

Abstract
Transition-metal catalysis is a powerful tool for the construction of chemical bonds. Here we show that Pseudomonas savastanoi ethylene-forming enzyme (PsEFE), a non-heme iron enzyme, can catalyze olefin aziridination and nitrene C–H insertion, and that these activities can be improved by directed evolution. The non-heme iron center allows for facile modification of the primary coordination sphere by addition of metal-coordinating molecules, enabling control over enzyme activity and selectivity using small molecules.
Graphical Abstract
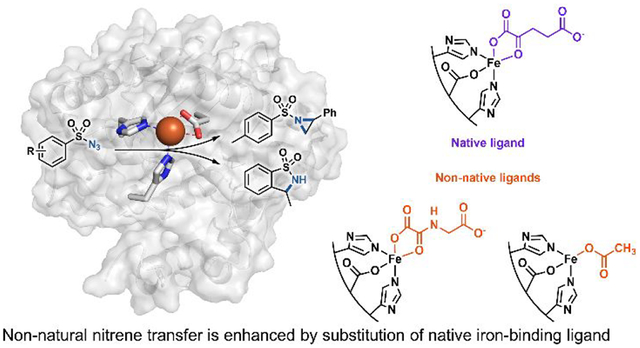
Over the last century, chemists have developed myriad synthetic transition-metal catalysts to access new chemical transformations and modes of reactivity. Nature has been developing catalysts for far longer: over billions of years, she has evolved a rich repertoire of proteins that perform most of the chemical reactions of life. But nature’s inventions do not include many of the best inventions of human chemists. Our efforts to merge abiological transition-metal chemistry with nature’s vast toolbox of metalloproteins have focused on heme-binding proteins1, as the heme cofactor and its analogues are well-studied in synthetic transition-metal chemistry. However, heme-binding proteins represent only a small fraction of the chemical diversity present in natural metalloproteins. Metalloproteins comprise greater than 30% of all proteins2 and are responsible for some of the most fundamental chemical reactions in biology, including nitrogen fixation, photosynthesis, and DNA synthesis. Natural metalloproteins bind a variety of metals in a wide range of metal-binding sites, either coordinating the metal ion itself or a more complex metal-containing cofactor. Nearly any heteroatom-containing side chain can coordinate to a metal, in addition to the peptide backbone, allowing myriad possible coordination environments3. Many coordination environments in non-heme metalloenzymes have multiple open coordination sites at the metal center, a key feature of numerous synthetic transition-metal catalysts. Expanding new-to-nature catalysis to non-heme metalloenzymes would open a new world of transition-metal biocatalysis. In this work we show that a non-heme iron enzyme can catalyze nitrene-transfer chemistry (Figure 1). This non-native activity is enhanced by binding of non-native small-molecule ligands and has been improved by directed evolution.
Figure 1.

Small-molecule activation of a non-heme iron center for nitrene transfer. Carboxylate-containing ligands α-ketoglutarate, N-oxalylglycine, and acetate modulate the nitrene-transfer activity of variants of P. savastanoi ethylene-forming enzyme.
To search for abiological catalytic promiscuity among natural metalloproteins, we looked at α-ketoglutarate (αKG)-dependent iron enzymes, a family of enzymes which features a conserved metal-binding active site with iron coordinated by two histidines and one aspartate or glutamate4. In nature, these enzymes perform similar chemistry to the heme-binding cytochrome P450 family, in which a high-valent iron-oxo intermediate performs C–H hydroxylation, olefin epoxidation, or other oxidative transformations5. Though members of this enzyme family have been reported to catalyze reactions beyond their native functions, all the reactions reported proceed through the native iron-oxo mechanism6. We hypothesized that non-heme iron enzymes might also be able to catalyze abiological transformations similar to heme-binding proteins through a non-natural mechanistic pathway.
