

Abstract
An age-associated increase in chronic, low-grade sterile inflammation termed “inflammaging” is a characteristic feature of mammalian aging that shows a strong association with occurrence of various age-associated diseases. However, the mechanism(s) responsible for inflammaging and its causal role in aging and age-related diseases are not well understood. Age-associated accumulation of damage-associated molecular patterns (DAMPs) is an important trigger in inflammation and has been proposed as a potential driver of inflammaging. DAMPs can initiate an inflammatory response by binding to the cell surface receptors on innate immune cells. Programmed necrosis, termed necroptosis, is one of the pathways that can release DAMPs, and cell death due to necroptosis is known to induce inflammation. Necroptosis-mediated inflammation plays an important role in a variety of age-related diseases such as Alzheimer’s disease, Parkinson’s disease, and atherosclerosis. Recently, it was reported that markers of necroptosis increase with age in mice and that dietary restriction, which retards aging and increases lifespan, reduces necroptosis and inflammation. Genetic manipulations that increase lifespan (Ames Dwarf mice) and reduce lifespan (Sod1−/− mice) are associated with reduced and increased necroptosis and inflammation, respectively. While necroptosis evolved to protect cells/tissues from invading pathogens, e.g., viruses, we propose that the age-related increase in oxidative stress, mTOR signaling, and cell senescence results in cells/tissues in old animals being more prone to undergo necroptosis thereby releasing DAMPs, which contribute to the chronic inflammation observed with age. Approach to decrease DAMPs release by reducing/blocking necroptosis is a potentially new approach to reduce inflammaging, retard aging, and improve healthspan.
Keywords: Necroptosis, Aging, Inflammation, Oxidative stress, Cell senescence, mTOR
Introduction
Chronic, low-grade sterile inflammation that occurs with age (inflammaging) has been observed in all mammalian species studied, e.g., rodents (Brubaker et al. 2011), rhesus monkeys (Didier et al. 2012), and humans (Franceschi and Campisi 2014), and has been identified as one of the “seven pillars of aging” (Kennedy et al. 2014). According to inflammaging theory, “physiological or pathological aging can be driven by the proinflammatory cytokines and substances produced by the innate immune system” (Franceschi et al. 2000; Goto 2008); therefore, inflammaging has been put forward as a mediator of reduced healthspan and unsuccessful aging in humans (Franceschi et al. 2007). Because inflammation is strongly associated with a variety of diseases (e.g., type 2 diabetes, cardiovascular disease, cancer, neurodegenerative diseases such as Alzheimer’s disease, and frailty), it has been argued that inflammaging is also an important factor in the etiology of most age-related diseases (Franceschi and Campisi 2014). In support of this argument are the studies showing that disease and environmental conditions that reduce lifespan (e.g., obesity, human immunodeficiency virus-infection, and exposure to cigarette smoke) are associated with increased inflammation (Iantorno et al. 2014; Deeks 2011; Lee et al. 2012), and interventions that increase lifespan in mice, e.g., dietary restriction (Spaulding et al. 1997), dwarfism (Masternak and Bartke 2012), and rapamycin treatment (Richardson et al. 2015) reduce inflammation. These data have led to the generally accepted view that inflammation plays an important role in the underlying mechanisms of aging (Franceschi and Campisi 2014). However, almost all the data in support of role of inflammation in aging are correlative. There are two studies that have directly tested the role of inflammation in aging. Zhang et al. (2013a) showed that inhibition of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway (which regulates the expression of various pro-inflammatory genes), in the hypothalamus of mice reduced brain inflammation, extended lifespan, and improved healthspan of mice. However, this study failed to test the effect of this genetic manipulation on systemic inflammation and it is possible that blocking NF-κB activation affected the brain and aging through processes other than inflammation. Second, Youm et al. (2013) reported that mice deficient in Nod-like receptor nucleotide-binding domain leucine rich repeat containing protein 3 (NLRP3) inflammasome (Nlrp3−/− mice), which is required for the caspase-1-dependent secretion of interleukin-1 beta (IL-1β) and IL-18, have reduced inflammation and improved measures of healthspan, such as cognition and memory, rotarod performance, bone loss, and glycemic control. However, no lifespan data for the Nlrp3−/− mice was presented.
One of the common features of inflammaging is the age-related increase in the level of circulating proinflammatory cytokines such as IL-6, tumor necrosis factor-α (TNF-α), and IL-1β (Hager et al. 1994; Pedersen et al. 2003; Ferrucci et al. 2005; Roubenoff et al. 1998). C-reactive protein (CRP) is yet another factor that has been shown to be associated with inflammaging in humans (Ahmadi-Abhari et al. 2013; Puzianowska-Kuźnicka et al. 2016). Increased levels of IL-6 and TNF-α in the serum of elderly are associated with disease, disability, and mortality (De Martinis et al. 2005) and elevated IL-1β levels are associated with the development of several age-related degenerative diseases, including type 2 diabetes and Alzheimer’s disease (Youm et al. 2011; Heneka et al. 2013).
