

Abstract
Reversible phosphorylation plays critical roles in cell growth, division, and signal transduction. Kinases which catalyze the transfer of γ-phosphate groups of nucleotide triphosphates to their substrates are central to the regulation of protein phosphorylation and are therefore important therapeutic targets. Top-down mass spectrometry (MS) presents unique opportunities to study protein kinases owing to its capabilities in comprehensive characterization of proteoforms that arise from alternative splicing, sequence variations, and post-translational modifications. Here, for the first time, we developed a top-down MS method to characterize the catalytic subunit (C-subunit) of an important kinase, cAMP-dependent protein kinase A (PKA). The recombinant PKA C-subunit was expressed in E. coli and successfully purified via his-tag affinity purification. By intact mass analysis with high resolution and high accuracy, four different proteoforms of the affinity-purified PKA C-subunit were detected and the most abundant proteoform was found containing seven phosphorylations with the removal of N-terminal methionine. Subsequently, the seven phosphorylation sites of the most abundant PKA C-subunit proteoform were characterized simultaneously using tandem MS methods. Four sites were unambiguously identified as Ser10, Ser11, Ser18, and Ser30 and the remaining phosphorylation sites were localized to Ser2/Ser3, Ser358/Thr368, and Thr[215-224]Tyr in the PKA C-subunit sequence with a 20mer 6xHis-tag added at the N-terminus. Interestingly, four of these seven phosphorylation sites were located at the 6xHis-tag. Furthermore, we have performed dephosphorylation reaction by Lambda protein phosphatase, and showed that all phosphorylations of the recombinant PKA C-subunit phosphoproteoforms were removed by this phosphatase.
Keywords: Post-translational modifications, Top-down mass spectrometry, Protein kinases
Graphical Abstract
Introduction
Reversible phosphorylation is one of the key biological processes that govern cellular events including cell cycle control, cell growth, and signal transduction [1, 2]. Aberrations in signaling events, such as up- and down-regulation of phosphorylation, are associated with the progress of human diseases [3-8]. Protein kinases are enzymes that catalyze the transfer of the γ-phosphate groups of nucleotide triphosphates to their substrates, and therefore are central to the regulation of protein phosphorylation [9, 10]. Dysregulation of kinase signaling networks is increasingly recognized as an underlying mechanism that contributes to human diseases [11-14]. Consequently, numerous kinases inhibitors are currently utilized or under development for use as therapeutics [15, 16].
Protein kinases are also modulated by phosphorylation [17]. Autophosphorylation of protein kinases, or phosphorylation by other protein kinases, results in their activation or deactivation due to changes in their secondary structures [18]. Structural changes affect the binding kinetics to kinase substrates such as adenosine triphosphate (ATP) and inhibitor peptides by altering the salt bridges and hydrogen bonding network at the active site [18, 19]. One of the important protein kinases is the cAMP-dependent protein kinase A (PKA), which partakes in many biological processes including mediating adrenergic stimulation in the heart and regulating the functions of skeletal muscle [20, 21]. This protein kinase is a heterotetramer composed of two catalytic subunits (C-subunits), and two different regulatory subunits [22, 23]. The PKA C-subunit has multiple phosphorylation sites displayed in the expressed proteins, which are associated with the physiochemical properties and enzymatic activity [18, 19, 24, 25].
The phosphate groups at the phosphorylation sites are removed by protein phosphatase through the biological process of dephosphorylation, which together with protein phosphorylation, constitutes the reversible phosphorylation [1]. The removal of a phosphate group at the phosphorylation sites by phosphatase largely depends on the substrate specificity of the phosphatase [26]. Additionally, structural information around the phosphorylation sites can be revealed as the efficacy of dephosphorylation also relies on the accessibility, which is based on the structural environment of the phosphorylation sites under physiological conditions [26]. To better understand the function of phosphorylations on PKA C-subunit, a comprehensive characterization of the phosphorylation sites and the analysis of the dephosphorylation reaction are necessary.
Top-down mass spectrometry (MS) presents unique opportunities to study protein kinases owing to its capabilities in analyzing alternative splicing, sequence variations and post-translational modifications (PTMs) [5, 27-34]. Compared to bottom-up MS, which analyzes digested peptides, top-down MS analysis provides a “bird’s eye” view of all proteoforms by analyzing proteins from the intact level [35-37]. In this study, we have developed a top-down MS strategy to characterize the recombinant PKA C-subunit. The affinity-purified PKA C-subunit, which was expressed in E. coli, was present with multiple proteoforms by intact mass analysis. The most abundant proteoform of PKA C-subunit was identified with seven phosphorylations along with the removal of N-terminal methionine. Using tandem MS (MS/MS) techniques including collision-induced dissociation (CID) and electron-capture dissociation (ECD), these seven phosphorylation sites were localized to specific amino acid residues or located to a region. Interestingly, four of these phosphorylation sites were located at the 6xHis-tag sequence. Dephosphorylation reactions using Lambda protein phosphatase (λPP) suggested that all phosphorylation sites were accessible to this particular phosphatase. Taken together, we have demonstrated that top-down MS has unique advantages in comprehensively characterizing protein kinases.
