

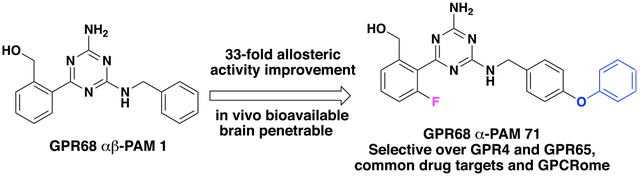
Abstract
G protein-coupled receptor 68 (GPR68) is an understudied orphan G protein-coupled receptor (GPCR). It is expressed most abundantly in the brain, potentially playing important roles in learning and memory. Pharmacological studies with GPR68 have been hindered by lack of chemical tools that can selectively modulate its activity. We previously reported the first small-molecule positive allosteric modulator (PAM), ogerin (1), and showed that 1 can potentiate proton activity at the GPR6n-Gs pathway. Here, we report the first comprehensive structure-activity relationship (SAR) study on the scaffold of 1. Our lead compound resulted from this study, MS48107 (71), displayed 33-fold increased allosteric activity compared to 1. Compound 71 demonstrated high selectivity over closely related proton GPCRs and 48 common drug targets, and was bioavailable and brain-penetrant in mice. Thus, our SAR study has resulted in an improved GPR68 PAM for investigating the physiological and pathophysiological roles of GPR68 in vitro and in vivo.
Graphical Abstract
INTRODUCTION
G protein-coupled receptor 68 (GPR68) was originally discovered as a clinically related receptor in an ovarian cancer cell line as “OGR1” for ovarian cancer G protein-coupled receptor.1 While it is widely expressed in most cells and tissues, including spleen, testes, small intestine, peripheral blood leukocytes, heart, lung, placenta, kidney, and bone,1,2 it is most abundantly expressed in the brain, particularly in the hippocampus3—the brain region that is critical for learning and memory. Despite being known for over two decades, GPR68 remains an understudied orphan G protein-coupled receptor (GPCR) according to IUPHAR.4 Generally known as a proton-sensing GPCR, it couples to multiple signaling pathways through various G proteins (Gs, Gq, Gi, and G12/13) in different cells.2,5–7 However, it remains to be established whether GPR68 couples to the β-arrestin signaling pathway. Consistent with its capacity to couple to multiple signaling pathways and its wide expression pattern, GPR68 has been implicated in many biological processes, including pH homeostasis,2,8 tumor growth and metastasis,6,7,9–11 inflammation,12–14 osteoclast development,9,15,16 insulin secretion,17,18 and epithelial barrier function.19,20 Interestingly, the receptor also functions as a mechanotransducer that senses blood flow (shear stress) in arteries, thus modulating vascular physiology.21 Thus, GPR68, a potentially important therapeutic target, is one of the National Institutes of Health (NIH)’s Illuminating the Druggable Genome GPCRs.22 Presently, the underlying molecular mechanisms have not been determined between proton-sensing and these physiological or pathological processes. Pharmacological studies of GPR68 have previously been hindered by the lack of small-molecule ligands. It is clear that potent and selective GPR68 ligands that can be used to activate or block its activity are urgently needed to elucidate GPR68 pharmacology and clarify its potential roles in the aforementioned biological processes.23
Previous studies have shown that receptor activity is reduced or completely lost if extracellular histidine residues are mutated, presumably by affecting protonation states responding to extracellular pH changes.2 Therefore, it was proposed that alkaline pH conditions stabilize GPR68 in an inactive conformation by hydrogen-bonding interactions involving extracellular histidine residues, while acidification and histidine protonation break hydrogen-bond interactions, initiating receptor activation.2 The putative histidine-binding sites for protons are scattered throughout different extracellular loops rendering challenges to directly activate or block this receptor with orthosteric ligands. This, in turn, makes allosteric binding site more attractive to target as with many other GPCRs.24,25 Allosteric modulators rely on endogenous protons to produce function and can preserve the spatial and temporal nature of the proton-mediated activation of GPR68. Allosteric modulators bind to GPR68 at a site different from that of protons and function by fine-tuning proton activity through increasing or reducing proton affinity (α-cooperativity), efficacy (β-cooperativity), or both.
For conventional GPCRs, such as metabotropic glutamate receptors (mGluRs), binding and functional assays can be carried out in biological systems that the concentration of the orthosteric ligand (i.e., glutamate) can be controlled. However, for GPR68, the orthosteric ligand is proton, which always presents in the receptor compartment. Therefore, the allosteric binding affinity (KB), defined as the dissociation equilibrium constant of allosteric modulator in the absence of orthosteric ligand (i.e., proton), does not have corresponding biological meaning and cannot be measured in binding assays. Instead, we can estimate the allosteric affinity cooperativity α, efficacy cooperativity β, and allosteric binding affinity KB from functional assays according to the standard allosteric operational model.26–28 The observed binding affinity of allosteric modulators can be calculated by the equation KB = (1 + [A]/KA)/(1 + α[A]/KA), where [A] is the proton concentration and KA is the equilibrium dissociation constant of protons at GPR68 in the absence of modulators. In structure-activity relationship (SAR) studies, we actually can optimize the potency of the modulators in proton-mediated signaling by optimizing α, β, and/or KB. In contrast to traditional orthosteric agonists or antagonists which rely on binding affinity (Ki) for selectivity, allosteric modulators achieve high selectivity through allosteric αβ cooperativity in addition to allosteric binding affinity (KB).29,30 The overall allosteric activity can be quantified with a characteristic allosteric index, Log(αβ/KB),31 which contains information about allosteric cooperativity and can be calculated and used as a guiding parameter for SAR studies and optimization.29,32
Development of small-molecule ligands for GPR68 remains a challenging task. A small number of isoxazole compounds have been reported to possess GPR68 agonist activity.33 However, several reports suggested that the weak activity of isoxazoles could be nonspecific.12,34 Divalent metal ions, such as Cu2+ and Zn2+, were initially used to inhibit GPR68 activity.2,19 Recently, these divalent ions, in addition to Co2+ and Mn2+, have been reported to be GPR68 agonists.35–37 However, these recent reports neither achieved full pH control nor used a broad enough pH range to address potential allosteric mechanisms. In addition, Zn2+ is a well-known allosteric modulator for the orphan receptor GPR39.38–40 Moreover, these divalent ions are biologically important metals or trace elements and play critical roles in many biological processes.41–43 Therefore, these divalent metals are unlikely GPR68 selective tools.
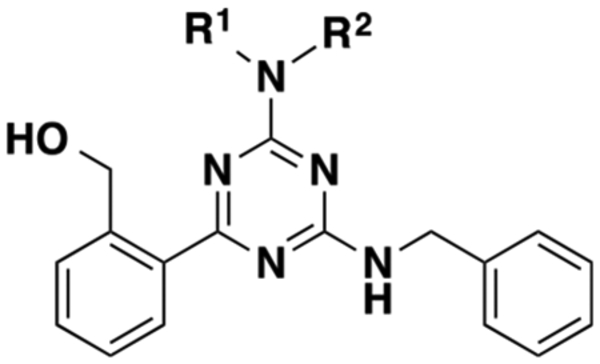
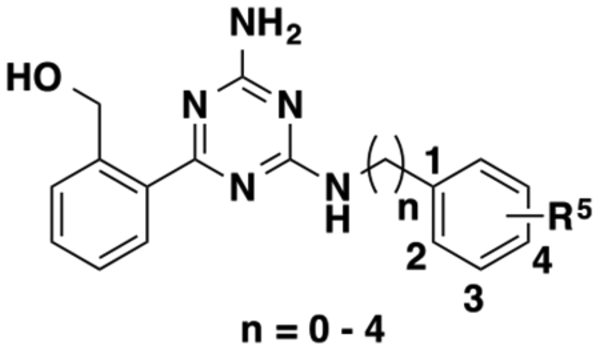
In 2015, we reported the discovery of ogerin (1) as the first positive allosteric modulator (PAM) of GPR68.44 Compound 1 potentiates proton activity in GPR68-mediated Gs−cAMP production, but has no activity at related proton-sensing GPCRs, including GPR4 and GPR65. This selective ligand led to the discovery that GPR68 activation suppresses contextual-but not cue-dependent learning and memory, and does so in wild-type but not GPR68 knockout mice.44 These data provide the first evidence for a role of GPR68 in cognition. Specifically, sustained hippocampal GPR68 activation may contribute to memory deficits. Compound 1, which is now commercially available, has been used by the research community, and its PAM activity has been confirmed by other groups.21,45 Compound 1 was identified from a ZINC sublibrary of about 3.1 million compounds through a virtual screening campaign targeting GPR68.44,46 Thorough SAR studies on the scaffold of compound 1 have not been reported to date. Here, we report a comprehensive SAR study at the following three regions of compound 1: (1) the aminotriazine core, (2) the benzyl alcohol moiety, and (3) the benzylamine moiety (Figure 1). We describe design, synthesis, and biological characterization of various analogues. This study has resulted in the identification of significantly improved GPR68 PAMs.
Figure 1.

Chemical structure of 1 and three highlighted regions for SAR study.
RESULTS
Biological Assay and Data Analysis.
We used a GloSensor cAMP assay to characterize all of the synthesized putative GPR68 PAMs. More specifically, HEK293T cells were transiently transfected with receptor and GloSensor cAMP plasmids for proton-mediated receptor activation of Gs−cAMP production. To determine allosteric modulator activity, proton concentration responses in the absence and presence of increasing concentrations of putative PAMs were quantified and results were analyzed using a standard allosteric operational model26–28 in GraphPad Prism. Several previously reported compounds44 (1, 20, 30, 55, 61, 62, and 63) were included in this study for comparison.
Modifications of the Aminotriazine Core.
To investigate the SAR of the aminotriazine core, we replaced the free amino group on the triazine core with a few small alkylamino groups. We also switched the triazine core to a pyrimidine core (Scheme 1 and Table 1). These designed compounds were prepared using the synthetic routes outlined in Scheme 1. Intermediates 4 and 9 were prepared according to the previously reported procedures44 from 2,4,6-trichloro-1,3,5-triazine (2) and 2,6-dichlor-pyrimidin-4-amine (8). Briefly, alkylamine substitution of one chloro group on trichlorotriazine 1 at low temperature provided alkylamino-substituted dichlorotriazines 3. Benzylamine substitution of one chloro group on dichlorotriazines 3 or dicholoropyrimidine 8 at an elevated temperature yielded intermediates 4 or 9, respectively. Suzuki coupling between intermediates 4 or 9 and commercially available (2-(hydroxymethyl)phenyl)boronic acid provided the designed compounds 5–7 and 10 (Scheme 1).44
Scheme 1. Synthesis of 5–7 and 10a.

aReagents and conditions: (a) tetrahydrofuran (THF), 0 °C to room temperature (rt), 48–67%; (b) dioxane, reflux, 1 h, 56–96%; (c) Pd(PPh3)4, K2CO3, dioxane/H2O = 5:3, 120 °C, microwave, 20 min, 42–98%.
Table 1.
Effects of Modifications to the Triazine Core on Allosteric Activityb
 |
|||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | Log (α) | Log (β) | −Log (KB) | Log (αβ/KB) | Δ |
| 1 | H | H | 0.60 ± 0.10 | 0.36 ± 0.07 | 4.35 ± 0.12 | 5.31 ± 0.05 | 0.00 |
| 5 | H | Me | 0.48 ± 0.10 | 0.37 ± 0.16 | 4.51 ± 0.07 | 5.36 ± 0.06 | 0.05 |
| 6 | H | Et | 0.47 ± 0.12 | 0.25 ± 0.13 | 4.63 ± 0.17 | 5.34 ± 0.09 | 0.03 |
| 7 | Me | Me | Inactive | ||||
| 10a | 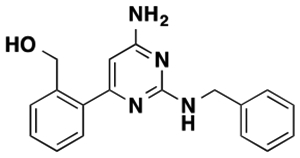 |
Weak PAM | |||||
The structure of 10 is shown.
Values represent means ± standard error of the mean (SEM) from a minimum of three independent assays, each in triplicate or quadruplicate. Δ stands for the difference of Log(αβ/KB) from that of 1, positive values indicated increased allosteric activity and negative values indicated reduced allosteric activity. Proton concentration-response curves are shown in Supporting Figure 1.
As indicated in Table 1, a small monosubstituted amino group on the triazine core is well tolerated. Either methylamino (5) or ethylamino (6) group has a minimal effect on allosteric parameters or binding affinity. However, dimethylamino group (7) completely eliminates the allosteric activity. Replacing the triazine core with a pyrimidine (10) also greatly reduces allosteric activity to barely detectable levels. These results suggest that the amino-substituted triazine core is critical for the PAM activity. Therefore, the aminotriazine core is maintained in the following SAR studies.
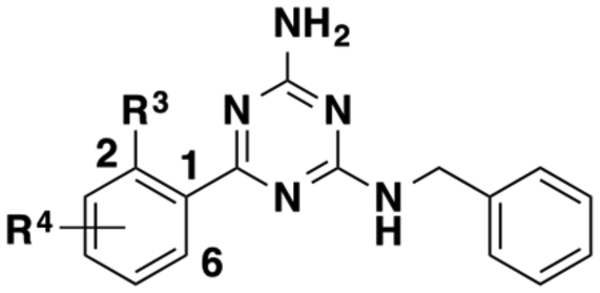
Modifications of the Benzyl Alcohol Moiety.
For the benzyl alcohol moiety, we explored a variety of substituents on the phenyl ring (Scheme 2 and Table 2). The synthesis of these analogues started with 2-amino-4,6-dichloro-1,3,5-triazine (11). Benzylamine substitution of one chloro group on 11 provided the common intermediate 12. Suzuki coupling between intermediate 12 and a set of commercially available phenylboronic acids provided compounds 13–15, 21, 22–29, 31, and 32 (Scheme 2A,D). Compound 16 was obtained by reducing acetophenone 15 (Scheme 2B). Compounds 18 and 19 were synthesized through reductive aminations of benzaldehyde 17 with different amines (Scheme 2C). Compounds 37 and 38 were prepared through Suzuki coupling between intermediate 12 and synthesized phenylboronic acids 36 bearing ethoxy and isopropoxy groups, respectively, followed by formal group reduction (Scheme 2E).
Scheme 2. Synthesis of 13–15, 21, 16, 18, 19, 22–29, 31, 32, 37, and 38a.

aReagents and conditions: (a) dioxane, reflux, 1 h, 78–94%; (b) Pd(PPh3)4, K2CO3, dioxane/H2O = 5:3, 120 °C, microwave, 20 min, 56–99%; (c) NaBH4, MeOH, 0 °C to rt, 42%; (d) NH3·H2O or NH2Me, MgSO4, NaBH4, THF, rt, 2 days, 45–67%. (e) Ethylene glycol, toluene, reflux, 4–8 h, 82–96%; (f) alkyl bromide, K2CO3, dimethylformamide, 85 °C, 34–78%; (g) i. n-BuLi, THF, −78 °C, 30 min; ii. B(OMe)3, −78 °C, 1 h, then rt, overnight; iii. HCl (3 N), rt, 1 h, 26–57%; (h) 12, Pd(PPh3)4, K2CO3, dioxane/H2O = 5:3, 120 °C, microwave, 20 min; (i) NaBH4, THF, 0 °C to rt, 23–97%.
