

Summary
Current approaches to reducing the latent HIV reservoir entail first reactivating virus-containing cells to become visible to the immune system. A critical second step is killing these cells to reduce reservoir size. Endogenous cytotoxic T-lymphocytes (CTLs) may not be adequate because of cellular exhaustion and the evolution of CTL-resistant viruses. We have designed a universal CAR-T cell platform based on CTLs engineered to bind a variety of broadly neutralizing anti-HIV antibodies. We show that this platform, convertibleCAR-T cells, effectively kills HIV-infected, but not uninfected CD4 T-cells from blood, tonsil or spleen, and only when armed with anti-HIV antibodies. ConvertibleCAR-T cells also kill within 48 hours more than half of the inducible reservoir found in blood of HIV-infected individuals on antiretroviral therapy. The modularity of convertibleCAR-T cell system, which allows multiplexing with several anti-HIV antibodies yielding greater breadth and control, makes it a promising tool for attacking the latent HIV reservoir.
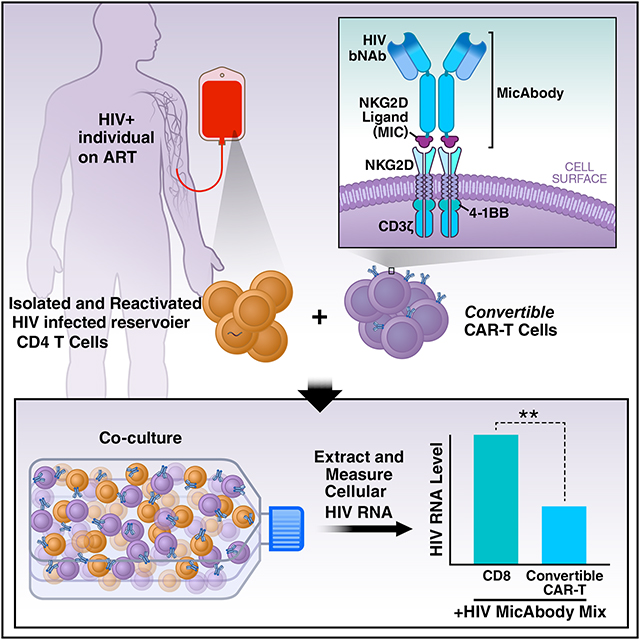
Graphical Abstract
eTOC Blurb
An adaptable CAR-T cell platform based on cytotoxic lymphocytes engineered to bind a variety of broadly neutralizing anti-HIV antibodies can effectively kill HIV-infected primary cells and reduce viral reservoirs in the blood of infected individuals on antiretroviral therapy.
Introduction
The main obstacle to curing HIV-infected individuals is the existence of a reservoir of latently infected cells that persists despite long term antiretroviral therapy (ART). These rare cells harbor an integrated HIV provirus but generally do not express viral proteins, making them invisible to the immune system and difficult to eliminate (Chun et al., 1997; Finzi et al., 1997; Richman et al., 2009; Wong et al., 1997). The establishment of latency may have various causes, including the infection of partially active cells transitioning to a resting state (Chun et al., 1995; Siliciano and Greene, 2011), the activation of a virus-encoded program (Razooky et al., 2015), or even the infection of resting CD4 T cells (Cameron et al., 2010). Regardless of the origin of the latent reservoir, the greater its size, the harder it is for the immune system to control viral levels (Lori et al., 1999), and the faster the virus rebounds in the event of ART interruption (Li et al., 2016). Although the advent of combination ART has revolutionized management of HIV infection and prevents development of AIDS, HIV-positive individuals must adhere to lifelong treatment, not without adverse side effects (Renju et al., 2017). Despite falling short of complete viral eradication, a consistent ability to reduce the size of the viral reservoir and control its activity to achieve a sustained viral remission in the absence of ART would radically alter approaches to the treatment of infection around the world (Goulder and Deeks, 2018; Lori et al., 1999).
Some HIV-positive individuals who received treatment early during acute infection exert long-lasting control negating viral rebound after withdrawal of ART. In these rare individuals, termed post-treatment controllers (PTCs) (Hocqueloux et al., 2010; Saez-Cirion et al., 2013), the immune system manages to keep the latent reservoir small, and sometimes even reduces the reservoir size and controls viremia without drug intervention (Saez-Cirion et al., 2013). The reasons for PTCs’ better control of HIV load are still under investigation, but levels of CD4 T cell activation are lower in these individuals than in non-controllers. CD4 T and NK cell responses are also higher and levels of inflammation are low (Saez-Cirion et al., 2013; Samri et al., 2016). We seek an intervention that routinely establishes post-treatment control in all HIV-infected people from both developed and developing countries.
One of the main approaches to reducing the size of the reservoir is “shock and kill” (Archin et al., 2012). This approach entails exposing cells to one or more latency reversing agents (LRAs) to induce viral gene expression, ideally with little or no toxicity to the host (Jean et al., 2019). Once successfully shocked, reservoir cells begin producing the viral protein Env, which is inserted on the cell surface. These cells may now be killed either due to a viral cytopathic effect, or by immune cells recognizing as foreign the viral Env protein/peptides expressed on the surface of the reservoir cells (Jones and Walker, 2016; Leonard et al., 1988). However, only strong LRAs lead to cell killing via viral-induced cytopathic effects, and their clinical utility is quite limited because of their toxic side-effects, including potential triggering of a cytokine storm (Chun et al., 1999; Prins et al., 1999). More subtle, non-toxic cell compounds would be desirable, but they may only stimulate a small fraction of the latent reservoir necessitating serial administration (Jean et al., 2019). Regardless of the way reservoir cells are reactivated, the success of the shock-and-kill strategy crucially depends on an efficient means of killing the reactivated cells.
HIV infection is commonly associated with the exhaustion of cytotoxic T lymphocytes (CTLs) manifested by loss of both effector function and proliferative capacity (Cella et al., 2010; Hersperger et al., 2010; Kalams et al., 1999; Shin and Wherry, 2007). In addition, CTL-resistant viral strains emerge, especially if treatment with ART is delayed beyond acute infection (Deng et al., 2015; Jain et al., 2013; Shan et al., 2012; Yang et al., 2002). Any attempt to boost the cell-mediated immune response of HIV-infected individuals must overcome these two hurdles.
Our work focuses on designing a more reliable way to kill the reactivated cells. Such killing is pivotal for reducing the size of the reservoir and critical for implementing a “reduce-and-control” strategy that, together with an immune intervention, might lead to post-treatment control. Two options for killing include: 1) improve the activity of the resident CTLs (Migueles et al., 2008), or 2) introduce new CTLs (Sung et al., 2018). Several broadly neutralizing antibodies (bNAbs) are now available that react with infected cells (Julg and Barouch, 2019; Mayer et al., 2017; McCoy, 2018; Sok et al., 2016). These bNAbs have the ability to recognize HIV-infected cells and contribute to their neutralization but still need a functional immune system to execute the bNAb-directed killing (Bar-On et al., 2018; Caskey et al., 2017). New CTLs have also been introduced by first removing patient T cells, inserting into these cells a chimeric antigen receptor (CAR) against a specific surface target along with costimulatory and signaling domains (e.g. 4–1BB and CD3ζ) (Guedan et al., 2019) and then reinfusing the CAR-T cells into the patient (Pule et al., 2003). In the most successful application of this CAR-T cell approach, cells were engineered to express an anti-CD20 antibody, which specifically recognizes the CD20 antigen (Onea and Jazirehi, 2016) expressed on malignant cells from patients with refractory B-cell lymphoma (Jacobson, 2019; Onea and Jazirehi, 2016; Zheng et al., 2018). Although successful, one of the problems of current CAR-T cells is that once administered, the cells are always in an “on-state” thus control is limited and there is little to no way to halt the activity of these cells should serious autoimmune side effects emerge (Bonifant et al., 2016; Zheng et al., 2018).
Similar strategies have been attempted to engineer CAR-T cells against HIV, by fusing single chain fragment variable (scFv) part of an anti-HIV Env antibody (Sung et al., 2018) (see Figure 1E) or the extracellular domains of the CD4 molecule that recognize HIV Env, to the zeta chain of the T cell receptor creating a CAR-T cell (Deeks et al., 2002). Although well-tolerated in humans, these CAR-T cells failed to produce clinical effects potentially due to lack of an adequate host response to LRAs (Wagner, 2018). The use of a single targeting motif on these CAR-Ts significantly limits the ability of these cells to address HIV’s diversity and high rate of viral epitope drift in patients. A more versatile platform that is inherently more responsive to viral fluidity is therefore needed to maximize the impact of a CAR-T based strategy.
Figure 1: Construction of MicAbody/ConvertibleCAR-T Platform.

(A) The MIC/ULBP-ligand family are natural ligands for NKG2D receptors present on NK cells and CTLs. NKG2D binds to the α1–α2 part of the ligands (B) Protein engineering of the α1–α2 ligand domain and NKG2D receptor to create a cognate ligand-receptor pair that no longer recognizes the natural ligand or receptor. (C) Protein engineering of bispecific antibody based on bNAb and mutated α1–α2 on the antibody (MicAbody), and a mutated NKG2D CAR fused to 4–1BB and CD3ζ as the signaling domains. (D) Construction of cCAR-T cell based on the mutated NKG2D. The convertibleCAR system allows specific binding of MicAbody to the mutated NKG2D-based CAR expressed on the T-cell. (E) Conventional scFv-based CAR-T cell. (F) ELISA binding assay of MicAbody to WT NKG2D receptor or to the mutated form. AU – arbitrary absorbance units. The figure represents one of three independent biological experiments yielding similar results. (G) Antibodies conjugated to Alexa flour (AF) fluorophore were assessed for selective binding of to cCAR-T cell with mutated NKG2D. Blue- MicAbody; Red- parental bNAb; Black-no Ab. 10,000 events were acquired and each dot represents number of cells with the same MFI. (H) MicAbody binds to HIV/GFP+ cells similarly to the parental bNAb. Red- parental bNAb; Blue- MicAbody; Gray- Isotype control. See also Tables S1 and S2. (I) In vivo killing by the cCAR-T platform. Comparison of the effectiveness of cCAR-T platform with the scFv conventional CAR-T platform in controlling Raji lymphoma cell growth in NSG mice. n=3 for each cohort.
CTLs and Natural killer (NK) cells express on their surface the NK group 2D receptors (NKG2D) which recognize a family of ligands overexpressed on cells stressed by viral infection or transformation (Cosman et al., 2001). The α1–α2 domains of these major histocompatibility complex class I chain-related (MIC) ligands – which include MICA, MICB, and UL16 binding proteins (ULBPs) 1–6 - bind to NKG2D which activates the cytolytic function of these cells (Bauer et al., 1999; Cosman et al., 2001; Steinle et al., 2001) (see Figure 1A). We leveraged the natural binding of MIC/ULBP ligands to NKG2D to develop an exclusive orthogonal ligand-receptor interaction to generate the components of a highly modular universal CAR-T cell platform (For more thorough biochemical characterization and application of the platform in other fields see U.S. Patent No. 10,259,858 and (Landgraf et al., 2019). The engineered extracellular domain of the NKG2D variant was tethered to the intracellular 4–1BB and CD3ζ co-signaling domains to generate the CAR while the mutant ligand domain was fused to an antibody to generate a bispecific molecule termed a MicAbody™. This two-part system, which we term convertibleCAR™-T (cCAR-T) cells, can readily be multiplexed with several distinct MicAbodies distinguished by their ability to engage unique epitopes. We reasoned that by combining bNAbs and cCAR-T, we might harness their individual benefits to expand the breadth of killing of HIV-infected targets while minimizing the emergence of viral resistance. We have explored the killing properties of these cCAR-T cells against both productively HIV-infected cells from human blood, tonsil and spleen and reactivated blood CD4 T cells obtained from ART-suppressed HIV-positive individuals. We describe our findings concluding this platform shows great potential as a robust approach to reduce the reservoir size.
