

Abstract
Methods that enable the direct C–H alkoxylation of complex organic molecules are significantly underdeveloped, particularly in comparison to analogous strategies for C–N and C–C bond formation. In particular, almost all methods for the incorporation of alcohols via C–H oxidation require the use of the alcohol component as solvent. This limits the practical scope of these reactions to simple, inexpensive alcohols. We report a photocatalytic protocol for the functionalization of benzylic C–H bonds with a wide range of oxygen nucleophiles. Our strategy merges the photoredox activation of arenes with copper(II)-mediated oxidation of the resulting benzylic radicals, which enables the introduction of benzylic C–O bonds with high site selectivity, chemoselectivity, and functional group tolerance using only two equivalents of the alcohol coupling partners. This method enables the late-stage introduction of complex alkoxy groups into bioactive molecules, providing a practical new tool with potential applications in synthesis and medicinal chemistry.
Keywords: C–H alkoxylation, copper, oxidation, photocatalysis, radicals
Graphical Abstract
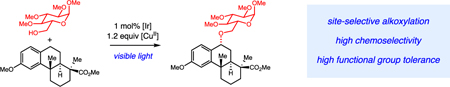
C–H alkoxylation: Photocatalysis offers a one-step strategy to selectively functionalize the benzylic positions of electron-rich arenes with alcohols in structurally complex organic molecules.
The development of methods for the site-selective functionalization of sp3-hybridized C–H bonds has profoundly influenced the way that organic chemists approach complex molecule synthesis.1 Late-stage functionalization strategies that enable one-step incorporation of new functional groups are particularly important because they can provide an efficient strategy to rapidly assess structural analogues of pharmaceutical candidates for new chemical and biological properties.2 Much of the recent activity in this field has centered on cross-coupling reactions that enable the site-selective incorporation of complex carbon- and nitrogen-centered fragments at aliphatic positions.3
Dialkyl ether linkages are ubiquitous in pharmaceuticals, natural products, and other bioactive compounds.4 There are numerous established methods for the oxidative functionalization of aliphatic C–H bonds in complex organic molecules using structurally simple alcohols, including electrochemical oxidations,5 chemical oxidations,6 and transition metal catalyzed C–H alkoxylation protocols.7 With a few notable exceptions,7ef however, none of these strategies are appropriate for couplings involving structurally complex alcohols. This is because in these methods, solvent quantities of the alcohol coupling partner are typically required for optimal reactivity, which limits the practical scope of these alkoxylation reactions to inexpensive simple alcohols that are produced on commodity chemical scales.8 A more synthetically useful method for C–H alkoxylation that would enable couplings with structurally much more complex alcohols, therefore, requires a new approach that operates at a more reasonable stoichiometry.
Recently, our laboratory has become interested in the design of photocatalytic oxidative functionalization reactions. We were inspired by the facility with which photoredox catalysis generates organoradical intermediates from readily available feedstocks.9 We proposed that Cu(II) salts could be ideal terminal oxidants for net-oxidative transformations whose use has been underexplored in photoredox reactions.10,11 Seminal studies by Kochi demonstrated that a variety of organic radicals undergo oxidation by Cu(II) at diffusion-controlled rates.12 Moreover, the use of Cu(II) oxidants avoids the intermediacy of reactive oxygen intermediates (e.g., oxygen-centered radicals, superoxide, singlet oxygen) that can be incompatible with organoradical intermediates or with highly functionalized complex organic molecules.
We imagined a strategy exploiting a photoredox catalyst in combination with a Cu(II) oxidant would enable the direct incorporation of a wide range of simple nucleophilic alcohols into Csp3–H bonds. A brief outline of our design strategy is summarized in Scheme 1. Photoinduced one-electron oxidation of an electron-rich arene (A) would afford a radical cation (B) whose benzylic C–H bonds are significantly acidified;13 facile deprotonation would afford benzylic radical intermediate C. Radicalophilic Cu(II) could transform C into the corresponding carbocation (D), which could be trapped by a nucleophilic alcohol. An analogous oxidation–deprotonation–oxidation sequence was proposed in the electrochemical benzylic methoxylation of estrone.5a This method, however, requires the use of an alcohol solvent because proton reduction at the counterelectrode drives this net-oxidative process. More recently, Pandey14 and Hong15 adopted an analogous oxidation-deprotonation-oxidation sequence in photoredox protocols for benzylic ether formation using oxygen as a terminal oxidant. These photocatalytic methods, however, are limited to intramolecular bond formations and thus cannot be applied to intermolecular cross-coupling reactions.
