for fraction purity. Lower panels: WCLs were probed with anti-RIG-I, anti-NS3 and anti-GAPDH.
(B) Upper panel: IFN-β luciferase activity in HEK 293T cells transfected for 40 h with a IFN-β luciferase reporter plasmid and GST (negative control), GST-NS3 from DENV (NGC; positive control) or increasing amounts of GST-NS3 from ZIKV (H/PF/2013), and then infected with SeV (50 HAU/mL) for 20 h. Luciferase activity was normalized to values for co-transfected β-galactosidase and presented relative to uninfected GST-expressing cells, set to 1.
Lower panel: IB analysis of WCLs with anti-GST and anti-Actin.
(C) IFN-β luciferase activity and IB analysis of HEK 293T cells transfected for 30 h with IFN-β luciferase reporter plasmid and RIG-I 2CARD together with GST (negative control), GSTNS3 from DENV (NGC; positive control), or increasing amounts of GST-NS3 from ZIKV (H/PF/2013), determined as in (B).
(D) IFN-β luciferase activity and IB analysis of HEK 293T cells transfected for 40 h with GST (negative control) or GST-NS3 from DENV (NGC; positive control) or the indicated ZIKV strains and then infected with SeV (50 HAU/mL) for 20 h, determined as in (B).
(E) Endogenous ISG (ISG15, ISG54 and RIG-I) or actin (loading control) protein abundances in HEK 293T cells that were transfected for 48 h with GST (negative control), or GST-NS3 from ZIKV (H/PF/2013) or DENV (NGC; positive control) and then infected with SeV (50 HAU/mL) for 24 h, determined by IB with the indicated antibodies.
(F) Endogenous MAVS filament formation in HEK 293T cells that were transfected with either GST or GST-NS3 (ZIKV) WT or KIKP for 24 h and then infected with SeV (100 HAU/mL) for 14 h, determined by semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) of mitochondrial extracts. WCLs were further analyzed by SDS-PAGE and IB with anti-MAVS, anti-GST, and anti-Actin (loading control).
(G) Upper panel: IFN-β luciferase activity of HEK 293T cells transfected for 30 h with GST (negative control) or GST-NS3 from ZIKV(H/PF/2013) and either empty vector, FLAG-MAVS or RIG-I 2CARD, determined as in (B).
Lower panel: IB analysis of WCLs with anti-GST and anti-Actin (loading control).
Data are expressed as means ± SD (n = 3). *p < 0.01, **p < 0.001, ***p < 0.0001 [ANOVA in (B–D)] or *p < 0.05, **p < 0.01, ***p < 0.001 [Student’s t-test in (G)]. ns, not statistically significant. Data are representative of three (A, D, F, G) or two (B, C, E) independent experiments.
ZIKV NS3 antagonizes MDA5 signaling through binding to 14–3-3η
As 14–3-3η was important for ZIKV-mediated cytokine induction (Figure 1C) and since our MS analysis also identified 14–3-3η as a ZIKV NS3-binding partner (Figures 2B and S2B), we hypothesized that ZIKV NS3 might antagonize the function of 14–3-3η in the MDA5 signaling pathway. Indeed, ZIKV NS3 expression effectively downregulated MDA5-mediated IFNB1, IFNL1, CCL5 and ISG (RSAD2, MX1, OAS1) transcript induction in a dose-dependent manner, although not as potently as the V protein of measles virus (MeV V), a well-characterized MDA5 antagonist (Davis et al., 2014) (Figure 4A). In Co-IP studies, ZIKV NS3 bound to 14–3-3η, but not 14–3-3σ (Figure 4B). NS3 also efficiently interacted with endogenous 14–3-3η in SVGA cells throughout a 72 h-time course of ZIKV infection (Figure 4C). The NS3–14-3–3η interaction was dependent on the 64-RLDP-67 motif in ZIKV NS3, as GST-NS3 WT, but not GST-NS3 KIKP, bound to 14–3-3η (Figure 4D). Finally, ZIKV NS3 suppressed the interaction between HA-14–3-3η and FLAG-MDA5 in a dose-dependent manner, suggesting a competitive binding mode (Figure 4E). Of note, the NS3 proteins of DENV and WNV also antagonized MDA5 2CARD-mediated signaling and efficiently bound to 14–3-3η, whereby the flavivirus NS3 proteins showed differential 14–3-3η-binding and MDA5-inhibitory capacities (Figures S3A and S3B). Together, these results indicate that through binding to 14–3-3η, ZIKV NS3 antagonizes MDA5-mediated antiviral gene expression. Furthermore, these data suggest that MDA5/14–3-3η antagonism is conserved also in DENV and WNV NS3, although further studies will be needed to determine the relevance of 14–3-3η targeting for efficient replication and IFN antagonism for DENV and WNV.
Figure 4. ZIKV NS3 antagonizes MDA5-mediated immune signaling by binding to 14–3-3η.

(A) qRT-PCR analysis of the indicated genes in HeLa cells that were transfected for 24 h with FLAG-MDA5 together with GST (negative control), increasing amounts of GST-NS3 from ZIKV (BRA/2015), or MeV V (positive control).
(B) Binding of NS3 in SVGA cells that were transfected for 40 h with empty vector or FLAG-tagged 14–3-3η or 14–3-3σ and then infected with ZIKV (BRA/2015; MOI 1) for 48 h, determined by FLAG-PD and IB with anti-NS3 and anti-FLAG. WCLs were further probed with anti-NS3 and anti-Actin (loading control).
(C) Interaction of endogenous 14–3-3η with ZIKV NS3 in SVGA cells infected with ZIKV (BRA/2015; MOI 1) for the indicated times, determined by IP with anti-NS3, followed by IB with anti-14–3-3η and anti-NS3. WCLs were further probed with anti-14–3-3η and anti-Actin (loading control).
(D) Binding of FLAG-14–3-3η to GST (negative control) or GST-fused ZIKV NS3 (BRA/2015) WT and KIKP in transiently transfected HEK 293T cells, determined by GST-PD and IB with anti-FLAG and anti-GST.
(E) Binding of HA-14–3-3η to FLAG-MDA5 in HEK 293T cells that were co-transfected with either vector or increasing amounts of GST-NS3 from ZIKV (BRA/2015) for 24 h and then treated with 5 μg/mL HMW-Poly(I:C)/Lyovec for 20 h, determined by HA-PD and IB with anti-FLAG and anti-HA. WCLs were immunoblotted with anti-FLAG and anti-GST.
Data are expressed as means ± SD (n = 3). *p < 0.01, **p < 0.001, ***p < 0.0001 (ANOVA). Data are representative of two (A, C, E) or three (B, D) independent experiments. See also Figure S3.