Although the exact cause of inflammaging is not known, cell senescence (Campisi and d'Adda di Fagagna 2007; Baker et al. 2011), immune senescence (Franceschi et al. 2000; McElhaney and Effros 2009), increased gut permeability or changes in oral microbiota (Fransen et al. 2017), and changes in the coagulation system (Biagi et al. 2011) have been proposed to play a role in inflammaging. Age-associated accumulation of damage-associated molecular patterns (DAMPs) is yet another important factor that has been proposed as a potential driver of inflammaging (Goldberg and Dixit 2015). In this review, we will discuss the potential role of DAMPs and a novel inflammatory cell death pathway, necroptosis in inflammaging and age-associated diseases and review the possible mechanisms of how necroptosis could potentially be activated with age.
Role of DAMPs in inflammaging
DAMPs are self-molecules that can initiate an inflammatory response through activation of the innate immune system (Seong and Matzinger 2004; Feldman et al. 2015). Under normal physiological conditions, DAMPs are sequestered inside the cell and are therefore hidden from recognition by the immune system. However, conditions that cause cell death or tissue injury, which result in the release of these molecules into the extracellular environment, can trigger activation of the innate immune system (Land 2015). The innate immune system is the first line of host defense against pathogens and is activated by unique microbial molecules called pathogen-associated molecular patterns or PAMPs through the binding to pattern recognition receptors (PRRs) (Albiger et al. 2007). DAMPs initiate the inflammatory response through the activation of the same group of PRRs, and sustained activation of the innate immune system by DAMPs can lead to chronic inflammation and tissue injury (Land 2015).
Because DAMPs are potent inducers of inflammation, it is possible that DAMPs might play a role in the age-related increase in chronic inflammation. Pinti et al. (2014) reported a strong association between the age-related increase in circulating mitochondrial DNA (mtDNA), a DAMP, and increase in inflammatory cytokines in humans. Circulating mtDNA levels gradually increase after 50 years of age, and subjects with the higher mtDNA plasma levels had the higher amounts of circulating TNF-α, IL-6, RANTES (regulated on activation, normal T cell expressed and secreted), and IL-1rα, and subjects with the lowest mtDNA levels had the lowest levels of the same cytokines. Importantly, they showed that treatment of monocytes with mtDNA also resulted in increased production of TNF-α, in vitro, providing direct evidence for the role of mtDNA in inflammation and possibly inflammaging. An age-associated increase in the circulating levels of the DAMP, high mobility group protein B1 (HMGB1) has been reported in mice (Davalos et al. 2013).
When cells rupture, DAMPs are released from different cellular compartments such as extracellular matrix (e.g., fibronectin, heparan sulfate), the nucleus (e.g., DNA, histones, HMGB1, and IL-1α), cytosol (e.g., S100 proteins, heat shock proteins (HSPs), amyloid beta, uric acid, ATP), mitochondria (e.g., mtDNA, Tfam), endoplasmic reticulum (e.g., calreticulin), and plasma membrane (e.g., syndecans and glypicans) (Schaefer 2014). A major source of DAMPs is necrosis, a non-regulated form of cell death. When the cell membrane ruptures in cells undergoing necrosis, there is a massive release of DAMPs into the extracellular space (Ellis and Horvitz 1986; Miura et al. 1993). Studies have shown that conditions that induce necrosis lead to the release of DAMPs (HMGB1, HSP70, ATP and IL-1α), and induce a strong inflammatory response both in vitro and in vivo (Kaczmarek et al. 2013).
Molecular mechanism of necroptosis and role of necroptosis in inflammation
Necrosis was initially thought to be an accidental (non-regulated) form of cell death because this cell death process is caspase-independent, in contrast to apoptosis. However, research over the past decade have shown that necrosis can also be programmed by a pathway called necroptosis (Degterev et al. 2005; Newton and Manning 2016; Pasparakis and Vandenabeele 2015). Cells undergoing necroptosis are characterized by cell swelling, loss of plasma membrane permeability, membrane rupture (Galluzzi et al. 2012), the release of DAMPs, such as HMGB1, S100 proteins, ATP, IL-33, IL-1α, HSP70, double stranded DNA (dsDNA) and mtDNA, and increased production of pro-inflammatory cytokines IL-6 and IL-1β (Moreno-Gonzalez et al. 2016). The key players in the necroptosis pathway are receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL) as shown in Fig. 1. Sequential activation of RIPK1 and RIPK3 is followed by phosphorylation and oligomerization of MLKL, then binding to and disruption of the cell membrane, and release of cellular components, such as DAMPs. The DAMPs bind to cell surface receptors on innate immune cells to trigger an inflammatory response.
Fig. 1.