Scheme 1.

Design Strategy for Photocatalytic Etherification of Benzylic C–H Bonds.
We reasoned that a combination of photoredox catalyst and Cu(II) oxidant might provide a highly effective method for performing benzylic C–H alkoxylation reactions without the need for solvent-quantity alcohol donors. To test this hypothesis, we examined the reaction of 4-ethylanisole (1) with 2 equiv of methanol (Table 1). [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 was selected as a photocatlyst because its excited-state reduction potential (+1.68 V vs SCE) 16 should be sufficiently positive to oxidize 1 (+1.52 V). The use of Cu(OAc)2 as a terminal oxidant indeed produced the desired product 2, but in only 8% yield (entry 1). Acetoxylation was the major oxidation pathway in this experiment. We hypothesized that less nucleophilic ligands on Cu(II) might decrease the formation of this undesired product. Indeed, Cu(TFA)2 provided a higher yield and no observable ester product (entry 2); however, benzylic alcohol was a significant byproduct, which we attributed to the presence of water in the hydrate form of the commercial salt. We thus prepared the anhydrous acetonitrile complex Cu(TFA)2(MeCN),17 the use of which prevented formation of the hydroxylated product and afforded the desired methanol adduct in 73% yield (entry 3). Cu(II) salts were unique in their ability to promote this alkoxylation: we screened a wide range of other common terminal oxidants, and none produced significant yields of 1 (entry 4), consistent with our guiding hypothesis. Alternate photocatalysts also afforded diminished reaction efficiency. Both less-oxidizing Ir photocatalysts18 (entry 5) and more-oxidizing organic photocatalysts19 (entry 6 and 7) afforded diminished yields. Finally, control experiments validated the photocatalytic nature of this reaction and the necessity of Cu(II) co-oxidant; no product was observed in the absence of photocatalyst, Cu(II) terminal oxidant, or light (entries 8–10).
Table 1.
Optimization Studies for Methoxylation of Benzylic C–H Bonds.
 | |||
|---|---|---|---|
| Entry[a] | Photocatalyst | Terminal oxidant | Yield[b] |
| 1 | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | Cu(OAc)2•H2O | 8% |
| 2 | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | Cu(TFA)2•H2O | 61% |
| 3 | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | Cu(TFA)2•MeCN | 73% |
| 4 | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | Air, PhI(OAc)2, TEMPO, MnO2, FeCl3, t-BuOOH, or benzoquinone |
<10% |
| 5 | [Ir(dF(CF3)ppy)2(bpy)]PF6 | Cu(TFA)2•MeCN | 23% |
| 6 | Fukuzumi acridinium (5.0 mol%) | Cu(TFA)2•MeCN | 10% |
| 7 | Triphenylpyrylium (2.5 mol%) | Cu(TFA)2•MeCN | 5% |
| 8 | none | Cu(TFA)2•MeCN | 0% |
| 9 | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | none | 0% |
| 10[c] | [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 | Cu(TFA)2•MeCN | 0% |
Unless otherwise noted, all reactions were conducted using 0.3 mmol of 1 in degassed MeCN and irradiated with a 427 nm Kessil lamp for 6 h.
Yields determined by 1H NMR analysis of the crude reaction mixtures using phenanthrene as an internal standard.
Reaction was conducted in the dark.
The scope of this benzylic alkoxylation protocol is summarized in Table 2. The reaction is most efficient at secondary benzylic positions; primary C–H bonds undergo competitive overoxidation that diminishes the yield of monoalkoxylation products somewhat (3), and tertiary sites undergo functionalization at significantly slower rates (4). The latter can be addressed by diluting the reaction to increase photon flux and by increasing the amount of MeOH to 8 equiv. The presence of alkoxy substituents on the arene that could stabilize the putative quinone methide intermediate is strictly required, but a variety of such groups along with additional substituents are well tolerated (5–9, 17–21). The functional group compatibility of this method is high and includes acidic heteroatoms (10 and 14), basic heterocycles (20), base-sensitive moieties (11 and 12), and boronate ester or azide groups that provide handles for further synthetic manipulation (13 and 15). In general, this reaction displayed a remarkable degree of site selectivity consistent with the predicted stability of the quinone methide intermediate (17–19). Thus functionalization of p-alkoxy-activated benzylic positions is highly selective even in the presence of weaker benzylic C–H bonds (20–22). Finally, the benzylic positions of a variety of electron-rich heterocycles that are also readily oxidized by the photocatalyst can also be functionalized using this protocol (23–25).