A recombinant ZIKV encoding a 14–3-3-binding-deficient NS3 protein shows diminished replication and IFN antagonism
To further define the relevance of 14–3-3ε/η binding in antagonism of the IFN response, we generated a recombinant ZIKV (strain MR 766), termed hereafter ZIKV(KIKP), by introducing the mutations R64→ K64, L65→I65, D66→K66 into NS3. We first compared the replication of ZIKV(KIKP) and the parental virus [ZIKV(WT)] in Vero cells, a cell line deficient in type I IFN genes (Desmyter et al., 1968), and found only minor differences between the growth of both viruses over a 72 hour-time course (Figure S4A), ruling out a general growth defect of ZIKV(KIKP). We next analyzed the growth of ZIKV(WT) and ZIKV(KIKP) in A549 cells, a human lung epithelial cancer cell line that has functional IFN signaling. We found ~100-fold difference in titers between ZIKV(WT) and ZIKV(KIKP) at 24 h post-infection. The difference in titers between ZIKV(WT) and ZIKV(KIKP) gradually decreased at later time points, likely due to saturation of ZIKV(WT) replication at these times (Figure 5A). Moreover, the percentage of ZIKV(KIKP)-infected cells was significantly lower than ZIKV(WT)-infected cells (Figure S4B). We observed that in A549 cells ZIKV(KIKP) infection led to greatly enhanced cytokine (IFNB1, IL6, IL8 and TNF), CCL5 and ISG (IFIT1, MX1 and RSAD2) transcript expression in comparison to infection with ZIKV(WT), despite the attenuated growth phenotype (Figure 5B). ZIKV(KIKP) also elicited higher cytokine, chemokine and/or ISG induction in SVGA (astrocytes) and HMC3 (microglia) cells than ZIKV(WT) (Figures 5C and S4C). To address whether ZIKV(KIKP), in contrast to ZIKV(WT), is unable to block the translocation of RIG-I and MDA5 to the mitochondria, we infected SVGA cells with ZIKV(WT) or ZIKV(KIKP) and assessed protein abundances for these two sensors in the cytosol and mitochondrial fractions by immunoblot analysis. Cells infected with ZIKV(WT) showed low amounts of RIG-I and MDA5 in the mitochondrial fraction, which were similar to mock-infected cells; in contrast, ZIKV(KIKP)-infected cells exhibited greater abundance of RIG-I and MDA5 at the mitochondria (Figures 5D and E). Finally, to confirm that the growth attenuation of ZIKV(KIKP) was due to greater activation of RLR signaling, we infected SVGA cells in which the RLR adapter MAVS was knocked-out using CRISPR-Cas9 gene editing (SVGA MAVS KO), or cells expressing a nontargeting (NT) guide RNA (Figures S4D and S4E). In NT control cells, ZIKV(KIKP) was growth-attenuated in comparison to ZIKV(WT), whereas there was no significant difference between the growth of the two viruses in MAVS KO SVGA cells (Figure 5F). This indicates that the major attenuating feature of ZIKV(KIKP) is its inability to antagonize RLR signaling, and further that the RLR-MAVS axis is a major sensing pathway of ZIKV infection in these cells.
Figure 5. A recombinant ZIKV expressing a 14–3-3-binding-deficient NS3 protein is growth-attenuated and shows diminished RLR antagonism.

(A) Viral titers in the supernatant of A549 cells infected with ZIKV(WT) or ZIKV(KIKP) (MOI 0.001 for each) for the indicated times, determined by plaque assay (limit of detection: 102) and presented as PFU/mL.
(B) qRT-PCR analysis of the indicated transcripts in A549 cells that were infected with ZIKV(WT) or ZIKV(KIKP) (MOI 0.5 for each) for the indicated times.
(C) qRT-PCR analysis of the indicated transcripts in SVGA cells that were infected with ZIKV(WT) or ZIKV(KIKP) (MOI 0.1, 0.5, or 1 for each) for 48 h.
(D) –(E) Upper panels: IB analysis of endogenous RIG-I (D) or MDA5 (E) in the cytosolic and mitochondrial fractions of SVGA cells that were mock-infected, or infected with ZIKV(WT) or ZIKV(KIKP) (MOI 0.1 for each) for 24 h. IB analysis of GAPDH (cytosolic protein) and MAVS (mitochondrial protein) served as controls of fraction purity. Lower panels: WCLs were probed with anti-RIG-I (D), anti-MDA5 (E), as well as anti-NS3 and anti-GAPDH (D, E). Asterisk denotes non-specific band (D).
(F) Viral titers in the supernatant of NT control and MAVS KO SVGA cells (clones 1 and 2) infected with ZIKV(WT) or ZIKV(KIKP) (MOI 0.01 for each) for 36 h, determined by plaque assay and presented as PFU/mL.
(G) Viral titers in the supernatant of NT control and MAVS KO SVGA cells (clone 2) transfected with the indicated siRNAs for 30 h and then infected with ZIKV(KIKP) (MOI 0.1) for 36 h, determined by plaque assay and presented as PFU/mL.
Data are expressed as means ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test). ns, not statistically significant. Data are representative of three (A), four (B) or two (C–G) independent experiments. See also Figures S4 and S5.
We reasoned that the use of ZIKV(KIKP), which is deficient in antagonism of RIG-I/14–3-3ε and MDA5/14–3-3η signaling, might allow us to reveal the relative contribution of the two antiviral pathways to ZIKV restriction. Individual depletion of RIG-I or MDA5 in SVGA cells enhanced ZIKV(KIKP) replication to a similar extent, as compared to transfection of nontargeting control siRNA (si.C), and silencing of both RIG-I and MDA5 had an additive effect on ZIKV(KIKP) replication (Figures S5A and S5B). Similarly, depletion of either 14–3-3ε or 14–3-3η detectably enhanced ZIKV(KIKP) replication, while their combined knockdown further increased ZIKV(KIKP) replication (Figures S5C and S5D). Together, these results indicate a critical and nonredundant role of RIG-I/14–3-3ε and MDA5/14–3-3η signaling in ZIKV restriction in astrocytes.
To corroborate that 14–3-3ε and 14–3-3η restrict ZIKV(KIKP) via the RLR-signaling pathway, we silenced both 14–3-3ε and 14–3-3η (si.14–3-3ε/η) in MAVS KO or NT control SVGA cells and infected with ZIKV(KIKP). Infection of cells that were transfected with si.C served as control. Whereas knockdown of both 14–3-3ε and 14–3-3η in NT cells enhanced ZIKV(KIKP) replication as compared to transfection of si.C, their combined knockdown did not lead to higher viral replication in MAVS KO cells (Figures 5G and S5E). Collectively, these results show that the RIG-I/14–3-3ε and MDA5/14–3-3η signaling axes restrict ZIKV infection; however, ZIKV NS3 effectively antagonizes both antiviral pathways. A recombinant ZIKV harboring a mutant NS3 protein that is deficient in 14–3-3ε/η binding induces elevated cytokine and ISG expression and has impaired growth kinetics.