Experimental
Chemicals and Reagents
All reagents were acquired from Sigma-Aldrich, Inc. (St. Louis, MO, USA), unless otherwise noted. Solvents, including HPLC grade water (H2O), acetonitrile (ACN) and ethanol (EtOH), were purchased from Fisher Scientific (Fair Lawn, NJ, USA).
Molecular Cloning
Commercial plasmid encoding the PKA C-subunit (plasmid # 14921) was purchased from Addgene (Watertown, MA, USA) in an agar gel piece [38]. A small agar piece was transferred in 5 mL TB media with 100 μg/mL ampicillin, and the mixture was allowed to grow for 9 h in a shaker. The cells were collected and the growth media was discarded. The plasmid DNA was extracted using QIAprep Spin Miniprep Kit (QIAGEN, Hilden, Germany) following the manufacturer recommended protocol. The plasmid product was transformed into ScarabXpress T7 E. coli cells (Scarab Genomics, Madison, WI, USA), and a glycerol stock was prepared.
Protein Expression and Purification
The protein expression and purification protocol was similar to that previously described [39]. Briefly, a starter LB broth culture with 100 μg/mL ampicillin was inoculated by glycerol stock of the E. coli and the starter culture was allowed to grow overnight. A small amount of starter culture was transferred to LB broth containing 100 μg/mL ampicillin. The culture was allowed to grow until the optical density of the culture reached 0.4 to 0.6. IPTG at a final concentration of 0.1 mM was introduced to induce protein expression, and the bacteria were cultured at 30 °C for 9 h. The cells were harvested by centrifugation and the cell pellets were stored at −80 °C prior to protein purification.
Unless stated otherwise, additives include 1 mM DTT and 0.25 mM PMSF. The cell pellets were lysed by sonication in 50 mM NaH2PO4 pH 7.4, 250 mM NaCl (10 mL/g pellet) buffer (Buffer A) with additives and protease inhibitor cocktail (Sigma-Aldrich Inc.). The cell debris were removed by centrifugation. For 2 mL of the supernatant, 250 μL of Dynabeads™ His-Tag Isolation and Pulldown (Invitrogen™, Carlsbad, CA, USA) was added, and the mixture was agitated at 4 °C for 30 min. The supernatant was removed and the Dynabeads were washed twice with Buffer A containing additives, once with 50 mM Tris pH 7.4, 50 mM NaCl buffer (Buffer B) with additives, and finally with Buffer B containing additives and 25 mM imidazole. The attached proteins were eluted with Buffer B with additives, 300 mM imidazole, and protease inhibitor cocktail, and was concentrated using a Pierce™ Protein Concentrators PES, 10K MWCO filter (Fisher Scientific). The efficacy of the protein purification was verified by SDS-PAGE analysis.
Dephosphorylation Reaction
The dephosphorylation reaction for the PKA C-subunit (~ 40 μg) was performed using ~ 150 units of λPP (New England Biolabs Inc., Ipswich, MA, USA) following the manufacturer recommended protocol. Briefly, the reaction was supplemented with 1 mM MnCl2 solution and 1X NEBuffer for PMP (New England Biolabs Inc.) and allowed to proceed for 2 h at 30 °C to achieve complete dephosphorylation.
Top-down Mass Spectrometry
For online MS analysis, the PKC C-subunit samples were separated using a homemade PLRP reversed-phase column (200 mm length × 500 μm i.d., 10 μm particle size, 1,000 Å pore size). PLRP-S particles were obtained from Agilent Technologies (Santa Clara, CA, USA). Mobile phase A (MPA) contained H2O with 0.1% formic acid (FA) and mobile phase B (MPB) contained 50:50 ACN:EtOH with 0.1% FA. Liquid chromatography (LC) was performed with a 60 min linear gradient which ran at 5% MPB from 0 to 5 min, followed by 5% to 65% MPB from 5 to 40 min, 65% to 95% MPB from 40-53 min, and back to 5% MPB at a flow rate of 12 μL/min. Five microliters (5 μL) of sample were injected for all experiments. The sample was analyzed either using a maXis II Q-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) coupled with an ACQUITY UPLC M-Class System (Waters Corporation, Milford, MA, USA), or using a 12T solariX FT-ICR mass spectrometer (Bruker Daltonics) coupled with a nanoACQUITY UPLC System (Waters Corporation). For online LC-MS/MS experiments with CID fragmentation using a maXis II Q-TOF mass spectrometer, the precursor ion was isolated and subjected to 15 - 20 eV energy for fragmentation.
For offline MS analysis, the fraction was collected using a nanoACQUITY UPLC System. The sample was introduced to a 12T solariX FT-ICR mass spectrometer using a TriVersa NanoMate® (Advion Bioscience, Ithaca, NY, USA) as previously described [5, 40]. The mass spectra were collected over a 200 to 3000 m/z range with 2 M transient size (1.2 s transient length) and a pulse at 28% excitation power. In MS/MS analysis, an isolation window of 1.8 – 2 m/z was used for the precursor ion. Mass spectra were accumulated for 500 to 750 scans. For CID experiments, an energy from 6 to 12 V was set to generate fragment ions. For ECD experiments, the parameters for ECD pulse length, ECD bias, and ECD lens were set to 0.020 s, 0.3 - 0.6 V, and 10 V, respectively.