Table 2.
Effects of Modifications of the Benzylmethanol Moiety on Allosteric Activitya
| compd | |||||||
|---|---|---|---|---|---|---|---|
| 1 | CH2OH | H | 0.60 ± 0.10 | 0.36 ± 0.07 | 4.35 ± 0.12 | 5.31 ± 0.05 | 0.00 |
| 13 | H | Inactive | |||||
| 14 | Me | Weak PAM | |||||
| 15 | COMe | Weak PAM | |||||
| 16 | CHOHMe | Weak PAM | |||||
| 18 | CH2NH2 | 0.86 ± 0.06 | 0.30 ± 0.14 | 3.86 ± 0.14 | 5.01 ± 0.06 | −0.30 | |
| 19 | CH2NHMe | Weak PAM | |||||
| 20 | H | 3-CH2OH | Inactive | ||||
| 21 | 3-COMe | Inactive | |||||
| 22 | CH2OH | 3-F | 0.59 ± 0.30 | 0.27 ± 0.13 | 4.01 ± 0.13 | 4.86 ± 0.06 | −0.45 |
| 23 | 4-F | 0.64 ± 0.13 | 0.24 ± 0.13 | 4.58 ± 0.06 | 5.46 ± 0.07 | 0.15 | |
| 24 | 5-F | 0.91 ± 0.07 | 0.20 ± 0.11 | 4.58 ± 0.08 | 5.69 ± 0.04 | 0.38 | |
| 25 | 6-F | 1.15 ± 0.14 | 0.35 ± 0.16 | 3.69 ± 0.12 | 5.19 ± 0.03 | −0.12 | |
| 26 | 4-CI | 1.01 ± 0.08 | 0.11 ± 0.04 | 4.76 ± 0.14 | 5.87 ± 0.14 | 0.56 | |
| 27 | 5-C1 | 1.20 ± 0.11 | 0.09 ± 0.02 | 4.49 ± 0.19 | 5.78 ± 0.09 | 0.47 | |
| 28 | 6-C1 | 1.25 ± 0.19 | −0.12 ± 0.03 | 4.43 ± 0.20 | 5.56 ± 0.04 | 0.25 | |
| 29 | 3-OMe | 0.40 ± 0.12 | 0.30 ± 0.06 | 4.16 ± 0.16 | 4.86 ± 0.10 | −0.45 | |
| 3044 | 4-OMe | 0.46 ± 0.03 | 0.14 ± 0.03 | 4.12 ± 0.11 | 4.97 ± 0.11 | −0.34 | |
| 31 | 5-OMe | 0.61 ± 0.03 | 0.18 ± 0.06 | 3.99 ± 0.07 | 4.77 ± 0.04 | −0.54 | |
| 32 | 6-OMe | Weak PAM | |||||
| 37 | 4-OEt | 0.87 ± 0.11 | 0.10 ± 0.10 | 4.37 ± 0.04 | 5.34 ± 0.03 | 0.03 | |
| 38 | 4–0 Pr | 0.56 ± 0.09 | 0.27 ± 0.07 | 4.91 ± 0.12 | 5.74 ± 0.07 | 0.43 | |
Values represent means ± SEM from a minimum of three independent assays, each in triplicate or quadruplicate. Δ stands for the difference of log(αβ/KB) from that of 1; positive values indicated increased allosteric activity, and negative values indicated reduced allosteric activity. Proton concentration-response curves are shown in Supporting Figure 1.
Switching the hydroxymethyl group (1) to hydrogen (13), methyl (14), methyl ketone (15), and 1-hydroxylethyl (16) groups is not tolerated and leads to either weak or inactive analogues. Replacing the hydroxymethyl group with an aminomethyl group (18) retains affinity cooperativity and efficacy cooperativity but with reduced allosteric binding affinity, rendering a less effective GPR68 PAM compared to 1. The N-methyl-substituted aminomethyl (19) group further reduces allosteric activities and results in a weak PAM. Previously, we have demonstrated the importance of the position of the hydroxymethyl group: the ortho-hydroxymethyl isomer was much more effective than the meta-hydroxymethyl isomer to activate GPR68 Gs−cAMP pathway.44 We have confirmed this trend by retesting 20 and by comparing the activities of 1 and 20. This trend has also been confirmed by comparing the activities of 15 and 21 (two methyl ketone analogues). Based on these SAR results, we kept the orthopositioned hydroxymethyl group in subsequent SAR studies and explored additional substituents, such as halides and alkoxy groups on the phenyl ring. First, we installed fluoro groups at 3- (22), 4- (23), 5- (24), and 6- (25) positions on the phenyl ring. Compared to 1, the 3-fluoro analogue 22 is a less effective GPR68 PAM due to the decreased efficacy cooperativity and allosteric binding affinity. The 4-fluoro analogue 23 shows slightly increased PAM activity compared to compound 1. Interestingly, the 6-fluoro analogue 25 has significantly improved affinity cooperativity (log(α) = 1.15 ± 0.14). However, its binding affinity (−log(KB) = 3.69 ± 0.12) is greatly reduced, resulting in an overall slightly worse PAM (log(αβ/KB) = 5.19 ± 0.03) than compound 1 (log(αβ/KB) = 5.31 ± 0.05). The 5-fluoro analogue 24 is a slightly better PAM (log(αβ/KB) = 5.69 ± 0.04) than 1 due to its slightly improved affinity cooperativity (log(α) = 0.91 ± 0.07) and allosteric binding affinity (−log(KB) = 4.58 ± 0.08). Next, we explored compounds with a chloro group at 4- (26), 5- (27), and 6- (28) positions on the phenyl ring. Similar to 25, all of these chloro analogues show improved affinity cooperativities (log(α) = 1.01 ± 0.08 (26); 1.20 ± 0.11 (27); and 1.25 ± 0.19 (28)). In addition, these compounds display improved allosteric binding affinities (−log(KB) = 4.76 ± 0.14 (26); 4.49 ± 0.19 (27); and 4.43 ± 0.20 (28)) albeit reduced efficacy cooperativities, resulting in slightly better PAMs (2- to 4-fold) than 1. We also explored analogues bearing a methoxy group at different substitution positions (29–32). A methoxy group in general leads to reduced allosteric index compared to 1. Among the four substitution positions, the 4-position (30) is more tolerated while the 6-position (32) shows least PAM activity. Consequently, we installed ethoxy (37) and isopropoxy (38) groups at the 4-position to explore steric effects. The 4-ethoxy group (37) slightly improves affinity cooperativity (log(α) = 0.87 ± 0.11), maintains allosteric binding affinities, and slightly decreases efficacy cooperativity, resulting in a comparable allosteric index to 1. The 4-isopropoxy group (38) shows slightly improved allosteric activity (log(αβ/KB) = 5.74 ± 0.07) compared to 1 due to the increased allosteric binding affinities (−log(KB) = 4.91 ± 0.12). These SAR results indicate that additional substituents on the benzyl alcohol moiety are tolerated, and appropriate substitutions on the phenyl ring could lead to enhanced allosteric activity. Several substituents were identified to improve the modulators’ affinity cooperativities and/or allosteric binding affinities. However, none of the substituents increased the modulators’ efficacy cooperativities.
Modifications of the Benzylamine Moiety.
To evaluate the effects of the benzylamine moiety on allosteric activity, we designed a variety of compounds with differentially substituted benzylamines. In addition, we also explored different carbon linker length between the phenyl and amino groups (Table 3). These compounds were prepared using nucleophilic aromatic substitution and Suzuki coupling reactions described in Scheme 3.
Table 3.
Effects of Modifications of the Benzylamine Side Chain on Allosteric Activitya
| compd | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 1 | H | 0.60 ± 0.10 | 0.36 ± 0.07 | 4.35 ± 0.12 | 5.31 ± 0.05 | 0.00 |
| 41 | 2-F | 0.98 ± 0.15 | 0.31 ± 0.10 | 4.27 ± 0.10 | 5.56 ± 0.07 | 0.25 | |
| 42 | 3-F | 0.70 ± 0.13 | 0.33 ± 0.10 | 4.52 ± 0.10 | 5.55 ± 0.05 | 0.24 | |
| 43 | 4-F | 1.09 ± 0.30 | 0.36 ± 0.02 | 3.97 ± 0.33 | 5.42 ± 0.03 | 0.11 | |
| 44 | 2-C1 | 0.64 ± 0.05 | 0.23 ± 0.04 | 5.07 ± 0.08 | 5.94 ± 0.07 | 0.63 | |
| 45 | 3-C1 | 0.48 ± 0.15 | 0.46 ± 0.06 | 4.97 ± 0.19 | 5.90 ± 0.05 | 0.59 | |
| 46 | 4-C1 | 0.49 ± 0.09 | 0.41 ± 0.07 | 5.22 ± 0.07 | 6.11 ± 0.02 | 0.80 | |
| 47 | 2-CFj | 0.60 ± 0.06 | 0.31 ± 0.05 | 5.05 ± 0.10 | 5.96 ± 0.05 | 0.65 | |
| 48 | 3-CF3 | 0.64 ± 0.19 | 0.32 ± 0.13 | 5.11 ± 0.12 | 6.07 ± 0.04 | 0.76 | |
| 49 | 4-CF3 | 0.78 ± 0.05 | 0.30 ± 0.06 | 5.03 ± 0.08 | 6.10 ± 0.06 | 0.79 | |
| 50 | 2-Me | 0.54 ± 0.15 | 0.33 ± 0.14 | 4.70 ± 0.08 | 5.57 ± 0.08 | 0.26 | |
| 51 | 3-Me | 0.92 ± 0.16 | 0.25 ± 0.09 | 4.40 ± 0.18 | 5.57 ± 0.08 | 0.26 | |
| 52 | 4-Me | 0.90 ± 0.06 | 0.32 ± 0.05 | 4.38 ± 0.17 | 5.60 ± 0.09 | 0.29 | |
| 53 | 2-OMe | 0.98 ± 0.17 | 0.06 ± 0.01 | 3.99 ± 0.20 | 5.03 ± 0.04 | −0.28 | |
| 54 | 3-OMe | 0.89 ± 0.18 | 0.23 ± 0.06 | 4.11 ± 0.18 | 5.22 ± 0.05 | −0.09 | |
| 55” | 4-OMe | 0.78 ± 0.11 | 0.49 ± 0.10 | 3.34 ± 0.13 | 4.61 ± 0.09 | −0.70 | |
| 56 | 4-OEt | 0.81 ± 0.05 | −0.01 ± 0.05 | 4.77 ± 0.03 | 5.57 ± 0.04 | 0.26 | |
| 57 | 4-O’Pr | 0.53 ± 0.09 | 0.17 ± 0.12 | 4.99 ± 0.09 | 5.69 ± 0.03 | 0.38 | |
| 58 | 4-Ocyclopropyl | 0.67 ± 0.07 | 0.04 ± 0.05 | 5.07 ± 0.12 | 5.77 ± 0.04 | 0.46 | |
| 59 | 4-OPh | 0.95 ± 0.09 | 0.00 ± 0.04 | 5.19 ± 0.14 | 6.14 ± 0.10 | 0.83 | |
| 60 | 0 | H | 0.72 ± 0.14 | 0.40 ± 0.09 | 3.92 ± 0.10 | 5.04 ± 0.06 | −0.27 |
| 6144 | 2 | 0.88 ± 0.15 | 0.24 ± 0.10 | 4.50 ± 0.21 | 5.62 ± 0.07 | 0.31 | |
| 6244 | 3 | 0.81 ± 0.05 | 0.08 ± 0.02 | 5.22 ± 0.04 | 6.11 ± 0.01 | 0.80 | |
| 6344 | 4 | 0.62 ± 0.05 | 0.21 ± 0.05 | 5.52 ± 0.12 | 6.35 ± 0.08 | 1.04 | |
| 64 | 2 | 4-OPh | 0.83 ± 0.14 | 0.03 ± 0.06 | 5.00 ± 0.11 | 5.90 ± 0.05 | 0.59 |
| 65 | 3 | 1.19 ± 0.12 | −0.01 ± 0.07 | 4.67 ± 0.10 | 5.84 ± 0.09 | 0.53 | |
| 66 | 4 | 1.03 ± 0.10 | 0.09 ± 0.06 | 4.81 ± 0.13 | 5.94 ± 0.11 | 0.62 |
Values represent means ± SEM from a minimum of three independent assays, each in triplicate or quadruplicate. Δ stands for the difference of log(αβ/KB) from that of 1; positive values indicate increased allosteric activity, and negative values indicate reduced allosteric activity. Proton concentration-response curves are shown in Supporting Figure 1.
Scheme 3. Synthesis of 41–54, 56–60, and 64–81a.

aReagents and conditions: (a) dioxane, reflux, 1 h, 75–96 %; (b) Pd(PPh3)4, K2CO3, dioxane/H2O = 5:3, 120 °C, microwave, 20 min, 16–99%.
Initially, a series of electron-withdrawing groups (EWGs) such as fluoro (41–43), chloro (44–46), and trifluoromethyl (47–49) were introduced to the phenyl moiety to explore their effects at different substitution positions. 2-, 3-, and 4-Fluoro groups lead to slightly improved allosteric indexes mainly due to the increased affinity cooperativity with log(α) values at 0.98 ± 0.15, 0.70 ± 0.13, and 1.09 ± 0.30, respectively. Compared to the fluoro substitutions, the chloro substitutions provide more significant allosteric activity improvement because of the higher allosteric binding affinities (−log(KB) = 5.07 ± 0.08 (44), 4.97 ± 0.19 (45), and 5.22 ± 0.07 (46)). Similarly, the trifluoromethyl analogues show improved allosteric index due to improved allosteric binding affinities (−log(KB) = 5.05 ± 0.10 (47), 5.11 ± 0.12 (48), and 5.03 ± 0.08 (49)). Thus, EWGs are able to improve the modulators’ allosteric activities by increased affinity cooperativities or allosteric binding affinities. However, the substitution positions only have moderate effects on the allosteric indexes. Next, we explored a couple of electron-donating groups: methyl (50–52) and methoxy (52–55). The methyl analogues show slightly improved allosteric indexes mainly due to slightly higher allosteric binding affinity (50) or affinity cooperativity (51 and 52). Although the 2-, 3-, and 4-methoxy groups improve the affinity cooperativities (log(α) = 0.98 ± 0.17 (53), 0.89 ± 0.18 (54), and 0.78 ± 0.11 (55)), they reduce the allosteric binding affinities, resulting in analogues with reduced allosteric indexes. We subsequently explored a few larger alkoxy groups, such as ethoxy (56), isopropoxy (57), cyclopropoxy (58), and phenoxy (59), at the 4-position. Interestingly, most of these larger ether substituents diminish the efficacy cooperativities, leading to GPR68 α-PAMs, positive allosteric modulators with affinity cooperativities (α) greater than 1 and efficacy cooperativities (β) equal or close to 1. Compared to 4-methoxy group (55), these larger alkoxy groups enhance allosteric binding affinities (−Log(KB) = 4.77 ± 0.03 (56), 4.99 ± 0.09 (57), 5.07 ± 0.12 (58), and 5.19 ± 0.14 (59)), leading to improved allosteric activities. In particular, the 4-phenoxy analogue (59) is an α-PAM with the most improved allosteric activity compared to compound 1. Previously, we have shown that longer carbon linkers between the phenyl and amino groups resulted in analogues with higher PAM activity.44 We have now confirmed this SAR trend by testing compounds 60–63. While a shorter linker (60) results in reduced allosteric index mainly due to the reduced allosteric binding affinity (−log(KB) = 3.92 ± 0.10 (60)), longer linkers (61–63) enhance allosteric binding affinities (−log(KB) = 4.50 ± 0.21 (61), 5.22 ± 0.04 (62), and 5.52 ± 0.12 (63)), leading to improved allosteric indexes. We also extended the carbon linker length of the 4-phenoxy-substituted benzylamine analogue 59, leading to three analogues 64–66. These analogues, however, do not show enhanced allosteric activities compared to 59. Instead, longer linkers lead to reduced allosteric activities. Interestingly, these analogues all retain high affinity cooperativity α values. Thus, extending the carbon linker length between the phenyl group and amino group does not always lead to better PAMs. Importantly, these SAR results suggest that similar to the benzyl alcohol moiety, modifications of the benzylamine moiety could improve the modulators’ affinity cooperativities and/or allosteric binding affinities, but not their efficacy cooperativities.