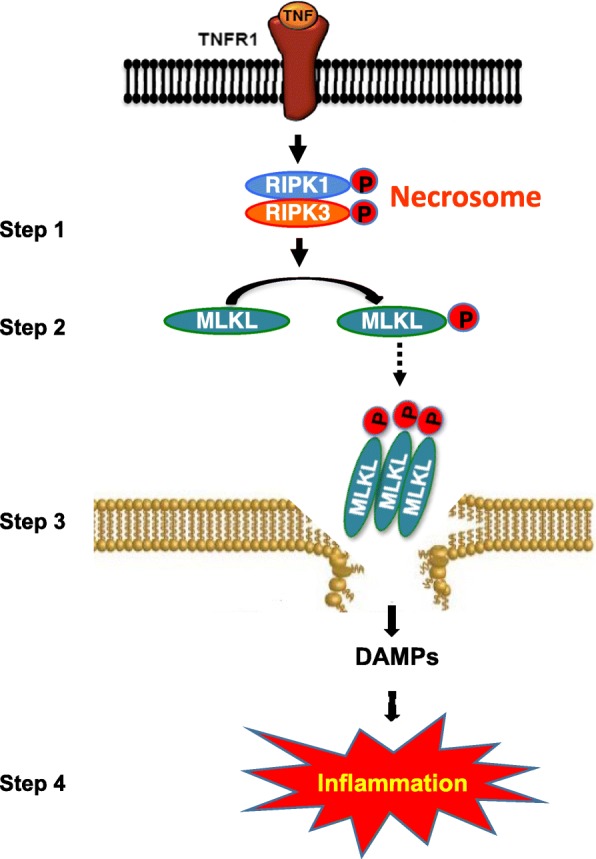
Schematic representation of TNF-α-induced necroptosis. Binding of TNF-α to its receptor, TNFR1, sequentially activates of RIPK1 and RIPK3 by phosphorylation, leading to the formation of necrosome, a complex of RIPK1 and RIPK3, which is a key event in necroptosis activation (step 1). This is followed by the phosphorylation of MLKL by active RIPK3, causing its oligomerization and membrane anchorage (step 2). Binding of oligomerized MLKL to the membrane causes its rapture and release of DAMPs (step 3). DAMPs bind to the cell surface receptors of innate immune cells, leading to increased transcription of proinflammatory cytokines and increased inflammation (step 4). RIPK, receptor-interacting protein kinase; MLKL, mixed lineage kinase domain-like protein
The term “necroptosis” was introduced by Degterev et al. 2005 when he observed that Necrostatin-1 [Nec-1, 5-(1H-indol-3-ylmethyl)-3-methyl-2-sulfanylidene-4-imidazolidinone] selectively inhibited TNF-α-induced necrosis. Later studies showed that Nec-1 blocks necroptosis by inhibiting the kinase activity of RIPK1, the first kinase in the necroptotic pathway (Degterev et al. 2008; Xie et al. 2013). Studies by Polykratis et al. (2014) have shown that mice expressing the kinase inactive form of Ripk1 (Ripk1D138N/D138N) are protected from TNF-induced necroptosis, in vivo, demonstrating that kinase activity of RIPK1 is essential for necroptosis. RIPK3 was identified as a crucial regulator of death receptor-induced necroptosis in 2009 (Cho et al. 2009; He et al. 2009; Zhang et al. 2009). Formation of necrosome, a complex of RIPK1 and RIPK3, was shown to be the key event in necroptosis activation (Linkermann and Green 2014; Vandenabeele et al. 2010) as shown in Fig. 1. Within the necrosome, the kinase-active conformation of RIPK1 is essential for the activation and autophosphorylation of RIPK3 (He et al. 2009). Similar to RIPK1, the kinase activity of RIPK3 is critical for necroptosis because kinase-inactive mutants of RIPK3 fail to reconstitute necroptosis in RIPK3-deficient cells (Cho et al. 2009; He et al. 2009; Zhang et al. 2009). In contrast to Ripk1−/− mice that die soon after birth (Kelliher et al. 1998), Ripk3−/− mice are viable and fertile and exhibit no obvious phenotype. While Ripk3−/− mice are resistant to TNF-induced hypothermia, mice expressing catalytically-inactive form of Ripk3 (RIPK3D161N) exhibit perinatal lethality due to apoptosis mediated by RIPK1 and caspase-8, suggesting that both necroptosis and apoptosis are held in balance by RIPK3 (Newton et al. 2014).
The RIPK3-mediated phosphorylation of the kinase-like domain of MLKL is the next key step in the necroptosis pathway (Wang et al. 2014) as shown in Fig. 1. Phosphorylation of MLKL results in its oligomerization, which exposes a motif in the N-terminus of MLKL that triggers the translocation of MLKL to the plasma membrane. Phosphatidylinositol phosphate within the plasma membrane interacts with the exposed motif on MLKL, leading to the disruption of plasma membrane through a yet to be identified mechanism (Murphy et al. 2013; Dondelinger et al. 2014). Similar to Ripk3−/− mice, Mlkl−/− mice are viable and exhibit no obvious phenotype. Cells derived from Mlkl−/− mice are resistant to TNF-induced necroptosis demonstrating that MLKL is a critical effector molecule in the execution of necroptosis (Murphy et al. 2013). As shown in Table 1, studies have shown that use of genetic and pharmacological manipulations that inhibit necroptosis can reduce inflammation induced in a variety of systems, in vivo, demonstrating the importance of necroptosis in inflammation.