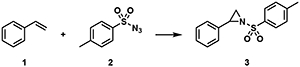
We screened a set of seven purified α-ketoglutarate (αKG)-dependent iron dioxygenases against the intermolecular aziridination reaction of styrene 1 and p-toluenesulfonyl azide 2. Aziridination7 and carbon–hydrogen (C–H) bond insertions of nitrenes8 have been reported using engineered heme-binding proteins and were subsequently proposed in a natural product biosynthetic pathway9. Chang and coworkers speculated the existence of a transient iron-nitrene intermediate in their report of the transformation of alkyl azides to nitriles by an αKG-dependent iron dioxygenase, but this reaction still proceeds through the canonical iron-oxo catalytic cycle6b. To date, no reported non-heme iron enzyme, natural or engineered, has been reported to catalyze productive nitrene transfer.
From the set of enzymes we tested, only Pseudomonas savastanoi ethylene-forming enzyme (PsEFE, UniProt ID P32021), formed aziridine 3 significantly above background (Supplementary Table S2). Compared to other members of the αKG-dependent iron dioxygenase family PsEFE is mechanistically and structurally distinct. While most enzymes of this family catalyze the oxidation of a substrate, often C–H hydroxylation, PsEFE natively catalyzes the fragmentation of the usual co-substrate α‑ketoglutarate to ethylene, as well as the hydroxylation of L-arginine10. Structurally, PsEFE possesses a hybrid fold, combining elements of both type I and type II αKG-dependent iron enzymes. It binds α-ketoglutarate in a strained conformation in an unusually hydrophobic pocket, which is likely responsible for the atypical catalytic activity11.
As the iron-binding site in PsEFE is quite unlike that of the heme-binding proteins that perform nitrene-transfer chemistry, we sought to characterize the necessary components of the reaction. Iron is required, with a single added equivalent of iron(II) sufficient to fully restore catalytic activity of the wild-type apoenzyme. PsEFE has three coordination sites filled by amino-acid side chains (two histidines and one aspartate), leaving up to three additional sites open for binding. In the native catalytic mechanism of PsEFE and other members of its family, α-ketoglutarate occupies two of these sites and is required for activity, as it is oxidatively decarboxylated to succinate to generate the reactive iron-oxo intermediate. PsEFE has been shown to catalyze arginine hydroxylation with α-ketoadipate instead of α-ketoglutarate, but with 500-fold lower activity. Other α-ketoacids were reported to give no activity12. Nitrene transfer, however, does not proceed through the native catalytic cycle and therefore does not require αKG as a co-substrate; the αKG is now more a ligand and as such could potentially be replaced by different small-molecule ligands. Intrigued by the possibility of modulating enzyme activity by changing the primary coordination sphere of the catalytic iron, we tested PsEFE for aziridination with a set of α-ketoglutarate mimics and related molecules as additives. We found that whereas addition of a carboxylate is beneficial for activity (though not required), the wild-type enzyme is significantly more active for aziridination with added acetate or N-oxalylglycine (NOG, a general αKG-dependent enzyme competitive inhibitor4) compared to α-ketoglutarate (Table 1).
Table 1.
Aziridination catalyzed by wild-type PsEFE
 | |
|---|---|
| Deviation from standard conditions1 | Relative activity |
| None | 1.00 |
| No iron | 0.08 |
| No αKG | 0.52 |
| Acetate2 instead of αKG | 6.72 |
| N-oxalylglycine3 instead of αKG | 7.75 |
| Acetate instead of αKG, no ascorbic acid | 6.01 |
Standard conditions: Reactions were performed in MOPS buffer (20 mM pH 7.0) with 5% ethanol co-solvent, with 20 μM apoenzyme, 1 mM Fe(NH4)2(SO4)2, 1 mM αKG (as disodium salt), 1 mM L-ascorbic acid, and 10 mM 1 and 2.
Sodium salt.
Free acid.