DISCUSSION
Multiple studies have demonstrated that the type I IFN response is crucial for the restriction of ZIKV and related flaviviruses in mammalian hosts. As a consequence, these viruses have evolved sophisticated mechanisms to delay or suppress both IFN production and signaling via the IFNα/β receptor. Our work showed that the sensors RIG-I and MDA5 are important for cytokine and ISG induction upon ZIKV infection of astrocytes, which is consistent with previous data obtained from other cell types (Esser-Nobis et al., 2019; Hertzog et al., 2018; Ma et al., 2018). Whereas in certain cell types (e.g., A549 cells) RIG-I has a more dominant role in antiviral gene induction in response to ZIKV infection, compared to MDA5 (Esser-Nobis et al., 2019; Hertzog et al., 2018), our work showed a nonredundant role of RIG-I and MDA5 in IFN/ISG induction and ZIKV restriction in SVGA astrocytes, which is similar to the roles of RIG-I and MDA5 in human trophoblast cells (Ma et al., 2018). This suggests that the relative contribution of these sensors during ZIKV infection is cell type-dependent. It is conceivable that differences in RLR protein abundance, RLR-regulatory mechanisms, and/or viral PAMP production may determine the relative contribution of RIG-I and MDA5 to anti-ZIKV responses in specific cell types, which warrants further investigation. Moreover, while our study demonstrated that 14–3-3ε and 14–3-3η are critical for RLR-mediated ZIKV restriction, it is possible that other cellular processes mediated by these two 14–3-3 proteins also contribute to ZIKV restriction.
Our work also showed that ZIKV effectively antagonizes antiviral signaling by both RNA sensors using its NS3 protein. This mechanism of inhibition is independent of the proteolytic activity of the NS3 protein as NS3, without its cofactor NS2B, potently blocked MDA5- and RIG-I-mediated antiviral signaling. This RLR-evasion strategy mirrors that of the NS3 protein of the closely-related DENV (Chan and Gack, 2016a), and varies from that of the distantly-related Flaviviridae member, HCV, which uses the proteolytic activity of NS3/NS4A to cleave MAVS and block RIG-I signal transduction (Meylan et al., 2005). Moreover, DENV and also ZIKV NS3 (together with NS2B) use their proteolytic activity to cleave human STING, thereby blunting type I IFN gene expression mediated by the DNA sensor cGAS after mitophagy (Aguirre et al., 2012; Ding et al., 2018). Future studies will need to determine if there are cell-type differences in NS3 antagonism of these innate immune pathways, or whether NS3 targets 14–3-3ε, 14–3-3η and STING in a spatio-temporal dependent manner during infection.
14–3-3 proteins are highly conserved (from yeast to mammals) scaffold and adapter proteins that regulate numerous signaling pathways via direct interaction with proteins that encode phosphorylated 14–3-3-binding motifs (Yaffe et al., 1997). Recently, 14–3-3ε and 14–3-3η have been shown to promote antiviral innate immunity by participating in a multi-protein complex called the ‘RLR translocon’ that facilitates the redistribution of RLRs from the cytoplasm to their signaling platforms on mitochondria (Lin et al., 2019; Liu et al., 2012; Tzivion et al., 2001). Our data indicate that the NS3 protein of ZIKV employs molecular mimicry of a cellular 14–3-3-binding motif, specifically Rxx(pS/pT)P, to interact with 14–3-3ε and 14–3-3η, which prevents their binding to RIG-I and MDA5, respectively. Similar to what we observed for DENV NS3 (Chan and Gack, 2016a), the motif in ZIKV NS3 is shorter than classical motifs found in cellular 14–3-3-interacting proteins and, harbors a central negatively-charged residue (D66 in ZIKV and E66 in DENV) instead of a phosphorylatable Ser or Thr residue as found in Rxx(pS/pT)P. Whereas the interactions of 14–3-3 with host proteins are dynamically regulated by kinases and phosphatases that (de-)phosphorylate the Rxx(pS/pT)P (or similar motifs) in mammalian 14–3-3-binding proteins, the phosphomimetic motif in ZIKV NS3 is expected to constitutively be in the ‘binding ON’ state and thus, as indicated by our data, can outcompete RIG-I and MDA5 for 14–3-3ε/η binding. Sequence alignment of flavivirus NS3 proteins showed that the phosphomimetic 64-RxE/DP-67 motif is conserved in all analyzed strains of ZIKV and WNV (both encoding RLDP), and almost all DENV strains (harboring RI/LEP) (Chan and Gack, 2016a); thus, 14–3-3 binding is widely conserved among these three mosquito-transmitted flaviviruses. However, NS3 from YFV did not encode a phosphomimetic motif at the corresponding site, nor bound to 14–3-3ε/η in our co-IP assays, suggesting that YFV has evolved a 14–3-3-independent immune evasion mechanism, although this requires further investigation. Along these lines, NS3 proteins of JEV and TBEV also did not encode an equivalent RxE/DP motif at the same position in NS3. Future studies will be required to determine whether these flaviviruses suppress RLR-mediated immunity by 14–3-3-dependent or independent mechanisms.
Another major question that arises from our study is: Why do ZIKV, DENV and WNV directly antagonize 14–3-3 proteins rather than RLRs? The ability to bind specific 14–3-3 proteins may allow these viruses to manipulate not only RLR signaling but also modulate other cellular processes regulated by 14–3-3 proteins. An advantage of targeting 14–3-3 proteins might be that these proteins are widely conserved, even in insects, and that 14–3-3 orthologs share high sequence homology (e.g. the protein sequence of human and mouse 14–3-3ε is identical). Thus, 14–3-3 targeting may allow these viruses to modulate 14–3-3 functions both in mammalian and insect hosts to suppress cell-intrinsic immune responses and/or regulate other pathways that could promote virus replication. Along these lines, it is also tempting to speculate that binding of NS3 to 14–3-3ε/η may alter the activity of NS3, its localization, and/or interaction with other proteins, which has been shown for cellular 14–3-3-interacting proteins (Tzivion et al., 2001).
Taken together, our study shows that ZIKV NS3 targets RLR-trafficking proteins of the 14–3-3 family to perturb innate immune signal transduction. Infection studies with a recombinant ZIKV encoding a 14–3-3-binding-deficient NS3 protein demonstrated that specific disabling of this immune evasion mechanism of ZIKV results in attenuation of the virus due to an enhanced antiviral response. As the 14–3-3-targeting mechanism is conserved among ZIKV, DENV and WNV, a deeper mechanistic understanding of this immune evasion mechanism may reveal targets for broad-spectrum therapies, or guide strategies for attenuating these viruses for the design of new vaccines.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michaela U. Gack (mgack@uchicago.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK 293T (embryonic, female, human) (ATCC), Vero (adult, female, African Green monkey) (ATCC), A549 (age 58, male, human) (ATCC) and HeLa (age 30, female, human) (ATCC) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) and 1% (v/v) penicillin-streptomycin (Gibco). SVGA cells (embryonic, human, sex unidentified) (Ellen Cahir-McFarland; Cambridge, MA; (Major et al., 1985)) and HMC3 cells (embryonic, human, sex unidentified) (ATCC) were cultured in Minimum Essential Medium (Gibco) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin. All cells were cultured under standard culture conditions—5% CO2 at 37°C. Purchased cell lines were authenticated by the ATCC and were not validated further in our laboratory. Cell lines obtained and validated by other groups were not further authenticated.