Results
HIV bNAbs fused to an orthogonal MIC ligand direct T cells expressing an inert NKG2D-based CAR construct to recognize HIV-1-infected primary cells
To combine bNAbs and CAR-T cells, we took advantage of a natural ligand-receptor system that normally participates in the body’s surveillance for malignant and virally infected cells. This system uses a ligand from the MIC/ULBP family, expressed on stressed cells, and its receptor NKG2D, expressed on CTL and NK cells (Figure 1A). The binding pocket of the NKG2D extracellular domain was mutated to render it inert and incapable of binding its natural ligands (Figure 1B). Compensatory mutations in the α1–α2 domain of a MIC ligand were introduced to eliminate binding to wild-type NKG2D and promote sub-nanomolar engagement of the mutant NKG2D (Figure 1F). This established a unique and exclusive orthogonal pairing system in which the mutant NKG2D was subsequently formatted as a CAR for expression on CD8 T cells (convertibleCAR-T cells, cCAR-T) and the mutant α1–α2 fused to HIV bNAbs (Figure 1C and STAR method) to generate bispecific agents called MicAbodies. The mutant α1–α2 exclusively engages the mutant NKG2D-CAR expressed on the cCAR-T cells (Figure 1G and Table S1) while the Fv domains engage the cognate antigen of the HIV-Env protein on the surface of tonsil-derived infected cells (Figure 1H). This high avidity and specific interaction generates an immunologic synapse that activates the cytotoxic effector functions of the cCAR-T cells to kill the targeted cell.
Four MicAbodies from two types of HIV-specific bNAbs were constructed. The first two bNAbs, 3BNC60 and 3BNC117, recognize the CD4 binding site on the virus envelope (Malbec et al., 2013; Scheid et al., 2016; Scheid et al., 2011); the other two, PGT121 and 10–1074, are HIV-V3 glycan loop domain-binding bNAbs (Malbec et al., 2013; Mouquet et al., 2012; Walker et al., 2011) (Table S2). Both types of bNAbs were shown to be potent in clinical trials when used alone or in combination (Bar-On et al., 2018; Caskey et al., 2019; Caskey et al., 2017; McCoy, 2018).
To test the specificity of cCAR-T killing, we constructed two additional MicAbodies as negative controls: one based on the B cell-specific, anti-CD20 monoclonal antibody, rituximab (Maloney et al., 1997) and the other on the breast cancer-specific monoclonal antibody, anti-HER2 or trastuzumab (Herbst and Hong, 2002) (Table S2). Using fluorophore-conjugated MicAbodies we demonstrated specific binding of MicAbodies to the effector cCAR-T (Figure 1G) and specific binding of the HIV MicAbodies to the HIV-infected (GFP positive) tonsil cells in a comparable manner to natural bNAb (Figure 1H).
Finally, to ensure that the effect of our system on infected cells would reflect specific binding to the cCAR-T, we introduced two mutations (D265A/N297A) in the MicAbody’s heavy chain (Table S2). These mutations abolish the MicAbodies’ ability to trigger cell killing via antibody-dependent cellular cytotoxicity (ADCC) often involving NK cells as effectors (Shields et al., 2001). Adding these mutations to the platform helps to ensure that killing is only triggered by the MicAbody-cCAR-T interaction, resulting in a safe, modular and regulatable killing platform.
We first tested the platform in vivo in subcutaneous lymphoma mouse model, where NSG mice were injected with Raji lymphoma cells. After tumor implantation, the mice were injected with Rituximab MicAbodies (20ug) every two days for 6 doses, and cCAR-T cells were injected one day after the first MicAbody dose. Tumor volumes were regularly monitored by caliper measurements. Conventional CAR-T (CD19 scFv) and cCAR-T armed with Rituximab MicAbodies displayed similar levels of control of tumor cell growth. Delivery of cCAR-T without MicAbody, produced no significant anti-tumor effects (Figure 1I).
cCAR-T cells combined with HIV MicAbodies specifically kill HIV-infected primary CD4 T cells
Although viral load is commonly tested in blood and a large number of latency studies have been performed using blood cells (Shacklett et al., 2019), it is important to explore HIV infection and latency in lymphoid tissues since most of the reservoir resides there and replication of the virus mainly occurs in these tissues (Chun et al., 2008; Haase et al., 1996; Pantaleo et al., 1993; Yukl et al., 2010). We have principally used tonsil-derived cells for infection with HIV-GFP in this study (Doitsh et al., 2010)(see STAR methods, and Figure S1) principal findings have also been confirmed in both spleen and peripheral blood cells.
To determine the optimal effector-to-target ratio, we combined cCAR-T cells and tonsil-derived cells at various effector: target (E:T) ratios in the presence of a mix of the four HIV-specific MicAbodies and assessed killing efficiency by measuring the depletion of HIV-infected CD4 T cells (HIV-positive cells were monitored by the expression of the GFP reporter). For each experiment, one million infected tonsil cells (containing ~1×104 GFP+, HIV-infected cells) were incubated for 48 hours with a range of cCAR-T cells from no-CAR-T to 2×105 CAR-T cells (In the no-CAR-T controls, untransduced CD8 T-cells from the same donor were added). All experiments were conducted in the presence of ART (saquinavir 5μM) to prevent a spreading infection that could potentially confound the killing results.
We found that the killing efficiencies of cells infected with HIV-1 CCR5 (R5) tropic virus (BaL/GFP) correlated with the number of effector cCAR-T cells present. These results were consistent regardless of the concentration of MicAbodies tested (Figure 2A–B; GFP+ bars). Although specific killing of HIV infected cells (GFP+) improved as we added more effector cells, by an E:T ratio of 20:1, the viability of the uninfected bystander CD4 T cells began to decrease both with high and low MicAbody concentrations (Figure 2A–B; GFP− bars). Based on these results, we selected an E:T ratio of 10:1 for all of subsequent experiments.
Figure 2: Specific killing of primary CD4 T cells infected with CCR5-tropic HIV by convertibleCAR-T combined with HIV Env-specific MicAbodies.

To determine the optimal effector to target cell ratio, one million tonsil derived cells (~1×104 HIV/GFP-infected target cells) were incubated with a range of cCAR-T effector cells from zero (0:1) to 2×105 (20:1) cCAR-T cells: target cells for 48 hours with the mix of four HIV Env-specific MicAbodies (Mix). In the absence of cCAR-T cells (0:1), the donor-matched untransduced CD8 T cells were present. GFP+ live GFP+/CD3+/CD8− cells were counted to assess reduction in target cells (GFP+) and live GFP−/CD3+/CD8− cells were counted to assess off target killing. HIV mix concentration was tested with high (0.5nM) (A) or low (10pM) (B) concentration of each individual MicAbody in the HIV MicAbody mix. Data derived from three independent experiments; mean + SEM. (C) Specific killing of R5 tropic HIV-1 (BaL) infected cells or (D) X4 tropic HIV-1 (NL4–3) used to infect tonsil cells followed by testing of individual HIV-specific MicAbody for arming of cCAR-T cells. One million tonsil derived cells (~1×104 infected cells) were incubated with 1×105 CAR-T cells for 48 hours, in the presence of different concentrations (0.1–10nM) of HIV Env-specific MicAbodies. B-cell specific MicAbody (Ritux) and anti-HER2 specific MicAbody (HER2) were used as negative control MicAbodies. Results are presented relative to the no cCAR-T control. For each individual MicAbody, an internal control of no cCAR-T supplemented with the highest MicAbody concentration tested is presented. To assess off target killing or generalized in-well toxicity, viability of GFP− CD4 T cells was assessed. This experiment was performed four times using cells from independent donors. Data are represented as mean + SEM. * = p≤0.05, ** = p≤0.01, *** = p≤0.001 (compared to no cCAR-T presence). See also Figures S1–3 and S5.
Next, a number of control experiments were performed to assess the specificity of cCAR-T killing HIV-infected cells. To confirm that the killing of target cells depended on HIV recognition, we tested unarmed cCAR-T or cCAR-T armed with the B cell-specific MicAbody rituximab or the anti-HER2 MicAbody, trastuzumab. No detectable killing of HIV-infected CD4 T-cells was observed with these control MicAbodies (Figure 2C left panel). Only when an HIV-specific MicAbody was added to the cCAR-T was a reduction in the number of HIV-infected, GFP-positive tonsil cells observed (Figure 2C, right panels). Infected CD4 T cell number was reduced by about 60% and specific killing was observed with all four HIV MicAbodies at doses spanning two orders of magnitude (0.1–10nM). We conclude that cCAR-T cells armed with HIV-specific MicAbodies can successfully kill HIV-infected tonsil cells.
To determine whether in-well toxicity or non-specific cCAR-T toxicity was contributing to reducing cell viability, we monitored the non-HIV-infected CD4 T cells (GFP− cells) in each well. No decrease in viability of these uninfected CD4 T cells was observed in any of the wells (Figures 2A–2C; gray bars, GFP−). We conclude that cCAR-T cells armed with an HIV-specific MicAbody selectively kills HIV-1-infected cells without affecting uninfected bystander cells. To test a second lymphoid tissue, we performed killing experiments on cells derived from the spleens of healthy donors, finding similar results (Figure S3).
In about 50% of AIDS patients, the R5 tropic virus converts to an X4 (CXCR4) tropic virus (Schuitemaker et al., 1992; Schuitemaker et al., 2011). We therefore tested the ability of cCAR-T cells to kill tonsil-derived CD4 T cells infected with X4-NL4–3, a GFP− tagged X4-tropic virus (Levy et al., 2004). We found that cCAR-T killed these cells efficiently and selectively when combined with the Env/CD4-binding MicAbodies (3BNC60/117), which are known to neutralize both R5-tropic and X4-tropic viruses (Bruel et al., 2016). However, when cCAR-T cells were combined with the Env/V3-loop binding MicAbodies, known to preferentially neutralize R5-tropic over X4-tropic viruses (Bruel et al., 2016), the killing was reduced (Figure 2D and Figure S5). We conclude that the type of bNAb selected for MicAbody construction is critical in the context of X4-tropic viral infections. These observations support the importance of being able to multiplex the arming of the cCAR-T cells.
Laboratory-adapted HIV strains, such as R5-BaL and X4-NL4–3 used above, are less predictive of in vivo outcomes than strains mediating transmission between HIV-positive individuals, also known as Transmitted/Founder (T/F) viruses, which consistently display CCR5 co-receptor tropism (Keele et al., 2008; Li and Chen, 2019; Parrish et al., 2013; Schwartz et al., 2018). To test our system in a more clinically relevant setting, we infected primary cells with a T/F virus 109FPB4 (F4) (Cavrois et al., 2017; Neidleman et al., 2017). Our previous experiments with the R5-BaL and X4-NL4–3 strains used MicAbodies in the nanomolar (nM) concentration range, all of which induced similar levels of killing (Figure 2). To identify the minimum amount of MicAbody sufficient to trigger CAR-T mediated killing, we assessed the killing of T/F infected cells in the presence of picomolar (pM) MicAbody concentrations. After 48 hours of incubation, dose-dependent killing of up to 65% of infected cells was observed with three of the four HIV-specific MicAbodies over a 10–500pM concentration range (Figure 3A). In the case of the 10–1074-based MicAbody, no detectable killing of T/F HIV-infected cells occurred at any of the MicAbody concentrations. These results show that the cCAR-T cell platform can recognize and efficiently kill primary cells infected with both laboratory-adapted and T/F HIV strains, but that depending on which virus the CD4 T-cell harbors, killing may vary depending on the specificity of the selected MicAbody. Moreover, our results show that picomolar MicAbody concentrations are sufficient to trigger robust killing of infected cells by the cCAR-T cell platform.
Figure 3: Arming cCAR-T cells with HIV Env-specific MicAbodies promotes effective killing of target cells infected with F4, a transmitted founder virus.