We then sought to improve PsEFE for aziridination via directed evolution, targeting active-site residues with site-saturation mutagenesis and screening for enhanced activity. Details of our engineering strategy are in the Supporting Information. Initial screening for the first round was performed in the presence of exogenous α-ketoglutarate, but for validation of this round and for all subsequent evolution we screened with exogenous acetate. Acetate was chosen as the preferred ligand because it enhanced the activity of the wild-type enzyme significantly more than the native α-ketoglutarate, it is biologically ubiquitous, and it is inexpensive. Although α-ketoglutarate is the native ligand and is naturally present at near-millimolar intracellular concentration in Escherichia coli13, we reasoned that by supplementing the reaction medium with acetate we could evolve PsEFE to be dependent on acetate instead, despite screening under whole cell or lysate conditions.
After two rounds of site-saturation mutagenesis and one round of recombination, we found a variant with five mutations from the wild type (T97M R171L R277H F314M C317M, PsEFE MLHMM) which catalyzed the formation of 3 with 120 total turnover number (TTN) and 88% enantiomeric excess (ee) favoring the (R)-enantiomer (Figure 2A). Four of the five introduced mutations are in the binding pocket of the native substrate arginine and presumably are involved in substrate binding. The fifth beneficial mutation is at Arg-277, the residue whose guanidino group natively binds the distal carboxylate of α-ketoglutarate (Figure 2B). The R277H mutation likely interferes with binding of the native ligand α-ketoglutarate; as a result, PsEFE MLHMM shows no significant increase in aziridination activity when α-ketoglutarate is added, but an 11-fold increase when acetate is added (Supplementary Table S4). Thus the evolved MLHMM variant is highly activated by acetate but is no longer activated by α-ketoglutarate at all, demonstrating the tunability of the ligand dependence of PsEFE.
Figure 2.

Directed evolution of PsEFE for aziridination. (A) Evolutionary lineage. Reactions were performed in triplicate anaerobically with acetate and quantified by analytical HPLC-UV. Full experimental details are given in the Supporting Information. (B) Structural representation of PsEFE with mutated sites highlighted in orange; metal-coordinating residues H189, D191, and H268 are represented in sticks and Mn (the metal with which the protein was crystallized) is represented as a purple sphere (PDB ID: 6CBA).
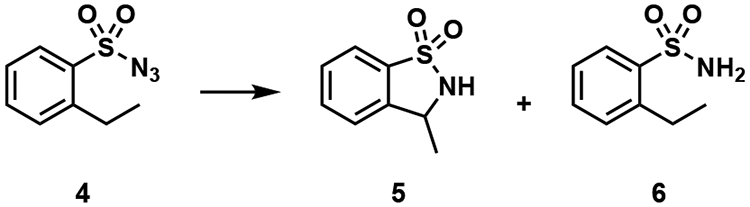
We reasoned that variants of PsEFE generated by directed evolution for aziridination could exhibit promiscuous activity for additional nitrene-transfer reactions beyond aziridination. Screening a panel of variants for the intramolecular C–H bond insertion reaction of 2-ethylbenzenesulfonyl azide 4 to form sultam 5, we identified multiple variants that perform this reaction with good TTN, excellent chemoselectivity, and moderate enantioselectivity. Remarkably, PsEFE R171V F314M C317M (PsEFE VMM) is significantly more active and more chemo- and enantioselective with N-oxalylglycine added than with either acetate or α-ketoglutarate, forming 5 with up to 730 TTN and greater than 100:1 selectivity for insertion over reduction (Table 2). This chemoselectivity is much higher than that of the heme proteins previously reported to catalyze similar reactions8a,b. To probe the specific effect of ligand binding on activity, we tested PsEFE R171V R227H F314M C317M (PsEFE VHMM), a variant in which αKG and NOG binding are disrupted by the R277H mutation. Whereas PsEFE VHMM’s activity is still enhanced nearly ten-fold with acetate, there is no significant difference between its activity with no additive, αKG, or NOG added to the reaction (Table 2). αKG or its analogues therefore appear to bind within the native αKG binding site, activating the protein through the primary metal coordination sphere.