Data Analysis
All reported masses are monoisotopic masses. For intact mass analysis, the spectra were analyzed using DataAnalysis 4.2 and deconvoluted using the Maximum Entropy deconvolution algorithm. The monoisotopic mass was calculated using the SNAP algorithm in DataAnalysis. For MS/MS analysis, the data were analyzed using MASH Suite Pro [41]. Peak extraction was performed using a signal-to-noise ratio of 3 and a minimum fit of 60%, and all peaks were subjected to manual validation. A 10-ppm mass tolerance was used to match the experimental fragment ions to the calculated fragment ions based on amino acid sequence.
Results and Discussion
We developed a top-down MS strategy for the comprehensive characterization of recombinant PKA C-subunit (Figure 1). The strategy started with obtaining a plasmid encoding the PKA C-subunit, and subsequently transforming the plasmid into a vector. Afterwards, the PKA C-subunit was overexpressed in E. coli, and the protein was purified by affinity purification. The protein was first subjected to intact mass analysis which reveals the sequence variations and PTMs by accurate mass measurements. These putative modifications were first assessed by online CID experiment for protein fragmentation analysis. Then the fraction containing the PKA C-subunit was collected after LC separation, and further subjected to offline characterization using both CID and ECD at various fragmentation settings for verification of the putative modifications.
Figure 1.

Workflow of expression, affinity purification, and top-down LC-MS analysis of the recombinant PKA C-subunit. a) The plasmid obtained from Addgene organization was transferred into a vector and expressed in E. coli cells. b) The PKA C-subunit was purified using affinity purification. c) The purified samples were subjected to online LC-MS and MS/MS analysis using a Q-TOF mass spectrometer and complemented with offline MS/MS analysis using a FT-ICR mass spectrometer. d) The modifications were characterized based on MS and MS/MS spectra.
PKA C-subunit expression and purification
The plasmid encoding the PKA C-subunit was kindly provided by Dr. Susan Taylor from UCSD through Addgene organization [38]. The plasmid includes a 20 amino acid 6xHis-tag sequence before the endogenous sequence of PKA C-subunit derived from mice [UniProtKB - P05132]. The mouse-derived C-subunit of PKA is composed of 351 amino acid residues. For overexpression of the PKA C-subunit, the plasmid was purified and transformed into the pET-28a(+) vector. To capture the 6xHis-tag on the PKA C-subunit, affinity purification using Dynabeads was employed, which is based on TALON technology (Figure S1a). The loading mixture, flow through, and elution fractions were evaluated by SDS-PAGE analysis (Figure S1b). The PKA C-subunit was determined to be successfully purified based on the presence of a dark band at around 42 kDa, which is consistent with the predicted protein mass (Mr: 42575.92 Da) from the encoding amino acid sequence. Although the PKA C-subunit was present as the most prominent band by SDS-PAGE analysis, other faint bands could also be observed in the elution lanes. In particular, some lower mass proteins might suppress the ionization and detection of the PKA C-subunit in the top-down MS analysis. Therefore, our strategy was to use reverse phase LC (RPLC) methods to separate the PKA C-subunit from other proteins for both online and offline characterization.
Online LC-MS/MS Profiling of Multiple Proteoforms
The affinity-purified PKA C-subunit was subjected to RPLC separation coupled online with high-resolution MS analysis using a Q-TOF instrument. Using H2O as MPA and 50:50 ACN:EtOH as MPB, the PKA C-subunit was separated and detected by MS with minimal impurities, and this was demonstrated by the charge state distribution envelope (Figure 2a and Figure S2). The deconvoluted spectra revealed the existence of multiple PKA C-subunit proteoforms but none of the masses of these proteoforms matched with the theoretical protein mass based on the predicted amino acid sequence (Figure 2a, inset). The mass difference between two neighboring peaks was 79.97 Da, indicating the occurrence of phosphorylation on these proteoforms. As it is common that the N-terminal methionine of recombinant proteins would be removed by methionyl-aminopeptidase after protein translation, the mass of methionine was first deducted from the theoretical protein mass [42]. The PKA C-subunit proteoforms were found to contain six to nine phosphorylations with N-terminal methionine removed based on the results from the deconvoluted spectra. With a mass shift of 559.49 Da, the most abundant proteoform was modified with seven phosphorylations in addition to the removal of N-terminal methionine, which accounted for ~ 45% of relative percentage of all PKA C-subunit proteoforms (Figure 2a, inset). Collectively, the affinity-purified PKA C-subunit was hyperphosphorylated from E. coli expression, which is consistent with previous studies [25, 43].
Figure 2.