One million tonsil-derived cells, including approximately 1×104 CD4 T cells infected with the F4 transmitted founder virus containing a GFP reporter (F4-GFP), were incubated with 1×105 CAR-T cells in the presence of different concentrations (10–500pM) of four different HIV-specific MicAbodies. After a 48-hour incubation, survival was assessed for the whole GFP-expressing population (A) and for cells gated on high (B) and low (C) GFP expression. Negative controls for the HIV Env-specific MicAbodies included B cell-specific MicAbody (Ritux) or anti-HER2 specific MicAbody (HER2). Additional controls for cCAR-T included No cCAR-T or CD8 cells supplemented with the highest MicAbody concentration tested (500nM). Ratios depicting fractional survival of GFP positive cells were determined by dividing the number of GFP positive cells found in the presence of MicAbody and cCAR-T by the GFP positive cells found in the MicAbody-only negative controls. Results are cumulated from four independent experiments. Data are represented as mean + SEM. N.S. = p > 0.05, * = p≤0.05, ** = p≤0.01, *** = p≤0.001. See also Figure S2.
cCAR-T cell-mediated killing correlates with HIV gene expression levels in target cells
Although highly specific, cCAR-T cell-mediated killing consistently plateaued at ~65% (Figure 3A). We sought to determine whether this plateau reflects a differential sensitivity of the infected cells to cCAR-T cells, possibly correlating with the levels of viral gene expression occurring in the infected cells. In these studies, cells were infected with HIV strains that express GFP as a multiply-spliced RNA under control of the viral LTR (Kutsch et al., 2002; Neidleman et al., 2017). In these viruses, GFP-IRES-Nef is inserted in the Nef coding region. Nef is expressed at physiological levels (Neidleman et al., 2017) and mediates CD4 downregulation of infected cells (Figure S2 panel C). Therefore, GFP expression is a quantitative marker of overall HIV gene expression. To assess a potential correlation between levels of HIV expression and sensitivity to cCAR-T killing, we gated HIV-infected cells based on their GFP fluorescence level (low or high) and tested their response to cCAR-T killing. While CD4 T cells with high GFP levels were readily eliminated by the cCAR-T system (killing efficiency up to ~90%; Figure 3B), many CD4 T cells with lower GFP levels survived exposure to the cCAR-T cells (killing efficiencies of ~30%; Figure 3C). We conclude that killing is correlated with the amount of HIV gene products the infected cells produce and most likely with the amount of Env protein they express on their cell surface. This is consistent with the observation that targeted antigen density is a major determinant in CAR-T cell efficacy (Walker et al., 2017; Watanabe et al., 2015).
Single-cell time-lapse microscopy shows a delay in bNAb-armed cCAR-T cell killing kinetics.
The convertibleCAR-T system requires a “three-body collision”: the cCAR-T cell, the MicAbody and the infected cell to create the cytolytic immunologic synapse. In order to monitor the killing kinetics, we used time-lapse microscopy. We incubated 3×106 primary cells infected with HIV-F4 with a mix of the four HIV specific MicAbodies in the presence of 3×105 cCAR-T cells on a μ-Dish slide. We then monitored the number of HIV-positive cells every 30 minutes for 48 hours. As observed in our end-point experiments (Figure 3 and Figure S4), cCAR-T cells killed 90% of the HIV-infected cells when the pool of MicAbodies was added (Figure 4). Interestingly, the killing did not commence until 10–15 hours after the initiation of the cultures. These findings suggest a requisite period of time for the effector cells to arm with MicAbodies and successfully collide with their target cells.
Figure 4: Time-lapse microscopy at single-cell resolution shows delay in killing initiation.

(A) Representative time-course of primary cells’ survival after infection with GFP-tagged F4-HIV and exposure to cCAR-T cells armed with a mix of the four HIV Env-specific MicAbodies (500pM each). Snapshots of 36 fields of view were taken every 30 minutes for 48 hours for bright field (cell borders), GFP (HIV+ cells) and RFP (cCAR-T cells) in X20 magnification on a confocal spinning disc microscope. Scale bar, 50 μm. See also Movie S1. (B) Quantitative analysis of HIV positive killing assay over 48 hours with cCAR-T and with or without MicAbodies. GFP quantification was made by ImageJ after reduction of background. The figure represents one of three independent biological experiments yielding similar results.
Multiplexing two MicAbody types results in specific killing of two different types of target cells
HIV-1 has proven to evade every single-drug or single-antibody therapy, regardless of their level of initial efficacy. Thus, the use of more than one bNAb-based MicAbody will likely prevent the emergence of resistant viruses over time. Since the convertibleCAR-T system is a two-part modular system, one of its advantages is the ability to multiplex with different MicAbodies, thus attacking the virus-infected cells with more than one type of bNAb. To confirm effective multiplexing with cCAR-T, we armed these cells with two different types of MicAbodies, one specific for B cells (rituximab) and the other corresponding to a mix of HIV-specific MicAbodies and assessed their killing properties in cultures containing a mixture of HIV-1-infected primary cells and ~10% B cells. The killing of B cells was not diminished by co-arming with HIV-specific MicAbodies (Figure 5A). Similarly, the presence of the B cell-specific MicAbody did not significantly impair killing of HIV-infected cells (Figure 5B). We conclude that multiplexing MicAbodies is possible with the cCAR-T system.
Figure 5: Multiplexing two MicAbodies targeting different cell types results in specific killing of the two different target cells.

(A) Killing of primary B cells and (B) CD4 T cells infected with F4-GFP strain of HIV (by cCAR-T complemented with a B cell-specific MicAbody (Ritux) or HIV-specific MicAbodies mix. Non-transduced cCAR-T parental CD8 cell, combined with HIV and B-cell MicAbodies (HIV+Ritux (CD8)) were tested as a negative control for MicAbody effect, and single MicAbody controls were tested to show maximal killing effect. Cell killing was normalized to the no cCAR-T controls. Data are the average of three independent experiments presented as mean + SEM. N.S. = p > 0.05, * = p≤0.05, ** = p≤0.01, *** = p≤0.001
Ex vivo cCAR-T cell killing of reactivated reservoir cells present in the blood of HIV-infected individuals on long term ART
We next turned to the effects of cCAR-T cells in the context of the latent HIV reservoir. In the prior in vitro primary-cell-based experiments, we used cells from the lymphoid tissues of healthy individuals productively infected ex-vivo with the various HIV strains. Since CD4 T cells from blood can only be productively infected after activation (Munoz-Arias et al., 2015), we compared cCAR-T cell killing of tonsil-derived and activated blood-derived CD4 T cells, and found that both were killed with a similar efficiency (Figure S4). To investigate potential effects of cCAR-T on the latent HIV reservoir, CD4 T cells were isolated from the blood of six HIV-positive individuals on suppressive ART(characteristics of the study participants are shown in Table S3). These cells were treated with a strong LRA (PMA + Ionomycin) and then cultured with cCAR-T cells armed with HIV MicAbodies at two concentrations. Since the specific virus genotype was not known, we used a mixture of the four HIV-specific MicAbodies. Two days after co-culture, RNA was extracted from the cells (caRNA) and HIV-RNA levels were assayed by Droplet Digital PCR (ddPCR) using an HIV-specific probe (Shan et al., 2013) (Figure 6A). We observed a significant reduction in the amount of HIV RNA in cultures including cCAR-T cells and MicAbodies, but not in cultures including MicAbodies and untransduced parental CD8 T cells or unarmed CAR-T cells (Figure 6B). Our results, showing superior killing with lower MicAbody concentration (0.1nM), compared to 1nM, are probably the result of the pro-zone effect reducing killing efficiency with higher antibody concentrations (Vaidya et al., 2017). These results indicate that it is possible to attack the inducible latent reservoir with bNAb-armed cCAR-T cells. The use of this ex vivo system provides a proof-of-concept validation for the ability of cCAR-T cells armed with HIV specific MicAbodies to reduce the reactivated latent reservoir.
Figure 6: Ex vivo killing of reactivated CD4 T-cells from HIV-1-infected individuals on ART by cCAR-T and MicAbodies.

(A) Experimental description. CD4 T-cells from HIV infected individuals on long term ART were isolated and activated for 72 hours with 50 ng/ml PMA and 1 μM ionomycin. After incubation for two days with cCAR-T cells or matched untransduced CD8 T-cells and a mix of four different HIV specific MicAbodies (either 0.1 or 1nM of each MicAbody), cell-associated RNA (caRNA) was extracted, and HIV RNA was quantitated by Droplet Digital PCR (ddPCR). (B) HIV RNA quantification results normalized to HIV RNA levels present in CD8 T cells and 1 nM HIV antibody mix. This experiment represents studies of 6 HIV-infected individual. Data are represented as mean ± SEM. N.S. = p > 0.05, * = p≤0.05, ** = p≤0.01 See also Table S3.
Discussion
A modular CAR-T cell platform armed with HIV bNAbs that recognizes and kills HIV-infected cells
Many attempts to cure HIV are focusing on “shock and kill” with the goal of activating virus expression in the latent reservoir followed by immune clearance of the reservoir cells, all performed under the cover of ART to prevent viral spread. Results with the first generation of LRAs, including histone deacetylase inhibitors (HDACi) and Protein kinase C (PKC) activators are disappointing due mainly to a lack of potency and/or unacceptable toxicity (Prins et al., 1999; Rasmussen and Lewin, 2016). The fact that CTLs present in HIV-infected individuals exhibit an exhausted phenotype and diminished killing capacity is also of concern as is the potential presence of CTL-resistant viruses within the latent reservoir. A delayed start of ART certainly sets the stage for the emergence of such resistance.
To ensure highly effective killing of reactivated reservoir cells, we developed and tested a platform, convertibleCAR-T cells, based on the binding of a mutant NKG2D receptor to an orthogonal MIC ligand fused to broadly neutralizing HIV antibodies (MicAbodies). The exclusive interaction engineered into this ligand-receptor pair ensures that on its own each component is functionally inert as was shown both in vitro and in vivo. As a consequence, neither the cCAR-T cells nor the MicAbody are able to kill target cells until they have bound to each other and the MicAbody has specifically engaged its epitope on an HIV-infected target cell to create an immunologic synapse. This system robustly incorporates many features that not only provide targeting flexibility and specificity but also enhanced controllability and safety. During HIV infection, soluble ligands (e.g. MICA and ULBP) for the natural NKG2D receptor are expressed but cleaved from the surface of infected cells, thereby compromising the ability of CTL and NK cells to kill infected cells (Matusali et al., 2013). The mutation of the MIC ligand and NKG2D receptor such that each is unable to bind their natural substrates circumvents this problem. Additionally, the MicAbody has been rendered ADCC-deficient with mutations in the Fc region that disrupt the binding to Fcγ receptors present on NK cells (Shields et al., 2001). In summary, these modular features ensure that the cCAR-T cells and the MicAbodies are unable to kill target cells until they are bound to each other and the MicAbodies have specifically contacted an HIV-infected target cell to create the lethal immunologic synapse.
We are able to infect different lymphoid tissues and blood CD4 T cells with a variety of HIV strains and then to reduce the number of productively infected cells using cCAR-T cells armed with an HIV-specific MicAbody. Importantly, we can also use these cCAR-T cells to attack latent reservoir cells reactivated ex vivo, removing approximately 60% of the inducible reservoir within 48 hours. Furthermore, killing by cCAR-T is specific for infected cells, and occurs only when an HIV-specific MicAbody is present. Although killing efficiency correlates with viral protein expression, we predict that levels of Env present in vivo will be sufficient for recognition by the cCAR-T platform since these reactivated reservoir cells where shown to be bound to and be enriched using the same HIV bNAbs we are using (Cohn et al., 2018).
