

Summary
Most of the gene expression systems available for Gram‐negative bacteria are afflicted by relatively high levels of basal (i.e. leaky) expression of the target gene(s). This occurrence affects the system dynamics, ultimately reducing the output and productivity of engineered pathways and synthetic circuits. In order to circumvent this problem, we have designed a novel expression system based on the well‐known XylS/Pm transcriptional regulator/promoter pair from the soil bacterium Pseudomonas putida mt‐2, in which the key functional elements are physically decoupled. By integrating the xylS gene into the chromosome of the platform strain KT2440, while placing the Pm promoter into a set of standard plasmid vectors, the inducibility of the system (i.e. the output difference between the induced and uninduced state) improved up to 170‐fold. We further combined this modular system with an extra layer of post‐translational control by means of conditional proteolysis. In this setup, the target gene is tagged with a synthetic motif dictating protein degradation. When the system features were characterized using the monomeric superfolder GFP as a model protein, the basal levels of fluorescence were brought down to zero (i.e. below the limit of detection). In all, these novel expression systems constitute an alternative tool to altogether suppress leaky gene expression, and they can be easily adapted to other vector formats and plugged‐in into different Gram‐negative bacterial species at the user's will.
This study describes the re‐purposing of the elements within the classical XylS/Pm system into a novel gene expression device which enables extremely tight expression control. The combination of the system with an extra layer of post‐translational regulation resulted in close‐to‐zero leaky expression of the target gene.
![]()
Introduction
The soil bacterium and platform strain Pseudomonas putida KT2440 has become the host of choice for metabolic engineering applications that require high levels of stress resistance and a robust, versatile metabolism (Nikel et al., 2016; Poblete‐Castro et al., 2017; Nikel and de Lorenzo, 2018). A dedicated synthetic biology toolbox is key to harness the full potential of this bacterium ― and strategies for tightly controlling gene expression are not only fundamental for understanding basic properties of the cell physiology and regulatory processes thereof, but they are also crucial for biotechnological applications (Gomez et al., 2012; Benedetti et al., 2016; Chavarría et al., 2016; Martínez‐García and de Lorenzo, 2017; Calero and Nikel, 2019). Gene expression systems offer the possibility to control when (and how much of) a target protein should be produced. Ideally, such systems should display two states (ON and OFF), enabling to temporally separate plasmid replication and maintenance (OFF) from an active state, when the gene of interest (GOI) is expressed and the encoded protein(s) is/are produced (ON) (Ter, 2006). In addition, since the accumulation of heterologous proteins is often accompanied by metabolic burden in the recombinant cells carrying the cognate gene construct (Wu et al., 2016), the ON and OFF states of the expression system should be clearly differentiated and externally controllable ― that is, the system should display very low levels (ideally, zero) of basal expression (leakiness) in the non‐induced, OFF state.
Transferring regulatory elements of gene expression devices across bacterial hosts is often accompanied by changes in the behaviour of the system, for example basal expression, fold change in expression levels between the induced and non‐induced state, and induction dynamics (Slusarczyk et al., 2012; Segall‐Shapiro et al., 2018). Expression systems traditionally used in the model host Escherichia coli (e.g. LacIQ/Ptrc), for instance, are known to display different characteristics when plugged‐in into a different bacterium ― with a much higher basal expression and lower induction fold in P. putida. Due to the versatile metabolism of Pseudomonas species, rich in degradation routes for alkane and aromatic compounds (Jiménez et al., 2002; Nikel et al., 2015a,2015b), several transcriptional regulators responsive to such substrates, along with the corresponding operator sites and other regulatory elements, have been sourced from these bacterial species. Transcriptional regulator/promoter pairs of this sort include, among many others, the (alkyl)benzoate(s)‐activated XylS/Pm system (de Lorenzo et al., 1993), the alkene(s)‐activated AlkS/PalkB system (Panke et al., 1999), the xylene(s)‐responsive XylR/Pu system (Marqués and Ramos, 1993), the phenol‐inducible DmpR regulator (Shingler and Moore, 1994) and the salicylate‐activated NahR/Psal system (Cebolla et al., 1996).
The XylS/Pm regulatory system originates from the catabolic megaplasmid pWW0 of P. putida mt‐2, where it controls the transcription of genes in the lower (meta) cleavage pathway for degradation of aromatic compounds (Ramos et al., 1997). Expression systems based on the XylS/Pm pair have been extensively adopted for both fundamental studies and metabolic engineering of Pseudomonas, because of its low basal expression, relatively high fold‐change induction, and the use of low‐cost inducers such as 3‐methylbenzoate (3‐mBz) and derivatives thereof (Gawin et al., 2017). In its native configuration, the XylS protein binds as a dimer to two operator sites (distal and proximal) closely upstream to the −35 motif of the Pm promoter, where it recruits the RNA polymerase with the σ32 or σ38 factors (Marqués et al., 1999; González‐Pérez et al., 2002). The dimerization is promoted by 3‐mBz and related aromatic molecules, but it can also occur at high intracellular concentrations of XylS (Ruíz et al., 2003) ― albeit at a rate lower than that triggered by 3‐mBz. Furthermore, xylS is under control of a constitutive promoter and a xylene‐inducible promoter (Gallegos et al., 1996), ensuring synchronic expression of the long upper and lower TOL degradation pathway genes through a metabolic amplification motif (Silva‐Rocha et al., 2011). While this transcriptional wiring is beneficial in its natural context, it hampers its application in a heterologous milieu. Zwick et al. (2013) showed that high xylS expression levels lead to increased basal Pm promoter activity and limited inducibility of the system by externally added effectors. Expression levels are likewise dependent on gene dosage: while the system is usually tightly controlled as a single‐copy genomic integration, basal expression dramatically increases with the copy number if the regulatory elements are plasmid‐borne. This situation helps explaining the significant discrepancies in the induction levels reported for the XylS/Pm system, ranging from ~10‐fold for medium copy number plasmids based on the origin of vegetative replication (oriV) pBBR1 in P. putida (Calero et al., 2016) to ~100‐fold for low copy number plasmids with oriV(RK2) in E. coli (Winther‐Larsen et al., 2000). The origins of replication commonly used in Pseudomonas display medium‐to‐high copy number levels [oriV(pBBR1) = 30 ± 7; oriV(RK2) = 20 ± 10; oriV(RSF1010) = 130 ± 40; and oriV(pRO1600/ColE1) ~30–40 (Jahn et al., 2016; Cook et al., 2018)].
In order to enable better control of gene expression in Gram‐negative bacteria (and, in particular, in Pseudomonas species), in this study, we have re‐wired the functional elements of the XylS/Pm regulatory system (Fig. 1) in combination with the recently described FENIX system for post‐translational control (Durante‐Rodríguez et al., 2018). By physically decoupling the xylS and Pm components, while rendering target protein(s) sensitive to conditional proteolysis, we have achieved a tightly controlled expression output of the system, with close‐to‐zero leakiness and in a plasmid copy number ― independent fashion.
Figure 1.