Table 1.
Use of genetic and pharmacological manipulations that inhibit necroptosis can reduce inflammation, in vivo
| Mouse model | Mode of inhibition | Outcome | Proinflammatory cytokines | Reference |
|---|---|---|---|---|
| Neonatal hypoxia-ischemia | Nec-1 | Reduced oxidative damage, inflammation, and neuronal cell death | IL-1β, IL-6, IL-12, and TNFα | Northington et al. 2011 |
| TNF-induced systemic inflammatory response syndrome |
Nec-1 or Ripk3−/− |
Protected against SIRS, reduced circulating DAMPs and inflammation | IL-1β and IL-1 | Duprez et al. 2011 |
| Dextran sulfate sodium (DSS)–induced colitis | Nec-1 |
Suppressed colitis symptoms, tumor growth associated with colitis, and inflammation |
IL-8, IL-1β, and Il-6 | Liu et al. 2015 |
| Chronic obstructive pulmonary disease | Nec-1 | Reduced DAMPs and neutrophils in bronchoalveolar fluid | IL-6 and IL-8 | Pouwels et al. 2016 |
| Ischemic brain injury | Nec-1 | Prevented cognitive impairment and reduced inflammatory response | TNFα, IFNγ, and IL-1β | Zhang et al. 2016 |
| Renal interstitial fibrosis | Nec-1 | Reduced inflammatory response and interstitial fibrosis | TNFα, IL-1β, and MCP-1 | Xiao et al. 2017 |
| Retinal degeneration model of rd1 mice | Nec-1 | Supress microglia-mediated inflammation, rescue retinal degeneration, and prevented neural injury | TNFα, Ccl2, IL-17 | Huang et al. 2018 |
| Post-operative cognitive impairments in d-galactose-induced aged mice | Nec-1 | Reduced cognitive impairment and alleviated postoperative amplified neuroinflammation in hippocampus | IL-1α, IL-1β, TNF-α | Duan et al. 2018 |
| Retinal infusion-reperfusion (IR) injury | Nec-1 | Reduced retinal damage after IR | IL-1β, Nos2, Ccl2, Ccl5, Cxcl10 | Dvoriantchikova et al. 2014 |
| Cuprizone-mediated demyelination, a model of multiple sclerosis | Nec-1s | Protected against oligodendrocyte cell death, demyelination, and inflammation | TNFα | Ofengeim et al. 2015 |
| Optn−/− mice, a model of ALS | Nec-1s or Ripk1D138N/D138N mice | Reduced dysmyelination, axonal degeneration, and inflammation | IL-1α, IL-1β, IL2, IL12, IFNγ and TNFα | Ito et al. 2016 |
| APP/PS1, a mouse model of AD | Nec-1s or Ripk1D138N/D138N mice | Reduces CNS Inflammation, AD pathology and improved behavioral deficits | TNFα and IL-1β | Ofengeim et al. 2017 |
| Controlled cortical impact traumatic brain injury | Ripk3−/− mice | Reduced inflammation, oxidative stress and improved cognitive function | TNFα, IL-6, and IL-1β | Liu et al. 2018 |
| Ethanol-induced steatosis | Ripk3−/− mice | Protected from steatosis, hepatocyte injury, and reduced inflammation | MCP-1, IL-6, and TNFα | Roychowdhury et al. 2013 |
| Methionine choline-deficient diet-induced liver steatosis | Ripk3−/− mice | Attenuated liver injury, steatosis, inflammation, fibrosis and oxidative stress. | TNFα, IL-1β | Afonso et al. 2015 |
| Atherosclerotic model, ApoE-knockout mice | Ripk3−/− mice | Delayed mortality, reduced atherosclerotic lesion and inflammation in atherosclerotic plaques | TNFα, IL-1α and IL2 | Meng et al. 2015 |
| Cerebral ischemia | Ripk3−/− and Mlkl−/− mice | Reduced lesion size in ischemia, locomotion recovery and reduced inflammation | TNFα, IL-18 | Yang et al. 2018 |
| Aging of mouse male reproductive system | Ripk3−/− and Mlkl−/− mice | Prevented age-associated decline of reproductive capacity and reduced inflammation in seminiferous tubules | TNFα | Li et al. 2017 |
| High fat diet–induced non-alcoholic fatty liver disease | Mlkl−/− mice | Protected against non-alcoholic steatohepatitis and reduced inflammation | TNFα, MCP1, IL6 | Saeed et al. 2019 |
| Dextran sulfate sodium-induced colitis | Mlkl−/− mice | Reduces colonic damage, inflammation and disruption of intestinal mucosal barrier integrity | TNFα, IL6, IL-1β and KC | Zhang et al. 2019a |
| Japanese encephalitis (JE) virus infection | Mlkl−/− mice | Alleviated the progression of JE and decreased the level of inflammatory cytokines | IL-1β, Ccl2, IFNγ, TNFα | Bian et al. 2017 |
| Ischemia-Reperfusion (IR) Injury of Steatotic Livers | Mlkl−/− mice | Reduced hepatic neutrophil infiltration and inflammation, and protected against hepatic IR injury | TNFα, IL-1β, IL6 | Ni et al. 2019 |
Role of necroptosis in aging and inflammaging