Up to 90% of primary human cells infected at high levels with HIV can be killed using the cCAR-T cell system. Killing is dose-dependent and requires the addition of only picomolar quantities of MicAbodies. This killing rate is similar or superior to that previously reported for in vitro CAR-T killing of HIV-infected cells including: 1) testing of CD4-based CAR-T on U1 human latent cell-line showing a reduction of 30–60% of reactivated cells (Zhen et al., 2017); 2) a CD3ζ based CAR-T with scFv derived PGT128 (which binds the V3 loop, like PGT121 in present study) showing killing of 43% of infected activated peripheral blood mononuclear cells (PBMCs) (Hale et al., 2017) and 3) testing of VRCO1 scFv-based CAR-T killing showing that more than 50% of CD4 T cells from three HIV-positive individuals were killed but this response required a 12-day incubation and the addition of large numbers of CAR-T cells (Liu et al., 2016). In the future, it will be interesting to compare directly the killing efficiencies of these various types of HIV-specific CAR-T cell platforms.
The cCAR-T cell platform increases the flexibility and safety
Treatment of HIV-infected individuals with a single anti-retroviral drug almost invariably leads to the development of drug resistance, an observation that prompted the introduction of combination antiviral therapy. Administering a single type of CAR-T cell (Sotillo et al., 2015) or a single bNAb (Caskey et al., 2017) could also select for emergence of resistant virus and loss of therapeutic benefit. To overcome this limitation, recent clinical trials with bNAb-based therapies are combining two strong bNAbs (10–1074 and 3BNC117) (Bar-On et al., 2018). However, classic CAR-T cells, as currently implemented, carry only a single anti-HIV scFv domain. Deploying a second targeting domain requires designing a second CAR-T cell, which is cumbersome and costly. However, in the convertibleCAR-T platform, multiplexing with two or more HIV-specific MicAbodies is easily accomplished. Indeed, our studies indicate that cCAR-T cells can be multiplexed with different antibodies promoting an effective attack on two different types of cells simultaneously.
cCAR-T containing the 10–1074 bNAb were unable to kill F4-infected cells but efficiently killed R5-BaL-infected cells. Our HIV-specific CAR-T platform is based on broadly neutralizing antibodies that can recognize and neutralize hundreds of strains, although a single antibody falls short of recognizing all strains (Eroshkin et al., 2014). This observation again strongly argues for arming cCAR-T cells with more than one bNAb-based MicAbody to increase the breadth of killing. The general ability to multiplex antibodies in the cCAR-T platform provides for great flexibility and an increased level of effectiveness. Additionally, the picomolar quantities needed for cytolysis supports the possibility of even higher levels of multiplexing if needed, leading to the possibility of implementing a single, defined cocktail of MicAbodies to achieve extremely broad coverage using a single CAR construct.
Another advantage of the cCAR-T system is that as a two-part system, it can be introduced into patients in an inert state. Only after the administration of the specific MicAbody or MicAbody mix will the cCAR-T start killing. This feature should allow clinicians to control both the dose and timing of administered HIV-specific MicAbody. By contrast, classical CAR-T cells are “on” all the time. Using a platform that is inert unless both parts are present promises to enhance safety and avoid some of the problems limiting the applicability of the current CAR-T systems in oncology, including increased cytokine production and CAR-T killing of unintended healthy host cells expressing low levels of the tumor target (Fedorov et al., 2013; Kochenderfer et al., 2012; Ma et al., 2019).
To further increase the safety of cCAR-T cells, one could envision specifically activating or suppressing the cCAR-T cells in vivo, taking advantage of their exclusive ligand-receptor interaction. In theory, any molecule fused to the mutated MIC-ligand binding domain will deliver the heterologous molecule specifically to its cognate NKG2D partner on cCAR-T cells. An example would be linking interleukins to the MIC ligand to preferentially promote cCAR-T growth and survival (McGill et al., 2010; Richer et al., 2015; Younes et al., 2016). Of note, CAR-T cells can persist in vivo in HIV-infected individuals for more than ten years after administration (Scholler et al., 2012). If cCAR-T cells prove equally durable, they could become a silent reservoir that clinicians could tap into when the need arises by simply delivering exclusively to the cCAR-T cells activation and proliferation signals along with the condition-specific MicAbody. Similarly, it should be possible to deliver specific suppressing molecules to rapidly or transiently silence the cCAR-T cells in case of adverse side effects, or deliver a kill signal if rapid termination of cCAR-T activity is desired.
Developing affordable and universal CAR-T cells
In spite of the great therapeutic potential of cCAR-T cells, its application in the clinic still faces important hurdles. The first one is the cost of such a treatment for a single patient. Whereas producing the MicAbodies is affordable and easily scalable, removing cells from a patient followed by isolation, expansion and transduction of these autologous cells with the mutated NKG2D-CAR receptor followed by their expansion and reinfusion is both expensive and requires a high degree of technical expertise (Sarkar et al., 2018). In addition, the long process from bedside to laboratory and then back to bedside makes deploying the current cCAR-T treatment difficult to envision in developing countries, many of which carry crippling burdens of HIV disease (Kharsany and Karim, 2016; Wang et al., 2016).
Several efforts are underway to reduce the cost of CAR-T therapies and make it more scalable. One high priority is to create allogeneic “universal donor” cells (Ruella and Kenderian, 2017; Torikai and Cooper, 2016) to provide off-the-shelf cells that can be used for all patients (Graham et al., 2018). To achieve such universal donor cells, the T-cell receptor (TCR) complex can be disabled to prevent graft-versus-host disease (Yang et al., 2015) and host versus graft elimination can be blocked by mutations within β2-microglobulin that prevents MHC-I surface expression (Ren et al., 2017). Modern techniques for cell editing, including CRISPR, transcription activator-like effector nuclease (Talen) and Zinc finger nucleases (ZFNs), offer potential ways to knock out those key cell-surface receptors responsible for graft versus host and host versus graft reactions (Osborn et al., 2016; Ren et al., 2017). Another promising approach involves the use of induced pluripotent stem cells (iPSCs) (Takahashi et al., 2007) as the source of the CAR-T cells. In this method, iPSCs are engineered to express the CAR prior to being differentiated into T cells (Themeli et al., 2013). These cells will act as effector cells in vivo, as has already been observed for hematopoietic stem/progenitor cells (HSPCs), which go through T cell development and selection in vivo (Zhen et al., 2017). It is now possible to convert iPSCs to various immune cells including T cells that contain chimeric antigen receptors (Lee, 2019; Themeli et al., 2013). Such cells can be engineered to be HLA-compatible for large populations thus dramatically reducing cost and increasing scalability of the CAR-T approach (Xu et al., 2019). Such universal donor cells could be readily adapted to the cCAR-T platform in view of its simplicity and modular design.
The urgent need for safe and effective LRAs
The other important hurdle to overcome before cCAR-T cells can be effectively tested in HIV-infected patients is the lack of safe and effective LRAs. To use cCAR-T in the kill phase of a “shock and kill” strategy, an effective shock must be delivered to render the latently infected cells visible to the immune system. The most commonly used LRAs include HDACi (Manson McManamy et al., 2014; Rasmussen et al., 2013) and PKC or PTEFb activators (Banerjee et al., 2012; Li et al., 2013; Mehla et al., 2010), which trigger latency activation in CD4 T cells. Unfortunately, these drugs showed either high toxicity or low efficiency in vivo (Prins et al., 1999; Rasmussen and Lewin, 2016). Furthermore, several types of LRAs, such as HDACi and PKC activators, exert suppressive effects on the cytolytic function of CTLs and by analogy would probably suppress activity of cCAR-T as well (Walker-Sperling et al., 2016). Nonetheless, approaches to improve shocking strategies, by lowering toxicity of some LRAs or administrating a weak LRA in multiple dosing, have shown some promise (Ke et al., 2018). Recently, combining new types of LRAs including toll-like receptor 7 (TLR-7) agonists (Jiang et al., 2018) and the PGT121 broadly neutralizing HIV antibody in SHIV-infected rhesus macaques resulted in prolonged times to rebound or in some cases no rebound (Borducchi et al., 2018). Of note, the TLR-7 agonist appears able to activate HIV-specific CTLs (Tsai et al., 2017), making it a preferable LRA for cCAR-T applications. Another emerging type of LRAs is the second mitochondria-derived activator of caspases (SMAC) mimetics that act through activation of the non-canonical NF-κB pathway and show promising LRA capabilities without the pleiotropic effect seen by other LRAs (Pache et al., 2015; Sampey et al., 2018). One apparent negative feature of all of these suggested LRAs is the small fractional response they elicit (Battivelli et al., 2018). In order to reach a robust level of reactivation, repetitive administration of the LRAs will be required. Using a controllable platform such as the cCAR-T could ensure high activity of the killer cells at the right time, and inert circulating killer cells between LRA and MicAbody administrations.
cCAR-T cells as a component of a reduce-and-control strategy
Absent a means for the safe and complete eradication of HIV in infected individuals, a reduce-and-control strategy seems to be an attractive option. This strategy involves both a reduction in the size of the reservoir and the introduction of an immune intervention that allows the infected host to control the virus despite the removal of ART. In its purest form, this strategy would imitate the natural, but rare state found in post-treatment controllers (PTCs). One of the hallmarks of post-treatment control is the presence of a small reservoir. A smaller reservoir size has been shown to be a good predictor of the ability of HIV-positive individuals to become PTCs (Saez-Cirion et al., 2013). Our ex vivo results show that we can reduce the reactivated reservoir size by 50% over the course of a two-day experiment. Introducing cCAR-T cells and refueling their killing activity with multiple, spaced doses of MicAbodies might provide a powerful way to prevent reservoir expansion in vivo, while maintaining the patients on ART would prevent the spread of infection by the reactivated cells. Future studies will concentrate on developing MicAbodies with different HIV-specific bNAbs, testing combinations of MicAbodies and evaluating the cCAR-T platform in vivo. Studies are being carried out now to test cCAR-T cells and MicAbodies in mice, and future experiments are envisioned in experimentally infected macaques prior to moving to human testing. In summary, the convertibleCAR-T cell system draws together exciting advances in both broadly neutralizing HIV antibody biology and CAR-T cell technology, to create a promising killing platform for attacking the latent HIV reservoir.
STAR ★ Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents should be directed to and will be fulfilled by Dr. Warner C. Greene (warner.greene@gladstone.ucsf.edu). Plasmid sequences for the convertibleCAR-T construct (which includes the mutant NKG2D receptor detailed in this manuscript) and the bNAb-MicAbodies (including the orthogonal ULBP2 variant) will be made available upon request. Purified MicAbodies can be generated upon execution of a material transfer agreement (MTA) with inquiries directed to Dr. Kaman Kim (kaman@xyphosinc.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Blood from HIV-infected individuals were obtained from volunteers participating in the SCOPE cohort (Hunt et al., 2003). Participants gave their informed consents as part of the SCOPE cohort. Specific characteristics of these participants and their ART regimens are summarized in Table S3.
Mice
Six-week old female NSG mice (Jackson Laboratories, Bar Harbor, ME) were housed and studied in strict accordance with the Institutional Animal Care and Use Committee (IACUC). Studies were performed by ProMab Biotechnologies, Inc. (Richmond, CA).
Primary-cell cultures
Human healthy tonsils and spleen were obtained from the Cooperative Human Tissue Network (CHTN, https://www.chtn.org). Human lymphoid aggregate culture (HLAC) prepared from tonsil or spleen were culture in HLAC medium: RPMI supplemented with 15% heat-inactivated fetal bovine serum (FBS), 100 μg/ml gentamicin, 200 μg/ml ampicillin, 1 mM sodium pyruvate, 1% nonessential amino acids, 2 mM L-glutamine, and 1% fungizone, at 37°C in 5% CO2 incubator.