Table 2.
Nitrene C–H insertion catalyzed by PsEFE
 | ||||
|---|---|---|---|---|
| PsEFE variant | Additive | TTN (5) | ee (%) | 5/6 |
| Wild type | Acetate | 12 | n.d.1 | 1.6 |
| VHMM | None | 27 | n.d.1 | 3.4 |
| VHMM | αKG | 31 | n.d.1 | 3.8 |
| VHMM | Acetate | 240 | 7.3 | 32 |
| VHMM | NOG | 33 | n.d.1 | 4.1 |
| VMM | None | 25 | n.d.1 | 0.9 |
| VMM | αKG | 130 | 61 | 9.0 |
| VMM | Acetate | 310 | 9.4 | 24 |
| VMM | NOG | 450 | 48 | 105 |
| VMM2 | NOG | 730 | 47 | 100 |
Reactions were performed anaerobically in MOPS buffer (20 mM pH 7.0) with 2.5% ethanol co-solvent, 20 μM apoenzyme, 1 mM Fe(NH4)2(SO4)2, 1 mM additive, 1 mM L-ascorbic acid, and 10 mM 4 (maximum 500 TTN). Reactions were quantified by analytical HPLC-UV. TTNs are shown for 5 only.
Not determined due to low conversion.
10 μM enzyme concentration (max. 1000 TTN).
Aziridination with PsEFE is reasonably oxygen tolerant, with the MLHMM variant maintaining 20% activity for aziridination when performed in air. C–H insertion, however, does not proceed detectably in aerobic conditions with any variant tested. These observations suggest that formation of the putative iron-nitrene intermediate is not significantly inhibited by oxygen, but that a subsequent mechanistic step in the C–H insertion reaction is inhibited aerobically. Unlike nitrene transfer with heme proteins8a, an additional reductant is not required when reactions are performed anaerobically (Supplementary Table S4).
PsEFE is highly expressed (>200 mg/L E. coli culture) and is catalytically active for nitrene transfer in whole E. coli cells and in cell lysate. We observe that in cell lysate the enzyme maintains high activity with no external additive; the enzyme presumably retains a ligand from the intracellular environment during lysis. Addition of N-oxalylglycine nevertheless enhances C–H insertion yield and chemoselectivity when PsEFE VMM is in cell lysate. In whole cells, however, there is no significant change upon addition of N-oxalylglycine (Supplementary Table S6). This is unsurprising as N-oxalylglycine is not reported to be cell-permeable. Future biochemical studies and further mutagenesis will likely enhance the selectivity for ligand analogues and impart in vivo activation to PsEFE variants.
In conclusion, we have discovered a non-heme iron enzyme capable of performing nitrene-transfer chemistry and enhanced that activity via directed evolution. This is the first example of enzymatic nitrene transfer catalyzed by a non-heme metalloprotein. PsEFE features a metal center whose primary coordination sphere can be altered by simple reaction additives, allowing for modulation of catalytic activity and selectivity. We anticipate that this biocatalytic system will lead to discovery of new metalloenzymatic transformations not possible with previously reported enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (NSF) Division of Molecular and Cellular Biosciences (grant MCB-1513007). N. W. G., A. M. K., and R. K. Z. acknowledge support from the NIH training grants NIH T32 GM07616 (N. W. G.) and NIH T32 GM112592 (A. M. K., R. K. Z.) and NSF Graduate Research Fellowship DGE-1144469 (A. M. K, R. K. Z.). We thank Sabine Brinkmann-Chen for critical reading of the manuscript and Noah P. Dunham, S. B. Jennifer Kan, and Benjamin J. Levin for helpful discussions. We thank Professor Hans Renata and Professor Harry Gray for generously sharing plasmids.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Materials and experimental methods, compound characterization data (PDF)
Full nucleotide and amino-acid sequences for all reported enzyme variants and sequences of all oligonucleotides used for mutagenesis (XLSX)
A provisional patent has been filed through the California Institute of Technology based on the results presented here.