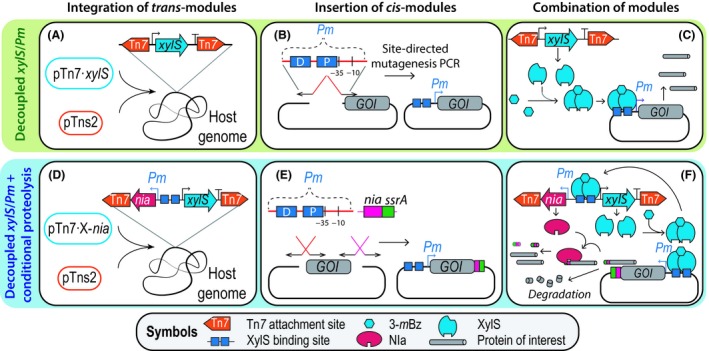
Scheme of tightly regulated expression systems for Pseudomonas based on physically decoupled XylS/Pm elements and conditional proteolysis.A. The trans‐module of the system, carrying the xylS regulator gene under transcriptional control of its native promoters, is flanked by the Tn7 recognitions sites borne by plasmid pTn7·xylS. The trans‐module is integrated into the host genome through the Tn7 transposase functions contained in the helper plasmid pTns2.B. Insertion of the cis‐module of the system into the vector of interest. The cis‐module is comprised of the distal and proximal XylS‐binding sites, followed by the −35 and −10 binding sites for the RNA polymerase. The whole module is 70‐bp long and can be easily introduced into any target vector through site‐directed mutagenesis PCR.C. A suitable plasmid carrying the cis‐module is delivered into a host Pseudomonas strain already containing the trans‐module integrated into the chromosome. The genomic integration of xylS in monocopy ensures low transcription levels of the regulator gene; upon addition of 3‐mBz as the inducer, XylS dimerizes and activates transcription from the Pm promoter.D. The trans‐module carrying the gene encoding the NIa protease under the transcriptional control of XylS/Pm is flanked by the Tn7 recognition sites borne by plasmid pTn7·X‐nia. This module is integrated into the host genome through the Tn7 transposase functions contained in the helper plasmid pTns2.E. Insertion of the Pm promoter and the 3′‐tag for conditional proteolysis to a gene of interest.F. The constructed plasmid (carrying the cis‐module) is delivered into a host Pseudomonas strain already containing the trans‐module in the chromosome. The inducer 3‐mBz promotes dimerization of XylS and, consequently, triggers transcription from the Pm promoter. Note that the Pm promoter drives the transcription of the gene of interest as well as that of nia. Only if both gene products are present in sufficient amounts, they will interact and the target protein is then relieved from the proteolysis tag – thereby enabling stable accumulation of the product. The abbreviations used in this scheme are as follows: 3‐mBz, 3‐methylbenzoate; GOI, gene of interest; NIa, nuclear inclusion protein A.
Protocol
Materials
Sucrose solution: 300 mM sucrose (cat. # 84100; Sigma‐Aldrich Corp., St. Louis, MO, USA) in double‐distiled water (ddH2O). Sterilized by filtration and kept at room temperature.
Induction solution: 0.5 M 3‐mBz (cat. # M29908; Sigma‐Aldrich Corp.) in ddH2O, pH adjusted to neutrality by dropwise addition of 5 M NaOH until no 3‐mBz precipitation is visible. Stored at room temperature.
Lysogeny broth (LB) medium: 10 g l−1 tryptone, 5 g l−1 yeast extract, and 5 g l−1 NaCl; dissolved in ddH2O and autoclaved. Bacteriological agar (cat. # A5306; Sigma‐Aldrich Corp.) was added at 15 g l−1 for LB agar plates before autoclaving.
Kanamycin (Km) stock solution: 50 mg ml−1 Km sulfate (cat. # T832.3; Carl Roth GmbH & Co. KG, Karlsruhe, Germany) dissolved in ddH2O, sterile filtered and stored at −20°C. Used at a final concentration of 50 μg ml−1.
Gentamycin (Gm) stock solution: 10 mg ml−1 Gm sulfate (cat. # G1264; Sigma‐Aldrich Corp.) in ddH2O, sterile filtered and stored at −20°C. Used at a final concentration of 10 μg ml−1.
General procedures
Plasmid purification: The Nucleospin Plasmid EasyPure Kit (cat. # 740727.250; Macherey‐Nagel GmbH & Co. KG, Düren, Germany) was used for plasmid purification according to the manufacturer's instructions.
Site‐directed mutagenesis: 45 μl of the PCR product is treated with DpnI (cat. # FD1703; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions to digest the template DNA used for the amplifications. An aliquot of 7 μl of digested PCR product is incubated with 1 μl of T4 polynucleotide kinase (cat. # EL0014; Thermo Fisher Scientific), 1 μl of T4 DNA ligase (cat. # EK0031; Thermo Fisher Scientific) and 1 μl of ligation buffer for 2 h at room temperature. The total amount of 10 μl is then used for transforming chemically competent E. coli DH5α cells (Boyer and Roulland‐Dussoix, 1969; Ruiz et al., 2006).
PCR reaction conditions: Phusion Hot Start II DNA polymerase (cat. # F549L; Thermo Fisher Scientific) was used according to the manufacturer's instructions with HF‐buffer in the presence of 3% (v/v) DMSO (cat. # F‐515; Thermo Fisher Scientific).
Agarose gel electrophoresis: 5 μl of the solution containing the DNA fragment(s) to be analysed was mixed with 1 μl of gel loading dye (cat. # R0611; Thermo Fisher Scientific) and analysed on a 1% (w/v) agarose gel (cat. # BN‐50004; BioNordika Denmark A/S, Herlev, Denmark) in TAE buffer [1 mM EDTA (cat. # E6758; Sigma‐Aldrich Corp.), 40 mM Tris (cat. # T1503; Sigma‐Aldrich Corp.) and 20 mM acetic acid (cat. # 33209; Sigma‐Aldrich Corp.)]. Fragment sizes were compared against a DNA ladder standard run in parallel (cat. # SM0313; Thermo Fisher Scientific).
Technical implementation
In this study, we have implemented two approaches to tightly control gene expression in Gram‐negative bacteria, and in particular P. putida, by rewiring the transcriptional machinery of the well‐characterized XylS/Pm expression system. Both procedures rely on the physical separation of the gene encoding the transcriptional regulator and the Pm promoter, as indicated below.