Generation of CRISPR Gene-Edited SVGA Cells
To generate SVGA MAVS KO and control cell lines, specific guide RNAs (gRNAs) were designed using the CRISPR MIT tool (Zhang Lab, MIT). Non-targeting (NT) gRNA, (5′-gtggaaaggacgaaacaccgACGGAGGCTAAGCGTCGCAAgttttagagctagaaatag-3′) and MAVS gRNA (5′-gtggaaaggacgaaacaccGAGGTGGCCCGCAGTCGATCCgttttagagctagaaatag-3′ containing 5′ and 3′ overhangs of the pSicoR-CRISPR-PuroR vector (van de Weijer et al., 2014; van Diemen et al., 2016) were cloned into the vector at the BsmBI restriction site using Gibson assembly. Briefly, the digested vector, gRNA oligonucleotide, and Gibson master mix (NEB Biolabs, E2611) were incubated at 50°C for 4 h, followed by transformation into TOP10 competent E. coli cells. Plasmids were sequenced to confirm correct insertion of gRNAs, then transfected along with the third-generation lentivirus packaging plasmids pmDC gag/pol, pRSV rev, and pmDC VSV-G (kindly provided by Jae Jung, University of Southern California) into HEK 293T cells to produce lentiviral particles. Forty-eight hours post-infection, lentiviral particles were collected and used to transduce SVGA cells. Positive clones were selected by incubation in MEM containing 1 μg/mL puromycin for two weeks, followed by expansion of single cell-derived clones. Knockout of MAVS expression was validated by immunoblotting to confirm absence of the protein.
Viruses
ZIKV strain Brazil Paraiba 2015 (BRA/2015) (Sapparapu et al., 2016) was propagated in Vero cells following a protocol described previously (Brien et al., 2013). Briefly, 5 × 106 Vero cells were seeded in a T-175 flask, then inoculated the next day with ZIKV at an MOI of 0.001 in 7 mL FBS-free DMEM. 8 mL of DMEM (supplemented with 10% FBS) was added 2 h after infection. Three days post-infection, viral supernatant was removed and clarified by centrifugation at 800 × g for 10 min. ZIKV strain MR 766 was generated by transfecting HEK 293T cells with ZIKV strain MR 766 cDNA (Matthew Evans, Icahn School of Medicine at Mount Sinai), followed by harvesting virus-containing supernatant 3 days post-transfection as previously described (Schwarz et al., 2016). Recombinant ZIKV(KIKP) was generated as described in detail below. ZIKV strains were propagated and titered in Vero cells as described below. SeV (strain Cantell) was purchased from Charles River Laboratories.
METHOD DETAILS
Plasmids and reagents
FLAG-NS3 (ZIKV, strain MR 766) and FLAG-NS3 (ZIKV, strain H/PF/2013) were custom-synthesized by Invitrogen in the pcDNA3.1+ vector. GST-NS3 (ZIKV strains MR 766 and H/PF/2013) were generated by subcloning the respective FLAG-NS3 cDNAs into the pEBG vector (encoding an N-terminal Glutathione S-transferase (GST)) using KpnI and NotI restriction sites. GST-NS3 (ZIKV, strain BRA/2015) was generated by introducing two silent mutations (nucleotide base positions 852 A → G, 1734 C → T) into GST-NS3 (ZIKV, strain H/PF/2013) by site-directed mutagenesis. FLAG-14–3-3η was generated by cloning human cDNA into pEFBOS together with an N-terminal FLAG tag. HA-14–3-3η was generated by subcloning human 14–3-3η from the FLAG-14–3-3η construct into pQCXIP together with an N-terminal HA tag using the BamHI and NotI restriction sites. FLAG-NS1 (ZIKV, H/PF/2013) was kindly provided by Tom Hobman (University of Alberta) (Kumar et al., 2016). Plasmids encoding GST (pEBG), GST-NS3 (DENV, strain NGC), GST-NS3 (WNV, strain NY99), GST-NS3 (YFV, strain 17D), FLAG-14–3-3σ, FLAG-14–3-3ε, HA-14–3-3ε, FLAG-MAVS (Chan and Gack, 2016a), GSTRIG-I 2CARD, GST-MDA5 2CARD (Gack et al., 2007), FLAG-MDA5, FLAG-RIG-I (Wies et al., 2013), HA-MeV V (Davis et al., 2014), IFN-β luciferase reporter plasmid (Lin et al., 2000), and β-galactosidase-expressing plasmid (pGK-β-gal) (Hatzivassiliou et al., 1997) have been described. All constructs were sequenced to verify the original sequence. Transfections were performed using the calcium phosphate method, Lipofectamine and Plus reagent (Life Technologies), linear polyethylenimine (1 mg/mL solution in 20 mM Tris pH 6.8; Polysciences), or TransIT-LT1 (Mirus Bio) following manufacturer’s instructions.
Anti-FLAG M2 magnetic beads (Sigma), Glutathione Sepharose 4B resin (GE Life Sciences), protein A/G agarose (Thermo Fisher), anti-HA magnetic beads (Thermo Scientific), or Dynabeads Protein G (Invitrogen) beads were used for respective pulldown or immunoprecipitation assays. Protease inhibitor cocktail (#P2714, Sigma) was added at a concentration of 1:500 to cell lysates for all immunoblot and immunoprecipitation experiments. IFNa2 was purchased from PBL Biomedical Laboratories. High-molecular weight (HMW)-poly(I:C) / LyoVec was purchased from InvivoGen.
Cell lysis and co-immunoprecipitation
HEK 293T or SVGA cells were lysed in Nonidet P-40 (NP-40) buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1% (v/v) NP-40, 1 mM EDTA, 1:500 protease inhibitor cocktail) and then centrifuged at 21,000 × g for 20 min at 4°C. GST or FLAG pull-downs, coimmunoprecipitations and western blot analyses were performed as previously described (Gack et al., 2007). Briefly, lysates were subjected to pull-down with GST or FLAG beads for 4 h at 4°C, or lysates were subjected to immunoprecipitation with 2 μg primary antibody overnight at 4°C followed by incubation with protein A/G agarose or Dynabeads Protein G beads for 2 h at 4°C. A portion of the cell lysate was taken prior to pull-down or immunoprecipitation for analysis as a whole-cell lysate (WCL) fraction. Precipitated proteins were eluted from beads by heating samples in Laemmli SDS sample buffer at 95°C for 5 minutes.
Immunoblot analysis and antibodies
Precipitated protein or cell lysates were resolved on 7% or 10% Bis-Tris SDS-PAGE gels (pH 6.4), and then transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) using a Novex Semi-Dry Blotter (Invitrogen). Membranes were blocked with 5% (w/v) non-fat dry milk in PBS-T (PBS with 0.05% (v/v) Tween-20) for 30 min, and then probed with the indicated primary antibody in 2.5% (w/v) non-fat dry milk in PBS-T at 4°C overnight. Following overnight incubation, membranes were probed with horseradish peroxidase (HRP)-conjugated secondary antibodies in 2.5% (w/v) non-fat dry milk in PBS-T for 1 h at room temperature. Proteins were visualized using SuperSignal West Pico or SuperSignal West Femto chemiluminescence reagents (Thermo Scientific) and detected by an ImageQuant LAS 4000 Chemiluminescent Image Analyzer (General Electric).