REFERENCES
- (1).Brandenberg OF; Fasan R; Arnold FH Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Degtyarenko K Metalloproteins. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics; Jorde L; Little P; Dunn M; Subramaniam S, Eds.; John Wiley & Sons, Ltd: New York, 2005. [Google Scholar]
- (3).Holm RH; Kennepohl P; Solomon EI Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev 1996, 96, 2239–2314. [DOI] [PubMed] [Google Scholar]
- (4).Hausinger RP Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- (5).Islam MD; Leissing TM; Chowdhury R; Hopkinson RJ; Schofield CJ 2-Oxoglutarate-Dependent Oxygenases. Annu. Rev. Biochem 2018, 87, 585–620. [DOI] [PubMed] [Google Scholar]
- (6)(a).Matthews ML; Chang W.-c.; Layne AP; Miles LA; Krebs C; Bollinger JM Jr. Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol 2014, 10, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davidson M; McNamee M; Fan R; Guo Y; Chang W.-c. Repurposing Nonheme Iron Hydroxylases To Enable Catalytic Nitrile Installation through an Azido Group Assistance. J. Am. Chem. Soc 2019, 141, 3419–3423. [DOI] [PubMed] [Google Scholar]; (c) Neugebauer ME; Sumida KH; Pelton JG; McMurry JL; Marchand JA; Chang MCY A family of radical halogenases for the engineering of amino-acid-based products. Nat. Chem. Biol 2019, 15, 1009–1016. [DOI] [PubMed] [Google Scholar]
- (7).Farwell CC; Zhang RK; McIntosh JA; Hyster TK; Arnold FH Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent. Sci 2015, 1, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8)(a).McIntosh JA; Coelho PS; Farwell CC; Wang ZJ; Lewis JC; Brown TR; Arnold FH Enantioselective Intramolecular C–H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew. Chem. Int. Ed 2013, 52, 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh R; Bordeaux M; Fasan R P450-Catalyzed Intramolecular sp3 C–H Amination with Arylsulfonyl Azide Substrates. ACS Catal 2014, 4, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme. Nat. Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tsutsumi H; Katsuyama Y; Izumikawa M; Takagi M; Fujie M; Satoh N; Shin-ya K; Ohnishi Y Unprecedented Cyclization Catalyzed by a Cytochrome P450 in Benzastatin Biosynthesis. J. Am. Chem. Soc 2018, 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- (10).Fukuda H; Ogawa T; Tazaki M; Nagahama K; Fujii T; Tanase S; Morino Y Molecular cloning in Escherichia coli, expression, and nucleotide sequence of the gene for the ethylene-forming enzyme of Pseudomonas syringae pv. phaseolicola PK2. Biochem. Biophys. Res. Commun 1992, 188, 483–489. [DOI] [PubMed] [Google Scholar]
- (11)(a).Zhang Z, Smart TJ; Choi H; Hardy F; Lohans CT; Abboud MI; Richardson MSW; Paton RS; McDonough MA; Schofield CJ Structural and stereoelectronic insights into oxygenasecatalyzed formation of ethylene from 2-oxoglutarate. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 4467–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martinez S; Fellner M; Ferr CQ; Ritchie A; Hu J; Hausinger RP Structures and Mechanisms of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Substrate Binding Creates a Twist. J. Am. Chem. Soc 2017, 139, 11980–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Martinez S; Hausinger RP Biochemical and Spectroscopic Characterization of the Non-Heme Fe(II)- and 2-Oxoglutarate-dependent Ethylene-Forming Enyzme from Pseudomonas syringae pv. phaseolicola PK2. Biochemistry 2016, 55, 5989–5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bennett BD; Kimball EH; Gao M; Osterhout R; Van Dien SJ; Rabinowitz JD Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol 2009, 5, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.