Genomic integration of a trans‐module encoding the XylS transcriptional regulator and the NIa protease in P. putida KT2440
The starting point for the construction of the novel gene expression device was to physically separate the gene encoding the XylS transcriptional regulator and its cognate promoter (Fig. 1). On this background, a trans‐module was defined herein as any genomic part that is physically separated from the GOI. Likewise, in the devices presented in this study, the trans‐modules are DNA segments integrated into the genome of P. putida KT2440 (i.e. xylS or xylS/Pm→nia). Note that this genetic architecture contrasts the traditional arrangement of most expression systems, that is, a plasmid‐based XylS/Pm system in which the parts are encoded in the same DNA molecule. Accordingly, cis‐modules are adjacent to the GOI (i.e. the Pm promoter and the synthetic nia/ssrA tag). The genomic integration of the xylS or xylS/Pm→nia modules was accomplished by co‐electroporating plasmids pTn7·xylS or pTn7·X‐nia (Table 1) respectively, together with the helper plasmid pTns2 (which encodes the Tn7 transposase functions) into wild‐type P. putida KT2440. For this, a 10‐ml overnight culture of P. putida KT2440, grown in LB medium in a 50‐ml tube (Corning™, cat. # 352070; Thermo Fisher Scientific), was centrifuged at 4000 g for 5 min at room temperature. The cell pellet was washed by resuspension in 1 ml of 300 mM sucrose and transferred to a 2‐ml reaction tube followed by centrifugation at 15 000 g for 1 min. The supernatant was carefully discarded, and the washing steps were repeated twice. In the final step, the cell pellet was resuspended in 0.4 ml of 300 mM sucrose. An aliquot of 0.1‐ml of the resulting suspension was mixed with 200 ng of each plasmid and transferred to a 0.2‐cm gap electroporation cuvette (cat. # 165‐2086; Bio‐Rad Corp., Hercules, CA, USA; Smith and Iglewski, 1989). A single 2.5‐kV electric pulse was applied for up to 6.0 ms in a microPulser electroporator (Bio‐Rad Corp.), followed by adding immediately 1 ml of LB medium. Cells were recovered by incubating the culture at 30°C with rotational agitation for 2 h. The biomass was then concentrated in 0.1 ml by centrifugation as indicated above and plated onto LB agar plates supplemented with Gm. Gm‐resistant (GmR) colonies were checked by colony PCR for the correct insertion of the Tn7 module with oligonucleotides Gm_check‐F and Gm_check‐R (Table 2), which anneal within the Tn7 integration site‐adjacent gene PP_5408 and in the left transposon‐flanking site (Tn7L) respectively. The correct insertion of the GmR‐determinant yields a 400‐bp amplification fragment (Zobel et al., 2015). Selected clones, which display the correct fragment, were further subjected to sequencing with the primer pair pS1 and pS2 (Table 2). Alternatively, plasmids can be delivered into P. putida by triparental mating; a detailed procedure is given by Zobel et al. (2015). These operations yielded strains KT·P and KT·PN, in which the xylS or xylS/Pm→nia modules respectively, are stably inserted in the att·Tn7 site in the chromosome of P. putida KT2440 (Fig. 1).
Table 1.
Plasmids used in this work
| Plasmid namea | Relevant characteristicsb | Source or reference |
|---|---|---|
| pTn7‐M | Tn7 integration vector; oriV(R6K); KmR, GmR | Choi et al. (2005) |
| pTn7·xylS | Derivative of vector pTn7‐M used for chromosomal integration of xylS | This work |
| pTn7·X‐nia | Derivative of vector pTn7‐M used for chromosomal integration of xylS/Pm→nia | This work |
| pTns2 | Helper plasmid constitutively expressing the tnsABCD genes encoding the Tn7 transposase; oriV(R6K); AmpR | Choi et al. (2005) |
| pSEVA238 | Expression vector; oriV(pBBR1), XylS/Pm expression system; KmR | Silva‐Rocha et al. (2013) |
| pSEVA248 | Expression vector; oriV(pRO1600/ColE1), XylS/Pm expression system; KmR | Silva‐Rocha et al. (2013) |
| pS238·NIa | Derivative of vector pSEVA238 used for regulated expression of nia, encoding the potyvirus NIa protease; XylS/Pm→nia; KmR | Durante‐Rodríguez et al. (2018); B. Calles and V. de Lorenzo (unpublished data) |
| pS238·GFP | Derivative of vector pSEVA238 used for regulated expression of msfGFP; XylS/Pm→msfGFP; KmR | This work |
| pS248·GFP | Derivative of vector pSEVA248 used for regulated expression of msfGFP; XylS/Pm→msfGFP; KmR | This work |
| pSEVA237M | Cloning vector; oriV(pBBR1); promoter‐less msfGFP; KmR | Silva‐Rocha et al. (2013) |
| pSEVA247M | Cloning vector; oriV(pRO1600/ColE1); promoter‐less msfGFP; KmR | Silva‐Rocha et al. (2013) |
| pS23·Pm‐GFP | Derivative of vector pSEVA237M bearing Pm→msfGFP; KmR | This work |
| pS24·Pm‐GFP | Derivative of vector pSEVA247M bearing Pm→msfGFP; KmR | This work |
| pS23·Pm‐GFP*c | Derivative of vector pSEVA237M bearing Pm→msfGFP*; KmR | This work |
| pS24·Pm‐GFP* | Derivative of vector pSEVA247M bearing Pm→msfGFP*; KmR | This work |
a. Plasmids can be obtained from Addgene (http://www.addgene.org) with the following deposit numbers: pTn7·xylS (122591), pTn7·X‐nia (122592), pS23·Pm‐GFP (122593) and pS24·Pm‐GFP (122594).
b. Antibiotic markers: Amp, ampicillin; Gm, gentamicin; Km, kanamycin.
c. Conditionally proteolizable variants of msfGFP are indicated by an asterisk (*) symbol.
Table 2.
Oligonucleotides used in this work.a
| Oligonucleotide | Sequence (5′→3′) | T m (°C) | Use |
|---|---|---|---|
| Pm_ins‐F | TAT CTC TAG TAA GGC CTA CCC CTT AGG CTT TAT GCA AGC TTA GGA GGA AAA ACA TAT GCG | 60 | Insertion of Pm into vector pSEVA237M by site‐directed mutagenesis PCR |
| Pm_ins‐R | GCC ATT TTT TGC ACT CCT GTA TCC GCT TCT TGC AAT TAA TTA AAG GCA TCA AAT AAA ACG AAA GGC TCA | 59 | |
| ssr_nia‐F | CGC TAA CGA CGA TAA CTA CGC CCT GGC TGC GTA AAC TAG TCT TGG ACT CCT GTT GAT AGA | 60 | Insertion of a nia/ssrA tag into pS23·Pm‐GFP by site‐directed mutagenesis PCR |
| ssr_nia‐R | GCA CGT TCA TCA GCT TGA TGC ACC ACG ACG TTG GAC TCG CCT TTG TAG AGT TCA TCC ATG CCG TGC | 57 | |
| xylS·pTn7‐F† | ACG TCT TAA UTA AAC GTT CGT AAT CAA GCC ACT T | 61 | Amplification of xylS to clone into vector pTn7‐M |
| xylS·pTn7‐R† | AGG ACA CUG CAC TTT ATG CTG GTT ATG C | 60 | |
| pTn7·xylS‐F† | AGT GTC CUA GGC CGC GGC CGC | 63 | Amplification of pTn7‐M to insert xylS or xylS/Pm→nia |
| pTn7·xylS‐R† | ATT AAG ACG UCT TGA CAT AAG CCT GTT CGG TTC | 62 | |
| pTn7·nia‐F† | ACT CAG GGU ACC CGG GGA TCC TCT AGA | 58 | |
| nia·pTn7‐R† | ACC CTG AGU GTA AAC AAA TTC CCC ATC AAG A | 57 | Cloning of xylS/Pm→nia |
| Gm_check‐F | AGT CAG AGT TAC GGA ATT GTA GG | 55 | Checking insertion of GmR into the chromosome |
| Gm_check‐R | ATT AGC TTA CGA CGC TAC ACC C | 56 | |
| gfp_RBS‐F | AAT CCT AGG CCG CGA CGC ATG TTT AGG AGG AAA AAC ATA TGC GTA AAG GTG AAG AAC TGT | 63 | Cloning of msfGFP |
| gfp‐R | AAG ACT AGT CAT TTA TTT GTA GAG TTC ATC CAT G | 54 | |
| pS1 | AGG GCG GCG GAT TTG TCC | 60 | Check SEVA plasmids cargo |
| pS2 | GCG GCA ACC GAG CGT TC | 59 |
a. Oligonucleotides designed for USER assembly are indicated with a † symbol.