The following antibodies were used for immunoblotting: anti-GST (1:2,000, GST-2, Sigma), anti-FLAG (1:2,000, M2, Sigma), anti-HA (1:2,000, HA-7, Sigma), anti-b-Actin (1:15,000, AC-15, Sigma), anti-RIG-I (1:2,000, Alme-1, Adipogen), anti-ISG15 (1:1000, F-9, Santa Cruz), anti-ISG54 (1:1,000, 25–735, ProSci), anti-14–3-3ε (1:1,000, 8C3, Santa Cruz), anti-14–3-3ε (1:1,500, N1C3, Genetex), anti-GAPDH (1:40,000, CS204254, Millipore), anti-MAVS (1:1,000, 3993σ, Cell Signaling Technology), anti-MB21D1/cGAS (1:1,000, HPA031700, Sigma), anti-STING (1:1,000, 723505, R&D Systems), and anti-TBK1 (1:1,000, D1B4, Cell Signaling Technology). A polyclonal anti-NS3 (ZIKV) antibody against the peptide CAALKSFKEFAAGKR was custom-generated by GenScript and was used for immunoblot analysis (1:1,500). A monoclonal anti-MDA5 antibody was purified from a mouse hybridoma cell line (1:500, Clone 17, provided by Jan Rehwinkel (Hertzog et al., 2018)). Mouse anti-4G2 antibody was purified from a mouse hybridoma cell line (1:50, D1–4G2–4-15, ATCC). Anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (both 1:2,000) were purchased from Cell Signaling Technology (7076S and 7074S, respectively). For immunoprecipitation of NS3, a polyclonal anti-NS3 (ZIKV) antibody against the peptide CRRGRIGRNPNKPGD was custom-generated by GenScript and was used at a concentration of 2 μg per sample. For immunoprecipitation of 14–3-3ε, anti-14–3-3ε (11648–2-AP, Proteintech) was used at a concentration of 2 μg per sample.
Large-scale pull-down and mass spectrometry
To identify cellular interaction partners for ZIKV NS3, twelve 10 cm-dishes of HEK 293T cells (~107 cells per dish) were each transfected with 20 μg of empty vector, FLAG-NS1 (ZIKV, strain H/PF/2013) or FLAG-NS3 (ZIKV, strain H/PF/2013). Forty-eight hours later, cells were lysed using RIPA buffer (150 mM NaCl, 1% (v/v) NP-40, 1% (w/v) deoxycholic acid, 0.01% (w/v) SDS, 20 mM Tris (pH 8.0), and protease inhibitor cocktail), followed by centrifugation at 21,000 × g for 20 min at 4°C. FLAG pulldown assay and SDS-PAGE were performed as described in (Full et al., 2019). To identify interacting proteins, samples were separated on a gradient gel (NuPAGE 4–12% Bis-Tris, Thermo Fisher) and then stained with GelCode Blue Stain Reagent (Thermo Fisher) overnight at room temperature, followed by destaining of the gel for 24 h in water at room temperature. Bands that were specifically present in the FLAG-NS3 sample, but not in the empty vector and FLAG-NS1 samples, were excised and analyzed by ion-trap MS at the Harvard Taplin Biological Mass Spectrometry Facility, Boston.
Luciferase reporter assay
HEK 293T cells were seeded into 12-well plates (~105 cells per well), and cells were transfected the following day with 300 ng of a β-galactosidase-expressing plasmid (pGK-β-gal), 200 ng of an IFN-β luciferase reporter plasmid, and 200 ng – 1.5 μg of plasmid encoding the indicated GST-NS3 constructs, or GST-expressing pEBG vector. To stimulate IFN-β luciferase activity, cells were infected with SeV (50 HAU/mL) ~42 h after transfection, or were co-transfected with 2.5 – 5 ng GST-RIG-I 2CARD, 100 ng of GST-MDA5 2CARD or 30 ng of FLAG-MAVS. Cells were harvested and luciferase activity (Promega) was measured at the indicated time points. Luciferase values were normalized to β-galactosidase activity to control for transfection efficiency, and fold luciferase activity was calculated relative to values for control cells, set to 1.
Cytosol-mitochondria fractionation assay
SVGA cells were infected with the indicated ZIKV strains or SeV at the indicated MOIs, or mock-infected. Twenty to twenty-four hours later, cells were harvested in an Isotonic Mitochondrial Buffer (MIT1000, Merck Millipore), and cytosolic and mitochondrial fractions were prepared. Briefly, cells were homogenized using a Dounce homogenizer, and WCLs were collected. Lysates were processed by low-speed centrifugation at 600 × g to pellet nuclei and unbroken cells. The supernatant was then centrifuged at 10,000 × g for 30 min at 4°C to pellet the mitochondrial fraction. The cytosolic (supernatant) and mitochondrial (pellet) fractions were subjected to a bicinchoninic acid (BCA) assay (Pierce), which was used to normalize protein amounts for SDS-PAGE and immunoblot analysis. Anti-GAPDH and anti-MAVS immunoblot analyses served as quality controls for relative cytosolic and mitochondrial fraction purity, respectively.
MAVS aggregation assay
To assess MAVS aggregation, HEK 293T cells (~2.5 × 106) were infected with SeV (100 HAU/mL) for 14 h and then lysed in a buffer containing 10 mM Tris-HCl (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 0.25 M D-mannitol, and EDTA-free protease inhibitor cocktail (Roche #11873580001) by repeated douncing. Cell lysates were centrifuged at 1000 × g for 5 min to remove cell debris, followed by a second centrifugation step at 10,000 × g for 10 min to separate the supernatant and the crude mitochondrial fraction, which was resuspended in 1x sample buffer (0.5x TBE, 10% (v/v) glycerol, 2% (w/v) SDS, and 0.0025% (w/v) bromophenol blue). Semidenaturing detergent agarose gel electrophoresis (SDD-AGE) was performed on the mitochondrial fraction using a 1.5% agarose gel (1x TBE, 0.1% SDS) at 100 V for 40 min at 4°C, followed by transfer onto a PVDF membrane (Bio-Rad) and immunoblotting with anti-MAVS antibody.
Generation of recombinant ZIKV(KIKP)
ZIKV(KIKP) (strain MR 766) was generated based on a ZIKV strain MR 766 cDNA clone (Schwarz et al., 2016). Site-directed mutagenesis by PCR was used to generate a mutant MR 766 cDNA clone encoding the mutations R64→K64, L65→I65, and D66→K66 in the gene encoding NS3. The WT and mutant infectious clone plasmids were transfected into HEK 293T cells using TransIT-LT1 transfection reagent (Mirus Bio) as previously described (Schwarz et al., 2016). Virus containing supernatant was collected 4 days post-transfection, and the WT and mutant viruses were further propagated in Vero cells. Supernatants containing amplified recombinant ZIKV(KIKP), or the parental virus, were serially diluted and titers were determined by standard plaque assay on Vero cells (as described below).