Because necroptosis is a major source of DAMPs, we were interested in determining whether the increased levels of circulating DAMPs with age could be due to an age-related increase in necroptosis. We measured the levels of the necroptosis marker, phosphorylated MLKL and MLKL in epididymal white adipose (eWAT) of mice, one of the major tissues involved in the production and secretion of proinflammatory cytokines. As shown in Fig. 2a, we found a 2.7-fold increase in the levels of phosphorylated MLKL and 3.5-fold increase in MLKL protein with age. The age-associated increase in necroptosis was paralleled by an increase in 14 inflammatory cytokines, including the proinflammatory cytokines IL-6, TNF-α, and IL-1β, and 11 chemokines in old mice. We next tested the effect of dietary restriction (DR) on necroptosis because DR increases the lifespan of a wide variety of species and has been shown to reduce inflammation in mammals (Spaulding et al. 1997). As shown in Fig. 2a, DR reduced necroptosis in eWAT of old mice, e.g., the levels of phosphorylated MLKL and MLKL were reduced to levels similar to young/adult mice. Importantly, DR also attenuated the expression of IL-6, TNF-α, and IL-1β as well as 85% of the other cytokines/chemokines induced with age. These were the first data showing that necroptosis increased with age (Deepa et al. 2018).
Fig. 2.

Changes in necroptosis and inflammation with age, in a mouse model of extended lifespan (Ames Dwarf mice), and in a mouse model of accelerated aging (Sod1−/− mice). a Left panel: graphical representation of quantified immunoblots of eWAT extracts from adult male (9-month-old, blue bar), old (25- to 29-month-old, red bar), and old-DR (25- to 29-month-old mice fed a DR diet starting at 4 months of age, green bar) mice for P-MLKL and MLKL, normalized to β-tubulin (n = 5–6/group). Right panel: graphical representation of transcript levels of IL-6, TNF-α, and IL-1β in eWAT, normalized to β-microglobulin (data taken from Deepa et al. 2018). b, c Left panel: immunoblots of eWAT extracts from 11-month-old male control mice (blue bar) and Ames Dwarf (Df/Df) mice (red bar) (b), and 9-month-old male control mice (blue bar) and Sod1−/− mice (red bar) (c) for P-MLKL, MLKL, and β-tubulin (n = 6/group). Middle panel: graphical representation of quantified blots normalized to β-actin. Right panel: graphical representation of transcript levels of IL-6, TNF-α and IL-1β in eWAT of Df/Df mice (b) and Sod1−/− mice (c), normalized to β-microglobulin. Data shown are mean ± SEM. p < 0.05 is taken as significant for the following: *adult vs old/control vs Df/Df/control vs Sod1−/− mice; ^old vs old-DR; #adult vs old-DR
We have recently measured necroptosis in two mouse models that have been genetically modified to alter aging: one that shows an extension in lifespan and retarded aging and another that shows a reduction in lifespan and accelerated aging. Ames dwarf mice, which lack growth hormone, prolactin, and thyroid-stimulating hormone, show a > 50% increase in lifespan (Brown-Borg et al. 1996). As shown in Fig. 2b phosphorylated MLKL and MLKL protein levels are reduced 54% and 42%, respectively, in eWAT from 11-month-old dwarf (Df/Df) mice compared with age-matched control mice. Transcript levels of Mlkl in eWAT of Df/Df mice are also reduced by 60% (data not shown), which is consistent with the reduction in necroptosis. Importantly, transcript levels of IL-6 and IL-1β are also reduced by 78% and 53% in the eWAT of Ames Dwarf mice (Fig. 2b).
Mice deficient in the antioxidant enzyme Cu/Zn superoxide dismutase (Sod1−/− mice) exhibit nearly 30% reduction in lifespan, increased levels of circulating proinflammatory cytokines and many phenotypes of accelerated aging (Zhang et al. 2013b; Deepa et al. 2019). As shown in Fig. 2c, phosphorylated MLKL was increased by 3-fold and MLKL protein expression was increased by 2-fold in the eWAT of 9-month-old Sod1−/− mice compared with age-matched WT mice. Consistent with the changes in MLKL protein levels, transcript levels of Mlkl were increased by 1.5-fold in the eWAT of Sod1−/− mice (data not shown). Similarly, transcript levels of proinflammatory cytokines IL-6 (2-fold), TNF-α (1.5-fold) and IL-1β (2.7-fold) were also increased in the eWAT of Sod1−/− mice (Fig. 2c).