Concentrated white blood cell preparations from healthy volunteers were obtained from Vitalant (www.vitalant.org) or from STEMCELL Technologies. PBMCs were cultured in RPMI supplemented with 10% FBS, 1000 U/ml Penicillin and 1 mg/ml Streptomycin and 2 mM L-glutamine, at 37°C in 5% CO2. cCAR-T and parental CD8 cells were cultured in T cell medium (X-Vivo 15 media, 5% human AB serum; 10 mM neutralized N-acetyl-LCysteine, 0.1% 2-mercaptoethanol, supplemented with 40U/ml IL-2). No personal identifiers were provided for either the uninfected lymphoid tissues or blood samples.
Cell line
Female HEK293T cells were transfected with various molecular clones of HIV to produce high titer virus preparations. Female Expi293™ cells were used for protein expression. Cells were cultured in DMEM supplemented with 10% FBS, 1000 U/ml Penicillin and 1 mg/ml Streptomycin and 2 mM L-glutamine, at 37°C in 5% CO2.
Virus strains
HIV molecular clones (pNL4–3-GFP, pBaL-GFP, and pF4-GFP) were purified from E.coli and used to transfect HEK293T cells.
METHOD DETAILS
Protein Expression and Purification
The orthogonal MIC variant was cloned as a C-terminal fusion to the human kappa light-chain via an APTSSSGGGGS linker. Additionally, D265A/N297A (Kabat numbering) mutations were introduced into the CH2 domain of the heavy chain of all antibody and MicAbody clones to reduce binding to all FcγR receptors in order to eliminate antibody-dependent cell cytotoxicity (ADCC) function. Cognate heavy- and light-chains for each bNAb clone were generated by swapping out the VH and VL domains (Table S2). Heavy- and light-chain plasmid DNAs (in the mammalian expression vector pD2610-V12, ATUM) for a given antibody clone were co-transfected into Expi293™ cells and purified by Protein A resin.
cCAR-T production
Leukopaks from healthy anonymous donors (STEMCELL Technologies) were used for primary T cell isolation. After a brief rinse in PBS + 2% FBS, cells were resuspended at 5×107 cells/ml in PBS + 2% FBS and CD8+ cells were enriched by negative selection (STEMCELL Technologies Human CD8 T Cell Isolation Kit). 50 μl of isolation cocktail was added per ml of cells and, after a 5 minute room temperature incubation, 50 μl of RapidSpheres™ (Stemcell) were added per ml of cells and total volume adjusted with PBS (to 35 ml total for each 21 ml of cells). Cells were isolated by applying an EasySep™ magnet for 10 min followed by transfer of buffer containing negatively enriched cells to new tubes for a second round of purification. Cells were then resuspended, counted, and cryopreserved at 10−15×106 cells/cryovial.
The extracellular domain of the mutant NKG2D was cloned with the CD8α signal sequence, hinge and transmembrane domains from CD8α, and the intracellular signaling domains from 4–1BB and CD3ζ into the pHR-PGK transfer plasmid for second generation Pantropic VSV-G pseudotyped lentivirus production along with packaging plasmids pCMVdR8.91 and pMD2.G as previously described (Roybal et al., 2016). For each batch of lentivirus produced, the three plasmids (pHR-PGK, 7.2 μg; pCMVdR8.91, 12.9 μg; and pMD2.G, 2.5 μg) were combined with 720 μl Opti-MEM™ (Fisher) then mixed with Fugene-HD (Promega) before adding to 6×106 Lenti-X 293T cells (Takara Bio) that had been seeded one day prior in a 10 cm dish. Two days post-transfection, supernatants were collected by centrifugation and passed through 0.22 μm filters. 5X concentrated PEG-6000 and NaCl are added to achieve final concentrations of 8.5% PEG-6000 (Hampton Research #HR2–533) and 0.3 M NaCl, incubated on ice for two hours, then spun at 3500 rpm at 4°C for 20 minutes. Concentrated viral particles were resuspended in 0.01 volume of PBS, and stored frozen at −80°C. Prior to lentiviral transduction, one vial of cryopreserved CD8 T cells was thawed and diluted into 10 ml of T cell medium “TCM” (X-Vivo 15 media, Lonza; 5% human AB serum, Corning; 10 mM neutralized N-acetyl-L-Cysteine, Sigma-Aldrich; 1X 2-mercaptoethanol, Thermo Fisher; 30 IU/ml human IL-2, R&D Systems). Cells were centrifuged at 400 × g for 5 minutes, resuspended in 10 ml TCM, adjusted to 1×106 cells/ml, 1 ml dispensed into each well of a 24-well plate, and allowed to rest overnight. Cells were then activated for 24 hours with Dynabeads™ Human T-Activator CD3/CD28 (Thermo Fisher) per manufacturer’s protocol. Concentrated lentiviral particles (50 μl) were added per well, cells incubated overnight, then transferred to T25 flasks with an added 6 ml TCM. After three days of expansion, Dynabeads were removed and cells back-diluted to 5×105 cells/ml with daily monitoring to ensure they did not exceed 4×106 cells/ml. Transduction efficiency was assessed by flow cytometry using a Rituximab-MicAbody that had been directly conjugated to Alexa Fluor 647 (Alexa Fluor Protein Labeling Kit, Thermo Fisher) per manufacturer’s protocol. Transduction efficiencies were above 70% for all cCAR-T tested.
Spinning disc time-lapse Microscopy
3×106 tonsil-derived cells were seeded in a 4-well imaging chamber and placed under the microscope for subsequent imaging. Imaging was performed using an Axiovert inverted fluorescence microscope (Carl Zeiss), equipped with a Yokogawa spinning disk, a CoolSNAP HQ2 14-bit camera (PhotoMetrics), and laser lines for 488 nm (40% laser power, 400-ms excitation) and 561 nm (40% laser power, 200-ms excitation). To facilitate time-lapse imaging, the microscope has a programmable stage with definite focus and a stage enclosure that maintains samples at 37 °C and 5% CO2 with humidity. Images were captured every 30 minutes for 48 hours. For each position a six-by-six X-Y grid was sampled. The objective used was 20× air, 1.3 N.A. Analysis was done using ImageJ by applying the following steps: First, a threshold for a positive GFP signal was set to 1000 arbitrary units to create a binary mask to distinguish HIV infected cells at a size range of 100–400 pixels from background fluorescence. Total area of infected cells (GFP positive area) was analyzed at each time point and normalized to the area of infected cells at the first two hours.
Culture and infection of primary cells
Spleen and tonsil tissues were minced into small pieces, then passed through a 70-μm cell strainer into FACS buffer (PBS supplemented with 2% FCS and 2% EDTA). The cell suspensions were then passed through 40-μm cell strainer to prepare a single cell culture. Preprocessed HLAC cells or blood, were mounted on top of Ficoll-Paque and centrifuged at 400 g for 30 minutes. The middle mononuclear cell layer was washed twice with FACS buffer and cells were resuspended in HLAC medium. Prior to HIV infection, PBMC cells were activated by 100 IU/ml IL-2 in the presence of 10 μg/ml Phytohemagglutinin (PHA) for 3 days.
HIV-1 infection
HEK293T cells were transfected using Fugene-HD in 24 well flat- bottom-plates with plasmid DNA (100 ng) corresponding to molecular clones of HIV-1 expressing a GFP reporter and Nef using an internal ribosomal entry site (Neidleman et al., 2017). The medium was replaced after 16 hours, and cells were tested for GFP expression. HLAC/PBMC were over-laid on the adherent GFP-expressing HEK293T cells for 24 hours. Suspension HLAC/PBMC cells were separated from the adherent cells after the 24-hour incubation, and spreading infection was allowed to proceed until 4–10% of the HLAC CD4 T cells were infected as determined by GFP epifluorescence measured by flow cytometry.
Reactivation of cells from HIV+ individuals
Leukopacks from HIV positive individuals on ART were processed using Ficoll-Paque gradients similar to that for healthy donor blood. CD4 T-cells were enriched by negative depletion with an EasySep Human CD4+ T-Cell Enrichment Kit (STEMCELL). Subsequently cells were activated with 50 ng/ml Phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin for 3 days to induce reactivation of latent proviruses within reservoir cells.
cCAR-T cell killing assay
Primary target cells were cultured until the level of infection rose above 4% in a Live/CD3+/CD8− gate as determined by FACS analysis. One million target cells (HLAC or PBMC) with ~104 infected cells were plated in a 96 V-bottom plate. 105 cCAR-T cells or donor-matched untransduced CD8 cells were incubated with different concentrations or types of MicAbodies for 5 minutes and then added to the target cells for 48 hours incubation. All experiments were performed in the presence of 5μM saquinavir to prevent spreading infection. For reactivated CD4 T-cells from HIV-positive individuals, 5×106 CD4 T-cells were plated in the presence of 5×105 cCAR-T cells or matched untransduced CD8 T-cells in the presence or absence of a mix of the four HIV-specific MicAbodies. Cultures were incubated for 48 hours.