Combined Modifications of Different Moieties.
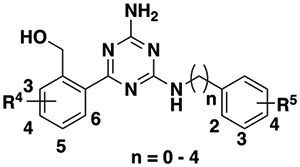
The SAR study described above indicates that several functional groups, such as 6-fluoro (25)- and 5-chloro (27)-substituted hydroxymethylphenyl moieties and 2-fluoro (41)-, 4-fluoro (43)-, 3-methyl (51)-, 2-methoxy (53)-, and 4-phenoxy (59)-substituted benzylamine, lead to high affinity cooperativities and/or improved allosteric binding affinities. Using these preferred moieties, we designed a number of combination compounds, to further improve the allosteric activity and obtain potent GPR68 α-PAMs. We also explored different linker lengths between the phenyl group and amino group (n = 1–4). Synthesis of these compounds is summarized in Scheme 3, and SAR results of these compounds are summarized in Table 4.
Table 4.
Effects of Combined Modifications on Allosteric Activitya
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | n | R4 | R5 | Log(α) | Log(β) | −Log(KB) | Log(αβ/KB) | Δ |
| 1 | H | H | 0.60 ± 0.10 | 0.36 ± 0.07 | 4.35 ± 0.12 | 5.31 ± 0.05 | 0.00 | |
| 67 | 2-F | 1.09 ± 0.08 | 0.09 ± 0.04 | 4.22 ± 0.10 | 5.40 ± 0.03 | 0.09 | ||
| 68 | 1 | 4-F | 1.01 ± 0.06 | 0.09 ± 0.03 | 4.45 ± 0.14 | 5.55 ± 0.04 | 0.24 | |
| 69 | 3-Me | 0.84 ± 0.08 | 0.14 ± 0.04 | 4.44 ± 0.12 | 5.42 ± 0.04 | 0.11 | ||
| 70 | 2-OMe | 1.12 ± 0.08 | −0.12 ± 0.03 | 4.11 ± 0.04 | 5.11 ± 0.02 | −0.19 | ||
| 71 | 6-F | 4-OPh | 0.90 ± 0.06 | 0.06 ± 0.04 | 5.85 ± 0.12 | 6.83 ± 0.08 | 1.52 | |
| 72 | 2 | 1.06 ± 0.15 | 0.03 ± 0.07 | 5.04 ± 0.09 | 6.13 ± 0.04 | 0.82 | ||
| 73 | 3 | H | 1.19 ± 0.10 | 0.02 ± 0.03 | 4.47 ± 0.04 | 5.67 ± 0.10 | 0.36 | |
| 74 | 4 | 1.24 ± 0.11 | 0.06 ± 0.04 | 4.04 ± 0.11 | 5.34 ± 0.04 | 0.03 | ||
| 75 | 2 | 4-OPh | 1.14 ± 0.09 | −0.06 ± 0.03 | 5.05 ± 0.14 | 6.14 ± 0.10 | 0.82 | |
| 76 | 3 | 1.15 ± 0.11 | 0.01 ± 0.05 | 4.94 ± 0.11 | 6.10 ± 0.11 | 0.81 | ||
| 77 | 2 | 1.09 ± 0.10 | 0.03 ± 0.05 | 5.14 ± 0.11 | 6.26 ± 0.09 | 0.95 | ||
| 78 | 3 | H | 1.03 ± 0.07 | 0.01 ± 0.04 | 5.35 ± 0.06 | 6.38 ± 0.05 | 1.07 | |
| 79 | 4 | 5-CI | 1.09 ± 0.13 | 0.06 ± 0.07 | 4.98 ± 0.10 | 6.14 ± 0.10 | 0.83 | |
| 80 | 1 | 4-OPh | 1.05 ± 0.09 | 0.03 ± 0.03 | 4.70 ± 0.14 | 5.78 ± 0.11 | 0.47 | |
| 81 | 2 | 1.39 ± 0.07 | −0.04 ± 0.08 | 4.59 ± 0.18 | 5.87 ± 0.14 | 0.56 | ||
Values represent means ± SEM from a minimum of three independent assays, each in triplicate or quadruplicate. Δ stands for the difference of Log(αβ/KB) from that of 1; positive values indicate increased allosteric activity, and negative values indicate reduced allosteric activity. Proton concentration-response curves are shown in Supporting Figure 1.
Compared to 25, 2-fluoro (67)-, 4-fluoro (68)-, 3-methyl (69)-, 2-methoxy (70)-, and 4-phenoxy (71)-substituted benzyl groups retain relatively high affinity cooperativity values and show low efficacy cooperativity values. Most strikingly, the 4-phenoxy analogue 71 displays greatly enhanced allosteric binding affinity (−log(KB) = 5.85 ± 0.12 vs 3.69 ± 0.12 for compound 25), resulting in a very potent GPR68 α-PAM. Compared to 1, Compound 71 has 33-fold better allosteric activity (log(αβ/KB) = 6.83 ± 0.08 vs 5.31 ± 0.05 for compound 1). We have shown that the linker length between the phenyl and amino groups has different effects on the allosteric activities, depending on different R5 groups (6–63 and 59, 64 and 65 (see above)). Extending the carbon linker length of 25 to 2–4 results in compounds 72–74. Compared to 25, all of these longer linker analogues maintain high affinity cooperativity values and show low efficacy cooperativity values. For allosteric binding affinity, the 2-carbon linker (72, −Log(KB) = 5.04 ± 0.09) is optimal among the 4 linker lengths explored. Extending the carbon linker length of 71 to 2–3 leads to α-PAMs 75 and 76 with retained high affinity cooperativities, low efficacy cooperativities, but reduced allosteric binding affinities.
To improve the allosteric activity of 27, we also increased the linker length between the phenyl group and the amino group. The longer linker analogues 77–79 maintain high affinity cooperativities and low efficacy cooperativities and enhance allosteric binding affinities (−log(KB) = 5.14 ± 0.11 (77), 5.35 ± 0.06 (78), and 4.98 ± 0.10 (79)) with the 3-carbon linker (78) being optimal. Finally, we replaced the benzylamine moiety of 27 with a 4-phenoxy substituted benzylamine moiety (80) or a 4-phenoxy-substituted homobenzylamine moiety (81). Compounds 80 and 81 preserve the α-PAM activity. Compared to 27, both compounds show similar allosteric binding affinities (−log(KB) = 4.70 ± 0.14 (80) and 4.59 ± 0.18 (81)) and similar overall allosteric activities (i.e., log(αβ/KB) values).
Our SAR study has identified several GPR68 PAMs with better allosteric activities than compound 1. For example, 46, an αβ-PAM (α and β > 1), has shown 6-fold higher allosteric activity (log(αβ/KB) value) than 1. α-PAM 71 has the highest log(αβ/KB) value, which is 33-fold higher than that of 1. Compounds 77 and 78, α-PAMs, have approximately 10-fold better allosteric activity than 1. We selected these four GPR68 PAMs for further characterization.
Selectivity Profiles of Selected GPR68 PAMs.
To determine selectivities of 46, 71, 77, and 78 for GPR68 over other closely related proton GPCRs, such as GPR4 and GPR65, we tested these GPR68 PAMs against GPR4 and GPR65 using GloSensor cAMP assays. As shown in Figure 2A–D, GPR4 and GPR65 have different response profiles to proton stimulation compared to GPR68 in the absence of modulators. The proton potency values (pEC50) were pH 8.04 (Hill slope of 5.25) for GPR4, pH 7.47 (Hill slope of 4.57) for GPR65, and pH 6.73 (Hill slope of 3.38) for GPR68. In the presence of the lead PAM 71 (Figure 2B) as well as 1 (Figure 2A), proton activity at GPR68 is potentiated in a PAM concentration-dependent manner (proton concentration-response curves were shifted leftward (affinity cooperativity α > 1) and upward (efficacy cooperativity β > 1)), while Compound 71 has no PAM activity at GPR4 (Figure 2C) and weak potentiation of proton activity at GPR65 only at high concentrations (Figure 2D). These data indicate that our lead PAM 71 is selective for GPR68 over the functionally related receptors GPR4 and GPR65. Similarly, as illustrated in Supporting Figure 2, compounds 46, 77, and 78 show no effect or weak activity only at high concentrations at GPR4 and GPR65.
Figure 2.

Selectivity of the GPR68 PAM 71. Proton concentration-response curves in the absence and presence of increasing concentrations of 1 at human GPR68 (A), 71 at human GPR68 (B), GPR4 (C), and GPR65 (D). Proton-mediated Gs−cAMP production was measured in HEK293T cells transiently transfected with GPR68, GPR4, or GPR65, and GloSensor cAMP plasmids. Results were normalized to the proton activity in the absence of modulator and represented means ± SEM from a minimum of three independent assays, each in quadruplicate. Curves were analyzed in Prism using the built-in four parameter logistic functions. (E) Selectivity of 71 over 48 common GPCRs, ion channels, and transporters in radioligand binding assays. Values represent average binding affinity (pKi) from a minimum of three independent assays, each in triplicate. Targets with less than 50% inhibition at 10 μM (in quadruplicate) are not shown. BZP for benzodiazepine receptor; DAT for dopamine transporter; DOR for delta (δ) opioid receptor; hERG for human ether-a-go-go-related potassium channel; KOR for kappa (κ) opioid receptor; MOR for mu (μ) opioid receptor; NET for noradrenaline transporter; PBR for peripheral benzodiazepine binding site; SERT for serotonin transporter. (F) Off-target agonist activity of 71 at human GPCRome. 71 was screened across the human GPCRome (318 GPCRs) for off-target agonist activity at 1 μM using the PRESTO-Tango screening platform. Results (fold of basal) represent means of four replicates. Dopamine receptor D2 with 100 nM quinpirole served as an assay control. The dashed lines indicate an arbitrary cutoff line at 3× fold of basal.
Compounds 46, 71, 77, and 78 were further screened in radioligand binding assays against a panel of 48 common drug targets, including GPCRs, neurotransmitter transporters, and hERG ion channels. As indicated in Figure 2E and Supporting Figure 3, most of these compounds have affinity values higher than 300 nM (pKi = 6.50) at all targets tested. Among the tested GPCRs, 5-HT2B has moderate binding affinity (pKi = 6.50–7.00) to 71, 77, and 78, with Ki values of 219, 209, and 295 nM, respectively. In addition, adenosine 2A (A2A) receptor has moderate binding affinity to 46 with Ki value of 300 nM. The compounds with binding affinities between 300 and 100 nM (pKi = 6.50–7.00) were further tested in functional assays to determine if they acted as agonists or antagonists. At 5-HT2B receptors, 71, 77, and 78 show no agonist activity but display weak antagonist activity (Supporting Figure 4A,B) with functional affinity (Ki) values of 310, 678, and 212 nM, respectively, which are comparable to their corresponding binding affinities. At A2A receptors (Supporting Figure 4C), 46 displays no agonist activity, but shows weak inverse agonist activity with potency of 1700 nM in the absence of reference agonist and antagonist activity with functional affinity of 6700 nM.
For lead PAMs with the potential to serve as molecular probes for chemical biology studies, it is also desirable to determine their off-target agonist activities at other human GPCRs. Therefore, 46, 71, 77, and 78 were tested against all human GPCRome in our PRESTO-Tango screening assays.47 As illustrated in Figure 2F, across the whole human GPCRome, Compound 71 has agonist activity only at the MT1 and MT2 receptors. Compound 46 shows no agonist activity, while compounds 77 and 78 display agonist activity at an atypical chemokine receptor 3 (ACKR3) (previously known as CXCR7)48,49 (Supporting Figure 5). Subsequent concentration-response assays indicate that 71 is a weak full agonist at the MT1 receptor (EC50 = 320 nM) and a weak partial agonist activity at the MT2 receptor (EC50 = 540 nM; Emax = 24%) (Supporting Figure 6). Consistently, 71 displays low binding affinities to MT1 (5900 nM) and MT2 (1100 nM) receptors. Compounds 77 and 78 show full agonist activities at ACKR3 with EC50 values of 1700 and 690 nM, respectively (Supporting Figure 6).
Characterization of Selected Lead Modulators at Mouse GPR68.
To assess whether selected human GPR68 PAMs also function as PAMs at mouse GPR68 receptor, we determined the PAM activity at mouse GPR68 in the absence and presence of lead modulators for proton-mediated Gs−cAMP production. As illustrated in Figure 3 and Supporting Figure 7, in the absence of modulators, mouse GPR68 responds to proton stimulation in a very similar way as human GPR68 with a potency of pH 6.68 (Hill slope of 2.95). As with human GPR68, the proton activity at mouse GPR68 is also potentiated by the lead PAMs 71 (Figure 3), 46, 77, and 78 (Supporting Figure 7) in a concentration-dependent manner—shifting proton response curves to the left with increased maxima. Extracted allosteric parameters determined by fitting the curves to the operational allosteric model are listed in Table 5. Compared to the allosteric activity of these PAMs at human GPCR68, the compounds display 2- to 3-fold less allosteric activity at mouse GPR68, suggesting a potential small species difference in sensitivity to GPR68 PAMs.
Figure 3.

Allosteric potentiation of proton activity by Compound 71 at mouse GPR68. Gs GloSensor cAMP assays were carried out in HEK293T cells transiently transfected with mouse GPR68 as with human GPR68. Proton concentration responses were measured in the absence and presence of increasing concentrations of modulator. Results were normalized to proton activity in the absence of modulator and represented means ± SEM from a minimum of three independent assays, each in quadruplicate. Curves were analyzed in Prism. Allosteric parameters are summarized in Table 5.
Table 5.