Design and construction of a set of standard vectors containing the Pm promoter
A synthetic cis‐module, carrying the essential Pm region (which includes the XylS‐ and σ factor‐binding sites), was constructed in such a way that it was made compatible with different cloning strategies ― so that the GOI can be inserted into any plasmid containing the module. This objective can be achieved, for example by traditional restriction/ligation cloning using the HindIII and SpeI sites present in plasmid pS23·Pm‐GFP (Table 1) to replace the gene encoding the monomeric superfolder GFP (msfGFP) with any other GOI. Otherwise, the synthetic cis‐module spanning the promoter region can be inserted into a different plasmid already harbouring the GOI. Such insertion can be achieved by site‐directed mutagenesis PCR with a set of primers containing the Pm motif (Fig. 2A). Forward and reverse oligonucleotides (Pm_ins‐F and Pm_ins‐R, Table 2) were designed so that they amplify the flanking DNA regions surrounding the locus intended for insertion of Pm via an extension added to their 5′‐termini. Thus, the 5′‐end of the forward primer anneals immediately in front of the intended insertion site, whereas the sequence complementary to the GOI is placed at its 3′‐end. For this purpose, the 5′‐terminus of the forward primer is appended with the synthetic sequence 5′‐CTA CCC CTT AGG CTT TAT GCA AGC TTA GGA GGA AAA ACA T‐3′. Following the same reasoning, the reverse primer is extended at its 5′‐end with the synthetic sequence 5′‐GCC TTA CTA GAG ATA GCC ATT TTT TGC ACT CCT GTA TCC GCT TCT TGC A‐3′ (Fig. 2B). Following the amplification of the target plasmid with these primers, a 5‐μl aliquot of the PCR product was analysed by gel electrophoresis. If the fragment had the correct size (i.e. length of the plasmid), the remaining PCR product was used according to the site‐directed mutagenesis PCR protocol as indicated in the previous section and then transformed into chemically competent E. coli DH5α cells (cat. # 18265017; Thermo Fisher Scientific). The efficiency of this procedure was very high, and the plasmid of three different colonies could be immediately sequenced without previous confirmation by colony PCR.
Figure 2.

Standard procedure used for easy insertion of the Pm promoter into any plasmid backbone.A. Insertion of the Pm element into a vector by site‐directed mutagenesis PCR. Each primer anneals to the plasmid backbone through its 3′‐end (shown in black), while the 5′‐end carries part of the Pm region (shown in red).B. Structure of the Pm promoter element. The promoter contains two XylS binding sites, depicted as distal and proximal binding site. The σ factors σ32 and σ38 bind and recruit the RNA polymerase into the –35 and –10 motifs of the promoter (González‐Pérez et al., 2002; Domínguez‐Cuevas et al., 2005). Direct cloning this region upstream of the gene of interest is achieved either by site‐directed mutagenesis PCR (necessary primers are shown in grey), restriction/ligation (relevant restriction sites, compatible with the Standard European Vector Architecture, are indicated), or a different cloning technique (e.g. USER or Gibson assembly).
Tagging proteins of interest with a synthetic NIa/SsrA degradation tag for conditional proteolysis
The hybrid nia/ssrA tag can be inserted at the 3′‐end of the GOI either through restriction/ligation cloning or site‐directed mutagenesis PCR. Alternatively, the proteolytic tag can be directly inserted into another plasmid by using the restriction sites BstXI and SpeI. To introduce the synthetic tag sequence via PCR, oligonucleotides were designed to include the nia/ssrA moiety. The reverse primer was designed against the 3′‐end of the GOI (but excluding the STOP codon), while the forward primer included both the STOP codon and a sequence complementary to the plasmid backbone (Fig. 3A). The 5′‐terminus of the forward primer was appended with the sequence 5′‐CGC TAA CGA CGA TAA CTA CGC CCT GGC TGC G‐3′, while the reverse primer was extended at its 5′ end with the sequence 5′‐GCA CGT TCA TCA GCT TGA TGC ACC ACG ACG TTG GAC TCG CC‐3′ (Fig. 3B). These sequences were added to insert the synthetic nia/ssrA tag, which contains the conserved NIa recognition site (NVVVHQ•A, the cleavage site is indicated with the • symbol; García et al., 1989) and the SsrA degradation tag, designed according to the conserved SsrA recognition sequence described for P. aeruginosa (Flynn et al., 2001). Following the amplification of the target plasmid with these primers, an aliquot of the PCR product (5 μl) was analysed by gel electrophoresis and, if the correct fragment size was observed, the remainder of the reaction was used for site‐directed mutagenesis PCR as indicated in the preceding section. Chemically competent E. coli DH5α cells were transformed with the reaction mix as indicated previously. The efficiency of this procedure resulted to be very high, and plasmid DNA from three different colonies can be immediately sequenced without previous confirmation by colony PCR.
Figure 3.
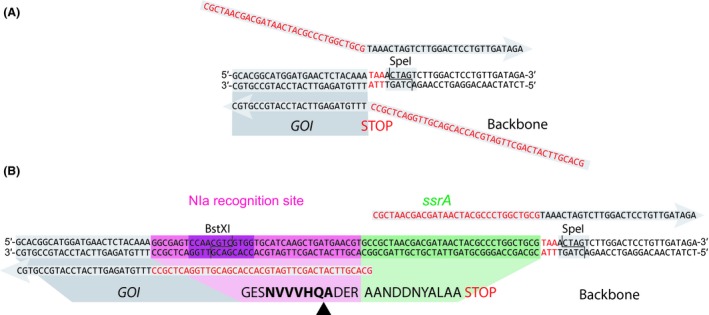
Introduction of the hybrid NIa/SsrA degradation tag into a target protein.A. The degradation tag can be fused to a gene of interest by site‐directed mutagenesis PCR. Primers used to fuse the tag to the monomeric superfolder GFP (msfGFP) in plasmid pS23·Pm‐GFP* are shown in dark grey. The 3′‐terminus of the primers anneals to the plasmid (black letters) and primes amplification of the fragment. The 5′‐ends carry the synthetic NIa/SsrA tag proper (red letters).B. After re‐circularization of the PCR product, the resulting plasmid contains the NIa/SsrA tag fused to the gene of interest. The tag includes the conserved NIa recognition site (indicated in boldface) with its cleavage site (black triangle) followed on the 3′‐terminal by the predicted SsrA degradation signal from P. aeruginosa (Flynn et al., 2001). Alternatively, other cloning strategies can be used to insert the degradation tag for conditional proteolysis, such as restriction/ligation (relevant restriction sites, compatible with the Standard European Vector Architecture, are indicated), or a different cloning technique (e.g. USER or Gibson assembly).