Thus, extension of lifespan in Ames Dwarf mice and DR mice is associated with reduction in necroptosis, and reduction in lifespan in the Sod1−/− mice is associated with an increase in necroptosis. Importantly, these changes in necroptosis were associated with either reduced or increased expression of proinflammatory cytokines, which would be predicted if the changes in necroptosis were playing a role in inflammaging. These data suggest that DAMPs released when cells undergo necroptosis with age in WT mice or accelerated aging in Sod1−/− mice leads to the age-related increase in the expression of cytokines. Conversely, reduction in necroptosis observed in Ames Dwarf mice and DR mice leads to reduced generation of DAMPs, resulting in a reduced expression of proinflammatory cytokines.
Role of necroptosis in age-related diseases
Over the past 5 years, several studies have explored the role of necroptosis in inflammation associated with various age-related diseases (e.g., neurodegenerative diseases, atherosclerosis, and cancer) using genetic and pharmacological manipulations that block necroptosis. Neuroinflammation is a hallmark of various neurodegenerative diseases, which increase with age (Yuan et al. 2019). Brain aging is also characterized by chronic activation of M1/pro-inflammatory microglia and this age-dependent activation of microglia is reported across different species (Cribbs et al. 2012; Holtman et al. 2015, Norden and Godbout 2013). It is widely accepted that inflammation mediated by microglia is a major contributor to the pathogenesis of various age-related neurodegenerative diseases. While factors that could activate microglia with age are not completely understood, DAMPs released by damaged neurons is one of the known activators of microglia leading to neuroinflammation (Katsumoto et al., 2018; Sarlus and Heneka 2017). Age-related accumulation of myelin defects and axon loss is also reported to activate microglia in rhesus monkey (Shobin et al. 2017).
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease characterized by the progressive degeneration of motor neurons in brain and spinal cord (Rowland and Shneider 2001). Neuroinflammation is a prominent pathological signature in ALS and increasing evidence suggests that the increase in inflammation in the central nervous system contributes to the pathogenesis of ALS (McGeer and McGeer 2002; Winkeler et al. 2010; Calvo et al. 2010). Using two mouse models of ALS [optineurin-deficient mice (Optn−/−) and SOD1G93A transgenic mice], Ito et al. (2016) showed that necroptosis was increased in oligodendrocytes in the ALS mice and that blocking necroptosis either genetically (Ripk3−/− and/or Ripk1D138N/D138N mice) or pharmacologically (using Necrostatin-1s (Nec-1s, 7-Cl-O-Nec1)) reduced oligodendrocyte death, microglial inflammation and axonal degeneration. In Optn−/− mice, blocking necroptosis using Nec-1s, also improved vertical rearing activity in mice (Ito et al. 2016). Importantly, in SOD1G93A transgenic mice, inhibition of necroptosis using Nec1-s or Ripk3 deficiency delayed the onset of motor dysfunction and extended survival of SOD1G93A mice as shown in Fig. 3a (Ito et al. 2016).
Fig. 3.
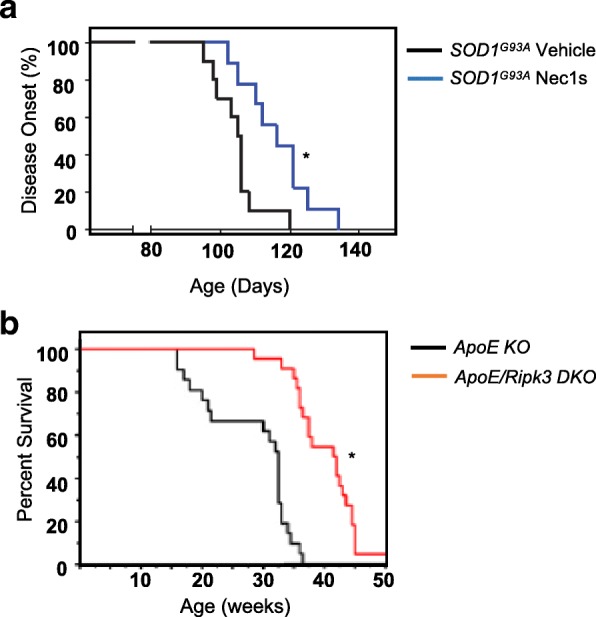
Effect of blocking necroptosis on lifespan. a Comparison of motor dysfunction onset in SOD1G93A mice treated with vehicle or Nec-1s for 1 month starting from 8 weeks of age. Data taken from Ito et al. 2016. b Survival of ApoE-single-knockout and Ripk3/ApoE-double-knockout mice fed high-cholesterol diet. Data taken from Meng et al. 2015
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline and by the presence of amyloid β plaques and neurofibrillary tangles. Chronic inflammation in the central nervous system is a hallmark of AD and is believed to play a central role in the progression of the neuropathological changes that are observed in AD (Mandrekar-Colucci and Landreth 2010). Caccamo et al. (2017) observed increased levels of necroptosis markers in postmortem human AD brains, and they showed that activation of necroptosis in an AD transgenic mouse model (APP/PS1 mice) exacerbated cognitive deficits as assessed by Morris water maze test (Caccamo et al. 2017). Other studies have shown that blocking necroptosis using Ripk1D138N/D138N mice or Nec-1s in APP/PS1 mice reduced amyloid burden, levels of inflammatory cytokines TNF-α and IL-1β, and memory deficits (Ofengeim et al. 2017; Degterev et al. 2013).