Measurement of cell-associated RNA by ddPCR
Two days after co-culture of reactivated CD4 T-cells from HIV-positive individuals with cCAR-T cells and MicAbody, cells were collected and centrifuged for 10 minutes and RLT lysis buffer (Qiagen) was immediately added to cell pellets. Cell associate RNA (caRNA) was extracted with the RNeasy kit (Qiagen) following manufacturer’s protocol. Extracted RNA was reverse transcribed and pre-amplified using previously described HIV-specific primers (Laird et al., 2015) (see also Key Resources Table) using the Superscript III One-Step RT-PCR system (Life Technologies) with 10μl purified RNA in 25μl final volume. RT-PCR was carried out using the following steps: reverse transcription at 50°C for 30 minutes, denaturation at 95°C for 2 minutes, 10 cycles of amplification (94°C 15 seconds, 55°C 30 seconds, 68°C 30 seconds) and a final amplification step at 68°C for 5 minutes on a ThermoFisher PCR instrument. Subsequently, ddPCR was applied to quantify pre-amplified cDNA. For ddPCR droplet generation, reactions were loaded into the Bio-Rad QX-100 emulsification device following the manufacturer’s instructions. Samples were transferred to a 96-well reaction plate and sealed with a pre-heated Eppendorf 96- well heat sealer (Bio-Rad). Finally, samples were amplified on a BioRad C1000 Thermocycler and analyzed on BioRad QX100 ddPCR Reader using QuantaSoft Software (Bio-Rad). Each 25μl ddPCR mix comprised the ddPCR Probe Supermix (no dUTP), 900nM primers, 250nM probe (Laird et al., 2015), and 5μl cDNA. The following conditions were used: 10 minutes at 95°C, and 40 amplification cycles (30 seconds denaturation at 94°C followed by 59.4°C extension for 60 seconds) and a final 10 minutes at 98°C.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cy7 anti-human CD3 [SK7] | Biolegend | 344818 |
| PE/Cy7 anti-human CD4 | Biolegend | 357410 |
| PE anti-human CD8a [HIT8a] | Biolegend | 300908 |
| APC anti-human CD19 [HIB19] | Biolegend | 302212 |
| HRP-conjugated mouse-anti-human kappa chain | Abcam | ab79115 |
| Bacterial and Virus Strains | ||
| NL4–3-GFP HIV-1 | Doitsh et al., 2010 | N/A |
| BaL-GFP HIV-1 | Neidleman et al., 2017 | N/A |
| F4-GFP HIV-1 | Neidleman et al., 2017 | N/A |
| One Shot Stbl3 Chemically Competent E. coli cells | Life Technologies | C7373–03 |
| Biological Samples | ||
| Leukopaks from HIV positive individuals | SCOPE cohort | See Table S3 for individuals’ details |
| Blood from Healthy donors | Vitalant | Vitalant.org |
| Human Peripheral Blood Leuko Pak | STEM CELL | 70500.1 |
| Tonsil and spleen from healthy donors | CHTN | chtn.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ionomycin | Sigma-Aldrich | I0634 |
| Phorbal 12-myristate 13-acetate (PMA) | Sigma-Aldrich | P1585 |
| Recombinant Human IL-2 Protein | R&D Systems | 202-IL-010/CF |
| Fugene HD – Transfection Reagent | Promega | E312 |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | 15710 |
| EDTA, pH 8.0 | Thermo-Fisher | AM9260G |
| RPMI | Fisher Scientific | MT10040CM |
| DMEM | Fisher Scientific | MT10013CM |
| X-Vivo 15 media | Lonza | 04–418Q |
| N-acetyl-L-Cysteine | Sigma-Aldrich | A9165 |
| Gibco™ 2-Mercaptoethanol | Thermo-Fisher | 21985023 |
| Human AB Serum | Corning | 35–060-CI |
| FBS | Gemini Bio-Products | 100–106 |
| PBS | Fisher Scientific | MT21031CV |
| Opti-MEM | Life Technologies | 31985–062 |
| PHA-LECTIN | Sigma-Aldrich | L1668 |
| Gentamicin Reagent Solution (10 mg/ml), Liquid | Thermo Fisher | 15710–072 |
| Ampicillin sodium salt | Sigma-Aldrich | A9518 |
| Sodium pyruvate solution 100 mM sterile-filtered Cell Culture Grade | Sigma-Aldrich | S8636 |
| Nonessential amino acids (MEM NEAA) | Life Technologies | 11140–050 |
| L-Glutamine: Penicillin: Streptomycin Solution | GEMINI Bio-products | 400–110 |
| Fungizone Amphotericin B 250UG/mL | Invitrogen | 15290–018 |
| AccuCount counting beads | Spherotech | ACFP-70–10 |
| MicAbody | Xyphos Inc | This paper and U.S. Patent No. 10,259,858 |
| PEG-6000 | Hampton Research | HR2–533 |
| Critical Commercial Assays | ||
| EasySep direct human CD4+ T cell kit | STEM CELL | 19662 |
| RosetteSEP™ system CD8 isolation kit | STEM CELL | 15023 |
| Dynabeads™ Human T-Activator CD3/CD28 | Thermo Fisher | 1131D |
| SuperScript® III One-Step RT-PCR System with Platinum® Taq DNA Polymerase | Thermo Fisher | 12574026 |
| ddPCR™ Supermix for Probes (No dUTP) | Bio Rad | 1863023 |
| Droplet generation oil for probes | Bio-Rad | 186–3005 |
| One-Step RT-ddPCR Advanced Kit for Probes | Bio-Rad | 1864021 |
| RNeasy Mini Kit | Qiagen | 74104 |
| Zeno Human IgG Labeling Kit | Thermo Fisher | Z25408 |
| Superdex 200 columns | GE life sciences | 28990944 |
| Pierce™ Protein A Agarose | Thermo Fisher | 20334 |
| Alexa Fluor Protein Labeling Kit | Thermo Fisher | A20173 |
| 1-Step Ultra TMB ELISA | Thermo Fisher | 34208 |
| Deposited Data | ||
| None | ||
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| Expi293™ | Thermo Fisher | A14635 |
| Lenti-X 293T | Takara | 632180 |
| Raji | ATCC | CCL-86 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) | The Jackson Laboratory | JAX: 005557 |
| Oligonucleotides | ||
| For ddPCR Forward primer CAGATGCTGCATATAAGCAGCTG | Thermo Fisher | Laird et al., 2015 |
| For ddPCR Reverse primer TTTTTTTTTTTTTTTTTTTTTTTTGAAGCAC | Thermo Fisher | Laird et al., 2015 |
| Probe for ddPCR FAM-CCTGTACTGGGTCTCTCTGG-MGB | Thermo Fisher | Laird et al., 2015 |
| Recombinant DNA | ||
| pNL4–3.GFP | Doitsh et al., 2010 | N/A |
| pBaL.GFP | Neidleman et al., 2017 | N/A |
| pF4.GFP | Neidleman et al., 2017 | N/A |
| pD2610-V12 | ATUM | D2610-v12–03 |
| pHR-PGK | Roybal et al., 2016 | N/A |
| pCMVdR8.91 | Roybal et al., 2016 | N/A |
| pMD2.G | Roybal et al., 2016 | N/A |
| Software and Algorithms | ||
| ImageJ | ImageJ/NIH | https://imagej.nih.gov/ij/ |
| FlowJo | FlowJo v10 | https://www.flowjo.com/ |
| QuantaSoft | Bio-Rad | http://www.bio-rad.com/en-us/sku/1864011-quantasoft-software-regulatory-edition?ID=1864011 |
| Other | ||
| None | ||
ELISA binding assay
The extracellular domain of wild-type NKG2D or mutant NKG2D was fused to the C-terminus of human IgG1-Fc, DNA constructs for Fc-NKG2D molecules were expressed in Expi293™ cells, secreted protein purified by Protein-A affinity chromatography and eluted material fractionated by size-exclusion chromatography (SEC) on an ÄKTA Pure system using Superdex 200 columns. For ELISA binding assays, 1 μg/mL of Fc-NKG2D reagents were coated onto microtiter plates and a dilution series of MicAbody introduced followed by detection with HRP-conjugated mouse-anti-human kappa chain antibody then developed with 1-Step Ultra TMB ELISA.
Flow cytometry
Equal volumes from all treatment wells were spun down and the cells were stained with fluorochrome-conjugated antibodies (see Key Resources Table). Fixable Viability Kit Zombie Violet (BioLegend) was used to exclude dead cells while simultaneously staining for surface markers (CD3/CD4/CD8/CD19). AccuCount counting beads (Spherotech) were added after the last wash to control for sampling errors and cells were fixed in 1% paraformaldehyde. Data were acquired on LSR-II (BD Bioscience), and FlowJo software was used for analysis (Treestar). Killing of HIV-infected cells was assessed by measuring the reduction in the number of GFP+ cells relative to control (see Figure S2 for gating strategy).
In vivo assay
Female NSG mice were implanted with 1×106 Raji-Luc cells subcutaneously and reached tumor volumes of <100 mm3 on day 12, at which point 20 μg of Rituximab-MicAbody were injected intraperitoneally in a 100 μl volume and repeated every two days for a total of six doses. 1×107 CAR-T cells, comprised of a 1:1 absolute CD4:CD8 ratio, was injected intravenously on day 13 in a 100 μl volume. Untreated animals received PBS. Tumor volumes were regularly monitored by caliper measurements and weights were also tracked to monitor overall health of the animals. n=3 mice per cohort.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of individual experiments, including number of independent donors, mean values, standard error of the mean (SEM), and p values derived from two-tailed t-tests are described in the figure legends and specified in the figures. Statistical analyses were performed using Microsoft Excel software. p values ≤ 0.05 were considered statistically significant. For comparison between two treatments, a Student’s two-tailed t-test was used. For the in vivo mice assays a two-way ANOVA test was used to assess statistically significance of the results (p values ≤ 0.05). Asterisk coding in figures is as follows: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Data are presented as means with error bars indicating SEM unless otherwise stated.
DATA AND CODE AVAILABILITY
This study did not generate datasets.
Supplementary Material
Figure S1: Lymphoid tissue-based killing assay. Related to Figure 2.
Tonsil or spleen cells were purchased from CHTN and processed (see STAR method). These cells were infected using HEK293T transfected with various HIV expression plasmid DNAs. The infected cells were then combined with cCAR-T cells and different MicAbodies for 48 hours. Killing was determined by reduction in the number of infected cells expressing GFP (live CD3+/CD8− GFP+) compared to the No cCAR-T cell control assessed by flow cytometry. In each experiment potential in-well toxicity or off-target killing by MicAbodies and cCAR-T cells was measured by tracking the number of live CD3+/CD8− GFP− cells.
Figure S2: Gating strategy for flow cytometric assessment of cCAR-T cell killing in presence of MicAbodies. Related to Figures 2 and 3.
Following antibody staining, cells were analyzed on an LSR2 flow cytometer (Becton Dickinson). (A) Cells in the lymphocyte gate were analyzed in two singlets gates (B). Next an exclusion live gate was made dividing the live cells into CD3+ or CD3− gates by using Fixable Viability Kit Zombie Violet. (C) Cells were further gated on CD3+/GFP+ (infected cells) or CD4+/GFP− (uninfected cells). Further gating based of GFP expression allowed separation into high and low GFP expressing cells. Relative viability of uninfected control cells (CD3+/CD4+/GFP−) in each well was assessed in parallel. (D) Gating on effector cells was made by a live CD3+/CD8+ gate. (E) For B cell killing, the gating was made on CD3−/CD19+ live cells.
Figure S3: Specific killing of HIV-infected primary spleen CD4 T-cells by cCAR-T cells combined with HIV Env-specific MicAbodies. Related to Figure 2.
Specific killing of R5 tropic HIV-1 (BaL)-infected spleen cells by cCAR-T cells armed with 4 different HIV Env-specific MicAbodies. One million splenocytes containing approximately 1×104 infected CD4 T-cells were incubated with 1×105 CAR-T cells for 48 hours, in the presence of different concentrations (10–500 pM) of HIV Env-specific MicAbodies. B-cell-specific MicAbody (Ritux) and anti-HER2 MicAbody (HER2) were incorporated as negative controls. Results are presented relative to the No cCAR-T cell control. For each individual MicAbody, an internal control of no cCAR-T cell supplemented with the highest MicAbody concentration tested is presented. Results are cumulated from four independent experiments. Data are represented as mean + SEM. * = p≤0.05, ** = p≤0.01, *** = p≤0.001, compared to no MicAbody.
Figure S4: Comparison of cCAR-T killing of HIV-infected cells present in activated PBMC versus tonsil derived cells. Related to Figure 6.
F4-HIV-infected cells from tonsil (HLAC) or activated blood cells (PBMC) were cultured with cCAR-T cells at a 10:1 effector-to-target ratio for 48 hours in the presence of a mix of four HIV MicAbodies, or HER-2 MicAbody. GFP+ cell number was measured by flow cytometry and data are presented relative to cCAR-T with no MicAbody present.
Figure S5: Specific killing of CXCR4 tropic HIV-infected primary CD4 T cells by cCAR-T cells combined with specific HIV MicAbodies. Related to Figure 2.
Specific killing of X4-tropic HIV-1 (NL4–3) infected tonsil (A), spleen (B), or blood cells (C) by 4 single HIV-specific MicAbodies with cCAR-T cells was assessed. One million primary cells (~1×104 infected cells) were incubated with 1×105 CAR-T cells for 48 hours in the presence of different concentrations (10–500 pM) of the HIV Env-specific MicAbodies. B cell-specific MicAbody (Ritux) and anti-HER2 MicAbody (HER2) were used as negative MicAbody controls. Results are presented for each individual MicAbody, no cCAR-T cells and donor-matched untransduced CD8 cells supplemented with the highest MicAbody concentration tested are included as controls. Results are cumulated from three independent experiments for each tissue. Data are represented as mean + SEM relative to the CD8 control. * = p≤0.05, ** = p≤0.01
Movie S1: Time lapse microscopy of cCAR-T cell killing assay. Related to figure 4.
Highlights.
Inert MICA-tagged Ab and convertibleCAR-T cells (cCAR-T) only kill when combined
cCAR-T decreases tumor size in a mouse lymphoma model as efficiently as scFv CAR-T
cCAR-T kills HIV-infected primary cells with high efficiency and specificity
cCAR-T reduces the inducible reservoir in blood of HIV+ individuals by 50% in 48h
Acknowledgments
We thank members of the Greene laboratory for helpful discussions, reagents and expertise. HIV-infected blood was obtained from the SCOPE cohort (), at HIV/AIDS Positive Health Program of the Zuckerberg San Francisco General Hospital, with the assistance of Rebecca Hoh. We thank Francoise Chanut for editorial assistance, John C.W. Carroll and Giovanni Maki for graphics arts, and Robin Givens for administrative assistance. Most importantly, we are grateful for the contributions made by the HIV-infected individuals who participated in these studies. This study was also supported by funding from the amfAR Institute for HIV Cure Research, the University of California San Francisco-Gladstone Institutes Center for AIDS Research (NIH P30 AI027763), and the James B. Pendleton Charitable Trust. L.S.W. acknowledges support from NIH award R01AI109593LSW and NIDCR award DP1DE024408.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
None of the Gladstone/UCSF scientists have a commercial relationship with Xyphos Biosciences, Inc. KCK is an employee of Xyphos Biosciences, SRW is an employee and shareholder of Xyphos Biosciences, KL and NK are members of the Xyphos Biosciences scientific advisory board and DWM is a founder of Xyphos Biosciences, a shareholder, and member of its scientific advisory board. A patent application has been filed related to this work.