ZIKV infection and titration
For infection experiments, Vero, A549, or SVGA cell monolayers were washed once with PBS, and virus diluted in FBS-free DMEM was incubated with cells at 37°C for 2 h using the indicated MOI. Viral supernatant was removed, cells were washed twice with PBS, and cells were then incubated in complete growth media at 37°C for the indicated time points. For plaque assay, virus-containing samples were serially diluted in DMEM with 1% (v/v) penicillin-streptomycin, then incubated on a confluent monolayer of Vero cells at 37°C for 2 h. Infectious supernatant was removed and replaced with low-melt agarose, and then incubated for 3 days until plaques formed. Cells were fixed in 1% (w/v) paraformaldehyde (PFA) (in PBS) for 1 h at room temperature, and then stained with crystal violet (VWR) in 20% (v/v) ethanol. Plaques were rinsed with water, counted, and virus titers calculated as described previously (Brien et al., 2013). Briefly, dilution wells with distinct plaque formations were counted, and a plaque-forming unit per mL (PFU/mL) was calculated as (plaque count/well) × (dilution factor)/(volume of inoculum).
qRT-PCR
Total RNA was extracted from cells using an RNA extraction kit (OMEGA Bio-Tek) following the manufacturer’s instructions. Equal amounts of RNA (25 – 500 ng) were used in a one-step qRT-PCR reaction using the SuperScript III Platinum One-Step qRT-PCR kit with ROX (Invitrogen) and commercially available FAM reporter dye primers (IDT) for the genes analyzed. Gene expression was normalized to that of GAPDH. The comparative CT method (ΔΔCT) was used to measure the induction of each target gene relative to its induction in mock-infected cells. ZIKV genomic RNA was analyzed using a previously described primer against mRNA encoding the ZIKV E protein: 5′-CCACTAACGTTCTTTTGCAGACAT-3′ (forward), 5′-CCGCTGCCCAACACAAG-3′ (reverse), 5′-/56-FAM/AGCCTACCT/ZEN/TGACAAGCAATCAGACACTCAA/3IABkFQ/-3′ (probe) (Lanciotti et al., 2008). All qRT-PCR reactions were performed using a 7500 FAST Real-time PCR machine (Applied Biosystems).
Flow cytometry analysis
To determine the percentage of ZIKV-infected cells, infected A549 cells (~1 × 105 cells per well) were fixed in 1% (w/v) PFA (in PBS) (Santa Cruz) for 30 min at room temperature and then permeabilized with PermWash (BD Scientific) for 15 min. Cells were incubated with anti-ZV2 mouse monoclonal primary antibody [1:10 (in PermWash); (Zhao et al., 2016)] (Figure 1B), or with anti-4G2 mouse monoclonal primary antibody [1:50 (in PermWash), purified from D1–4G2–4-15 hybridoma cells (ATCC)] (Figure S4B) for 1 h at room temperature. Cells were washed twice with PBS-T and stained with a goat anti-mouse secondary Alexa Fluor 488 antibody (1:5,000, A-10667, Invitrogen) for 1 h at room temperature, washed again with PBS-T, and then resuspended in PBS for flow cytometry using a BD LSRFortessa. Analysis of flow cytometry data was performed using FlowJo software (Tree Star).
Gene silencing using siRNA
To transiently knockdown endogenous RIG-I, MDA5, 14–3-3ε, 14–3-3η, or 14–3-3σ expression in SVGA cells, cells were transfected in 12-well plates (~1 × 105 per well) with 50 nM of gene-specific siGENOME SMARTpool siRNAs (Dharmacon) using RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions. As a control, non-targeting siRNA (siGENOME Non-Targeting siRNA Pool #2, D-001206–14-05) was transfected. The following siRNAs were used: siRNAs targeting RIG-I (siGENOME SMARTpool M-012511–01-0005), MDA5 (siGENOME SMARTpool M-013041–00-0005), 14–3-3ε (siGENOME SMARTpool M-017302–03-005), 14–3-3η (siGENOME SMARTpool M-010626–01-0005), or 14–3-3σ (siGENOME SMARTpool M-005180–00-0005). Knockdown efficiency of these genes was determined by measuring transcript abundance at indicated times by qRT-PCR using specific, predesigned primers (IDT).
Flavivirus NS3 sequence analysis
The amino acid sequences of Flaviviridae family members were aligned using tools at viprbrc.org, and their NS3 sequences analyzed for the presence or absence of a potential 14–3-3 binding motif RxE/DP at the region corresponding to amino acids 64–67 in ZIKV NS3. All sequences were gathered from the NIAID Virus Pathogen Database and Analysis Resource (ViPR). Furthermore, the cladogram shown in Figure 2A was generated using anvi’o (version 5.5).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were presented as means ± SD and analyzed using GraphPad Prism software (version 7). One-way ANOVA with Dunnett’s multiple comparisons (P < 0.01 was considered statistically significant), or an unpaired two-tailed Student’s t-test (P < 0.05 was considered statistically significant) was used as indicated in the legends. Pre-specified effect sizes were not assumed, and in general three biological replicates (n) for each condition were used within independent experiments. Data were reproduced in independent experiments as indicated in the legends.
DATA AND CODE AVAILABILITY
No new data/code was generated in this study.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Glutathione-S-Transferase (GST) (clone GST-2) | Sigma-Aldrich | Cat# G1160, RRID:AB_259845 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| Mouse monoclonal anti-HA (clone HA-7) | Sigma-Aldrich | Cat# H9658, RRID:AB_260092 |
| Mouse monoclonal anti-β-Actin | GeneTex | Cat# GTX629630, RRID:AB_2728646 |
| Mouse monoclonal anti-RIG-I (clone alme-1) | Adipogen | Cat# AG-20B-0009, RRID:AB_2490189 |
| Mouse monoclonal anti-ISG15 (clone F-9) | Santa Cruz | Cat# sc-166755, RRID:AB_2126308 |
| Rabbit polyclonal anti-ISG54 | ProSci | Cat# 25–735, RRID:AB_10913015 |
| Mouse monoclonal anti-14–3-3ε (clone 8C3) | Santa Cruz | Cat# sc-23957, RRID:AB_626619 |
| Rabbit polyclonal anti-14–3-3ε (clone N1C3) | GeneTex | Cat# GTX109090, RRID:AB_1952629 |
| Rabbit polyclonal anti-MAVS | Cell Signaling Technology | Cat# 3993, RRID:AB_823565 |