Table 2.
Scope of C–H Oxygenation via Photoredox Catalysisa
 |
Reaction condition: substrate (0.6 mmol, 1 equiv.), [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 (1 mol%), K2HPO4 (3 equiv), Cu(TFA)2(MeCN) (1.2 equiv), methanol (2 equiv), MeCN (0.2 M) unless otherwise noted. Yields represent the averaged isolated yields from two experiments.
Yield determined by 1H NMR analysis of the crude reaction mixtures using phenanthrene as an internal standard.
Reaction conducted at 0.1 M using 8 equiv methanol.
Reaction conducted using 8 equiv alcohol.
The scope of the reaction with respect to the heteroatomic nucleophile was also explored. Water, unfortunately, is not a suitable nucleophile, and overoxidation to the ketone was the major product of this reaction (26). Alcohols of increased steric bulk react more slowly, although the yields of these reactions can be increased using 8 equiv of alcohol (27–29). A variety of diverse functional groups were readily tolerated (30–40), including those containing easily oxidizable moieties (30–33, and 37) and potentially base-sensitive groups (36, 38, and 39). An unsymmetrical 1,3-diol reacted exclusively through the less-sterically-bulky primary alcohol (40). Carboxylic acids were also readily incorporated, including both simple aliphatic and more functionalized acids (41–45). Nucleophiles that are more readily oxidized than the anisole unit (47) or that bear strongly coordinating heterocycles (48) were unfortunately not suitable reaction partners. Finally, although N-Boc carbamate reacted with reasonable efficiency (46), other nitrogen nucleophiles were generally not effective (49), suggesting that alternate conditions will be required for the general introduction of C–N bonds using this photocatalytic strategy.
The combination of high site selectivity and significant functional group compatibility suggested to us that this method might be applicable to the diversification of highly complex bioactive organic compounds. Table 3 outlines the functionalization of a range of commercially available natural product scaffolds, all of which are selectively functionalized at the unique activated benzylic C–H position. No diastereoselectivity is observed when the substrate lacks significant stereochemical bias (50), but six-membered rings with a strong preference for a single chair conformation undergo highly selective functionalization of the axial C–H bond (51 and 52).
Table 3.
Late-Stage Functionalization via Oxidative Photoredox Catalysis[a]
 |
Reaction condition: substrate (0.3 mmol, 1 equiv), Ir[dF(CF3)ppy]2(5,5’-dCF3bpy)]PF6 (1 mol%), K2HPO4 (3 equiv.), Cu(TFA)2(MeCN) (1.2 equiv.), alcohol (2 equiv.), MeCN (0.2 M), 9.5 h–10 h unless otherwise noted. Yields represent the averaged isolated yields from two experiments.
Reaction conducted at 0.05 M MeCN:CH2Cl2 using 8 equiv methanol. RSM yield is provided in parentheses.
Reaction conducted at 0.05 M using 8 equiv methanol.
The observation that synthetically useful yields can be obtained using only two equivalents of the alcohol nucleophile also suggests that this intermolecular C–H functionalization might operate as a method to couple two high-value reaction partners. Thus, the ability to conjugate O-methylpodocarpate with a variety of structurally complex nucleophilic partners was explored. The alcohol moiety of a protected serine can readily be installed in good yield (53) without competitive attack of the Boc carbamate. The reaction also incorporates protected hexose and pentose moieties (54 and 55) as well as primary and secondary terpene alcohols (56 and 57) in synthetically useful yields.