Top-down MS analysis of the PKA C-subunit. a) Mass spectra with charge state distribution and deconvoluted (inset) spectra of the PKA C-subunit. The most abundant proteoform purified from E. coli expression contained seven phosphorylations with removal of N-terminal methionine. b) The precursor at charge state 49+ was subjected to online LC-MS/MS with CID fragmentation. 560 – 850 m/z is zoomed in to show a variety of b and y fragment ions (inset).
For the initial PTM site characterization, the PKA C-subunit was subjected to online LC-MS/MS with CID fragmentation on the precursor ion corresponding to the most abundant proteoform with seven phosphorylation sites. Since phosphorylation is the only PTM being considered, the mass list was matched with the theoretical fragment ion list by adding the mass of phosphorylation modification. Asides from the precursor ion (M49+), several abundant fragment ions were observed (Figure 2b). The masses of these abundant fragment ions were identical and could be identified as y114 ions at different charge states after accounting for the mass of one phosphorylation. Less abundant ions at 560 – 850 m/z afforded additional information regarding the phosphorylation sites (Figure 2b, inset). Several low mass y ions (y20, y19, and y15) suggested a phosphorylation site located after Arg356 at the C-terminus. A series of b ions (b54, b55, b56, and b57) could also be identified with mass difference equivalent to five phosphorylations, indicating five phosphorylation sites located before Lys54 at the N-terminus. Lastly, a b254 ion was identified with mass difference equivalent to six phosphorylation, suggesting that a phosphorylation site was located in the middle of the recombinant PKA C-subunit sequence. Using online CID characterization, fragment ions from MS/MS spectra localized five phosphorylation site before Lys54 at the N-terminus, one phosphorylation site after Arg356 at the C-terminus, and one phosphorylation site in the middle of the sequence for the most abundant PKA C-subunit proteoform.
Characterization of PKA C-subunit phosphorylation sites by high-resolution MS MS Analysis
We sought to localize all phosphorylation sites present in the most abundant proteoform of the PKA C-subunit using offline MS analysis combining different fragmentation methods. The fraction containing the recombinant PKA C-subunit was collected after LC separation, and the samples were analyzed on an ultrahigh-resolution FT-ICR mass spectrometer. In this study, when referring the amino acid residue in the endogenous sequence, note that the reference is to the UniProt sequence [UniProtKB - P05132] with N-terminal methionine removed to be consistent with the previous reports [19, 24]. As shown from the online CID results, there were five phosphorylation sites located near the N-terminus. As a result, ECD fragmentation method was used for site localization, which is known to preserve labile modifications such as phosphorylation [44]. ECD was able to effectively fragment most of the bonds at the N-terminus, and a plethora of fragment ions was observed in the raw spectra from the ECD experiment (Figure 3a and Figure S3).
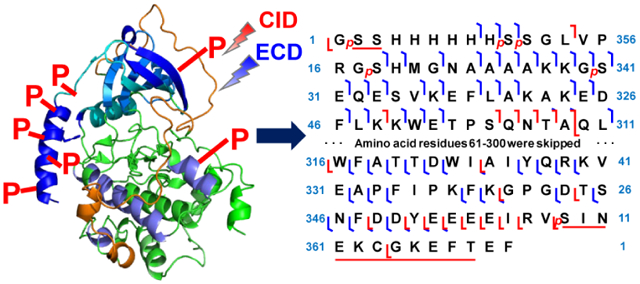
Figure 3.

Identification of five phosphorylation sites at the N-terminal region using ECD. a) ECD fragment ion mapping for the first 45 amino acid residues. b) Representative fragment ions from ECD fragmentation. Five phosphorylation sites, Ser2/Ser3, Ser10, Ser11, Ser18, and Ser30 were confirmed by phosphorylated c ions.
For the first phosphorylation site at the N-terminus, both c8 and c9 differed from the theoretical mass by 79.97 Da, indicative of the occurrence of phosphorylation (Figure 3b). Ser2 and Ser3 are the only two amino acid residues that can be phosphorylated; however, the site could not be definitively localized to either Ser2 or Ser3 without additional fragment ions. Three additional phosphorylation sites were localized at Ser10, Ser11, and Ser18, which were confirmed by c9, c10, and c19 ions (Figure 3b). Ser11 and Ser18 were confirmed with only two c ions, c10, and c19, as these are the only two sites which could be phosphorylated. Intriguingly, all of these four phosphorylation sites were located at the added 6xHis-tag sequence. Hyperphosphorylation at the 6xHis-tag sequence was also observed in other case using E. coli for kinase expression such as that for Aurora A [45]. Iakoucheva et al. suggested that protein phosphorylation predominantly occurred at disordered regions [46]. The structure of the 6xHis-tag sequence along with the first 12 amino acid residues of the PKA C-subunit was found to be disordered from previous X-ray crystallography study, which supported our observation that the four phosphorylations took place at the disordered 6xHis-tag sequence [38]. This 20mer 6xHis-tag sequence (MGSSHHHHHHSSGLVPRGSH) is a common sequence added at the N-terminus due to its dual functionality [47, 48]. This tag includes a 6xHis-tag for affinity purification and a thrombin cleavage site (LVPR/GS). Proteins with only Gly-Ser-His added at the N-terminus of the endogenous protein sequence could be yielded after reacting the affinity-purified protein with thrombin [49]. In the case of the PKA C-subunit, the thrombin cleavage site was not utilized as the 20mer 6xHis-tag did not affect the structure and enzymatic activity of this protein [19, 38].