References
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, et al. (2012). Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, and Montano M (2012). BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol 92, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On Y, Gruell H, Schoofs T, Pai JA, Nogueira L, Butler AL, Millard K, Lehmann C, Suarez I, Oliveira TY, et al. (2018). Safety and antiviral activity of combination HIV-1 broadly neutralizing antibodies in viremic individuals. Nat Med 24, 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Tojal Da Silva I, Cohn LB, Gramatica A, Deeks S, Greene WC, Pillai SK, et al. (2018). Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, and Spies T (1999). Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727–729. [DOI] [PubMed] [Google Scholar]
- Bonifant CL, Jackson HJ, Brentjens RJ, and Curran KJ (2016). Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 3, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu WH, Fischinger S, Broge T, Abbink P, Mercado NB, Chandrashekar A, et al. (2018). Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel T, Guivel-Benhassine F, Amraoui S, Malbec M, Richard L, Bourdic K, Donahue DA, Lorin V, Casartelli N, Noel N, et al. (2016). Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat Commun 7, 10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA, Boucher G, Haddad EK, Sekaly RP, Harman AN, et al. (2010). Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc Natl Acad Sci U S A 107, 16934–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Klein F, and Nussenzweig MC (2019). Broadly neutralizing anti-HIV-1 monoclonal antibodies in the clinic. Nat Med 25, 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Schoofs T, Gruell H, Settler A, Karagounis T, Kreider EF, Murrell B, Pfeifer N, Nogueira L, Oliveira TY, et al. (2017). Antibody 10–1074 suppresses viremia in HIV-1-infected individuals. Nat Med 23, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrois M, Banerjee T, Mukherjee G, Raman N, Hussien R, Rodriguez BA, Vasquez J, Spitzer MH, Lazarus NH, Jones JJ, et al. (2017). Mass Cytometric Analysis of HIV Entry, Replication, and Remodeling in Tissue CD4+ T Cells. Cell Rep 20, 984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Presti R, Vermi W, Lavender K, Turnbull E, Ochsenbauer-Jambor C, Kappes JC, Ferrari G, Kessels L, Williams I, et al. (2010). Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur J Immunol 40, 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Engel D, Mizell SB, Hallahan CW, Fischette M, Park S, Davey RT Jr., Dybul M, Kovacs JA, Metcalf JA, et al. (1999). Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat Med 5, 651–655. [DOI] [PubMed] [Google Scholar]
- Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, and Siliciano RF (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1, 1284–1290. [DOI] [PubMed] [Google Scholar]
- Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, Kottilil S, Moir S, Mican JM, Mullins JI, et al. (2008). Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis 197, 714–720. [DOI] [PubMed] [Google Scholar]
- Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, and Fauci AS (1997). Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94, 13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn LB, da Silva IT, Valieris R, Huang AS, Lorenzi JCC, Cohen YZ, Pai JA, Butler AL, Caskey M, Jankovic M, et al. (2018). Clonal CD4(+) T cells in the HIV-1 latent reservoir display a distinct gene profile upon reactivation. Nat Med 24, 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, and Chalupny NJ (2001). ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C, June CH, et al. (2002). A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther 5, 788–797. [DOI] [PubMed] [Google Scholar]
- Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, et al. (2015). Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 517, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, and Greene WC (2010). Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroshkin AM, LeBlanc A, Weekes D, Post K, Li Z, Rajput A, Butera ST, Burton DR, and Godzik A (2014). bNAber: database of broadly neutralizing HIV antibodies. Nucleic Acids Res 42, D1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VD, Themeli M, and Sadelain M (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 5, 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300. [DOI] [PubMed] [Google Scholar]
- Goulder P, and Deeks SG (2018). HIV control: Is getting there the same as staying there? PLoS Pathog 14, e1007222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham C, Jozwik A, Pepper A, and Benjamin R (2018). Allogeneic CAR-T Cells: More than Ease of Access? Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan S, Calderon H, Posey AD Jr., and Maus MV (2019). Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang ZQ, et al. (1996). Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274, 985–989. [DOI] [PubMed] [Google Scholar]
- Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, and Wagner TA (2017). Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther 25, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, and Hong WK (2002). IMC-C225, an anti-epidermal growth factor receptor monoclonal antibody for treatment of head and neck cancer. Semin Oncol 29, 18–30. [DOI] [PubMed] [Google Scholar]
- Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, et al. (2010). Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog 6, e1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocqueloux L, Prazuck T, Avettand-Fenoel V, Lafeuillade A, Cardon B, Viard JP, and Rouzioux C (2010). Long-term immunovirologic control following antiretroviral therapy interruption in patients treated at the time of primary HIV-1 infection. AIDS 24, 1598–1601. [DOI] [PubMed] [Google Scholar]
- Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, and Deeks SG (2003). T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis 187, 1534–1543. [DOI] [PubMed] [Google Scholar]
- Jacobson CA (2019). CD19 Chimeric Antigen Receptor Therapy for Refractory Aggressive B-Cell Lymphoma. J Clin Oncol 37, 328–335. [DOI] [PubMed] [Google Scholar]
- Jain V, Hartogensis W, Bacchetti P, Hunt PW, Hatano H, Sinclair E, Epling L, Lee TH, Busch MP, McCune JM, et al. (2013). Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J Infect Dis 208, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean MJ, Fiches G, Hayashi T, and Zhu J (2019). Current Strategies for Elimination of HIV-1 Latent Reservoirs Using Chemical Compounds Targeting Host and Viral Factors. AIDS Res Hum Retroviruses 35, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Nguyen D, Archin NM, Yukl SA, Mendez-Lagares G, Tang Y, Elsheikh MM, Thompson GR 3rd, Hartigan-O’Connor DJ, Margolis DM, et al. (2018). HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest 128, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RB, and Walker BD (2016). HIV-specific CD8(+) T cells and HIV eradication. J Clin Invest 126, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julg B, and Barouch DH (2019). Neutralizing antibodies for HIV-1 prevention. Curr Opin HIV AIDS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalams SA, Goulder PJ, Shea AK, Jones NG, Trocha AK, Ogg GS, and Walker BD (1999). Levels of human immunodeficiency virus type 1-specific cytotoxic T-lymphocyte effector and memory responses decline after suppression of viremia with highly active antiretroviral therapy. J Virol 73, 6721–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke R, Conway JM, Margolis DM, and Perelson AS (2018). Determinants of the efficacy of HIV latency-reversing agents and implications for drug and treatment design. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al. (2008). Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharsany AB, and Karim QA (2016). HIV Infection and AIDS in Sub-Saharan Africa: Current Status, Challenges and Opportunities. Open AIDS J 10, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119, 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsch O, Benveniste EN, Shaw GM, and Levy DN (2002). Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 76, 8776–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, and Siliciano RF (2015). Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest 125, 1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf K, Williams SR, Steiger D, Gebhart D, Lok S, Martin DW, Roybal KT, and Kim KC (2019). convertibleCAR-T cells provide a modular universal system for dose control of activity, flexible targeting, and versatile maintenance of CAR-cells. bioRxiv, 696401. [Google Scholar]
- Lee JM (2019). When CAR Meets Stem Cells. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard R, Zagury D, Desportes I, Bernard J, Zagury JF, and Gallo RC (1988). Cytopathic effect of human immunodeficiency virus in T4 cells is linked to the last stage of virus infection. Proc Natl Acad Sci U S A 85, 3570–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Aldrovandi GM, Kutsch O, and Shaw GM (2004). Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101, 4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Chen BK (2019). Variable infectivity and conserved engagement in cell-to-cell viral transfer by HIV-1 Env from Clade B transmitted founder clones. Virology 526, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Etemad B, Ahmed H, Aga E, Bosch RJ, Mellors JW, Kuritzkes DR, Lederman MM, Para M, and Gandhi RT (2016). The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 30, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, and Zhou Q (2013). The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res 41, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zou F, Lu L, Chen C, He D, Zhang X, Tang X, Liu C, Li L, and Zhang H (2016). Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4+ T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive Combined Antiretroviral Therapy. J Virol 90, 9712–9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori F, Jessen H, Lieberman J, Finzi D, Rosenberg E, Tinelli C, Walker B, Siliciano RF, and Lisziewicz J (1999). Treatment of human immunodeficiency virus infection with hydroxyurea, didanosine, and a protease inhibitor before seroconversion is associated with normalized immune parameters and limited viral reservoir. J Infect Dis 180, 1827–1832. [DOI] [PubMed] [Google Scholar]
- Ma G, Shen J, Pinz K, Wada M, Park J, Kim S, Togano T, and Tse W (2019). Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. [DOI] [PubMed] [Google Scholar]
- Malbec M, Porrot F, Rua R, Horwitz J, Klein F, Halper-Stromberg A, Scheid JF, Eden C, Mouquet H, Nussenzweig MC, et al. (2013). Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med 210, 2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, et al. (1997). IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 90, 2188–2195. [PubMed] [Google Scholar]
- Manson McManamy ME, Hakre S, Verdin EM, and Margolis DM (2014). Therapy for latent HIV-1 infection: the role of histone deacetylase inhibitors. Antivir Chem Chemother 23, 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusali G, Tchidjou HK, Pontrelli G, Bernardi S, D’Ettorre G, Vullo V, Buonomini AR, Andreoni M, Santoni A, Cerboni C, et al. (2013). Soluble ligands for the NKG2D receptor are released during HIV-1 infection and impair NKG2D expression and cytotoxicity of NK cells. FASEB J 27, 2440–2450. [DOI] [PubMed] [Google Scholar]
- Mayer KH, Seaton KE, Huang Y, Grunenberg N, Isaacs A, Allen M, Ledgerwood JE, Frank I, Sobieszczyk ME, Baden LR, et al. (2017). Safety, pharmacokinetics, and immunological activities of multiple intravenous or subcutaneous doses of an anti-HIV monoclonal antibody, VRC01, administered to HIV-uninfected adults: Results of a phase 1 randomized trial. PLoS Med 14, e1002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy LE (2018). The expanding array of HIV broadly neutralizing antibodies. Retrovirology 15, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill J, Van Rooijen N, and Legge KL (2010). IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med 207, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, and Chauhan A (2010). Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One 5, e11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, et al. (2008). Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouquet H, Scharf L, Euler Z, Liu Y, Eden C, Scheid JF, Halper-Stromberg A, Gnanapragasam PN, Spencer DI, Seaman MS, et al. (2012). Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci U S A 109, E3268–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, and Greene WC (2015). Blood-Derived CD4 T Cells Naturally Resist Pyroptosis during Abortive HIV-1 Infection. Cell Host Microbe 18, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidleman JA, Chen JC, Kohgadai N, Muller JA, Laustsen A, Thavachelvam K, Jang KS, Sturzel CM, Jones JJ, Ochsenbauer C, et al. (2017). Mucosal stromal fibroblasts markedly enhance HIV infection of CD4+ T cells. PLoS Pathog 13, e1006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onea AS, and Jazirehi AR (2016). CD19 chimeric antigen receptor (CD19 CAR)-redirected adoptive T-cell immunotherapy for the treatment of relapsed or refractory B-cell Non-Hodgkin’s Lymphomas. Am J Cancer Res 6, 403–424. [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, McElroy AN, Starker CG, Lee C, Merkel S, et al. (2016). Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther 24, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pache L, Dutra MS, Spivak AM, Marlett JM, Murry JP, Hwang Y, Maestre AM, Manganaro L, Vamos M, Teriete P, et al. (2015). BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 18, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, and Fauci AS (1993). HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362, 355–358. [DOI] [PubMed] [Google Scholar]
- Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, et al. (2013). Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110, 6626–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins JM, Jurriaans S, van Praag RM, Blaak H, van Rij R, Schellekens PT, ten Berge IJ, Yong SL, Fox CH, Roos MT, et al. (1999). Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 13, 2405–2410. [DOI] [PubMed] [Google Scholar]
- Pule M, Finney H, and Lawson A (2003). Artificial T-cell receptors. Cytotherapy 5, 211–226. [DOI] [PubMed] [Google Scholar]
- Rasmussen TA, and Lewin SR (2016). Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents? Curr Opin HIV AIDS 11, 394–401. [DOI] [PubMed] [Google Scholar]
- Rasmussen TA, Tolstrup M, Winckelmann A, Ostergaard L, and Sogaard OS (2013). Eliminating the latent HIV reservoir by reactivation strategies: advancing to clinical trials. Hum Vaccin Immunother 9, 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, Pai A, Aull K, Rouzine IM, and Weinberger LS (2015). A hardwired HIV latency program. Cell 160, 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu X, Fang C, Jiang S, June CH, and Zhao Y (2017). Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 23, 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renju J, Moshabela M, McLean E, Ddaaki W, Skovdal M, Odongo F, Bukenya D, Wamoyi J, Bonnington O, Seeley J, et al. (2017). ‘Side effects’ are ‘central effects’ that challenge retention in HIV treatment programmes in six sub-Saharan African countries: a multicountry qualitative study. Sex Transm Infect 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, and Harty JT (2015). Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest 125, 3477–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, and Pomerantz RJ (2009). The challenge of finding a cure for HIV infection. Science 323, 1304–1307. [DOI] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016). Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruella M, and Kenderian SS (2017). Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 31, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, et al. (2013). Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog 9, e1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampey GC, Irlbeck DM, Browne EP, Kanke M, McAllister AB, Ferris RG, Brehm JH, Favre D, Routy J-P, Jones CD, et al. (2018). The SMAC Mimetic AZD5582 is a Potent HIV Latency Reversing Agent. bioRxiv, 312447. [Google Scholar]
- Samri A, Bacchus-Souffan C, Hocqueloux L, Avettand-Fenoel V, Descours B, Theodorou I, Larsen M, Saez-Cirion A, Rouzioux C, Autran B, et al. (2016). Polyfunctional HIV-specific T cells in Post-Treatment Controllers. AIDS 30, 2299–2302. [DOI] [PubMed] [Google Scholar]
- Sarkar RR, Gloude NJ, Schiff D, and Murphy JD (2018). Cost-Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Pediatric Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. J Natl Cancer Inst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu CL, Lorenzi JC, Feldmann A, Braunschweig M, Nogueira L, Oliveira T, et al. (2016). HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 535, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, et al. (2011). Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, et al. (2012). Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4, 132ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, and Tersmette M (1992). Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol 66, 1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuitemaker H, van ‘t Wout AB, and Lusso P (2011). Clinical significance of HIV-1 coreceptor usage. J Transl Med 9 Suppl 1, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JA, Zhang H, Ende Z, Deymier MJ, Lee T, Singer J, Mazzulli T, Hunter E, and Ostrowski MA (2018). Characterization of the Plasmacytoid Dendritic Cell Response to Transmitted/Founder and Nontransmitted Variants of HIV-1. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacklett BL, Ferre AL, and Kiniry BE (2019). Tissue issues: mucosal T-cell responses in HIV-1 infection. Curr Opin HIV AIDS 14, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, and Siliciano RF (2012). Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Rabi SA, Laird GM, Eisele EE, Zhang H, Margolick JB, and Siliciano RF (2013). A novel PCR assay for quantification of HIV-1 RNA. J Virol 87, 6521–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. (2001). High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 276, 6591–6604. [DOI] [PubMed] [Google Scholar]
- Shin H, and Wherry EJ (2007). CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol 19, 408–415. [DOI] [PubMed] [Google Scholar]
- Siliciano RF, and Greene WC (2011). HIV latency. Cold Spring Harb Perspect Med 1, a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok D, Pauthner M, Briney B, Lee JH, Saye-Francisco KL, Hsueh J, Ramos A, Le KM, Jones M, Jardine JG, et al. (2016). A Prominent Site of Antibody Vulnerability on HIV Envelope Incorporates a Motif Associated with CCR5 Binding and Its Camouflaging Glycans. Immunity 45, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. (2015). Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, and Spies T (2001). Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 53, 279–287. [DOI] [PubMed] [Google Scholar]
- Sung JA, Patel S, Clohosey ML, Roesch L, Tripic T, Kuruc JD, Archin N, Hanley PJ, Cruz CR, Goonetilleke N, et al. (2018). HIV-Specific, Ex Vivo Expanded T Cell Therapy: Feasibility, Safety, and Efficacy in ART-Suppressed HIV-Infected Individuals. Mol Ther 26, 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, and Sadelain M (2013). Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol 31, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, and Cooper LJ (2016). Translational Implications for Off-the-shelf Immune Cells Expressing Chimeric Antigen Receptors. Mol Ther 24, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A, Irrinki A, Kaur J, Cihlar T, Kukolj G, Sloan DD, and Murry JP (2017). Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya KS, Oleksijew A, Tucker LA, Pappano WN, Anderson MG, Grinnell CM, Zhang Q, Heighton SJ, Mitten MJ, Mishra S, et al. (2017). A “Prozone-Like” Effect Influences the Efficacy of the Monoclonal Antibody ABT-700 against the Hepatocyte Growth Factor Receptor. Pharmacology 100, 229–242. [DOI] [PubMed] [Google Scholar]
- Wagner TA (2018). Quarter Century of Anti-HIV CAR T Cells. Curr HIV/AIDS Rep 15, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, Lopomo P, Vigny M, Fry TJ, Orentas RJ, et al. (2017). Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther 25, 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, et al. (2011). Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker-Sperling VE, Pohlmeyer CW, Tarwater PM, and Blankson JN (2016). The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 8, 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wolock T, Carter A, Nguyen G, Kyu H, Gakidou E, Hay S, Mills E, Trickey A, Msemburi W, et al. (2016). Estimates of global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2015: the Global Burden of Disease Study 2015. Lancet HIV 3, e361–e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, Sakemura R, Goto T, Hanajiri R, Imahashi N, et al. (2015). Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol 194, 911–920. [DOI] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, and Richman DD (1997). Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295. [DOI] [PubMed] [Google Scholar]
- Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, et al. (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell. [DOI] [PubMed] [Google Scholar]
- Yang OO, Nguyen PT, Kalams SA, Dorfman T, Gottlinger HG, Stewart S, Chen IS, Threlkeld S, and Walker BD (2002). Nef-mediated resistance of human immunodeficiency virus type 1 to antiviral cytotoxic T lymphocytes. J Virol 76, 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jacoby E, and Fry TJ (2015). Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol 22, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes SA, Freeman ML, Mudd JC, Shive CL, Reynaldi A, Panigrahi S, Estes JD, Deleage C, Lucero C, Anderson J, et al. (2016). IL-15 promotes activation and expansion of CD8+ T cells in HIV-1 infection. J Clin Invest 126, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl SA, Gianella S, Sinclair E, Epling L, Li Q, Duan L, Choi AL, Girling V, Ho T, Li P, et al. (2010). Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J Infect Dis 202, 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen A, Peterson CW, Carrillo MA, Reddy SS, Youn CS, Lam BB, Chang NY, Martin HA, Rick JW, Kim J, et al. (2017). Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog 13, e1006753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng PP, Kros JM, and Li J (2018). Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today 23, 1175–1182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Lymphoid tissue-based killing assay. Related to Figure 2.
Tonsil or spleen cells were purchased from CHTN and processed (see STAR method). These cells were infected using HEK293T transfected with various HIV expression plasmid DNAs. The infected cells were then combined with cCAR-T cells and different MicAbodies for 48 hours. Killing was determined by reduction in the number of infected cells expressing GFP (live CD3+/CD8− GFP+) compared to the No cCAR-T cell control assessed by flow cytometry. In each experiment potential in-well toxicity or off-target killing by MicAbodies and cCAR-T cells was measured by tracking the number of live CD3+/CD8− GFP− cells.
Figure S2: Gating strategy for flow cytometric assessment of cCAR-T cell killing in presence of MicAbodies. Related to Figures 2 and 3.
Following antibody staining, cells were analyzed on an LSR2 flow cytometer (Becton Dickinson). (A) Cells in the lymphocyte gate were analyzed in two singlets gates (B). Next an exclusion live gate was made dividing the live cells into CD3+ or CD3− gates by using Fixable Viability Kit Zombie Violet. (C) Cells were further gated on CD3+/GFP+ (infected cells) or CD4+/GFP− (uninfected cells). Further gating based of GFP expression allowed separation into high and low GFP expressing cells. Relative viability of uninfected control cells (CD3+/CD4+/GFP−) in each well was assessed in parallel. (D) Gating on effector cells was made by a live CD3+/CD8+ gate. (E) For B cell killing, the gating was made on CD3−/CD19+ live cells.
Figure S3: Specific killing of HIV-infected primary spleen CD4 T-cells by cCAR-T cells combined with HIV Env-specific MicAbodies. Related to Figure 2.
Specific killing of R5 tropic HIV-1 (BaL)-infected spleen cells by cCAR-T cells armed with 4 different HIV Env-specific MicAbodies. One million splenocytes containing approximately 1×104 infected CD4 T-cells were incubated with 1×105 CAR-T cells for 48 hours, in the presence of different concentrations (10–500 pM) of HIV Env-specific MicAbodies. B-cell-specific MicAbody (Ritux) and anti-HER2 MicAbody (HER2) were incorporated as negative controls. Results are presented relative to the No cCAR-T cell control. For each individual MicAbody, an internal control of no cCAR-T cell supplemented with the highest MicAbody concentration tested is presented. Results are cumulated from four independent experiments. Data are represented as mean + SEM. * = p≤0.05, ** = p≤0.01, *** = p≤0.001, compared to no MicAbody.
Figure S4: Comparison of cCAR-T killing of HIV-infected cells present in activated PBMC versus tonsil derived cells. Related to Figure 6.
F4-HIV-infected cells from tonsil (HLAC) or activated blood cells (PBMC) were cultured with cCAR-T cells at a 10:1 effector-to-target ratio for 48 hours in the presence of a mix of four HIV MicAbodies, or HER-2 MicAbody. GFP+ cell number was measured by flow cytometry and data are presented relative to cCAR-T with no MicAbody present.
Figure S5: Specific killing of CXCR4 tropic HIV-infected primary CD4 T cells by cCAR-T cells combined with specific HIV MicAbodies. Related to Figure 2.
Specific killing of X4-tropic HIV-1 (NL4–3) infected tonsil (A), spleen (B), or blood cells (C) by 4 single HIV-specific MicAbodies with cCAR-T cells was assessed. One million primary cells (~1×104 infected cells) were incubated with 1×105 CAR-T cells for 48 hours in the presence of different concentrations (10–500 pM) of the HIV Env-specific MicAbodies. B cell-specific MicAbody (Ritux) and anti-HER2 MicAbody (HER2) were used as negative MicAbody controls. Results are presented for each individual MicAbody, no cCAR-T cells and donor-matched untransduced CD8 cells supplemented with the highest MicAbody concentration tested are included as controls. Results are cumulated from three independent experiments for each tissue. Data are represented as mean + SEM relative to the CD8 control. * = p≤0.05, ** = p≤0.01
Movie S1: Time lapse microscopy of cCAR-T cell killing assay. Related to figure 4.
Data Availability Statement
This study did not generate datasets.