Allosteric Parameters of Selected PAMs at Mouse GPR68a
| compd | Log(α) | Log(β) | −Log(KB) | Log(αβ/KB) | Δ |
|---|---|---|---|---|---|
| 1 | 0.80 ± 0.11 | 0.04 ± 0.05 | 4.13 ± 0.09 | 4.97 ± 0.02 | −0.34 |
| 46 | 1.41 ± 0.08 | −0.0 2 ± 0.06 | 4.30 ± 0.03 | 5.68 ± 0.05 | −0.43 |
| 63 | 0.91 ± 0.18 | 0.08 ± 0.03 | 4.91 ± 0.17 | 5.89 ± 0.02 | −0.40 |
| 71 | 1.26 ± 0.14 | 0.09 ± 0.03 | 5.12 ± 0.19 | 6.46 ± 0.05 | −0.43 |
| 77 | 1.06 ± 0.12 | 0.13 ± 0.09 | 4.72 ± 0.02 | 5.92 ± 0.02 | −0.34 |
| 78 | 0.99 ± 0.06 | 0.13 ± 0.06 | 4.86 ± 0.08 | 5.98 ± 0.04 | −0.40 |
Values represent means ± SEM from a minimum of three independent assays, each in quadruplicate. Δ stands for the difference of Log(αβ/KB) from the corresponding values at human GPR68 (Tables 1–4); positive values indicate increased allosteric activity, and negative values indicate reduced allosteric activity. Proton concentration-response curves are presented in Figure 3 and Supporting Figure 7.
In Vivo Pharmacokinetic (PK) Properties of Compound 71.
After obtaining the promising in vitro potency and selectivity results for Compound 71, we assessed in vivo pharmacokinetic properties of this lead compound in mice. Since GPR68 is most abundantly expressed in the brain and is associated with learning and memory, it is necessary to test whether this compound can penetrate the blood-brain barrier (BBB) in animals. A single intraperitoneal injection at the dose of 25 mg/kg led to high exposure levels (above 10 μM) in both plasma and brain at 0.5 h in mice (Figure 4). The high compound exposure levels in both plasma and brain were maintained for 2 h. These results have demonstrated that Compound 71 is bioavailable and can readily cross the BBB in mice. Furthermore, there were no clinical signs observed in test mice. Taken together, these results suggest that Compound 71 is a valuable chemical tool for investigating biological function of GPR68 in vivo.
Figure 4.

Mouse pharmacokinetic profile of Compound 71. Compound concentrations were quantified in both plasma (red curve) and brain (blue curve) at 0.5, 1.0, and 2.0 h after a single 25 mg/kg intraperitoneal (IP) administration. Compound concentrations in plasma and brain at each time point are average values from three test mice. The error bars represent ± SEM.
Characterization of Selected Inactive Analogues.
Compounds 7, 20 (a known inactive analogue of 1),44 and 21 do not show PAM activity at GPR68, GPR4, and/or GPR65 (Supporting Figure 8), proton responses are left-shifted by 71 as expected and are minimally affected by the presence of the inactive analogues, suggesting that the inactive analogues do not bind to the allosteric site of GPR68 to antagonize the PAM activity of 71. In addition, inactive analogues 7 and 21 show minimal binding affinities to common drug targets (Supporting Figure 3). Moreover, 7 and 21 display no off-target agonist activity across the human GPCRome (Supporting Figure 5). Therefore, compounds 7, 20, and 21 are potentially useful inactive controls for studying GPR68 pharmacology.
DISCUSSION AND CONCLUSIONS
It is worth noting the unique nature of GPR68 pharmacology as the co-binding ligand for the allosteric effect, namely, hydrogen ion, is always present. Therefore, GPR68 allosteric modulators will likely have direct consequences to cellular signaling: PAMs will produce direct agonism, while negative allosteric modulators will produce inverse agonism. In addition, differences exist for PAMs depending on whether the allosterism primarily affects affinity (α-effect) or efficacy (β-effect). Specifically, α-PAMs (α > 1 and β ≈ 1) will produce greater agonism at neutral pH conditions while β-PAMs (α ≈ 1 and β > 1) will produce relatively more agonism in acidic environments. The differences in signaling produced by α- versus β-modulators are described in detail in the Supporting Information. In the present study, we are able to achieve potent and selective GPR68 α-PAMs (e.g., compounds 71, 77, and 78) and αβ-PAMs (e.g., compound 46), but not β-PAMs.50–53
There are examples that precipitous changes in allosteric activity may come from apparently small changes in molecular structure, known specifically as “activity switching” in the PAMs for mGluRs.54–56 In this SAR study, with compound 1 as a scaffold, we also observed large changes in allosteric activity (from active PAMs to inactive analogues) with small structural modifications to the aminotriazine core and benzyl alcohol group. Further SAR and mutational studies are needed to elucidate underlying molecular mechanisms, which will aid in designing better modulators.
Depending on the pKa values, the physiochemical properties of modulators, such as net charges and H-bonding potential, could change under different pH conditions. Correspondingly, binding interactions of modulators with GPR68 could be pH-dependent, leading to different levels of receptor activation in both pH- and concentration-dependent manner. Therefore, our functional assay is designed to use a wide pH range and is done in the absence and presence of increasing concentrations of modulators to generate at least six sets of 16-point proton concentration-response curves (each in quadruplicate) in a single 384-well plate for proper comparison.
To achieve novel brain-penetrant GPR68 PAMs, we tried to maintain low tPSA values by utilizing lipophilic substituents on the triazine scaffold. While this strategy successfully led to BBB permeable Compound 71, these lipophilic groups could also reduce the aqueous solubility of the compounds. Indeed, we have observed bell-shaped proton concentration-response curves at high compound concentrations (10 and/or 30 μM) for compounds 57–59, 63–66, 75–78, and 81. The assay signals waned when modulators were tested at pH < 7.40. This is likely due to the reduced aqueous solubility of these compounds under acidic conditions or aggregation.57,58 Addition of 1 mg/ mL bovine serum albumin (BSA) in assay buffers may have improved solubility but did not affect the bell-shaped curves. Under these circumstances, we may have slightly under-estimated the allosteric parameters of these compounds, especially with regard to the efficacy cooperativity β. In any case, this effect prevented high concentrations of modulators from being included in the functional assays. This, in turn, prevented observation of the characteristic saturation (termed the “ceiling effect”) of allosteric modulation, further leading to increased levels of variation in estimated allosteric parameters.
In summary, we have performed a comprehensive SAR study by exploring three regions of the scaffold represented by compound 1. While structural changes on the aminotriazine core did not improve the allosteric activity, modifications to the benzyl alcohol and benzylamino groups did lead to slightly more active GPR68 PAMs with increased affinity cooperativities and allosteric binding affinities. The subsequently designed combination compounds with the substituents showing best α and KB values from each region ultimately resulted in much improved GPR68 α-PAMs, including Compound 71, which has 33-fold increased allosteric activity compared to 1. We also identified a significantly improved αβ-PAM, compound 46. We further determined that compounds 71 and 46 and two additional GPR68 PAMs were selective for GPR68 over GPR4 and GPR65, two closely related proton GPCRs. In addition, these four compounds had no or low off-target agonist activity across the human GPCRome and had weak to moderate binding affinity to 48 common GPCRs, ion channels, and transporters. Moreover, we demonstrated that our lead compound, 71, is bioavailable in mice and has excellent brain penetration. Finally, we identified several potentially useful inactive controls. These novel GPR68 PAMs and inactive controls are valuable chemical tools for studying GPR68 biology and pharmacology. Furthermore, our study paved the way for developing the next generation of GPR68 PAMs and allosteric modulators for other GPCRs.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All chemical reagents were purchased from commercial vendors and used without further purification. Dry solvents were purchased from commercial vendors with a verified anhydrous standard. Microwave-heated reactions were performed with a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, U.K.). EMD Millipore 210–270 μm 60-F254 silica gel plates were used to perform analytical thin-layer chromatography (TLC). The plates were examined under a UV Lamp. The flash column chromatography was conducted using a Teledyne ISCO CombiFlash Rf+ instrument. This instrument was also equipped with a variable-wavelength UV detector and a fraction collector. RediSep Rf normal phase silica columns were used for purification. Preparative high-performance liquid chromatography (HPLC) was used to purify all final compounds. HPLC was conducted on an Agilent Prep 1200 series with the UV detector set to 254 nm. Samples were injected into a Phenomenex Luna 750 × 30 mm2, 5 μm C18 column. Separation was performed at room temperature with a flow rate of 40 mL/min. The linear gradient program was set as follows: 10% methanol (A) in H2O containing 0.1% trifluoroacetic acid (B) to 100% methanol (A). All compounds for biological evaluation were determined to have >95% purity after the purification using the aforementioned prep-HPLC methods. An Agilent 1200 series system with diode array detector and a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column for chromatography were used to obtain HPLC spectra for all compounds. Samples (0.5 μL) were injected onto a C18 column at room temperature. The column flow rate was set to 0.4 mL/min. Chromatography was performed with water containing 0.1% formic acid as Solvent A and acetonitrile-containing 0.1% formic acid as solvent B. The linear gradient was set as follows: 1% B (0–1 min), 1– 99% B (1–4 min), and 99% B (4–8 min). An Agilent G1969A atmospheric pressure interface time of flight with an electrospray ionization source was used to acquire high-resolution mass spectra data in positive-ion mode. All compounds were also characterized using a Bruker (Billerica, MA) DRX-600 nuclear magnetic resonance (NMR) spectrometer (600 MHz, 1H NMR). Chemical shifts are reported in parts per million (δ) relative to residual solvent peaks. 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant, and integration.
General Procedures for Preparation of Compounds 5–7.
(2–4-(Benzylamino)-6-(methylamino)-1,3,5-triazin-2-yl)phenyl)-methanol (5).59
Intermediate 3 was prepared according to previously published procedures starting with 4,6-dichloro-N-methyl-1,3,5-triazin-2-amine (2) and benzylamine. The white solid 3 (24.9 mg, 0.1 mmol) was suspended in dioxane (0.8 mL) and water (0.2 mL) in a microwave tube. Then, tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 5 mol %), potassium carbonate (27.6 mg, 0.2 mmol), and (2-(hydroxymethyl)phenyl)boronic acid (30.4 mg, 0.2 mmol) were added. The mixture was heated to 120 °C for 20 min under microwave irradiation. The resulting mixture was filtered through Celite with ethyl acetate as eluent solvent. The filter was washed with water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in 1.5 mL of methanol and purified by preparative HPLC to yield 7 as a white solid (27 mg, 84%). 1H NMR (600 MHz, CD3OD) δ 7.81 (d, J = 7.8 Hz, 1H), 7.60 (dd, J = 20.0, 7.4 Hz, 3H), 7.50 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.8 Hz, 2H), 7.31–7.24 (m, 1H), 4.81 (s, 2H), 4.72 (d, J = 5.1 Hz, 2H), 3.05 (d, J = 5.3 Hz, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O+ 322.1662, found 322.1678.
(2-(4-(Benzylamino)-6-(ethylamino)-1,3,5-triazin-2-yl)phenyl)-methanol (6).
White solid (15.2 mg, 98%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.8 Hz, 1H), 7.69–7.57 (m, 2H), 7.51 (t, J = 7.6 Hz, 1H), 7.35 (ddt, J = 41.4, 34.0, 7.3 Hz, 5H), 4.81 (s, 2H), 4.70 (s, 2H), 3.54 (q, J = 7.2 Hz, 2H), 1.22 (t, J = 7.2 Hz, 3H). HRMS m/z [M + H]+ calcd for C19H22N5O+ 336.1819, found 336.1873.
(2-(4-(Benzylamino)-6-(dimethylamino)-1,3,5-triazin-2-yl)-phenyl)methanol (7).
White solid (32 mg, 42%). 1H NMR (600 MHz, CD3OD) δ 7.90 (d, J = 7.8 Hz, 1H), 7.62 (dd, J = 24.7, 7.5 Hz, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.42 (d, J = 7.5 Hz, 2H), 7.35 (t, J = 7.4 Hz, 2H), 7.28 (t, J = 7.3 Hz, 1H), 4.84 (s, 2H), 4.70 (s, 2H), 3.32 (d, J = 24.3 Hz, 6H). HRMS m/z [M + H]+ calcd for C19H22N5O+ 336.1819, found 336.1832.
(2-(6-Amino-2-(benzylamino)pyrimidin-4-yl)phenyl)methanol (10).
Compound 10 was synthesized following the same procedure for preparing 5 starting with 2,6-dichloropyrimidin-4-amine 8 and benzylamine. The title compound 10 was obtained as a white solid (27.2 mg, 92%). 1H NMR (600 MHz, CD3OD) δ 7.57 (d, J = 5.2 Hz, 2H), 7.53–7.43 (m, 2H), 7.39 (d, J = 7.6 Hz, 2H), 7.34 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.3 Hz, 1H), 6.20 (s, 1H), 4.66 (s, 2H), 4.62 (s, 2H). HRMS m/z [M + H]+ calcd for C18H19N4O+ 307.1553, found 307.1542.
General Procedures for Preparation of Compounds 13–15.
Intermediate 12 was prepared according to previously published procedures.44
N2-Benzyl-6-phenyl-1,3,5-triazine-2,4-diamine (13).
To a dried microwave tube were added intermediate 12 (23.6 mg, 0.1 mmol), phenylboronic acid (24.4 mg, 0.2 mmol), tetrakis(triphenylphosphine) palladium (5.8 mg, 5 mol %), potassium carbonate (27.6 mg, 0.2 mmol) dioxane (0.8 mL), and water (0.2 mL). The mixture was heated to 120 °C for 20 min under microwave irradiation. The resulting mixture was filtered through Celite with ethyl acetate as eluent solvent. The filter was washed with water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in 1.5 mL of methanol and purified by preparative HPLC to yield 13 as a white solid (26 mg, 94%). 1H NMR (600 MHz, CD3OD) δ 8.15 (t, J = 6.3 Hz, 2H), 7.69 (q, J = 8.9, 8.2 Hz, 1H), 7.57 (dt, J = 11.2, 7.5 Hz, 2H), 7.42–7.30 (m, 4H), 7.27 (q, J = 6.9 Hz, 1H), 4.69 (d, J = 3.8 Hz, 2H). HRMS m/z [M + H]+ calcd for C16H N + 16 5 278.1400, found 278.1415.
N2-Benzyl-6-(o-tolyl)-1,3,5-triazine-2,4-diamine (14).
White solid (29 mg, 98%). 1H NMR (600 MHz, CD3OD) δ 7.60 (dd, J = 12.1, 7.8 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.43–7.31 (m, 6H), 7.28 (q, J = 7.2, 5.7 Hz, 1H), 4.69 (s, 2H), 2.52 (s, 3H). HRMS m/z [M + H]+ calcd for C17H18N5+ 292.1557, found 292.1539.
1-(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)phenyl)ethan-1-one (15).
White solid (48 mg, 54%). 1H NMR (600 MHz, CD3OD) δ 8.06 (d, J = 7.7 Hz, 1H), 7.89 (t, J = 7.9 Hz, 1H), 7.80 (dd, J = 7.7, 3.2 Hz, 1H), 7.73 (dt, J = 11.0, 7.6 Hz, 1H), 7.44–7.23 (m, 5H), 4.71 (s, 2H), 2.01 (d, J = 2.4 Hz, 3H). HRMS m/z [M + H]+ calcd for C18H18N5O+ 320.1506, found 320.1531.
1-(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)phenyl)ethan-1-ol (16).