The last of the five phosphorylation sites at the N-terminus was localized at Ser30, which was confirmed by c18 and c32 ions (Figure 3b). This phosphorylation site is equivalent to Ser10 in the endogenous sequence. Previously, Tholey et al. suggested that phosphorylation at Ser10 altered the structure at the N-terminus, resulting in the amplified extent of electrostatic interaction [50]. Yonemoto et al. argued that Ser10 could be autophosphorylated in vitro, and that this site was significant for protein solubility [19]. Mutation at Ser10 significantly impaired the solubility of protein in aqueous solution. Therefore, the phosphorylation at Ser10 was shown to be important for protein structure and solubility.
Next, we sought to identify the phosphorylation site at the C-terminus. Since ECD did not generate sufficient fragment ions, the site was instead characterized primarily by CID fragment ions. The phosphorylation site was localized at Ser358 or Thr368 by y13 and a series of y ions from y15 to y21 (Figure 4a and Figure 4b). Although an y7 ion without phosphorylation was observed in the CID experiment, this ion could not be used for confident identification of phosphorylation site at Ser358 due to the possibility that this ion was present after the loss of the phosphorylation at Thr368 (Figure 4b) [44]. One of the potential phosphorylation sites, Ser358, equivalent to Ser338 in the endogenous sequence, has been reported previously [19, 51]. Yonemoto et al. suggested that this phosphorylation site was relevant to catalytic activity and protein stability. Mutations of recombinant PKA C-subunit with S338A or S338E either disrupted the catalytic activity or altered the binding kinetics for inhibitor peptide and ATP [19].
Figure 4.

Phosphorylation site mapping at the C-terminus and in the middle region by CID and ECD. a) CID fragment ion mapping at the C-terminus. b) Representative fragment ions from CID experiment. A phosphorylation site was localized at Ser358 or Thr368. c) CID and ECD fragment ion mapping for the middle sequence of PKA C-subunit. d) Representative fragment ions from the CID and ECD experiments. A phosphorylation site was located at Thr[215-224]Tyr.
The localization of phosphorylation site in the middle of the sequence required fragment ions generated from both CID and ECD fragmentation methods. The phosphorylation site was narrowed down to T[215-224]Y by b224 with six phosphorylations, c210 with five phosphorylations and z•157 with two phosphorylations (Figure 4c and Figure 4d). One of the potential phosphorylation sites was Thr217, which is equivalent to Thr197 in the endogenous sequence. Phosphorylation at Thr197 is crucial to catalytic activity, as it allows PKA C-subunit to change from an inactive state to an active state [18]. It does so by forming salt bridges with amino acid residues from other parts of the PKA C-subunit, such as C-helix, catalytic loop, β9, and activation loop.
In previous studies, phosphorylation sites on PKA C subunit were usually identified by bottom-up MS based on the detection of phosphopeptides, in which the identified phosphorylation sites are from a mixture of multiply-phosphorylated proteoforms [24, 25]. Compared to the bottom-up MS strategy, our top-down MS strategy analyzes intact proteins, giving a bird’s eye view of all proteoforms present. This approach not only shows the stoichiometry of different proteoforms in a single sample, but also provides a comprehensive analysis of all phosphorylation sites present in a single proteoform.
Top-down MS MS Sequencing of the PKA C-subunit
By combining five CID spectra and three ECD spectra, 191 of 369 possible bonds were cleaved, providing a 52% sequence coverage for the recombinant PKA C-subunit (Figure 5). A series of CID fragment ions was observed at Ser[54-58]Gln, Ser[134-145]Gly, Gly[246-265]Gln, and Tyr[350-357]Arg (Figure S4). Interestingly, although loss of phosphorylation would sometimes occur in CID, a series of b ions with all phosphorylations intact was observed from Ser[54-58]Gln, Ser[134-145]Gly, and Gly[246-255]Tyr. Compared to peptide fragmentation, CID of intact proteins often could retain a portion of the labile modifications in the top-down approach [44]. Fragmentation at the amide backbone was preferred over PTM ejection, which is likely due to the higher-order structure of gas phase ions that are larger than ~8 kDa [52]. By contrast, only a few b ions were observed for the first 50 amino acid residues in the N-terminus, likely due to the loss of phosphate group(s) as a result of their smaller size. ECD fragmentation yielded bond cleavages unique to CID fragmentation due to the difference in the dissociation mechanism [53]. This method provided good sequence coverage at both the N- and C-terminus; however, the fragmentation efficiency was suboptimal for bonds in the middle of the sequence, despite some larger ECD fragment ions being observed (Figure S5 and Figure S6). For this study, utilizing both CID and ECD fragmentation methods, the phosphorylation sites of the PKA C-subunit proteoform with seven phosphorylations were characterized. Current development in fragmentation methods, such as UVPD, will be beneficial to achieve a higher sequence coverage due to additional generated ion species such as a and x ions, in addition to b, c, y and z• ions [54].