Parallel integration of the Pm promoter and a synthetic NIa/SsrA degradation tag for conditional proteolysis
In order to obtain a construct containing the Pm promoter and the GOI fused to the tag in one step, the gene encoding msfGFP can be replaced by the GOI in plasmid pS23·Pm‐GFP* or pS24·Pm‐GFP* (which differ in the oriV and hence in plasmid copy number; Table 1) through traditional restriction/ligation using the restriction sites HindIII and BstXI. In order to introduce both sites in another plasmid, USER assembly (Cavaleiro et al., 2015) or Gibson assembly (Gibson et al., 2009) can be likewise used as desired.
Application example
After observing high basal expression stemming from the XylS/Pm system in plasmid‐based systems, we set out to characterize this expression system on different standard vectors (i.e. carrying different oriV and hence displaying diverse copy numbers) to gain insight in the regulation of the transcriptional response. In order to investigate the consequences of copy number on the behaviour of the XylS/Pm system, plasmids with different copy numbers were constructed (Table 1), containing the gene encoding msfGFP under the transcriptional control of XylS/Pm. Plasmids pS238·GFP and pS248·GFP were constructed following the Standard European Vector Architecture (SEVA; Silva‐Rocha et al., 2013; Martínez‐García et al., 2014) by amplification of msfGFP from pSEVA237M (Table 1) with the primer pair gfp_RBS‐F and gfp‐R (Table 2) and ligation of the DNA fragment into the SpeI and AvrII‐digested vectors pSEVA238 and pSEVA248. The msfGFP fluorescence (λ excitation = 485 nm, λ emission = 516 nm) was determined during cultivation of the cells in M9 minimal medium (Nikel et al., 2015a,2015b) supplemented with 0.2% (w/v) citrate at 30°C in a 96‐well plate (Synergy H1, BioTek). The expression strength of msfGFP was estimated from the change of fluorescence divided by the change of optical density at 600 nm.
Interestingly, higher copy number correlated with elevated maximum expression of msfGFP under the XylS/Pm system, but inducibility (i.e. the fold‐change difference between the induced and uninduced state) also decreased drastically. In particular, we observed a nine and threefold change in the ratio of induced/uninduced states when P. putida was expressing msfGFP from plasmid pS238·GFP or pS248·GFP respectively. This result is in accordance to the functional model of the XylS regulator proposed by Zwick et al. (2013), indicating that the formation of XylS dimers depends on inducer concentration at low XylS concentration, while at higher XylS availability, active regulator dimers can be formed even without inducer. Furthermore, this model points that the active XylS dimer concentration seems to be limited by its low solubility in the cell cytoplasm.
Against this background, and in order to uncouple the copy number of xylS (hence, the intracellular concentration of XylS) from msfGFP under the transcriptional control of Pm, xylS was integrated into the chromosome by using the Tn7‐bearing plasmid pTn7·xylS. For the construction of plasmid pTn7·xylS, the fragment contain xylS was amplified from plasmid pS238·nia with the primer pair xylS·pTn7‐F and xylS·pTn7‐R (Table 2), and vector pTn7‐M was amplified with the primer pair pTn7·xylS‐F and pTn7·xylS‐R. These DNA fragments were then merged in a USER assembly reaction. Furthermore, plasmids pS23·Pm‐GFP and pS24·Pm‐GFP (which contain msfGFP under the control of Pm, but do not carry xylS) were constructed in parallel. This set of plasmids were obtained by site‐directed mutagenesis PCR of plasmids pSEVA237M and pSEVA247M with the primers Pm_ins‐F and Pm_ins‐R (Table 2). A P. putida strain, carrying xylS integrated into the genome, and transformed with either plasmid pS23·Pm‐GFP (in which the oriV is pBBR1) or pS24·Pm‐GFP (in which the oriV is RO1600/ColE1) showed inducibility levels of 170 and 84‐fold when compared to control experiments without 3‐mBz respectively. In these uncoupled systems, the basal expression strength decreased more than the induced expression strength. On the other hand, it was observed that the maximum expression strength decreased, likely because XylS is produced at subsaturation concentrations with respect to activation of Pm. It is also possible that the basal expression of the GOI decreased due to a lower amount of available XylS (which would dimerize without inducer if its concentration reaches a certain threshold). Even though the uncoupling strategy led to a drastic improvement in the inducibility of the XylS/Pm expression system, some degree of basal (i.e. leaky) expression was still detected.
In order to reduce basal expression further, we set to design a system in which not only xylS and Pm copy numbers are controlled by physical uncoupling, but also expression of the GOI was further controlled by post‐translational degradation control. Plasmids pS23·Pm‐GFP* and pS24·Pm‐GFP* were constructed through site‐directed mutagenesis PCR using plasmids pS23·Pm‐GFP and pS24·Pm‐GFP respectively, as the template and primers ssr_nia_F and ssr_nia‐R (Table 2). The msfGFP used in our experiments appears to be an excellent reporter for post‐translational control, as it is very stable and therefore tends to accumulate in the cells (Pedelacq et al., 2006). The gene encoding the nuclease NIa was integrated through the use of the plasmid pTn7·X‐nia. For the construction of plasmid pTn7·X‐nia, the fragment containing xylS/Pm→nia was amplified from plasmid pS238·NIa with the primer pair xylS·pTn7‐F and nia·pTn7‐R (Table 2), and plasmid pTn7‐M was amplified with the primer pair pTn7·nia‐F and pTn7·xylS‐R. These fragments were then merged in a USER assembly reaction as indicated above.
The expression system employing transcriptional and post‐translational control over the expression of msfGFP showed lower expression levels when induced as compared with the expression system without post‐translational control (Fig. 4). This phenomenon could be caused by the presence of additional XylS binding sites (in the Pm promoter of the integration), which recruit active XylS dimers and hence lowers the actual concentration of free XylS dimers, which can then bind to the plasmid‐borne XylS binding sites. Interestingly, no basal expression was observed with either plasmid, which leads to close‐to‐zero levels of leaky expression in the absence of 3‐mBz and, at the same time, yielded the highest (indicated as ∞ in Fig. 4B) inducibility of the system.
Figure 4.