Compound 15 (15 mg, 0.05 mmol) was dissolved in THF (mL). After the mixture was cooled to 0 °C, sodium borohydride (4 mg, 0.1 mmol) was added slowly. The reaction was monitored by TLC until 15 disappeared. Methanol (1 mL) was added slowly to quench the reaction. The mixture was filtered and concentrated in vacuo. The resulting residue was dissolved in 1.5 mL of methanol and purified by preparative HPLC to yield 16 as a white solid (16 mg, 51%). 1H NMR (600 MHz, CD3OD) δ 7.76–7.59 (m, 3H), 7.52–7.44 (m, 2H), 7.43– 7.31 (m, 4H), 7.28 (dd, J = 9.2, 5.7 Hz, 1H), 5.27–5.18 (m, 1H), 4.70 (s, 2H), 1.53 (d, J = 6.5 Hz, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O+ 322.1662, found 322.1669.
6-(2-(Aminomethyl)phenyl)-N2-benzyl-1,3,5-triazine-2,4-diamine (18).
Intermediate 17 (123 mg, 86%) was prepared following the same procedures that were used to prepare 13 starting with 2-formylphenyl boronic acid and intermediate 12. To a suspension of intermediate 17 (47 mg, 0.15 mmol), magnesium sulfate (200 mg) and NH3·H2O (0.75 mmol, 107 μL) in THF (2 mL) was added sodium borohydride (5.7 mg, 0.15 mmol) slowly. After stirring for 2 days at room temperature, methanol (1 mL) was added slowly to quench the reaction. The mixture was filtered and concentrated in vacuo. The resulting residue was dissolved in 1.5 mL of methanol and purified by preparative HPLC to yield 18 as yellow oil (21 mg, 45%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.8 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.60 (d, J = 7.5 Hz, 1H), 7.51 (q, J = 7.0, 6.5 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.37–7.31 (m, 3H), 7.29 (q, J = 7.9, 7.2 Hz, 1H), 4.82 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H N + 19 6 307.1666, found 307.1676.
N2-Benzyl-6-(2-((methylamino)methyl)phenyl)-1,3,5-triazine-2,4-diamine (19).
Compound 19 was prepared following the general procedure for preparing compound 18 from intermediate 17 and methylamine (2 M in THF). Yellow oil (22 mg, 67%). 1H NMR (600 MHz, CD3OD) δ 7.69 (dd, J = 7.6, 3.7 Hz, 1H), 7.66–7.49 (m, 3H), 7.39–7.15 (m, 5H), 4.62 (s, 2H), 4.39 (s, 2H), 2.79 (s, 3H). HRMS m/ z [M + H]+ calcd for C18H N + 21 6 321.1882, found 321.1877.
General Procedures for Preparation of Compounds 21–29, 31, and 32.
Compounds 21–19, 31 and 32 were prepared following the general procedure for preparing compound 13 from intermediate 12 and commercially available substituted phenylboronic acids.
1-(3-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)phenyl)ethan-1-one (21).
White solid (28 mg, 87%). 1H NMR (600 MHz, CD3OD) δ 8.80 (s, 1H), 8.44 (d, J = 7.8 Hz, 1H), 8.32–8.20 (m, 1H), 7.78–7.63 (m, 1H), 7.47–7.19 (m, 5H), 4.69 (s, 2H), 2.67 (s, 3H). HRMS m/z [M + H]+ calcd for C18H18N5O+ 320.1506, found 320.1522.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-6-fluorophenyl)-methanol (22).
White solid (29 mg, 89%). 1H NMR (600 MHz, CD3OD) δ 7.63 (d, J = 7.8 Hz, 1H), 7.53 (td, J = 8.0, 5.1 Hz, 1H), 7.44–7.32 (m, 5H), 7.28 (t, J = 7.5 Hz, 1H), 4.83 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1435.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-5-fluorophenyl)-methanol (23).
White solid (30 mg, 91%). 1H NMR (600 MHz, CD3OD) δ 7.86 (dt, J = 10.6, 5.3 Hz, 1H), 7.43–7.30 (m, 5H), 7.30–7.15 (m, 2H), 4.84 (d, J = 5.1 Hz, 2H), 4.67 (d, J = 5.0 Hz, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1426.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-4-fluorophenyl)-methanol (24).
White solid (28 mg, 86%). 1H NMR (600 MHz, CD3OD) δ 7.66–7.57 (m, 2H), 7.42–7.31 (m, 5H), 7.30–7.24 (m, 1H), 4.78 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1407.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-3-fluorophenyl)-methanol (25).
White solid (31 mg, 94%). 1H NMR (600 MHz, CD3OD) δ 7.63–7.54 (m, 2H), 7.43–7.31 (m, 5H), 7.28–7.23 (m, 1H), 4.76 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1419.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-5-chlorophenyl)methanol (26).
White solid (16 mg, 64%). 1H NMR (600 MHz, CD3OD) δ 7.81 (dd, J = 8.4, 2.0 Hz, 1H), 7.65 (s, 1H), 7.48 (dd, J = 21.4, 8.4 Hz, 1H), 7.41–7.30 (m, 4H), 7.27 (t, J = 7.5 Hz, 1H), 4.81 (s, 2H), 4.68 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1111.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (27).
White solid (27 mg, 93%). 1H NMR (600 MHz, CD3OD) δ 7.86 (d, J = 2.3 Hz, 1H), 7.65–7.56 (m, 2H), 7.42–7.32 (m, 4H), 7.28 (t, J = 7.3 Hz, 1H), 4.79 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1126.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-3-chlorophenyl)methanol (28).
White solid (6.8 mg, 78%). 1H NMR (600 MHz, CD3OD) δ 7.42–7.30 (m, 3H), 7.26–7.09 (m, 5H), 4.53 (s, 2H), 4.49 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1123.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-6-methoxyphenyl)methanol (29).
White solid (13 mg, 84%). 1H NMR (600 MHz, CD3OD) δ 7.48 (t, J = 8.1 Hz, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.37–7.31 (m, 4H), 7.29 (dd, J = 7.8, 3.5 Hz, 2H), 4.82 (s, 2H), 4.69 (s, 2H), 3.91 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O2+ 338.1612, found 338.1618.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-4-methoxyphenyl)methanol (31).
White solid (28 mg, 86%). 1H NMR (600 MHz, CD3OD) δ 7.47 (dd, J = 8.6, 5.9 Hz, 1H), 7.44 (d, J = 2.8 Hz, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.35 (t, J = 7.6 Hz, 3H), 7.31–7.24 (m, 1H), 7.17 (dd, J = 8.5, 2.7 Hz, 1H), 4.72 (d, J = 2.8 Hz, 2H), 4.69 (d, J = 2.9 Hz, 2H), 3.86 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O2+ 338.1612, found 338.1614.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-3-methoxyphenyl)methanol (32).
White solid (16 mg, 48%). 1H NMR (600 MHz, CD3OD) δ 7.54 (td, J = 8.2, 3.0 Hz, 1H), 7.42–7.32 (m, 4H), 7.29 (tt, J = 8.5, 4.8 Hz, 1H), 7.13 (dd, J = 7.9, 5.6 Hz, 2H), 4.68 (s, 2H), 4.68 (s, 2H), 3.88 (d, J = 1.6 Hz, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O2+ 338.1612, found 338.1625.
General Procedures for Preparation of Compounds 37 and 38.
Intermediate 36 was prepared according to previously published procedures.60
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-5-ethoxyphenyl)methanol (37).
To a dried microwave tube were added intermediate 12 (23.6 mg, 0.1 mmol), intermediate 36 (39.2 mg, 0.2 mmol), tetrakis(triphenylphosphine)palladium(0) (5.8 mg, 5 mol %), potassium carbonate (27.6 mg, 0.2 mmol %), dioxane (0.8 mL), and water (0.2 mL). The mixture was heated to 120 °C for 20 min under microwave. The resulting mixture was filtered through Celite with ethyl acetate as eluent solvent. The filter was washed with water and extracted with ethyl acetate. The combined organic layers were washed brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in 5 mL of THF. After the mixture was cooled to 0 °C, sodium borohydride (11 mg, 0.3 mmol) was added slowly. The reaction was warmed to room temperature and stirred for 10 min before methanol was added slowly to quench the reaction. The mixture was washed with water and extracted with ethyl acetate. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in 1.5 mL of methanol and purified by preparative HPLC to yield 37 as a white solid (3.7 mg, 23%). 1H NMR (600 MHz, CD3OD) δ 7.68 (d, J = 7.9 Hz, 2H), 7.60 (d, J = 8.1 Hz, 2H), 7.35–7.30 (m, 3H), 6.29 (s, 1H), 4.10 (s, 2H), 3.65 (s, 2H), 2.65–2.51 (q, J = 7.2 Hz, 2H), 1.49 (t, J = 7.2 Hz, 3H). HRMS m/z [M + H]+ calcd for C19H22N5O2+ 352.1768, found 352.1762.
(2-(4-Amino-6-(benzylamino)-1,3,5-triazin-2-yl)-5-isopropoxyphenyl)methanol (38).
White solid (32 mg, 88%). 1H NMR (600 MHz, CD3OD) δ 7.86 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 7.3 Hz, 1H), 7.37–7.30 (m, 3H), 7.27 (t, J = 7.3 Hz, 1H), 7.14 (d, J = 2.6 Hz, 1H), 7.00 (dd, J = 8.8, 2.7 Hz, 1H), 4.81 (s, 2H), 4.78–4.72 (m, 1H), 4.68 (s, 2H), 1.35 (d, J = 6.0 Hz, 6H). HRMS m/z [M + H]+ calcd for C20H24N5O2+ 366.1925, found 366.1934.
General Procedures for Preparation of Compounds 41–54, 56–60, and 64–81.
Intermediate 40 was prepared from intermediates 3 and 11 and commercially available amines (intermediate 39) according to the previously reported procedures.44 Compounds 41–54, 56–60, and 64–66 were prepared following the general procedure for preparing compound 13 from intermediate 40 and (2-(hydroxymethyl)phenyl)boronic acid. Compounds 67–81 were prepared following the general procedure for preparing compound 13 from intermediate 40 and substituted (2-(hydroxymethyl)phenyl)-boronic acids.
(2-(4-Amino-6-((2-fluorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (41).
White solid (27 mg, 82%). 1H NMR (600 MHz, CD3OD) δ 7.82 (d, J = 7.7 Hz, 1H), 7.65–7.54 (m, 2H), 7.47 (dt, J = 26.2, 7.6 Hz, 2H), 7.33–7.25 (m, 1H), 7.13 (t, J = 7.7 Hz, 1H), 7.07 (t, J = 9.3 Hz, 1H), 4.79 (d, J = 3.9 Hz, 2H), 4.74 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1443.
(2-(4-Amino-6-((3-fluorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (42).
White solid (27 mg, 83%). 1H NMR (600 MHz, CD3OD) δ 7.82 (d, J = 7.8 Hz, 1H), 7.65–7.56 (m, 2H), 7.49 (t, J = 7.9 Hz, 1H), 7.32 (td, J = 8.0, 5.7 Hz, 1H), 7.21–7.10 (m, 2H), 6.98 (td, J = 8.5, 2.6 Hz, 1H), 4.81 (s, 2H), 4.68 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1435.
(2-(4-Amino-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (43).
White solid (26 mg, 80%). 1H NMR (600 MHz, CD3OD) δ 7.81 (dt, J = 7.9, 4.7 Hz, 1H), 7.72–7.53 (m, 3H), 7.49 (dd, J = 7.9, 4.8 Hz, 1H), 7.39 (dt, J = 9.7, 5.0 Hz, 1H), 7.03 (tt, J = 8.7, 4.5 Hz, 2H), 4.80 (t, J = 4.9 Hz, 2H), 4.64 (d, J = 4.9 Hz, 2H). HRMS m/z [M + H]+ calcd for C17H17FN5O+ 326.1412, found 326.1438.
(2-(4-Amino-6-((2-chlorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (44).
White solid (32 mg, 94%). 1H NMR (600 MHz, CD3OD) δ 7.84 (d, J = 7.7 Hz, 1H), 7.68–7.58 (m, 2H), 7.54–7.47 (m, 2H), 7.41 (dd, J = 7.2, 2.0 Hz, 1H), 7.29 (ddt, J = 9.4, 6.9, 4.0 Hz, 2H), 4.83 (s, 2H), 4.79 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1113.
(2-(4-Amino-6-((3-chlorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (45).
White solid (26 mg, 78%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.4 Hz, 1H), 7.70–7.57 (m, 2H), 7.51 (td, J = 7.5, 1.5 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H), 7.36–7.19 (m, 3H), 4.83 (s, 2H), 4.69 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1102.
(2-(4-Amino-6-((4-chlorobenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (46).
White solid (18 mg, 52%). 1H NMR (600 MHz, CD3OD) δ 7.83 (dd, J = 7.9, 1.2 Hz, 1H), 7.67–7.58 (m, 2H), 7.51 (td, J = 7.5, 1.5 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 4.82 (s, 2H), 4.67 (s, 2H). HRMS m/z [M + H]+ calcd for C17H17ClN5O+ 342.1116, found 342.1108.
(2-(4-Amino-6-((2-(trifluoromethyl)benzyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (47).
White solid (35 mg, 93%). 1H NMR (600 MHz, CD3OD) δ 7.86 (d, J = 7.7 Hz, 1H), 7.72 (t, J = 6.7 Hz, 1H), 7.67–7.55 (m, 4H), 7.53 (td, J = 7.5, 1.5 Hz, 1H), 7.47 (td, J = 7.4, 2.2 Hz, 1H), 4.93 (s, 2H), 4.85 (s, 2H). HRMS m/z [M + H]+ calcd for C18H17F3N5O+ 376.1380, found 376.1388.
(2-(4-Amino-6-((3-(trifluoromethyl)benzyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (48).
White solid (35 mg, 94%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.6 Hz, 1H), 7.73–7.46 (m, 7H), 4.83 (s, 2H), 4.78 (s, 2H). HRMS m/z [M + H]+ calcd for C18H17F3N5O+ 376.1380, found 376.1367.
(2-(4-Amino-6-((4-(trifluoromethyl)benzyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (49).
White solid (36 mg, 96%). 1H NMR (600 MHz, CD3OD) δ 7.83 (dd, J = 7.8, 1.3 Hz, 1H), 7.67–7.55 (m, 3H), −6.76 (m, 1H), 4.82 (s, 2H), 4.66 (s, 2H), 3.78 (s, 3H). HRMS m/z [M + H]+ calcd for C18H17F3N5O+ 376.1380, found 376.1374.
(2-(4-Amino-6-((2-methylbenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (50).
White solid (28 mg, 88%). 1H NMR (600 MHz, CD3OD) δ 7.82 (d, J = 7.7 Hz, 1H), 7.64–7.54 (m, 2H), 7.54–7.44 (m, 1H), 7.35–7.22 (m, 1H), 7.19–7.12 (m, 3H), 4.80 (s, 2H), 4.68 (s, 2H), 2.35 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O+ 322.1662, found 322.1654.
(2-(4-Amino-6-((3-methylbenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (51).
White solid (29 mg, 92%). 1H NMR (600 MHz, CD3OD) δ 7.81 (d, J = 7.7 Hz, 1H), 7.68–7.55 (m, 2H), 7.50 (td, J = 7.5, 1.6 Hz, 1H), 7.25–7.13 (m, 3H), 7.08 (d, J = 7.3 Hz, 1H), 4.81 (s, 2H), 4.65 (s, 2H), 2.31 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O+ 322.1662, found 322.1657.
(2-(4-Amino-6-((4-methylbenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (52).