Figure 5.

Top-down MS/MS sequencing of the PKA C-subunit. The map combined three ECD spectra and five CAD spectra. 191 of 369 possible bonds were cleaved, providing a 52% sequence coverage.
Dephosphorylation of hyperphosphorylated PKA C-subunit
Dephosphorylation, which removes phosphate groups on their substrates by phosphatases, is complimentary to phosphorylation. We were interested in how the recombinant PKA C-subunit proteoforms react to a common phosphatase, λPP. The dephosphorylation reaction of the affinity-purified PKA C-subunit was performed and the reaction product was analyzed by top-down MS. A drastic shift in peaks was observed in each charge state due to the loss of multiple phosphorylations (Figure 6a and Figure S7). From the deconvoluted spectra, all phosphoproteoforms collapsed into a single unphosphorylated proteoform after the dephosphorylation reaction (Figure 6b). This suggested that all phosphorylation sites were accessible by λPP and were subsequently dephosphorylated. Byrne et al. observed that after dephosphorylation, the most abundant proteoform still possessed two phosphorylations detected by low-resolution top-down MS analysis [25]. The reaction conditions of the dephosphorylation reaction between our studies and the study done by Byrne et al. were different. In our protocol, we followed the manufacturer recommended conditions from the New England Biolabs and performed the dephosphorylation reaction at 30 °C. In comparison, Byrne et al. conducted the dephosphorylation reaction at 37 °C using bacterially expressed λPP. At elevated temperature, the phosphatase might not reach its maximum kinetics and might denature after prolonged incubation. Conclusively, our result showed that all phosphorylations on the phosphoproteoform of the PKA C-subunit were removed by λPP. The discrepancy between our result and result from Byrne et al. might be due to the difference in reactions conditions.
Figure 6.

Analysis of the dephosphorylation of the PKA C-subunit using Lambda protein phosphatase (λPP). a) Charge states 48+, 47+, and 46+ were shown for PKA C-subunit before (top) and after (bottom) the dephosphorylation reaction. b) Deconvoluted spectra were shown for the analysis of the dephosphorylation reaction. All phosphorylations were removed, resulting in a single unphosphorylated proteoform in the spectra.
Conclusion
For the first time, a top-down MS strategy was developed to achieve a comprehensive characterization of the recombinant PKA C-subunit. The PKA C-subunit was overexpressed in E. coli and the expressed protein with 6xHis-tag was successfully purified by affinity purification. The affinity-purified PKA C-subunit was subjected to intact mass analysis and proteoforms with six to nine phosphorylations were observed. The most abundant proteoform was identified with seven phosphorylations and removal of N-terminal methionine. Using CID and ECD fragmentation methods, all seven phosphorylation sites were characterized simultaneously for the first time. Four of the phosphorylation sites were unambiguously localized to Ser10, Ser11, and Ser18, which were located at the 20mer 6xHis-tag sequence, as well as Ser30, which corresponded to Ser10 in the endogenous sequence Three other phosphorylation sites were localized to Ser2/Ser3, Thr[215-224]Tyr, and Ser358/Thr368. By combining five CID and three ECD experiments, a 52% sequence coverage was achieved for the PKA C-subunit with seven phosphorylations. Finally, dephosphorylation experiments showed that all phosphorylations of the PKA C-subunit phosphoproteoform were removed by λPP.
Supplementary Material
Acknowledgement
The authors would like to thank Dr. Wenxuan Cai for making the construct for PKA C-subunit for bacterial expression. Financial support was provided by NIH R01 GM117058 (to S. J. and Y. G.) and R01 GM125085 (to Y. G.). Y. G. also would like to acknowledge the NIH grants, R01 HL096971, R01 HL109810, and S10 OD018475.