Parkinson’s disease (PD) is a neurodegenerative disease caused by the degeneration of dopaminergic neurons of the substantia nigra, leading to severe motor deficits, and inflammation is proposed to play a key role in the pathogenesis of PD (Deleidi and Gasser 2013). A recent study by Iannielli et al. (2018) showed that pharmacological inhibition of necroptosis using Nec-1s protected dopaminergic neurons from cell death in a mouse model of PD, optic atrophy type 1 (OPA1)–deficient mice. However, this study did not determine if blocking necroptosis reduced inflammation in the PD mouse model.
Age-related macular degeneration (AMD) is a leading cause of blindness, and chronic inflammation is reported to play a central role in this retinal degenerative disease. AMD is characterized by progressive degeneration of retinal pigment epithelium (RPE) and studies by Murakami et al. (2014) showed that necroptosis and inflammation are increased in a mouse model of dsDNA-induced retinal degeneration. They also showed that blocking necroptosis using Ripk3−/− mice reduced retinal degeneration and inflammation in this mouse model.
Atherosclerosis is also a disease of aging, as age is a major risk factor for the development of atherosclerosis (Wang and Bennett 2012). Apolipoprotein E–knockout (ApoE KO) mice is a model of cholesterol metabolic dysfunction and are widely used to study atherosclerosis because these mice show chronic inflammation and develop atherosclerotic plaques when fed a high-cholesterol diet (Libby 2002; Breslow 1996). Meng et al. (2015) reported that necroptosis and inflammation are increased in aortic plaques of ApoE-KO mice and blocking necroptosis genetically (ApoE/Ripk3-double-KO (DKO) mice) reduced inflammation as assessed by the reduction in the transcript levels of 10 inflammatory cytokines in the plaque regions of ApoE/Ripk3-DKO mice. In addition, lymphocyte infiltration was reduced in the adipose tissue. More importantly, the survival of the ApoE/Ripk3-DKO mice was significantly increased compared with ApoE-KO mice as shown in Fig. 3b.
Necroptosis also appears to play a role in the reproductive aging in mice. Aging of the reproductive system results in the reduction of reproductive capacity (Zirkin and Tenover 2012; Wang et al. 2017). Blocking necroptosis using Ripk3−/− and Mlkl−/− mice reduced inflammation and delayed aging of mice testes, both morphologically and functionally, suggesting a role of necroptosis in the aging of testes in mice (Li et al. 2017). Interestingly, induction of necroptosis in testes of young, wild type mice by local application of the necroptosis inducer TSZ (a combination of TNF-α, Smac mimetic, and caspase inhibitor z-VAD-FMK) resulted in a phenotype of male reproductive system aging, which was characterized by reduced fertility rate and depletion of cells in the seminiferous tubules. However, Ripk3−/− and Mlkl−/− mice were protected from such effects, suggesting a potential role of necroptosis and possibly inflammation in testis in male reproductive system aging.
Aging is also the major risk factor for cancer development and inflammation is a hallmark of both aging and cancer (Leonardi et al. 2018). Inflammation is the key driver of cancer growth and metastasis, and increased expression of TNF-α in the tumor microenvironment is a characteristic feature of many malignant tumors (Wu and Zhou 2009). In support of the role of necroptosis-induced inflammation in cancer, Liu et al. (2015) showed that Nec-1 treatment reduced inflammation and colitis-associated tumorigenesis in a mouse model of DSS-induced colitis. Similarly, blocking necroptosis by Ripk3 deletion or Nec1-s treatment protected mice from pancreas oncogenesis that was driven by the chemokine CXCL1 (Seifert et al. 2016). Tumor cell–induced necroptosis of endothelial cells has been shown to promote metastasis, and endothelial cell-specific deletion of Ripk3 or Mlkl or Nec-1 treatment reduced tumor cell-induced endothelial necroptosis, tumor cell extravasation and metastasis (Strilic et al. 2016). Chronic liver inflammation is the most important risk factor for the development of primary liver cancer that comprises hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC). Seehawer et al. (2018) showed that a necroptosis-associated hepatic cytokine microenvironment shifts HCC to ICC development and blocking necroptosis genetically or pharmacologically reverted the necroptosis-dependent cytokine microenvironment and changed ICC to HCC. (Seehawer et al. 2018). Sod1−/− mice that have a significantly shorter lifespan and exhibit various accelerated aging phenotypes are also characterized by a dramatic increase in HCC. Sod1−/− mice developed enlarged livers as early as 3 months of age with many of the mice developing HCC (Elchuri et al. 2005). We found significantly elevated transcript levels of necroptosis markers Ripk3 (1.5-fold) and Mlkl (3-fold), and proinflammatory cytokines IL-6 (2-fold), IL-1β (3.1-fold), and IL-1α (2.2-fold) in the liver of 5-month-old male Sod1−/− mice (Fig. 4a). Reducing the expression of Ripk3 by 50% in Sod1−/− mice (Sod1−/−Ripk3+/−) significantly attenuated the expression of IL-6, IL-1β and IL-1α and were comparable to the expression in age-matched wild type mice (Fig. 4b).