To better understand the mechanism of this transformation, we first examined the nature of the initial photoinduced electron-transfer step. The excited-state reduction and oxidation potentials of the optimal photocatalyst (+1.68 V and –0.43 V vs SCE, respectively) are sufficient to either oxidize arene substrate 1 (Ep/2 = +1.52 V) or reduce Cu(II) (E1/2 = +0.38 V). Indeed, a Stern–Volmer analysis indicates that both can quench the photocatalyst excited state at similar rates (Scheme 2A). Thus the arene radical cation might be generated either by the excited Ir*(III) photocatalyst or the Ir(IV) complex (E1/2 = +1.94 V vs SCE) resulting from oxidative quenching by Cu(II). Both mechanisms might be operative. Consistent with this hypothesis, a less strongly oxidizing photocatalyst ([Ir(dF(CF3)ppy)2(bpy)]PF6, *E1/2 = +1.31 V) that is not capable of directly oxidizing 1 nevertheless provides the methoxylation product, albeit in diminished yield (Table 1, entry 4). We also examined the photocatalytic reaction of 1 with CD3OD, which resulted in the formation of d-2 in 83% yield with no incorporation of deuterium at the benzylic position. This result indicates that the deprotonation step is essentially irreversible under the optimized conditions, as might be expected from Kochi’s observation that the oxidation of electron-rich organoradicals by Cu(II) occurs at diffusion-controlled rates.12
Scheme 2.

Development of a Mechanistic Hypothesis.
A mechanistic proposal consistent with these data is outlined in Scheme 2C. Photoexcitation of the Ir(III) catalyst affords a triplet excited state that is oxidatively quenched by Cu(II) to afford a strongly oxidizing Ir(IV) complex. Subsequent oxidation of the arene affords an arene radical cation that undergoes benzylic deprotonation and rapid oxidation by Cu(II). The resulting quinone methide undergoes nucleophilic attack by the alcohol coupling partner to afford the observed benzylic ether products. The reaction proceeds to completion with only 1.2 equiv of Cu(II). Thus both oxidizing equivalents of Cu(II) are consumed in this reaction, which could be attributed to the disproportionation of the Cu(I) byproduct of either oxidation step to Cu(II) and Cu(0), the latter of which we observe precipitating from solution during the reaction.
In summary, we have developed a new photocatalytic strategy to introduce diverse alkoxide functionalities into complex organic molecules by functionalization of benzylic C–H bonds. The site selectivity and broad functional group compatibility of this method renders it amenable to the late-stage functionalization of complex bioactive compounds. Importantly, the efficiency of the reaction provides synthetically useful yields using only a two-fold excess of the alcohol reaction partner, which enables the formation of new benzylic ether compounds from the coupling of two structurally complex units. From a broader perspective, these results are intriguing because they demonstrate that the organoradical intermediates that are readily generated by photoredox activation can be diverted towards cationic reactivity via in situ oxidation by Cu(II). This combination thus provides a promising new platform to design new bond-forming oxidative functionalization reactions with broad utility in synthetic and medicinal chemistry.
Supplementary Material
Acknowledgements
We gratefully acknowledge the NIH (GM095666) for funding this work. We thank Prof. Shannon Stahl and Sijie Chen (UW–Madison) for sharing unpublished data.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) McMurray L, O’Hara F, Gaunt MJ, Chem. Soc. Rev 2011, 40, 1885–1898. [DOI] [PubMed] [Google Scholar]; b) Gutekunst WR, Baran PS, Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]; c) Yamaguchi J, Yamaguchi AD, Itami K Angew. Chem. Int. Ed 2012, 51, 8960–9009; Angew. Chem. 2012,124, 9092–9142. [DOI] [PubMed] [Google Scholar]
- [2].a) Wencel-Delord J, Glorius F, Nature Chem 2013, 5, 369–375. [DOI] [PubMed] [Google Scholar]; b) Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- [3].a) Ma J, Li S, Org. Chem. Front 2014, 1, 712–715. [Google Scholar]; b) Yu J-Q, Shi Z, Eds. CH Activation: Topics in Current Chemistry, Vol. 292; Springer-Verlag: Berlin, 2010. [Google Scholar]
- [4].Roughley SD, Jordan AM, J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- [5].a) Weinberg NL, Brown EA, J. Org. Chem 1966, 31, 4058–4061. [Google Scholar]; b) Ponsold K, Kasch H, Tetrahedron Lett 1977, 46, 4463–4464. [Google Scholar]; c) Shono T, Matsumura Y, Onomura O, Yamada Y, Synthesis 1987, 1099–1100.; d) Ginzel K-D, Steckhan E, Degner D, Tetrahedron 1987, 43, 5797–5805. [Google Scholar]; e) Frankowski KJ, Liu R, Milligan GL, Moeller KD, Aubé J, Angew. Chemie Int. Ed 2015, 54, 10555–10558; Angew. Chem. 2015, 127, 10701–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Moriarty RM, Hu H, Tetrahedron Lett 1981, 22, 2747–2750. [Google Scholar]; b) Moriarty RM, Vaid RK, Ravikumar VT, Vaid BK, Hopkins TE, Tetrahedron 1988, 44, 1603–1607. [Google Scholar]; c) Zhu C, Zhang Y, Zhao H, Huang S, Zhang M, Su W, Adv. Synth. Catal 2015, 357, 331–338. [Google Scholar]; d) Yu H, Xu Y, Fang Y, Dong R, Eur. J. Org. Chem 2016, 5257–5262.; c) Kotagiri R, Adepu R, Eur. J. Org. Chem 2018, 4556–4564.