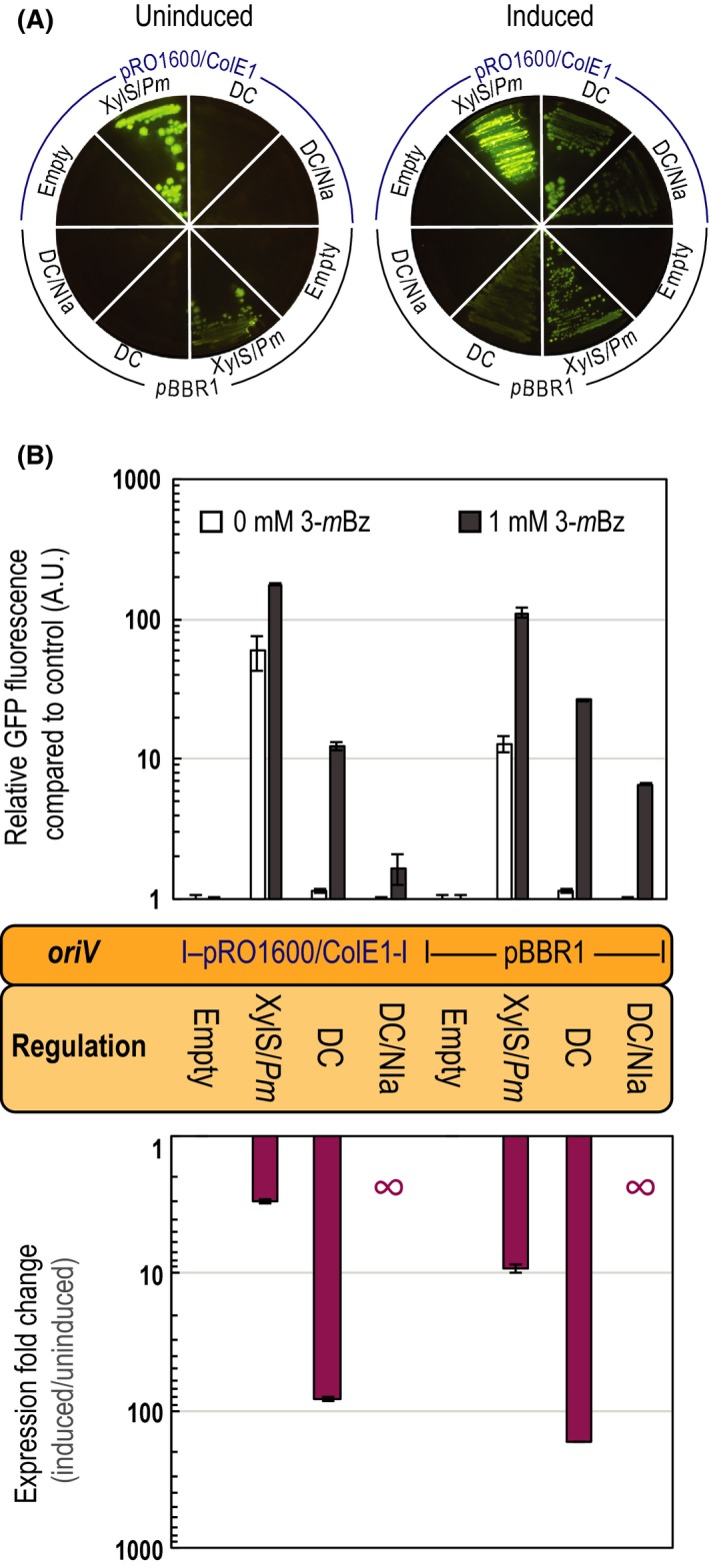
Characterization of the tightly regulated expression systems for Pseudomonas based on physically decoupled XylS/Pm elements and conditional proteolysis.A. M9 minimal medium plates with 0.2% (w/v) citrate, containing 0 mM (left) or 1 mM (right) 3‐methylbenzoate (3‐mBz) were seeded with P. putida with an empty Tn7 module integrated into the chromosome and transformed with either vector pSEVA231 or vector pSEVA241, which served as negative controls (indicated as Empty in the figure). The same strain was transformed either with plasmid pS238·GFP or pS248·GFP carrying the XylS/Pm expression system in its original configuration (indicated as XylS/Pm in the figure). Strain KT·P, containing a chromosomal copy of xylS, was transformed either with plasmid pS23·Pm‐GFP or pS24·Pm‐GFP, in which the xylS/Pm system has been physically decoupled (indicated as DC in the figure). Strain KT·PN, containing a single xylS/Pm→nia integration in the chromosome, was transformed either with plasmid pS23·Pm‐GFP* or pS24·Pm‐GFP* harbouring the decoupled xylS/Pm system and equipped with conditional proteolytic control of GFP due to inducible nia expression (indicated as DC/NIa in the figure). Strains harbouring plasmids pS24x (x represents any of the variants described above) carrying the origin of vegetative replication (oriV) pRO1600/ColE1 were seeded in the upper part of the plate, while strains harbouring the plasmids pS23x, carrying oriV(pBBR1), were seeded in the lower part. Plates were incubated at 30°C for 18 h. The plates were visualized under blue light (Safe‐Imager, Invitrogen) and photographed.B. The increase in msfGFP fluorescence per unit of biomass (estimated as the optical density at 600 nm) relative to the negative control was measured during cultivation in M9 minimal medium containing 0.2% (w/v) citrate supplemented or not with 3‐mBz at 1 mM (upper part) and the resulting expression fold changes (lower part). Depicted in the diagram are the genetic elements either integrated into the single Tn7 integration site in the chromosome or plasmid‐borne. Bars represent the mean values of the corresponding parameter ± standard deviation (n ≥ 3 in all cases), and a Student t‐test revealed significant differences between all groups. Note that in some cases the level of msfGFP fluorescence of control experiments was below the limit of detection; in these experiments (DC/NIa), the ratio between induced and uninduced conditions is indicated with a ∞ symbol.
Discussion
In this work, we have demonstrated how the repurposing of a classical gene expression system by means of the physical separation of xylS and Pm can lead to much higher induction fold changes compared with the systems carrying xylS and Pm on the same vector. The phenomenon underlying the results observed in our uncoupled system could be connected to the data reported by Goñi‐Moreno et al. (2017), indicating that the proximity of a source (i.e. XylS) and target (i.e. Pm promoter) elements dictates noise patterns of the expression system. Furthermore, the dual transcriptional and post‐translational control of the gene expression flow in our system reduced basal expression down to undetectable amounts of msfGFP caused by leaky expression of the GOI thereof. Since the expression of xylS might be too low to reach saturation of the Pm promoter by active XylS dimers upon addition of the chemical inducer, the final output of the system was lower than in the plasmid‐based counterpart. If stronger gene expression is desired, the system could be further improved through the implementation of engineered xylS and/or variants of the Pm promoter (Bakke et al., 2009; Vee Aune et al., 2010; Zwick et al., 2012). In any case, for applications where tight control over gene expression is crucial, the modular expression systems presented here offer a solid alternative that can be easily implemented into virtually any combination of vectors and GOIs (and Gram‐negative bacterial hosts). In particular, the synthetic systems discussed in this article constitute a valuable tool for maintaining low leaky expression of genes involved in pathways leading to toxic intermediates or products (a classical problem in metabolic engineering), and for the design of circuits in which the transcriptional output of a given system is ‘cascaded’ into another signal, thereby multiplying the tightness of the transcriptional regulation thereof.