White solid (22 mg, 69%). 1H NMR (600 MHz, CD3OD) δ 7.81 (d, J = 7.7 Hz, 1H), 7.70–7.57 (m, 2H), 7.55–7.47 (m, 1H), 7.27 (d, J = 7.8 Hz, 2H), 7.15 (d, J = 7.7 Hz, 2H), 4.81 (s, 2H), 4.64 (s, 2H), 2.29 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O+ 322.1662, found 322.1672.
(2-(4-Amino-6-((2-methoxybenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (53).
White solid (27 mg, 79%). 1H NMR (600 MHz, CD3OD) δ 7.80 (d, J = 7.8 Hz, 1H), 7.66–7.58 (m, 2H), 7.49 (t, J = 7.6 Hz, 1H), 7.38–7.18 (m, 2H), 7.04–6.74 (m, 2H), 4.80 (s, 2H), 4.67 (s, 2H), 3.85 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O2 + 338.1612, found 338.1623.
(2-(4-Amino-6-((3-methoxybenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (54).
White solid (29 mg, 87%). 1H NMR (600 MHz, CD3OD) δ 7.83 (dd, J = 7.8, 1.3 Hz, 1H), 7.67–7.55 (m, 3H), −6.76 (m, 1H), 4.82 (s, 2H), 4.66 (s, 2H), 3.78 (s, 3H). HRMS m/z [M + H]+ calcd for C18H20N5O2+ 338.1612, found 338.1618.
(2-(4-Amino-6-((4-ethoxybenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (56).
White solid (32 mg, 93%). 1H NMR (600 MHz, CD3OD) δ 7.81 (t, J = 6.4 Hz, 1H), 7.69–7.55 (m, 2H), 7.55–7.46 (m, 1H), 7.31 (td, J = 7.8, 7.0, 3.6 Hz, 1H), 6.95–6.79 (m, 3H), 4.81 (s, 2H), 4.60 (s, 2H), 4.00 (q, J = 6.7 Hz, 2H), 1.36 (t, J = 6.7 Hz, 3H). HRMS m/z [M + H]+ calcd for C19H22N5O2+ 352.1768, found 352.1765.
(2-(4-Amino-6-((4-isopropoxybenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (57).
White solid (3.2 mg, 52%). 1H NMR (600 MHz, CD3OD) δ 7.83 (dd, J = 7.8, 1.3 Hz, 1H), 7.70–7.55 (m, 2H), 7.51 (td, J = 7.6, 1.5 Hz, 1H), 7.36–7.16 (m, 1H), 6.98–6.73 (m, 3H), 4.81 (s, 2H), 4.61 (s, 2H), 4.60–4.53 (m, 1H), 1.29 (d, J = 6.0 Hz, 6H). HRMS m/z [M + H]+ calcd for C20H24N5O2+ 366.1925, found 366.1928.
(2-(4-Amino-6-((4-cyclopropoxybenzyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (58).
White solid (30 mg, 84%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.8 Hz, 1H), 7.62 (dt, J = 21.8, 7.4 Hz, 2H), 7.51 (t, J = 7.5 Hz, 1H), 7.33 (d, J = 8.2 Hz, 2H), 7.02 (d, J = 8.2 Hz, 2H), 4.81 (s, 2H), 4.62 (s, 2H), 3.80–3.71 (m, 1H), 0.77 (t, J = 6.3 Hz, 2H), 0.66 (t, J = 2.9 Hz, 2H). HRMS m/z [M + H]+ calcd for C20H22N5O2+ 364.1768, found 364.1776.
(2-(4-Amino-6-((4-phenoxybenzyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (59).
White solid (13.4 mg, 84%). 1H NMR (600 MHz, CD3OD) δ 7.82 (dd, J = 7.8, 1.2 Hz, 1H), 7.65–7.56 (m, 3H), 7.56–7.46 (m, 2H), 7.40 (d, J = 8.6 Hz, 1H), 7.32 (dd, J = 8.6, 7.3 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 6.95 (d, J = 8.5 Hz, 3H), 4.81 (s, 2H), 4.65 (s, 2H). HRMS m/z [M + H]+ calcd for C23H22N5O2+ 400.1768, found 400.1743.
(2-(4-Amino-6-(phenylamino)-1,3,5-triazin-2-yl)phenyl)-methanol (60).
White solid (28 mg, 96%). 1H NMR (600 MHz, CD3OD) δ 7.88 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.9 Hz, 2H), 7.64 (dt, J = 17.2, 7.7 Hz, 2H), 7.53 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.9 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 4.87 (s, 2H). HRMS m/z [M + H]+ calcd for C16H16N5O+ 294.1349, found 294.1341.
(2-(4-Amino-6-((4-phenoxyphenethyl)amino)-1,3,5-triazin-2-yl)-phenyl)methanol (64).
White solid (32 mg, 77%). 1H NMR (600 MHz, CD3OD) δ 7.82 (dd, J = 7.7, 1.3 Hz, 1H), 7.67–7.57 (m, 3H), 7.51 (td, J = 7.6, 1.4 Hz, 1H), 7.35–7.19 (m, 4H), 7.08 (t, J = 7.3 Hz, 1H), 6.93 (dd, J = 8.6, 6.8 Hz, 3H), 4.79 (s, 2H), 3.73 (t, J = 7.3 Hz, 2H), 2.94 (t, J = 7.4 Hz, 2H). HRMS m/z [M + H]+ calcd for C24H24N5O2+ 414.1925, found 414.1945.
(2-(4-Amino-6-((3-(4-phenoxyphenyl)propyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (65).
White solid (18 mg, 87%). 1H NMR (600 MHz, CD3OD) δ 7.83 (d, J = 7.8 Hz, 1H), 7.70–7.56 (m, 3H), 7.51 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 6.97–6.78 (m, 4H), 4.81 (s, 2H), 3.52 (t, J = 7.0 Hz, 2H), 2.71 (t, J = 6.1 Hz, 2H), 2.08–1.86 (m, 2H). HRMS m/z [M + H]+ calcd for C25H26N5O2+ 428.2081, found 428.2076.
(2-(4-Amino-6-((4-(4-phenoxyphenyl)butyl)amino)-1,3,5-triazin-2-yl)phenyl)methanol (66).
White solid (17 mg, 73%). 1H NMR (600 MHz, CD3OD) δ 7.82 (d, J = 7.8 Hz, 1H), 7.63 (dt, J = 22.8, 7.7 Hz, 3H), 7.52 (t, J = 7.5 Hz, 1H), 7.30 (q, J = 6.2, 4.7 Hz, 1H), 7.19 (d, J = 8.2 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 6.91 (dd, J = 23.1, 8.2 Hz, 4H), 4.81 (s, 2H), 3.52 (t, J = 6.4 Hz, 2H), 2.66 (q, J = 6.4 Hz, 2H), 1.70 (dt, J = 7.9, 3.9 Hz, 4H). HRMS m/z [M + H]+ calcd for C26H28N5O2+ 442.2238, found 442.2234.
(2-(4-Amino-6-((2-fluorobenzyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (67).
White solid (33 mg, 96%). 1H NMR (600 MHz, CD3OD) δ 7.54 (td, J = 8.0, 5.5 Hz, 1H), 7.45 (td, J = 7.6, 1.7 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.34–7.26 (m, 1H), 7.23–7.13 (m, 2H), 7.13–7.04 (m, 1H), 4.71 (s, 2H), 4.68 (s, 2H). HRMS m/z [M + H]+ calcd for C17H16F2N5O+ 344.1317, found 344.1321.
(2-(4-Amino-6-((4-fluorobenzyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (68).
White solid (32 mg, 92%). 1H NMR (600 MHz, CD3OD) δ 7.58 (td, J = 8.0, 5.5 Hz, 1H), 7.44–7.31 (m, 3H), 7.25–7.19 (m, 1H), 7.08 (t, J = 8.6 Hz, 2H), 4.72 (s, 2H), 4.65 (s, 2H). HRMS m/z [M + H]+ calcd for C17H16F2N5O+ 344.1317, found 344.1323.
(2-(4-Amino-6-((3-methylbenzyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (69).
White solid (32 mg, 94%). 1H NMR (600 MHz, CD3OD) δ 7.61–7.53 (m, 1H), 7.39 (d, J = 7.7 Hz, 1H), 7.26–7.15 (m, 4H), 7.14–7.05 (m, 1H), 4.73 (s, 2H), 4.64 (s, 2H), 2.34 (s, 3H). HRMS m/z [M + H]+ calcd for C18H19FN5O+ 340.1568, found 340.1562.
(2-(4-Amino-6-((2-methoxybenzyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (70).
White solid (32 mg, 91%). 1H NMR (600 MHz, CD3OD) δ 7.62–7.54 (m, 1H), 7.42–7.37 (m, 1H), 7.33 (dd, J = 7.4, 2.2 Hz, 1H), 7.31–7.26 (m, 1H), 7.26–7.20 (m, 1H), 6.98 (dd, J = 8.4, 2.1 Hz, 1H), 6.96–6.88 (m, 1H), 4.73 (s, 2H), 4.66 (s, 2H), 3.86 (s, 3H). HRMS m/z [M + H]+ calcd for C18H19FN5O2+ 356.1517, found 356.1523.
(2-(4-Amino-6-((4-phenoxybenzyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (71).
White solid (16 mg, 75%). 1H NMR (500 MHz, CD3OD) δ 7.59 (td, J = 8.1, 5.6 Hz, 1H), 7.43–7.37 (m, 3H), 7.36–7.30 (m, 2H), 7.27–7.21 (m, 1H), 7.13–7.07 (m, 1H), 6.99–6.95 (m, 4H), 4.73 (s, 2H), 4.65 (s, 2H); 13C NMR (126 MHz, CD3OD) δ 163.70, 163.17, 162.58, 160.59, 158.72, 158.44, 144.72, 134.40, 133.79, 131.03, 130.77, 125.63, 124.64, 120.03, 120.01, 116.34, 116.17, 62.66, 45.33. HRMS m/z [M + H]+ calcd for C23H21FN5O2+ 418.1674, found 418.1665.
(2-(4-Amino-6-(phenethylamino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (72).
White solid (30 mg, 89%). 1H NMR (600 MHz, CD3OD) δ 7.60 (td, J = 8.2, 5.5 Hz, 1H), 7.41 (t, J = 8.8 Hz, 1H), 7.35–7.14 (m, 6H), 4.73 (d, J = 2.9 Hz, 2H), 3.71 (t, 2H), 2.95 (t, J = 7.5 Hz, 2H). HRMS m/z [M + H]+ calcd for C18H19FN5O+ 340.1568, found 340.1577.
(2-(4-Amino-6-((3-phenylpropyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (73).
White solid (25 mg, 71%). 1H NMR (600 MHz, CD3OD) δ 7.60 (td, J = 8.1, 5.6 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.29–7.23 (m, 2H), 7.23–7.10 (m, 4H), 4.73 (s, 2H), 3.50 (t, J = 7.2 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H), 2.04–1.89 (m, 2H). HRMS m/z [M + H]+ calcd for C19H21FN5O+ 354.1725, found 354.1735.
(2-(4-Amino-6-((4-phenylbutyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (74).
White solid (23 mg, 64%). 1H NMR (600 MHz, CD3OD) δ 7.65–7.56 (m, 1H), 7.40 (t, J = 6.6 Hz, 1H), 7.24 (ddt, J = 8.9, 5.1, 2.6 Hz, 3H), 7.21–7.17 (m, 2H), 7.14 (tt, J = 7.2, 1.5 Hz, 1H), 4.74 (s, 2H), 3.50 (t, 2H), 2.67 (t, J = 7.0 Hz, 2H), 1.81–1.53 (m, 4H). HRMS m/z [M + H]+ calcd for C20H23FN5O+ 368.1881, found 368.1883.
(2-(4-Amino-6-((4-phenoxyphenethyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (75).
White solid (19 mg, 88%). 1H NMR (600 MHz, CD3OD) δ 7.75–7.49 (m, 4H), 7.41 (t, J = 8.4 Hz, 1H), 7.32 (ddd, J = 8.6, 6.0, 2.1 Hz, 1H), 7.29–7.18 (m, 2H), 7.07 (t, J = 7.4 Hz, 1H), 6.99–6.85 (m, 3H), 4.73 (s, 2H), 3.72 (t, J = 7.4 Hz, 2H), 2.94 (t, J = 7.3 Hz, 2H). HRMS m/z [M + H]+ calcd for C24H23FN5O2+ 432.1830, found 432.1832.
(2-(4-Amino-6-((3-(4-phenoxyphenyl)propyl)amino)-1,3,5-triazin-2-yl)-3-fluorophenyl)methanol (76).
White solid (16 mg, 70%). 1H NMR (600 MHz, CD3OD) δ 7.64–7.46 (m, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.31 (ddd, J = 8.7, 7.4, 1.5 Hz, 2H), 7.26–7.13 (m, 4H), 7.07 (t, J = 7.4 Hz, 1H), 7.01–6.77 (m, 3H), 4.68 (s, 2H), 3.48 (t, J = 7.1 Hz, 2H), 2.70 (t, J = 7.7 Hz, 2H), 2.06–1.83 (m, 2H). HRMS m/z [M + H]+ calcd for C25H25FN5O2+ 446.1987, found 446.1978.
(2-(4-Amino-6-(phenethylamino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (77).
White solid (15 mg, 43%). 1H NMR (600 MHz, CD3OD) δ 7.84 (d, J = 2.2 Hz, 1H), 7.70–7.52 (m, 3H), 7.36–7.08 (m, 4H), 4.77 (s, 2H), 3.72 (t, J = 7.5 Hz, 2H), 2.95 (t, J = 7.6 Hz, 2H). HRMS m/z [M + H]+ calcd for C18H19ClN5O+ 356.1273, found 356.1278.
(2-(4-Amino-6-((3-phenylpropyl)amino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (78).
White solid (7 mg, 19%). 1H NMR (600 MHz, CD3OD) δ 7.86 (t, J = 1.7 Hz, 1H), 7.68–7.54 (m, 3H), 7.35–7.05 (m, 4H), 4.77 (s, 2H), 3.50 (t, J = 7.0 Hz, 2H), 2.71 (t, J = 7.7 Hz, 2H), 2.06–1.86 (m, 2H). HRMS m/z [M + H]+ calcd for C19H21ClN5O+ 370.1429, found 370.1423.
(2-(4-Amino-6-((4-phenylbutyl)amino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (79).
White solid (12 mg, 32%). 1H NMR (600 MHz, CD3OD) δ 7.86 (t, J = 3.4 Hz, 1H), 7.69–7.52 (m, 3H), 7.33–7.00 (m, 4H), 4.76 (s, 2H), 3.58–3.44 (m, 2H), 2.67 (q, J = 7.5, 6.9 Hz, 2H), 1.70 (tdd, J = 9.8, 5.3, 2.7 Hz, 4H). HRMS m/z [M + H]+ calcd for C20H23ClN5O+ 384.1586, found 384.1589.
(2-(4-Amino-6-((4-phenoxybenzyl)amino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (80).
White solid (8 mg, 18%). 1H NMR (600 MHz, CD3OD) δ 7.91–7.87 (m, 1H), 7.63–7.53 (m, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.35–7.30 (m, 2H), 7.10 (t, J = 7.6 Hz, 1H), 6.99–6.93 (m, 4H), 4.75 (s, 2H), 4.64 (s, 2H). HRMS m/z [M + H]+ calcd for C23H21ClN5O2+ 434.1387, found 434.1376.