- [7].a) Dick AR, Hull KL, Sandford MS, J. Am. Chem. Soc 2004, 126, 2300–2301. [DOI] [PubMed] [Google Scholar]; b) Zhang SY, Gang H, Zhao Y, Wright K, Nack WA, Chen G, J. Am. Chem. Soc 2012, 134, 7313–7316. [DOI] [PubMed] [Google Scholar]; c) Chen F-J, Zhao S, Hu F, Chen K, Zhang Q, Zhang S-Q, Shi B-F, Chem. Sci 2013, 4, 4187–4192. [Google Scholar]; d) Shan G, Yang X, Zong Y, Rao Y , Angew. Chemie Int. Ed 2013, 52, 13606–13610; Angew. Chem. 2013, 125, 13851–13855. [DOI] [PubMed] [Google Scholar]; e) Nelson TAF, Blakey SB, Angew. Chem. Int. Ed 2018, 57, 14911–14915; Angew. Chem. 2018, 130, 15127–15131. [DOI] [PubMed] [Google Scholar]; f) Hu H, Chen S-J, Krska S, Stahl S ChemRxiv 2019, 10.26434/chemrxiv.8159645.v1. [DOI]
- [8].Stahl has articulated the characteristics of what might be considered a cross-coupling reaction to afford C–O bonds. See ref. 7f.
- [9].a) For recent reviews of photoredox catalysis, see: Narayanam JMR, Stephenson CRJ, Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; b) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DWC, Nature Rev. Chem 2017, 1, 0052. [Google Scholar]; d) Zou YQ, Hörmann FM, T. Bach T. Chem. Soc. Rev 2018, 47, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Silvi M, Melchiorre P, Nature 2018, 554, 41–49. [DOI] [PubMed] [Google Scholar]
- [10].Reed NL, Herman MI, Miltchev VP, Yoon TP, Org. Lett 2018, 20, 7345–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For a related photoredox strategy to accomplish decarboxylative elimination, see: Cartwright KC, Lang SB, Tunge JA, J. Org. Chem 2019, 84, 2933–2940. [DOI] [PubMed] [Google Scholar]
- [12].a) Kochi JK, Bemis A, J. Am. Chem. Soc 1968, 90, 4038–4051. [Google Scholar]; b) Kochi JK, Bemis A, Jenkins CL, J. Am. Chem. Soc 1968, 90, 4616–4625. [Google Scholar]; c) Jenkins CL, Kochi JK, J. Am. Chem. Soc 1972, 94, 856–865. [Google Scholar]
- [13].a) de P. Nicholas AM, Arnold DR, Can. J. Chem 1982, 60, 2165–2179. [Google Scholar]; b) Bordwell FG, Cheng J-P, J. Am. Chem. Soc 1989, 111 1792–1795. [Google Scholar]; c) Wayner DDM, Parker VD, Acc. Chem. Res 1993, 26, 287–294. [Google Scholar]
- [14].G. Pandey, S. Pal, R. Laha, Angew Pandey G, Pal S, Laha R Angew. Chem. Int. Ed 2013, 52, 5146–5149; Angew. Chem. 2013, 125, 5250–5253. [DOI] [PubMed] [Google Scholar]
- [15].Im H, Kang D, Choi S, Shin S, Hong S, S. Org. Lett 2018, 20, 7437–7441. [DOI] [PubMed] [Google Scholar]
- [16].Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR, Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Karpova EV, Boltalin AI, Zakharov MA, Sorokina NI, Korenev YM, Troyanov SI, Anorg. Allg. Chem 1998, 624, 741–744. [Google Scholar]
- [18].Hanss D, Freys JC, Bernardinelli GR, Wenger OS, Eur. J. Inorg. Chem 2009, 4850–4859.
- [19].Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.