Conflict of interest
None declared.
Acknowledgements
We are indebted to Belén Calles (CNB‐CSIC, Madrid, Spain) for sharing research materials and fruitful discussions. The financial support from The Novo Nordisk Foundation (NNF10CC1016517) and from the European Union's Horizon2020 Research and Innovation Programme under grant agreement No. 814418 (SinFonia) to P.I.N. is gratefully acknowledged. J.T. and V.M. are the recipients of a fellowship from the Novo Nordisk Foundation as part of the Copenhagen Bioscience Ph.D. Programme, supported through grant NNF 18CC0033664. The responsibility of this article lies with the authors. The NNF and the European Union are not responsible for any use that may be made of the information contained therein.
Microbial Biotechnology (2020) 13(1), 222–232
Funding InformationThe financial support from The Novo Nordisk Foundation (NNF10CC1016517) and from the European Union's Horizon2020 Research and Innovation Programme under grant agreement No. 814418 (SinFonia) to P.I.N. is gratefully acknowledged. J.T. and V.M. are the recipients of a fellowship from the Novo Nordisk Foundation as part of the Copenhagen Bioscience Ph.D. Programme, supported through grant NNF 18CC0033664.
References
- Bakke, I. , Berg, L. , Aune, T.E. , Brautaset, T. , Sletta, H. , Tøndervik, A. , and Valla, S. (2009) Random mutagenesis of the Pm promoter as a powerful strategy for improvement of recombinant‐gene expression. Appl Environ Microbiol 75: 2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, I. , de Lorenzo, V. , and Nikel, P.I. (2016) Genetic programming of catalytic Pseudomonas putida biofilms for boosting biodegradation of haloalkanes. Metab Eng 33: 109–118. [DOI] [PubMed] [Google Scholar]
- Boyer, H.W. , and Roulland‐Dussoix, D. (1969) A complementation analysis of the restriction and modification of DNA in Escherichia coli . J Mol Biol 41: 459–472. [DOI] [PubMed] [Google Scholar]
- Calero, P. , and Nikel, P.I. (2019) Chasing bacterial chassis for metabolic engineering: a perspective review from classical to non‐traditional microorganisms. Microb Biotechnol 12: 98–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , Jensen, S.I. , and Nielsen, A.T. (2016) Broad‐host‐range ProUSER vectors enable fast characterization of inducible promoters and optimization of p‐coumaric acid production in Pseudomonas putida KT2440. ACS Synth Biol 5: 741–753. [DOI] [PubMed] [Google Scholar]
- Cavaleiro, A.M. , Kim, S.H. , Seppälä, S. , Nielsen, M.T. , and Nørholm, M.H.H. (2015) Accurate DNA assembly and genome engineering with optimized uracil excision cloning. ACS Synth Biol 4: 1042–1046. [DOI] [PubMed] [Google Scholar]
- Cebolla, A. , Guzmán, C. , and de Lorenzo, V. (1996) Nondisruptive detection of activity of catabolic promoters of Pseudomonas putida with an antigenic surface reporter system. Appl Environ Microbiol 62: 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarría, M. , Goñi‐Moreno, A. , de Lorenzo, V. and Nikel, P.I. (2016) A metabolic widget adjusts the phosphoenolpyruvate‐dependent fructose influx in Pseudomonas putida . mSystems 1, e00154‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.‐H. , Gaynor, J.B. , White, K.G. , López, C. , Bosio, C.M. , Karkhoff‐Schweizer, R.R. , and Schweizer, H.P. (2005) A Tn7‐based broad‐range bacterial cloning and expression system. Nat Methods 2: 443. [DOI] [PubMed] [Google Scholar]
- Cook, T.B. , Rand, J.M. , Nurani, W. , Courtney, D.K. , Liu, S.A. , and Pfleger, B.F. (2018) Genetic tools for reliable gene expression and recombineering in Pseudomonas putida . J Ind Microbiol Biotechnol 45: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez‐Cuevas, P. , Marín, P. , Ramos, J.L. , and Marqués, S. (2005) RNA polymerase holoenzymes can share a single transcription start site for the Pm promoter: critical nucleotides in the –7 to –18 region are needed to select between RNA polymerase with σ38 or σ32 . J Biol Chem 280: 41315–41323. [DOI] [PubMed] [Google Scholar]
- Durante‐Rodríguez, G. , de Lorenzo, V. , and Nikel, P.I. (2018) A post‐translational metabolic switch enables complete decoupling of bacterial growth from biopolymer production in engineered Escherichia coli . ACS Synth Biol 7: 2686–2697. [DOI] [PubMed] [Google Scholar]
- Flynn, J.M. , Levchenko, I. , Seidel, M. , Wickner, S.H. , Sauer, R.T. , and Baker, T.A. (2001) Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA 98: 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos, M.T. , Marqués, S. , and Ramos, J.L. (1996) Expression of the TOL plasmid xylS gene in Pseudomonas putida occurs from a σ70‐dependent promoter or from σ70‐ and σ54‐dependent tandem promoters according to the (aromatic) compound used for growth. J Bacteriol 178: 2356–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García, J.A. , Riechmann, J.L. , and Lain, S. (1989) Artificial cleavage site recognized by plum pox potyvirus protease in Escherichia coli . J Virol 63: 2457–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawin, A. , Valla, S. , and Brautaset, T. (2017) The XylS/Pm regulator/promoter system and its use in fundamental studies of bacterial gene expression, recombinant protein production and metabolic engineering. Microb Biotechnol 10: 702–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D.G. , Young, L. , Chuang, R.Y. , Venter, J.C. , Hutchison, C.A. , and Smith, H.O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Gomez, J.G.C. , Méndez, B.S. , Nikel, P.I. , Pettinari, M.J. , Prieto, M.A. , and Silva, L.F. (2012) Making green polymers even greener: towards sustainable production of polyhydroxyalkanoates from agroindustrial by‐products In Advances in Applied Biotechnology. Petre M. (ed). Rijeka, Croatia: InTech, pp. 41–62. [Google Scholar]
- Goñi‐Moreno, A. , Benedetti, I. , Kim, J. , and de Lorenzo, V. (2017) Deconvolution of gene expression noise into spatial dynamics of transcription factor‐promoter interplay. ACS Synth Biol 6: 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Pérez, M.M. , Marqués, S. , Domínguez‐Cuevas, P. , and Ramos, J.L. (2002) XylS activator and RNA polymerase binding sites at the Pm promoter overlap. FEBS Lett 519: 117–122. [DOI] [PubMed] [Google Scholar]
- Jahn, M. , Vorpahl, C. , Hübschmann, T. , Harms, H. , and Müller, S. (2016) Copy number variability of expression plasmids determined by cell sorting and droplet digital PCR. Microb Cell Fact 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez, J.I. , Miñambres, B. , García, J.L. , and Díaz, E. (2002) Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ Microbiol 4: 824–841. [DOI] [PubMed] [Google Scholar]