| Mouse monoclonal anti-MDA5 (clone 17) | Jan Rehwinkel (Hertzog et al., 2018) | N/A |
| Rabbit polyclonal anti-NS3 (ZIKV) “CAALKSFKEFAAGKR” | GenScript; this paper | N/A |
| Rabbit polyclonal anti-NS3 (ZIKV) “CRRGRIGRNPNKPGD” | GenScript; this paper | N/A |
| Rabbit polyclonal anti-14–3-3ε | Proteintech | Cat# 11648–2-AP, RRID:AB_2217787 |
| Mouse monoclonal anti-14–3-3η (clone 6A12) | Santa Cruz | Cat# sc293464 |
| Mouse monoclonal anti-ZV-2 | Michael Diamond (Zhao et al., 2016) | N/A |
| Mouse monoclonal anti-GAPDH | Millipore | Cat# CS204254 |
| Rabbit polyclonal anti-MB21D1/cGAS | Sigma-Aldrich | Cat# HPA031700, RRID:AB_10601693 |
| Mouse monoclonal anti-STING/TMEM173 | R&D Systems | Cat# MAB7169 |
| Rabbit monoclonal anti-TBK1 | Cell Signaling Technology | Cat# 3504, RRID:AB_2255663 |
| Mouse monoclonal anti-flavivirus antibody (4G2) purified from hybridoma (D1–4G2–4-15) | ATCC | Cat# HB-112, RRID:CVCL J890 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Bacterial and Virus Strains | ||
| ZIKV Brazil Paraiba 2015 | Michael Diamond (Sapparapu et al., 2016) | N/A |
| MR 766 cDNA | Matthew Evans (Schwarz et al., 2016) | N/A |
| ZIKV(KIKP) MR 766 | This paper | N/A |
| Sendai Virus (Cantell) | Charles River | Cat# 10100773 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| SuperSignal West Pico chemiluminescent reagent | Thermo Scientific | Cat# 34580 |
| SuperSignal West Femto chemiluminescent reagent | Thermo Scientific | Cat# 34094 |
| Lipofectamine | Life Technologies | Cat# 18324020 |
| Linear polyethylene | Polysciences | Cat# 09829–1 |
| TransIT-LT1 | Mirus Bio | Cat# MIR 2304 |
| Anti-FLAG M2 beads | Sigma-Aldrich | Cat# M8823, RRID:AB_2637089 |
| Protein A/G agarose beads | Thermo Fisher | Cat# 20421 |
| Glutathione Sepharose 4B resin | GE Life Sciences | Cat# 17075601 |
| Dynabeads Protein G | Invitrogen | Cat# 10009D |
| Anti-HA Magnetic Beads | Thermo Scientific | Cat# 88837 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P2714 |
| HMW-Poly(I:C) / LyoVec | InvivoGen | Cat# tlrl-piclv |
| IFNα2 | PBL Biomedical Laboratories | Cat# 11105–1 |
| Lipofectamine 2000 | Life Technologies | Cat# 11668027 |
| Critical Commercial Assays | ||
| Luciferase Assay System | Promega | Cat# E1500 |
| Mitochondria/Cytosol Fractionation Kit | Millipore | Cat# MIT1000 |
| RNA extraction kit | OMEGA Bio-Tek | Cat# R6834–02 |
| SuperScript III Platinum One-Step qRT-PCR Kit | Invitrogen | Cat# 11732088 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HEK 293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| Vero | ATCC | Cat# CCL-81, RRID:CVCL_0059 |
| A549 | ATCC | Cat# CCL-185, RRID:CVCL_0023 |
| SVGA | Ellen Cahir-McFarland; Major et al., 1985 | N/A |
| HMC3 | ATCC | Cat# CRL-3304, RRID:CVCL_II76 |
| HeLa | ATCC | Cat# CRM-CCL-2, RRID:CVCL_0030 |
| D1–4G2–4-15 | ATCC | Cat# HB-112, RRID:CVCL_J890 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Non-targeting gRNA: 5′-gtggaaaggacgaaacaccgACGGAGGCTAAGCGTCGCAAgttttagagctagaaatag-3 | This paper | N/A |
| MAVS gRNA: 5′-gtggaaaggacgaaacaccGAGGTGGCCCGCAGTCGATCCgttttagagctagaaatag-3′ | This paper | N/A |
| Non-targeting control siRNA (si.C) | Dharmacon | D-001206–14-05 |
| si.RIG-I | Dharmacon | M-012511–01-0005 |
| si.MDA5 | Dharmacon | M-013041–00-0005 |
| si.14–3-3ε | Dharmacon | M-017302–03-005 |
| si.14–3-3η | Dharmacon | M-010626–01-0005 |
| si.14–3-3σ | Dharmacon | M-005180–00-0005 |
| Recombinant DNA | ||
| Plasmid: GST-NS3 (ZIKV, strain MR 766) | This paper | N/A |
| Plasmid: GST-NS3 (ZIKV, strain H/PF/2013) | This paper | N/A |
| Plasmid: GST-NS3 (ZIKV, strain BRA/2015) | This paper | N/A |
| Plasmid: GST-NS3 KIKP (ZIKV, strain BRA/2015) | This paper | N/A |
| Plasmid: FLAG-14–3-3σ | Chan and Gack, 2016 | N/A |
| Plasmid: FLAG-14–3-3ε | Chan and Gack, 2016 | N/A |
| Plasmid: HA-14–3-3ε | Chan and Gack, 2016 | N/A |
| Plasmid: FLAG-14–3-3η | This paper | N/A |
| Plasmid: HA-14–3-3η | This paper | N/A |
| Plasmid: GST-RIG-I 2CARD | Gack et al., 2007 | N/A |
| Plasmid: pEBG | Gack et al., 2007 | N/A |
| Plasmid: GST-NS3 (DENV, strain NGC) | Chan and Gack, 2016 | N/A |
| Plasmid: GST-NS3 (WNV, strain NY99) | Chan and Gack, 2016 | N/A |
| Plasmid: GST-NS3 (YFV, strain 17D) | Chan and Gack, 2016 | N/A |
| Plasmid: FLAG-RIG-I | Wies et al., 2013 | N/A |
| Plasmid: FLAG-MAVS | Chan and Gack, 2016 | N/A |
| Plasmid: IFN-β luciferase reporter plasmid | Lin et al., 2000 | N/A |
| Plasmid: pGK-β-gal | Hatzivassiliou et al., 1997 | N/A |
| Plasmid: GST-MDA5 2CARD | Gack et al., 2007 | N/A |
| Plasmid: FLAG-MDA5 | Wies et al., 2013 | N/A |
| Plasmid: FLAG-NS1 (ZIKV, strain H/PF/2013) | Tom Hobman, (Kumar et al., 2016) | N/A |
| Plasmid: HA-MeV V | Davis et al., 2014 | N/A |
| Software and Algorithms | ||
| FlowJo | T ree Star | https://www.flowjo.com/ |
| GraphPad Prism 7 | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Novex Semi-Dry Blotter | Invitrogen | SD1000 |
| 7500 FAST Real-time PCR machine | Applied Biosystems | |
| LAS 4000 ImageQuant Chemiluminescent Image Analyzer | General Electric | |
Highlights:
The NS3 protein of Zika virus encodes a highly-conserved 14–3-3-binding motif.
NS3 interacts with 14–3-3ε and h to inhibit RIG-I and MDA5 signaling, respectively.
NS3 blocks 14–3-3-mediated translocation of RLRs from the cytosol to mitochondria.
A mutant virus deficient in 14–3-3 binding is attenuated due to elevated immunity.
ACKNOWLEDGEMENTS
We greatly thank Matthew Evans (Mount Sinai), Jae Jung (University of Southern California), Tom Hobman (University of Alberta) and Jan Rehwinkel (Oxford University) for providing reagents. We are grateful to Andrea Watson (University of Chicago) for help with generating the cladogram. This study was supported by the US National Institutes of Health grants R01 AI127774 (to M.U.G. and M.S.D.) and R01 AI087846 (to M.U.G). W.R. and T.S. received support from NIH training grant T32 GM007183.