Footnotes
Conflict of Interest Disclosure
The authors declare no compete for financial interest.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Reference
- 1.Hunter T: Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 80, 225–236 (1995) [DOI] [PubMed] [Google Scholar]
- 2.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M: Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 127, 635–648 (2006) [DOI] [PubMed] [Google Scholar]
- 3.Grimes M, Hall B, Foltz L, Levy T, Rikova K, Gaiser J, Cook W, Smirnova E, Wheeler T, Clark NR, Lachmann A, Zhang B, Hornbeck P, Ma'ayan A, Comb M: Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci Signal. 11, 531 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanger DP, Anderton BH, Noble W: Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 15, 112–119 (2009) [DOI] [PubMed] [Google Scholar]
- 5.Peng Y, Gregorich ZR, Valeja SG, Zhang H, Cai W, Chen YC, Guner H, Chen AJ, Schwahn DJ, Hacker TA, Liu X, Ge Y: Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction. Mol Cell Proteomics. 13, 2752–2764 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong XT, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP, Ge Y: Augmented Phosphorylation of Cardiac Troponin I in Hypertensive Heart Failure. Journal of Biological Chemistry. 287, 848–857 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Guy MJ, Norman HS, Chen YC, Xu QG, Dong XT, Guner H, Wang SJ, Kohmoto T, Young KH, Moss RL, Ge Y: Top-Down Quantitative Proteomics Identified Phosphorylation of Cardiac Troponin I as a Candidate Biomarker for Chronic Heart Failure. J Proteome Res. 10, 4054–4065 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen IH, Xue L, Hsu CC, Paez JSP, Pan L, Andaluz H, Wendt MK, Iliuk AB, Zhu JK, Tao WA: Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. P Natl Acad Sci USA. 114, 3175–3180 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S: The protein kinase complement of the human genome. Science. 298, 1912–1934 (2002) [DOI] [PubMed] [Google Scholar]
- 10.Hanks SK, Quinn AM, Hunter T: The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 241, 42–52 (1988) [DOI] [PubMed] [Google Scholar]
- 11.Sacco F, Silvestri A, Posca D, Pirro S, Gherardini PF, Castagnoli L, Mann M, Cesareni G: Deep Proteomics of Breast Cancer Cells Reveals that Metformin Rewires Signaling Networks Away from a Pro-growth State. Cell Syst. 2, 159–171 (2016) [DOI] [PubMed] [Google Scholar]
- 12.Dhillon AS, Hagan S, Rath O, Kolch W: MAP kinase signalling pathways in cancer. Oncogene. 26, 3279–3290 (2007) [DOI] [PubMed] [Google Scholar]
- 13.Chatterjee K: Neurohormonal activation in congestive heart failure and the role of vasopressin. Am J Cardiol. 95, 8b–13b (2005) [DOI] [PubMed] [Google Scholar]
- 14.Vlahos CJ, McDowell SA, Clerk A: Kinases as therapeutic targets for heart failure. Nat Rev Drug Discov. 2, 99–113 (2003) [DOI] [PubMed] [Google Scholar]
- 15.Zhang JM, Yang PL, Gray NS: Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 9, 28–39 (2009) [DOI] [PubMed] [Google Scholar]
- 16.Ferguson FM, Gray NS: Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 17, 353–376 (2018) [DOI] [PubMed] [Google Scholar]
- 17.Roskoski R Jr.: ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 66, 105–143 (2012) [DOI] [PubMed] [Google Scholar]
- 18.Steichen JM, Iyer GH, Li S, Saldanha SA, Deal MS, Woods VL Jr., Taylor SS: Global consequences of activation loop phosphorylation on protein kinase A. J Biol Chem. 285, 3825–3832 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yonemoto W, McGlone ML, Grant B, Taylor SS: Autophosphorylation of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. Protein Eng. 10, 915–925 (1997) [DOI] [PubMed] [Google Scholar]
- 20.Wheeler-Jones CP: Cell signalling in the cardiovascular system: an overview. Heart. 91, 1366–1374 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruehr ML, Russell MA, Ferguson DG, Bhat M, Ma J, Damron DS, Scott JD, Bond M: Targeting of protein kinase A by muscle A kinase-anchoring protein (mAKAP) regulates phosphorylation and function of the skeletal muscle ryanodine receptor. J Biol Chem. 278, 24831–24836 (2003) [DOI] [PubMed] [Google Scholar]
- 22.Bauman AL, Scott JD: Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat Cell Biol. 4, E203–E206 (2002) [DOI] [PubMed] [Google Scholar]
- 23.Taylor SS, Knighton DR, Zheng JH, Teneyck LF, Sowadski JM: Structural Framework for the Protein-Kinase Family. Annu Rev Cell Biol. 8, 429–462 (1992) [DOI] [PubMed] [Google Scholar]
- 24.Yonemoto W, Garrod SM, Bell SM, Taylor SS: Identification of phosphorylation sites in the recombinant catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 268, 18626–18632 (1993) [PubMed] [Google Scholar]
- 25.Byrne DP, Vonderach M, Ferries S, Brownridge PJ, Eyers CE, Eyers PA: cAMP-dependent protein kinase (PKA) complexes probed by complementary differential scanning fluorimetry and ion mobility-mass spectrometry. Biochem J. 473, 3159–3175 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy J, Cyert MS: Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal. 2, re9 (2009) [DOI] [PubMed] [Google Scholar]