(2-(4-Amino-6-((4-phenoxyphenethyl)amino)-1,3,5-triazin-2-yl)-4-chlorophenyl)methanol (81).
White solid (16 mg, 36%). 1H NMR (600 MHz, CD3OD) δ 7.86 (d, J = 2.2 Hz, 1H), 7.57 (dt, J = 10.6, 8.4 Hz, 2H), 7.38–7.19 (m, 4H), 7.13–7.04 (m, 1H), 7.01–6.81 (m, 4H), 4.74 (s, 2H), 3.78–3.65 (m, 2H), 2.93 (t, J = 7.5 Hz, 2H). HRMS m/z [M + H]+ calcd for C24H23ClN5O2+ 448.1535, found 448.1537.
Calcium Release Assays.
Calcium release assays for Gq-GPCRs were performed according to published protocol61 with minor modifications. In brief, HEK293 cells stably expressing 5-HT2B receptors62 were plated in 384-well black clear bottom plates (coated with poly-l-Lys) at a density of 10 000 cells/well in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% dialyzed fetal bovine serum (dFBS) overnight (16–20 h). The cells were loaded with Fluo-4 Direct Calcium dye (20 μL/well) for 1 h at 37 °C in FLIPR buffer [1× Hank’s balanced salt solution (HBSS), 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES), and 2.5 mM probenecid, pH 7.40, room temperature]. Drug working dilutions (3× of final) were prepared in the FLIPR buffer supplemented with 1 mg/ mL BSA and 0.1 mg/mL ascorbic acid. The FLIPRTETRA was programmed to transfer 10 μL/well of drug solution, and fluorescence was recorded for a total of 2 min with 1 read/s and 10 s before drug addition. The maximum fluorescence intensity (fold of basal) within the first minute of drug addition was exported and analyzed in Prism.
GloSensor cAMP Assays.
Proton-mediated GPR68 Gs activation was determined by measuring cAMP production using GloSensor cAMP assays as reported previously44 with modifications as detailed below. Calcium- and magnesium-free Hank’s balanced salt solution (1× HBSS) was used as the base for all pH buffers. Different zwitterionic organic buffer agents were used for different pH ranges, 1× HBSS with 20 mM 2-(N-morpholino)ethanesulfonic acid for pH 5.60–6.70, 1× HBSS with 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) for pH 6.80–8.20, and 1× HBSS with 20 mM (tris(hydroxymethyl)methylamino)propanesulfonic acid (TAPS) for pH 8.30–8.60. The pH was adjusted with KOH and fresh buffers were made in the morning to use within 24 h. The phosphodiesterase (PDE) inhibitor Ro 20–1724 (10 μM) was included in assay buffer to eliminate potential effect of PDE inhibition. BSA (1 mg/mL) was added to buffer for samples with solubility issues. Addition of BSA does not affect GloSensor cAMP assays and is routinely applied in cell-based GPCR functional assays.63,64 Briefly, HEK293T cells were co-transfected with a modified polyethylenimine (PEI) transfection method.65 For a 10 cm dish of HEK293T cells with 12 mL growth medium, 4 μg of receptor DNA and 4 μg of GloSensor DNA were mixed in 0.4 mL of Opti-MEM medium, followed by addition of 40 μL PEI reagent (increase proportionally when larger dishes are used). The mixture was incubated for 20 min at room temperature and then added to cells for incubation overnight (16–20 h). Transfected cells were plated in poly-l-Lys-coated 384-well white clear bottom plates using DMEM + 1% dFBS for a minimum of 6 h (up to 24 h) before being used for assays. After culture medium was removed, the cells were incubated with 20 μL/well of 3.5 mM luciferin prepared in assay buffer (1× HBSS, 20 mM TAPS, pH 8.40) for 1 h at 37 °C. Ligand stock was made in dimethyl sulfoxide at 10 mM and diluted with pH buffers to desired concentrations. To stimulate cells with premixed drug solutions at desired pH conditions, luciferin-loading solution was removed before addition of drug solution (25 μL/well). Luminescence was counted at 20–30 min after drug application, and data were analyzed in the GraphPad Prism.
GPCRome Off-Target Agonist Assays.
Off-target agonist activity was carried out according to previously published procedure using the PRESTO-Tango assay platform47 with modifications as outlined below. The assay was designed and modified such that 40 receptors were tested in each 384-well plate, each in quadruplicate wells for basal (with medium vehicle) and in quadruplicate for sample. Each plate also contained 16 wells of dopamine D2 receptors stimulated with 100 nM quinpirole as assay controls and 16 wells of D2 with vehicle alone as D2 basal activity. In addition, there were two columns of wells (32 wells) of untransfected cells as negative controls. The whole procedure takes 3 days from plating cells to reading plates. In brief, HTLA cells were plated in poly-L-Lys-coated 384-well white clear bottom plates in DMEM supplemented with 2% dFBS (10 000 cells per 40 μL per well) overnight, transfected with receptor Tango constructs (15 ng/well) overnight using an in-plate PEI transfection method,65 in which receptor DNA was resuspended in Opti-MEM and hybridized with PEI reagent prior to dilution and distribution to 384-well plates and subsequent addition to cells. After overnight incubation, cells received drugs without replacement of the medium and were incubated overnight. Drug solutions and medium were removed and Bright-Glo reagent (20 μL/well) were added. Luminescence was counted on a luminescence reader after 20 min incubation at room temperature. Results were presented in the form of fold of basal for each receptor and analyzed in the Prism.
For Tango arrestin recruitment concentration responses with individual GPCRs, HTLA cells were transfected with receptor DNA overnight in DMEM with 10% FBS and plated into poly-L-Lys-coated 384-well plates as above using DMEM supplemented with 1% dFBS. Drugs were added 6 h later and incubated overnight (16–20 h). Medium and drug solutions were removed, and Bright-Glo reagent (20 μL/well) was added. Luminescence was counted and results were analyzed in Prism.
Radioligand Binding Assays.
Ligand binding profiling was performed by the NIMH PDSP. The NIMH PDSP is directed by Bryan L Roth, MD, PhD, at the University of North Carolina at Chapel Hill, North Carolina, and Program Officer Jamie Driscoll at NIMH, Bethesda, MD. Details about binding assay procedures and protocols are available online at: https://pdspdb.unc.edu/pdspWeb/?site= assays.
Data Analysis.
Proton-mediated GPR68 Gs activity, as measured as relative luminescence unit, in the absence and presence of increasing concentrations of ligands was fitted to the standard allosteric operational model to obtain allosteric parameters, affinity cooperativity α and efficacy cooperativity β, and allosteric ligand binding affinity KB.26–28 Briefly, affinity cooperativity α defines the reciprocal effect between allosteric modulator and orthosteric agonist (protons in this case) with α > 1 for increasing affinity and 0 < α < 1 for reducing affinity. Efficacy cooperativity β defines the effect of allosteric modulator on the efficacy of the orthosteric agonist (protons in this case) with β > 1 for increasing proton efficacy and 0 < β < 1 for reducing proton efficacy. The KB defines the equilibrium binding affinity of test allosteric ligands in the absence of orthosteric agonist (H+). Since orthosteric agonist protons always exist in the receptor compartment, KB in this case has no physical meaning. We applied the comprehensive index log(αβ/KB) to quantify the overall allosteric activity of test allosteric ligand and the difference from that of reference ligand, ΔLog(αβ/KB), to represent the net change of allosteric activity.31 For summary, results were normalized to the percentages, pooled together, and analyzed in the Prism. Normalization was performed using the built-in normalization function in the Prism, where proton basal activity in the absence of test allosteric ligand was set as 0% and the maximum proton activity in the absence of test allosteric ligand was set as 100%. In functional antagonist assays, functional affinity (Ki) values were calculated from IC50 values using the Cheng-Prusoff equation Ki = IC50/(1 + L/ EC50),66 where IC50 is the antagonist concentration when 50% inhibition is achieved against reference agonist at concentration [L], EC50 is the potency of the reference agonist.
Mouse Pharmacokinetic Study.
A standard PK study was performed. Compound 71 was dissolved in 5% N-methyl-2-pyrrolidone, 5% Solutol HS-15, and 90% normal saline. Nine male Swiss Albino mice were treated with Compound 71 intraperitoneally at the 25 mg/kg dose. Plasma and brain samples were collected at 0.5, 1.0, and 2.0 h. Compound concentrations in plasma and brain at each time point are average values from three test mice. The error bars represent ± SEM.
Supplementary Material
ACKNOWLEDGMENTS
The research described here was in part supported by the grants R56MH111789 (to X.-P.H. and T.P.K.) and U24DK116195 (to J.J.) from the U.S. National Institutes of Health, and by the PDSP contract HHSN-271-2013-00017-C from the U.S. National Institute of Mental Health. M.L.M. acknowledges the support by the NIGMS-funded Integrated Pharmacological Sciences Training Program T32GM062754 and the MSTP training grants at the Icahn School of Mount Sinai (NIH T32GM007280).
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- SAR
structure-activity relationship
- GS
stimulatory G protein
- cAMP
cyclic adenosine monophosphate
- PAM
positive allosteric modulator
- MT1
melatonin receptor 1
- MT2
melatonin receptor 2
- ACKR3
atypical chemokine receptor 3
- 5-HT2B
5-hydroxytryptamine receptor 2B
- mGLuR5
metabotropic glutamate receptor 5
- D2
dopamine D2 receptor
- TFA
trifluoroacetic acid
- Na2SO4
sodium sulfate
- dioxane
1,4-dioxane
- THF
tetrahydrofuran
- K2CO3
potassium carbonate
- Pd(PPh3)4
tetrakis(triphenylphosphine)palladium(0)
- MgSO4
magnesium sulfate
- NaBH4
sodium borohydride
- nBuLi
n-butyllithium solution
- B(OMe)3
trimethyl borate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00869.
1H NMR spectra of compounds 46, 71, 77, and 78; 13C NMR spectra of Compound 71; proton concentration- response curves at human GPR68 in the absence and presence of increasing concentrations of test compounds; proton concentration-response curves of compounds 20, 46, 77, and 78 at GPR4 and GPR65; heat map showing off-target binding activities of selected lead compounds at 48 common GPCRs, ion channels, and transporters; functional activities of compounds with binding affinity values lower than 300 nM (pKi > 6.50); off-target agonist activity at human GPCRome; functional characterization of the compounds identified from the GPCRome screening; proton concentration-response curves of compounds 1, 46, 63, 71, 77, and 78 at mouse GPR68; effects of inactive analogues on the GPR68 PAM 71; allosteric modulation of GPR68 proton activity: and implications for pH-dependent agonism and inverse agonism (PDF)
Molecular formula strings for all compounds (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Xu Y; Casey G Identification of Human OGR1, a Novel G Protein-Coupled Receptor That Maps to Chromosome 14. Genomics 1996, 35, 397–402. [DOI] [PubMed] [Google Scholar]
- (2).Ludwig M-G; Vanek M; Guerini D; Gasser JA; Jones CE; Junker U; Hofstetter H; Wolf RM; Seuwen K Proton-Sensing G-Protein-Coupled Receptors. Nature 2003, 425, 93–98. [DOI] [PubMed] [Google Scholar]
- (3).Regard JB; Sato IT; Coughlin SR Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell 2008, 135, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Alexander SP; Christopoulos A; Davenport AP; Kelly E; Marrion NV; Peters JA; Faccenda E; Harding SD; Pawson AJ; Sharman JL; Southan C; Davies JA CGTP Collaborators. The Concise Guide to Pharmacology 2017/18: G Protein-Coupled Receptors. Br. J. Pharmacol. 2017, 174, S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mogi C; Tomura H; Tobo M; Wang J-Q; Damirin A; Kon J; Komachi M; Hashimoto K; Sato K; Okajima F Sphingosyl-phosphorylcholine Antagonizes Proton-Sensing Ovarian Cancer G-Protein-Coupled Receptor 1 (OGR1)-Mediated Inositol Phosphate Production and cAMP Accumulation. J. Pharmacol. Sci 2005, 99, 160–167. [DOI] [PubMed] [Google Scholar]
- (6).Singh LS; Berk M; Oates R; Zhao Z; Tan H; Jiang Y; Zhou A; Kirmani K; Steinmetz R; Lindner D; Xu Y Ovarian Cancer G Protein-Coupled Receptor 1, a New Metastasis Suppressor Gene in Prostate Cancer. JNCI, J. Natl. Cancer Inst. 2007, 99, 1313–1327. [DOI] [PubMed] [Google Scholar]
- (7).Li J; Guo B; Wang J; Cheng X; Xu Y; Sang J Ovarian Cancer G Protein Coupled Receptor 1 Suppresses Cell Migration of MCF7 Breast Cancer Cells via a Gα12/13-Rho-Rac1 Pathway. J. Mol. Signal 2013, 8, No. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Damaghi M; Wojtkowiak JW; Gillies RJ pH Sensing and Regulation in Cancer. Front. Physiol. 2013, 4, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li H; Wang D; Singh LS; Berk M; Tan H; Zhao Z; Steinmetz R; Kirmani K; Wei G; Xu Y Abnormalities in Osteoclastogenesis and Decreased Tumorigenesis in Mice Deficient for Ovarian Cancer G Protein-Coupled Receptor 1. PLoS One 2009, 4, No. e5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ren J; Zhang L Effects of Ovarian Cancer G Protein Coupled Receptor 1 on the Proliferation, Migration, and Adhesion of Human Ovarian Cancer Cells. Chin. Med. J 2011, 124, 1327–1332. [PubMed] [Google Scholar]
- (11).Wiley SZ; Sriram K; Liang W; Chang SE; French R; McCann T; Sicklick J; Nishihara H; Lowy AM; Insel PA GPR68, a Proton-Sensing GPCR, Mediates Interaction of Cancer-Associated Fibroblasts and Cancer Cells. FASEB J. 2018, 32, 1170–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Okajima F Regulation of Inflammation by Extracellular Acidification and Proton-Sensing GPCRs. Cell. Signal 2013, 25, 2263–2271. [DOI] [PubMed] [Google Scholar]