- de Lorenzo, V. , Fernández, S. , Herrero, M. , Jakubzik, U. , and Timmis, K.N. (1993) Engineering of alkyl‐ and haloaromatic‐responsive gene expression with mini‐transposons containing regulated promoters of biodegradative pathways of Pseudomonas . Gene 130: 41–46. [DOI] [PubMed] [Google Scholar]
- Marqués, S. , and Ramos, J.L. (1993) Transcriptional control of the Pseudomonas putida TOL plasmid catabolic pathways. Mol Microbiol 9: 923–929. [DOI] [PubMed] [Google Scholar]
- Marqués, S. , Manzanera, M. , González‐Pérez, M.M. , Gallegos, M.T. , and Ramos, J.L. (1999) The XylS‐dependent Pm promoter is transcribed in vivo by RNA polymerase with σ38 or σ32 depending on the growth phase. Mol Microbiol 31: 1105–1113. [DOI] [PubMed] [Google Scholar]
- Martínez‐García, E. , and de Lorenzo, V. (2017) Molecular tools and emerging strategies for deep genetic/genomic refactoring of Pseudomonas . Curr Opin Biotechnol 47: 120–132. [DOI] [PubMed] [Google Scholar]
- Martínez‐García, E. , Aparicio, T. , Goñi‐Moreno, A. , Fraile, S. , and de Lorenzo, V. (2014) SEVA 2.0: an update of the Standard European Vector Architecture for de‐/re‐construction of bacterial functionalities. Nucleic Acids Res 43: D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , and de Lorenzo, V. (2018) Pseudomonas putida as a functional chassis for industrial biocatalysis: from native biochemistry to trans‐metabolism. Metab Eng 50: 142–155. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Chavarría, M. , Fuhrer, T. , Sauer, U. , and de Lorenzo, V. (2015a) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner‐Doudoroff, Embden‐Meyerhof‐Parnas, and pentose phosphate pathways. J Biol Chem 290: 25920–25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , Romero‐Campero, F.J. , Zeidman, J.A. , Goñi‐Moreno, A. and de Lorenzo, V. (2015b) The glycerol‐dependent metabolic persistence of Pseudomonas putida KT2440 reflects the regulatory logic of the GlpR repressor. mBio 6, e00340‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , Chavarría, M. , Danchin, A. , and de Lorenzo, V. (2016) From dirt to industrial applications: Pseudomonas putida as a Synthetic Biology chassis for hosting harsh biochemical reactions. Curr Opin Chem Biol 34: 20–29. [DOI] [PubMed] [Google Scholar]
- Panke, S. , Meyer, A. , Huber, C.M. , Witholt, B. , and Wubbolts, M.G. (1999) An alkane‐responsive expression system for the production of fine chemicals. Appl Environ Microbiol 65: 2324–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq, J.D. , Cabantous, S. , Tran, T. , Terwilliger, T.C. , and Waldo, G.S. (2006) Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24: 79–88. [DOI] [PubMed] [Google Scholar]
- Poblete‐Castro, I. , Borrero de Acuña, J.M. , Nikel, P.I. , Kohlstedt, M. , and Wittmann, C. (2017) Host organism: Pseudomonas putida In Industrial Biotechnology: Microorganisms. Wittmann C., and Liao J.C. (eds). Weinheim: Wiley‐VCH Verlag GmbH & Co. KGaA. [Google Scholar]
- Ramos, J.L. , Marqués, S. , and Timmis, K.N. (1997) Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid‐encoded regulators. Annu Rev Microbiol 51: 341–373. [DOI] [PubMed] [Google Scholar]
- Ruiz, J.A. , Fernández, R.O. , Nikel, P.I. , Méndez, B.S. , and Pettinari, M.J. (2006) dye (arc) Mutants: insights into an unexplained phenotype and its suppression by the synthesis of poly(3‐hydroxybutyrate) in Escherichia coli recombinants. FEMS Microbiol Lett 258: 55–60. [DOI] [PubMed] [Google Scholar]
- Ruíz, R. , Marqués, S. , and Ramos, J.L. (2003) Leucines 193 and 194 at the N‐terminal domain of the XylS protein, the positive transcriptional regulator of the TOL meta‐cleavage pathway, are involved in dimerization. J Bacteriol 185: 3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall‐Shapiro, T.H. , Sontag, E.D. , and Voigt, C.A. (2018) Engineered promoters enable constant gene expression at any copy number in bacteria. Nat Biotechnol 36: 352–358. [DOI] [PubMed] [Google Scholar]
- Shingler, V. , and Moore, T. (1994) Sensing of aromatic compounds by the DmpR transcriptional activator of phenol‐catabolizing Pseudomonas sp. strain CF600. J Bacteriol 176: 1555–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Rocha, R. , de Jong, H. , Tamames, J. , and de Lorenzo, V. (2011) The logic layout of the TOL network of Pseudomonas putida pWW0 plasmid stems from a metabolic amplifier motif (MAM) that optimizes biodegradation of m‐xylene. BMC Syst Biol 5: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Rocha, R. , Martínez‐García, E. , Calles, B. , Chavarría, M. , Arce‐Rodríguez, A. , de las Heras, A. , et al (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41: D666–D675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk, A.L. , Lin, A. , and Weiss, R. (2012) Foundations for the design and implementation of synthetic genetic circuits. Nat Rev Genet 13: 406–420. [DOI] [PubMed] [Google Scholar]
- Smith, A.W. , and Iglewski, B.H. (1989) Transformation of Pseudomonas aeruginosa by electroporation. Nucleic Acids Res 17: 10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter, K. (2006) Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72: 211–222. [DOI] [PubMed] [Google Scholar]
- Vee Aune, T.E. , Bakke, I. , Drablos, F. , Lale, R. , Brautaset, T. , and Valla, S. (2010) Directed evolution of the transcription factor XylS for development of improved expression systems. Microb Biotechnol 3: 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther‐Larsen, H.C. , Josefsen, K.D. , Brautaset, T. , and Valla, S. (2000) Parameters affecting gene expression from the Pm promoter in Gram‐negative bacteria. Metab Eng 2: 79–91. [DOI] [PubMed] [Google Scholar]
- Wu, G. , Yan, Q. , Jones, J.A. , Tang, Y.J. , Fong, S.S. , and Koffas, M.A.G. (2016) Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol 34: 652–664. [DOI] [PubMed] [Google Scholar]
- Zobel, S. , Benedetti, I. , Eisenbach, L. , de Lorenzo, V. , Wierckx, N. , and Blank, L.M. (2015) Tn7‐Based device for calibrated heterologous gene expression in Pseudomonas putida . ACS Synth Biol 4: 1341–1351. [DOI] [PubMed] [Google Scholar]
- Zwick, F. , Lale, R. , and Valla, S. (2012) Strong stimulation of recombinant protein production in Escherichia coli by combining stimulatory control elements in an expression cassette. Microb Cell Fact 11: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick, F. , Lale, R. , and Valla, S. (2013) Regulation of the expression level of transcription factor XylS reveals new functional insight into its induction mechanism at the Pm promoter. BMC Microbiol 13: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]