Footnotes
DECLARATION OF INTERESTS
M.U.G. and Y.K.C. are co-inventors on a patent application for use of the RxD/EP motif and KIKP mutant Zika virus and related viruses.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, et al. (2012). DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathogens 8, e1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, and Osorio JE (2016). Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl Trop Dis 10, e0004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachboard DC, and Horner SM (2016). Innate immune evasion strategies of DNA and RNA viruses. Current Opinion in Microbiology 32, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O'Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, and Suthar MS (2017). Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathogens 13, e1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD, Lazear HM, and Diamond MS (2013). Propagation, quantification, detection, and storage of West Nile virus. Current Protocols in Microbiology 31, 15D 13 11–15D 13 18. [DOI] [PubMed] [Google Scholar]
- Chan YK, and Gack MU (2016a). A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nature Immunology 17, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YK, and Gack MU (2016b). Viral evasion of intracellular DNA and RNA sensing. Nature Reviews Microbiology 14, 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazal M, Beauclair G, Gracias S, Najburg V, Simon-Loriere E, Tangy F, Komarova AV, and Jouvenet N (2018). RIG-I Recognizes the 5' Region of Dengue and Zika Virus Genomes. Cell Reports 24, 320–328. [DOI] [PubMed] [Google Scholar]
- Davis ME, Wang MK, Rennick LJ, Full F, Gableske S, Mesman AW, Gringhuis SI, Geijtenbeek TB, Duprex WP, and Gack MU (2014). Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host & Microbe 16, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmyter J, Melnick JL, and Rawls WE (1968). Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). Journal of Virology 2, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Gaska JM, Douam F, Wei L, Kim D, Balev M, Heller B, and Ploss A (2018). Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proceedings of the National Academy of Sciences of the United States of America 115, E6310–E6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser-Nobis K, Aarreberg LD, Roby JA, Fairgrieve MR, Green R, and Gale M Jr. (2019). Comparative analysis of African and Asian lineage-derived Zika Virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. Journal of Virology 93 (13) e00640–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson NM (2018). Challenges and opportunities in controlling mosquito-borne infections. Nature 559, 490–497. [DOI] [PubMed] [Google Scholar]
- Full F, van Gent M, Sparrer KMJ, Chiang C, Zurenski MA, Scherer M, Brockmeyer NH, Heinzerling L, Sturzl M, Korn K, et al. (2019). Centrosomal protein TRIM43 restricts herpesvirus infection by regulating nuclear lamina integrity. Nature Microbiology 4, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, et al. (2016). Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host & Microbe 19, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan SS, Sevvana M, Kuhn RJ, and Rossmann MG (2018). Structural biology of Zika virus and other flaviviruses. Nat Struct Mol Biol 25, 13–20. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou E, Cardot P, Zannis VI, and Mitsialis SA (1997). Ultraspiracle, a Drosophila retinoic X receptor alpha homologue, can mobilize the human thyroid hormone receptor to transactivate a human promoter. Biochemistry 36, 9221–9231. [DOI] [PubMed] [Google Scholar]
- Hertzog J, Dias Junior AG, Rigby RE, Donald CL, Mayer A, Sezgin E, Song C, Jin B, Hublitz P, Eggeling C, et al. (2018). Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol 48, 1120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Keller TH, and Luo D (2017). Zika Virus Protease: An Antiviral Drug Target. Trends in Microbiology 25, 797–808. [DOI] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, and Hobman TC (2016). Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17, 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, and Duffy MR (2008). Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerging Infectious Diseases 14, 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, and Diamond MS (2016). A Mouse Model of Zika Virus Pathogenesis. Cell Host & Microbe 19, 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JP, Fan YK, and Liu HM (2019). The 14–3-3εta chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathogens 15, e1007582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Genin P, Mamane Y, and Hiscott J (2000). Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Molecular and Cellular Biology 20, 6342–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Loo YM, Horner SM, Zornetzer GA, Katze MG, and Gale M Jr. (2012). The mitochondrial targeting chaperone 14–3-3εpsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host & Microbe 11, 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ketkar H, Geng T, Lo E, Wang L, Xi J, Sun Q, Zhu Z, Cui Y, Yang L, et al. (2018). Zika Virus Non-structural Protein 4A Blocks the RLR-MAVS Signaling. Frontiers in Microbiology 9, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JS, Gubler DJ, and Petersen LR (2004). Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med 10, S98–109. [DOI] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, and Sever J (1985). Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proceedings of the National Academy of Sciences of the United States of America 82, 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, and Tschopp J (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. [DOI] [PubMed] [Google Scholar]
- Miner JJ, and Diamond MS (2017). Zika Virus Pathogenesis and Tissue Tropism. Cell Host & Microbe 21, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kuhn RJ, and Rossmann MG (2005). A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3, 13–22. [DOI] [PubMed] [Google Scholar]
- Neufeldt CJ, Cortese M, Acosta EG, and Bartenschlager R (2018). Rewiring cellular networks by members of the Flaviviridae family. Nat Rev Microbiol 16, 125–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olagnier D, Muscolini M, Coyne CB, Diamond MS, and Hiscott J (2016). Mechanisms of Zika Virus Infection and Neuropathogenesis. DNA and Cell Biology 35, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TC, and Diamond MS (2018). The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581. [DOI] [PubMed] [Google Scholar]
- Sapparapu G, Fernandez E, Kose N, Bin C, Fox JM, Bombardi RG, Zhao H, Nelson CA, Bryan AL, Barnes T, et al. (2016). Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz MC, Sourisseau M, Espino MM, Gray ES, Chambers MT, Tortorella D, and Evans MJ (2016). Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 1 (5) e00246–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Balasubramaniam VR, Brown JA, Mena I, Grant A, Bardina SV, Maringer K, Schwarz MC, Maestre AM, Sourisseau M, et al. (2017). A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathogens 13, e1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G, and Avruch J (2002). 14–3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. The Journal of Biological Chemistry 277, 3061–3064. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Shen YH, and Zhu J (2001). 14–3-3 proteins; bringing new definitions to scaffolding. Oncogene 20, 6331–6338. [DOI] [PubMed] [Google Scholar]
- van de Weijer ML, Bassik MC, Luteijn RD, Voorburg CM, Lohuis MA, Kremmer E, Hoeben RC, LeProust EM, Chen S, Hoelen H, et al. (2014). A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nature Communications 5, 3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diemen FR, Kruse EM, Hooykaas MJ, Bruggeling CE, Schurch AC, van Ham PM, Imhof SM, Nijhuis M, Wiertz EJ, and Lebbink RJ (2016). CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathogens 12, e1005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, and Gack MU (2013). Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, and Cui J (2017). Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov 3, 17006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, and Cantley LC (1997). The structural basis for 14–3-3:phosphopeptide binding specificity. Cell 91, 961–971. [DOI] [PubMed] [Google Scholar]
- Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, Hastings AK, Fikrig E, Kong Y, Horvath TL, et al. (2018). Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol 3 (19) e1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, et al. (2016). Structural Basis of Zika Virus-Specific Antibody Protection. Cell 166, 1016–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data/code was generated in this study.