- (13).Chen Y-J; Huang C-W; Lin C-S; Chang W-H; Sun W-H Expression and Function of Proton-Sensing G-Protein-Coupled Receptors in Inflammatory Pain. Mol. Pain 2009, 10, S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).de Vallière C; Wang Y; Eloranta JJ; Vidal S; Clay I; Spalinger MR; Tcymbarevich I; Terhalle A; Ludwig M-G; Suply T; Fried M; Kullak-Ublick GA; Frey-Wagner I; Scharl M; Seuwen K; Wagner CA; Rogler G G Protein-Coupled pH-Sensing Receptor OGR1 Is a Regulator of Intestinal Inflammation. Inflammatory Bowel Dis. 2015, 21, 1269–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yang M; Mailhot G; Birnbaum MJ; MacKay CA; Mason-Savas A; Odgren PR Expression of and Role for Ovarian Cancer G-Protein-Coupled Receptor 1 (OGR1) During Osteoclastogenesis. J. Biol. Chem 2006, 281, 23598–23605. [DOI] [PubMed] [Google Scholar]
- (16).Pereverzev A; Komarova SV; Korcok J; Armstrong S; Tremblay GB; Dixon SJ; Sims SM Extracellular Acidification Enhances Osteoclast Survival through an NFAT-Independent, Protein Kinase C-Dependent Pathway. Bone 2008, 42, 150–161. [DOI] [PubMed] [Google Scholar]
- (17).Aoki H; Mogi C; Hisada T; Nakakura T; Kamide Y; Ichimonji I; Tomura H; Tobo M; Sato K; Tsurumaki H; Dobashi K; Mori T; Harada A; Yamada M; Mori M; Ishizuka T; Okajima F Proton-Sensing Ovarian Cancer G Protein-Coupled Receptor 1 on Dendritic Cells is Required for Airway Responses in a Murine Asthma Model. PLoS One 2013, 8, No. e79985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nakakura T; Mogi C; Tobo M; Tomura H; Sato K; Kobayashi M; Ohnishi H; Tanaka S; Wayama M; Sugiyama T; Kitamura T; Harada A; Okajima F Deficiency of Proton-Sensing Ovarian Cancer G Protein-Coupled Receptor 1 Attenuates Glucose-Stimulated Insulin Secretion. Endocrinology 2012, 153, 4171–4180. [DOI] [PubMed] [Google Scholar]
- (19).Mohebbi N; Benabbas C; Vidal S; Daryadel A; Bourgeois S; Velic A; Ludwig M-G; Seuwen K; Wagner CA The Proton-Activated G Protein Coupled Receptor OGR1 Acutely Regulates the Activity of Epithelial Proton Transport Proteins. Cell. Physiol. Biochem 2012, 29, 313–324. [DOI] [PubMed] [Google Scholar]
- (20).de Vallière C; Vidal S; Clay I; Jurisic G; Tcymbarevich I; Lang S; Ludwig M-G; Okoniewski M; Eloranta JJ; Kullak-Ublick GA; Wagner CA; Rogler G; Seuwen K The pH-Sensing Receptor OGR1 Improves Barrier Function of Epithelial Cells and Inhibits Migration in an Acidic Environment. Am. J. Physiol.: Gastrointest. Liver Physiol 2015, 309, G475–G490. [DOI] [PubMed] [Google Scholar]
- (21).Xu J; Mathur J; Vessières E; Hammack S; Nonomura K; Favre J; Grimaud L; Petrus M; Francisco A; Li J; Lee V; Xiang F-L; Mainquist JK; Cahalan SM; Orth AP; Walker JR; Ma S; Lukacs V; Bordone L; Bandell M; Laffitte B; Xu Y; Chien S; Henrion D; Patapoutian A GPR68 Senses Flow and Is Essential for Vascular Physiology. Cell 2018, 173, 762.e16–775.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Illuminating the Druggable Genome | NIH Common Fund, 2019. https://www.commonfund.nih.gov/idg/ (accessed May 7, 2019). [Google Scholar]
- (23).Wiley SZ; Sriram K; Salmerón C; Insel PA GPR68: An Emerging Drug Target in Cancer. Int. J. Mol. Sci 2019, 20, No. 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lu S; Zhang J Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug-Target Interactions. J. Med. Chem 2019, 62, 24–45. [DOI] [PubMed] [Google Scholar]
- (25).Coughlin Q; Hopper AT; Blanco M-J; Tirunagaru V; Robichaud AJ; Doller D Allosteric Modalities for Membrane-Bound Receptors: Insights from Drug Hunting for Brain Diseases. J. Med. Chem 2019, 5979–6002. [DOI] [PubMed] [Google Scholar]
- (26).Kenakin T New Concepts in Drug Discovery: Collateral Efficacy and Permissive Antagonism. Nat. Rev. Drug Discovery 2005, 4, 919–927. [DOI] [PubMed] [Google Scholar]
- (27).Ehlert FJ Analysis of Allosterism in Functional Assays. J. Pharmacol. Exp. Ther 2005, 315, 740–754. [DOI] [PubMed] [Google Scholar]
- (28).Price MR; Baillie GL; Thomas A; Stevenson LA; Easson M; Goodwin R; McLean A; McIntosh L; Goodwin G; Walker G; Westwood P; Marrs J; Thomson F; Cowley P; Christopoulos A; Pertwee RG; Ross RA Allosteric Modulation of the Cannabinoid CB1 Receptor. Mol. Pharmacol 2005, 68, 1484–1495. [DOI] [PubMed] [Google Scholar]
- (29).Mistry SN; Valant C; Sexton PM; Capuano B; Christopoulos A; Scammells PJ Synthesis and Pharmacological Profiling of Analogues of Benzyl Quinolone Carboxylic Acid (BQCA) as Allosteric Modulators of the M1 Muscarinic Receptor. J. Med. Chem 2013, 56, 5151–5172. [DOI] [PubMed] [Google Scholar]
- (30).Bradley SJ; Bourgognon J-M; Sanger HE; Verity N; Mogg AJ; White DJ; Butcher AJ; Moreno JA; Molloy C; Macedo-Hatch T; Edwards JM; Wess J; Pawlak R; Read DJ; Sexton PM; Broad LM; Steinert JR; Mallucci GR; Christopoulos A; Felder CC; Tobin AB M1 Muscarinic Allosteric Modulators Slow Prion Neurodegeneration and Restore Memory Loss. J. Clin. Invest 2017, 127, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kenakin T A Scale of Agonism and Allosteric Modulation for Assessment of Selectivity, Bias, and Receptor Mutation. Mol. Pharmacol 2017, 92, 414–424. [DOI] [PubMed] [Google Scholar]
- (32).Mistry SN; Jörg M; Lim H; Vinh NB; Sexton PM; Capuano B; Christopoulos A; Lane JR; Scammells PJ 4-Phenylpyridin-2-one Derivatives: A Novel Class of Positive Allosteric Modulator of the M1 Muscarinic Acetylcholine Receptor. J. Med. Chem 2016, 59, 388–409. [DOI] [PubMed] [Google Scholar]
- (33).Russell JL; Goetsch SC; Aguilar HR; Coe H; Luo X; Liu N; van Rooij E; Frantz DE; Schneider JW Regulated Expression of pH Sensing G Protein-Coupled Receptor-68 Identified through Chemical Biology Defines a New Drug Target for Ischemic Heart Disease. ACS Chem. Biol 2012, 7, 1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mogi C; Nakakura T; Okajima F Role of Extracellular Proton-Sensing OGR1 in Regulation of Insulin Secretion and Pancreatic to Cell Functions. Endocr. J 2014, 61, 101. [DOI] [PubMed] [Google Scholar]
- (35).Abe-Ohya R; Ishikawa T; Shiozawa H; Suda K; Nara F Identification of Metals from Osteoblastic ST-2 Cell Supernatants as Novel OGR1 Agonists. J. Recept. Signal Transduction 2015, 35, 485–492. [DOI] [PubMed] [Google Scholar]
- (36).Negishi J; Omori Y; Shindo M; Takanashi H; Musha S; Nagayama S; Hirayama J; Nishina H; Nakakura T; Mogi C; Sato K; Okajima F; Mochimaru Y; Tomura H Manganese and Cobalt Activate Zebrafish Ovarian Cancer G-protein-Coupled Receptor 1 but Not GPR4. J. Recept. Signal Transduction 2017, 37, 401–408. [DOI] [PubMed] [Google Scholar]
- (37).Mochimaru Y; Negishi J; Murakami S; Musha S; Sato K; Okajima F; Tomura H Metals Differentially Activate Ovarian Cancer G Protein-Coupled Receptor 1 in Various Species. Zool. Sci 2018, 35, 109–114. [DOI] [PubMed] [Google Scholar]
- (38).Yasuda S; Miyazaki T; Munechika K; Yamashita M; Ikeda Y; Kamizono A Isolation of Zn2+ as an Endogenous Agonist of GPR39 from Fetal Bovine Serum. J. Recept. Signal Transduction 2007, 27, 235–246. [DOI] [PubMed] [Google Scholar]
- (39).Holst B; Egerod KL; Schild E; Vickers SP; Cheetham S; Gerlach L-O; Storjohann L; Stidsen CE; Jones R; Beck-Sickinger AG; Schwartz TW GPR39 Signaling Is Stimulated by Zinc Ions but Not by Obestatin. Endocrinology 2007, 148, 13–20. [DOI] [PubMed] [Google Scholar]
- (40).Sato S; Huang X-P; Kroeze WK; Roth BL Discovery and Characterization of Novel GPR39 Agonists Allosterically Modulated by Zinc. Mol. Pharmacol 2016, 90, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ayton S; Lei P; Bush AI Biometals and Their Therapeutic Implications in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nordberg M; Nordberg GF Trace Element Research-Historical and Future Aspects. J. Trace Elem. Med. Biol 2016, 38, 46–52. [DOI] [PubMed] [Google Scholar]
- (43).Prakash A; Dhaliwal GK; Kumar P; Majeed ABA Brain Biometals and Alzheimer’s Disease - Boon or Bane? Int. J. Neurosci 2017, 127, 99–108. [DOI] [PubMed] [Google Scholar]
- (44).Huang X-P; Karpiak J; Kroeze WK; Zhu H; Chen X; Moy SS; Saddoris KA; Nikolova VD; Farrell MS; Wang S; Mangano TJ; Deshpande DA; Jiang A; Penn RB; Jin J; Koller BH; Kenakin T; Shoichet BK; Roth BL Allosteric Ligands for the Pharmacologically Dark Receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McAleer JP; Fan J; Roar B; Primerano DA; Denvir J Cytokine Regulation in Human CD4 T Cells by the Aryl Hydrocarbon Receptor and Gq-Coupled Receptors. Sci. Rep 2018, 8, No. 10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG ZINC: a Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kroeze WK; Sassano MF; Huang X-P; Lansu K; McCorvy JD; Giguère PM; Sciaky N; Roth BL PRESTO-Tango as an Open-Source Resource for Interrogation of the Druggable Human GPCRome. Nat. Struct. Mol. Biol 2015, 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bachelerie F; Ben-Baruch A; Burkhardt AM; Combadiere C; Farber JM; Graham GJ; Horuk R; Sparre-Ulrich AH; Locati M; Luster AD; Mantovani A; Matsushima K; Murphy PM; Nibbs R; Nomiyama H; Power CA; Proudfoot AEI; Rosenkilde MM; Rot A; Sozzani S; Thelen M; Yoshie O; Zlotnik A International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol. Rev 2014, 66, 1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bachelerie F; Graham GJ; Locati M; Mantovani A; Murphy PM; Nibbs R; Rot A; Sozzani S; Thelen M An Atypical Addition to the Chemokine Receptor Nomenclature: IUPHAR Review 15. Br. J. Pharmacol 2015, 172, 3945–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nickols HH; Conn PJ Development of Allosteric Modulators of GPCRs for Treatment of CNS Disorders. Neurobiol. Dis 2014, 61, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Burford NT; Clark MJ; Wehrman TS; Gerritz SW; Banks M; O’Connell J; Traynor JR; Alt A Discovery of Positive Allosteric Modulators and Silent Allosteric Modulators of the μ-Opioid Receptor. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Livingston KE; Stanczyk MA; Burford NT; Alt A; Canals M; Traynor JR Pharmacologic Evidence for a Putative Conserved Allosteric Site on Opioid Receptors. Mol. Pharmacol 2018, 93, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Haas LT; Salazar SV; Smith LM; Zhao HR; Cox TO; Herber CS; Degnan AP; Balakrishnan A; Macor JE; Albright CF; Strittmatter SM Silent Allosteric Modulation of mGluR5 Maintains Glutamate Signaling While Rescuing Alzheimer’s Mouse Phenotypes. Cell Rep. 2017, 20, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sharma S; Rodriguez AL; Conn PJ; Lindsley CW Synthesis and SAR of a mGluR5 Allosteric Partial Antagonist Lead: Unexpected Modulation of Pharmacology with Slight Structural Modifications to a 5-(Phenylethynyl)pyrimidine Scaffold. Bioorg. Med. Chem. Lett 2008, 18, 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wood MR; Hopkins CR; Brogan JT; Conn PJ; Lindsley CW Molecular Switches” on mGluR Allosteric Ligands That Modulate Modes of Pharmacology. Biochemistry 2011, 50, 2403–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Gregory KJ; Conn PJ Molecular Insights into Metabotropic Glutamate Receptor Allosteric Modulation. Mol. Pharmacol 2015, 88, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sassano MF; Doak AK; Roth BL; Shoichet BK Colloidal Aggregation Causes Inhibition of G Protein-Coupled Receptors. J. Med. Chem 2013, 56, 2406–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Owen SC; Doak AK; Ganesh AN; Nedyalkova L; McLaughlin CK; Shoichet BK; Shoichet MS Colloidal Drug Formulations Can Explain “Bell-shaped” Concentration-Response Curves. ACS Chem. Biol 2014, 9, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Vijayaraghavan S; Mahajan S Docking, Synthesis and Antimalarial Activity of Novel 4-Anilinoquinoline Derivatives. Bioorg. Med. Chem. Lett 2017, 27, 1693–1697. [DOI] [PubMed] [Google Scholar]
- (60).Yu X; Lu X Cationic Palladium Complex Catalyzed Diastereo- and Enantioselective Tandem Annulation of 2-Formylarylboronic Acids with Allenoates. Org. Lett 2009, 11, 4366–4369. [DOI] [PubMed] [Google Scholar]
- (61).Huang X-P; Setola V; Yadav PN; Allen JA; Rogan SC; Hanson BJ; Revankar C; Robers M; Doucette C; Roth BL Parallel Functional Activity Profiling Reveals Valvulopathogens Are Potent 5-Hydroxytryptamine(2B) Receptor Agonists: Implications for Drug Safety Assessment. Mol. Pharmacol 2009, 76, 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).McCorvy JD; Wacker D; Wang S; Agegnehu B; Liu J; Lansu K; Tribo AR; Olsen RHJ; Che T; Jin J; Roth BL Structural Determinants of 5-HT2B Receptor Activation and Biased Agonism. Nat. Struct. Mol. Biol 2018, 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Stauch B; Johansson LC; McCorvy JD; Patel N; Han GW; Huang X-P; Gati C; Batyuk A; Slocum ST; Ishchenko A; Brehm W; White TA; Michaelian N; Madsen C; Zhu L; Grant TD; Grandner JM; Shiriaeva A; Olsen RHJ; Tribo AR; Yous S; Stevens RC; Weierstall U; Katritch V; Roth BL; Liu W; Cherezov V Structural Basis of Ligand Recognition at the Human MT1 Melatonin Receptor. Nature 2019, 569, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Johansson LC; Stauch B; McCorvy JD; Han GW; Patel N; Huang X-P; Batyuk A; Gati C; Slocum ST; Li C; Grandner JM; Hao S; Olsen RHJ; Tribo AR; Zaare S; Zhu L; Zatsepin NA; Weierstall U; Yous S; Stevens RC; Liu W; Roth BL; Katritch V; Cherezov V XFEL Structures of the Human MT2 Melatonin Receptor Reveal the Basis of Subtype Selectivity. Nature 2019, 569, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Longo PA; Kavran JM; Kim M-S; Leahy DJ Transient Mammalian Cell Transfection with Polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Cheng C; Prusoff WH Relationship Between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.