- 27.Toby TK, Fornelli L, Kelleher NL: Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem (Palo Alto Calif). 9, 499–519 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai WX, Tucholski TM, Gregorich ZR, Ge Y: Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev Proteomic. 13, 717–730 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen B, Brown KA, Lin Z, Ge Y: Top-Down Proteomics: Ready for Prime Time? Anal Chem. 90, 110–127 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li MX, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL: Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 480, 254–U141 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown KA, Chen B, Guardado-Alvarez TM, Lin Z, Hwang L, Ayaz-Guner S, Jin S, Ge Y: A photocleavable surfactant for top-down proteomics. Nat Methods. 16, 417–420 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen B, Hwang L, Ochowicz W, Lin Z, Guardado-Alvarez TM, Cai W, Xiu L, Dani K, Colah C, Jin S, Ge Y: Coupling functionalized cobalt ferrite nanoparticle enrichment with online LC/MS/MS for top-down phosphoproteomics. Chem Sci. 8, 4306–4311 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts DS, Chen BF, Tiambeng TN, Wu ZJ, Ge Y, Jin S: Reproducible large-scale synthesis of surface silanized nanoparticles as an enabling nanoproteomics platform: Enrichment of the human heart phosphoproteome. Nano Res. 12, 1473–1481 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ge Y, Rybakova IN, Xu QG, Moss RL: Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. P Natl Acad Sci USA. 106, 12658–12663 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Major LL, Denton H, Smith TK: Coupled Enzyme Activity and Thermal Shift Screening of the Maybridge Rule of 3 Fragment Library Against Trypanosoma brucei Choline Kinase; A Genetically Validated Drug Target. In: El-Shemy HA (ed.). Rijeka (HR), (2013) [PubMed] [Google Scholar]
- 36.Smith LM, Kelleher NL: Proteoforms as the next proteomics currency. Science. 359, 1106–1107 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, Ge Y, Gunawardena J, Hendrickson RC, Hergenrother PJ, Huber CG, Ivanov AR, Jensen ON, Jewett MC, Kelleher NL, Kiessling LL, Krogan NJ, Larsen MR, Loo JA, Loo RRO, Lundberg E, MacCoss MJ, Mallick P, Mootha VK, Mrksich M, Muir TW, Patrie SM, Pesavento JJ, Pitteri SJ, Rodriguez H, Saghatelian A, Sandoval W, Schluter H, Sechi S, Slavoff SA, Smith LM, Snyder MP, Thomas PM, Uhlen M, Van Eyk JE, Vidal M, Walt DR, White FM, Williams ER, Wohlschlager T, Wysocki VH, Yates NA, Young NL, Zhang B: How many human proteoforms are there? Nat Chem Biol. 14, 206–214 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narayana N, Cox S, Shaltiel S, Taylor SS, Xuong N: Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5–24) and adenosine. Biochemistry. 36, 4438–4448 (1997) [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Tiambeng TN, Cai W, Chen B, Lin Z, Gregorich ZR, Ge Y: Impact of Phosphorylation on the Mass Spectrometry Quantification of Intact Phosphoproteins. Anal Chem. 90, 4935–4939 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YC, Ayaz-Guner S, Peng Y, Lane NM, Locher M, Kohmoto T, Larsson L, Moss RL, Ge Y: Effective top-down LC/MS+ method for assessing actin isoforms as a potential cardiac disease marker. Anal Chem. 87, 8399–8406 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai W, Guner H, Gregorich ZR, Chen AJ, Ayaz-Guner S, Peng Y, Valeja SG, Liu X, Ge Y: MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol Cell Proteomics. 15, 703–714 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S: Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci U S A. 86, 8247–8251 (1989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herberg FW, Bell SM, Taylor SS: Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng. 6, 771–777 (1993) [DOI] [PubMed] [Google Scholar]
- 44.Siuti N, Kelleher NL: Decoding protein modifications using top-down mass spectrometry. Nat Methods. 4, 817–821 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haydon CE, Eyers PA, Aveline-Wolf LD, Resing KA, Maller JL, Ahn NG: Identification of novel phosphorylation sites on Xenopus laevis Aurora A and analysis of phosphopeptide enrichment by immobilized metal-affinity chromatography. Mol Cell Proteomics. 2, 1055–1067 (2003) [DOI] [PubMed] [Google Scholar]
- 46.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK: The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barraud P, Banerjee S, Mohamed WI, Jantsch MF, Allain FH: A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc Natl Acad Sci U S A. 111, E1852–1861 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Comstock MJ, Whitley KD, Jia H, Sokoloski J, Lohman TM, Ha T, Chemla YR: Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science. 348, 352–354 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waugh DS: An overview of enzymatic reagents for the removal of affinity tags. Protein Expres Purif. 80, 283–293 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tholey A, Pipkorn R, Bossemeyer D, Kinzel V, Reed J: Influence of myristoylation, phosphorylation, and deamidation on the structural behavior of the N-terminus of the catalytic subunit of cAMP-dependent protein kinase. Biochemistry. 40, 225–231 (2001) [DOI] [PubMed] [Google Scholar]
- 51.Toner-Webb J, van Patten SM, Walsh DA, Taylor SS: Autophosphorylation of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 267, 25174–25180 (1992) [PubMed] [Google Scholar]
- 52.Meng FY, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL: Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 19, 952–957 (2001) [DOI] [PubMed] [Google Scholar]
- 53.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW: Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 72, 563–573 (2000) [DOI] [PubMed] [Google Scholar]
- 54.Holden DD, Sanders JD, Weisbrod CR, Mullen C, Schwartz JC, Brodbelt JS: Implementation of Fragment Ion Protection (FIP) during Ultraviolet Photodissociation (UVPD) Mass Spectrometry. Anal Chem. 90, 8583–8591 (2018) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.