Fig. 4.
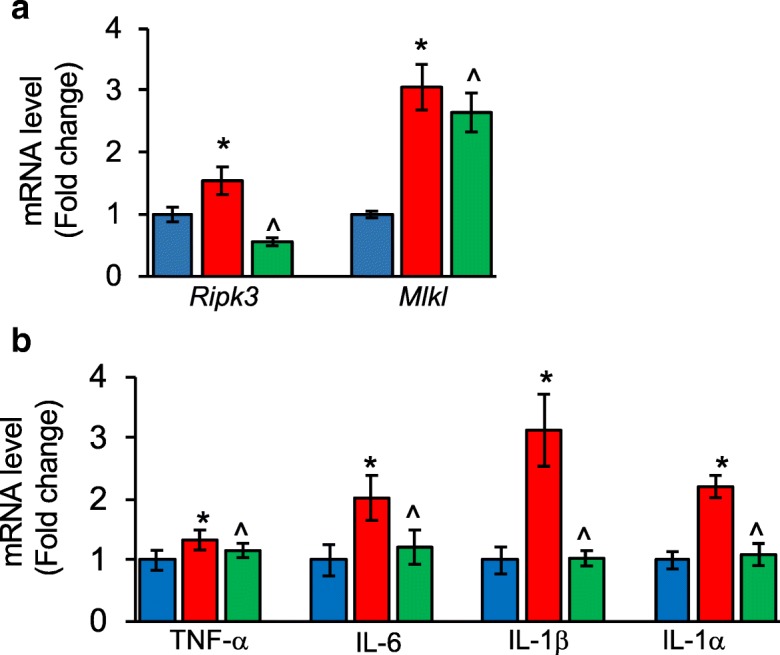
Necroptosis and inflammation in the liver of Sod1−/− mice. Transcript levels of Ripk3 and Mlkl (a), and transcript levels of TNF-α, IL-6, IL-1β and IL-1α (b) in the liver of 5-month-old control mice (blue bar), Sod1−/− mice (red bar), and Sod1−/−Ripk3+/− mice (green bar), normalized to β-actin. Data shown are mean ± SEM of 5 to 8 mice per group. p < 0.05 is taken as significant for the following: *control vs Sod1−/−; ^Sod1−/− vs Sod1−/−Ripk3+/−
In summary, research over the past 10 years has demonstrated that necroptosis-mediated inflammation plays an important role in a variety of age-related diseases based on the activation of necroptosis in each of these diseases. Importantly, blocking necroptosis during these age-related conditions resolved inflammation and reduced the progression of the disease, suggesting a potential role of necroptosis-mediated inflammation in disease development and progression.
Possible mechanism(s) responsible for age-related increase in necroptosis
Based on the current literature, the increase in necroptosis could arise through three pathways that have been shown to increase with age: oxidative stress, mTOR activation, and cell senescence. There is evidence that oxidative stress can induce necroptosis in certain conditions, and there is a great deal of data showing that oxidative stress, assessed by the levels of oxidative damage to lipid, DNA, and protein increase with age in a wide variety of tissues and animal models (Bokov et al. 2004). In support of the role of oxidative stress in necroptosis, in vitro studies have shown that high concentrations of hydrogen peroxide can induce necroptosis in RPE cells (Hanus et al. 2015), and oxidative stress induced by paraquat leads to necroptosis in cardiomyocytes (Zhang et al. 2018). Deletion of the antioxidant enzyme glutathione peroxidase 4 in hematopoietic cells resulted in increased ROS generation and necroptosis in erythroid precursor cells (Canli et al. 2016). Similarly, excessive acetaminophen treatment–induced ROS production and necroptosis in mice (Takemoto et al. 2014). Increased oxidative stress due to hyperoxia exposure also led to necroptotic cell death in the lung tissue of rats (Han et al. 2017). As shown in Fig. 2c we found that markers of necroptosis are increased in Sod1−/− mice, which show a dramatic increase in oxidative stress (Muller et al. 2006; Zhang et al. 2016). Based on the current data that oxidative stress can induce necroptosis, the age-related increase in oxidative stress is a prime candidate for the age-associated increase in necroptosis. Reduced efficiency of Nrf2, master regulator of redox homeostasis, could be one of the reasons for increased oxidative stress with age (Zhang et al. 2015; Schmidlin et al. 2019). In support of this, interventions that extend lifespan (CR and rapamycin) is reported to increase the expression of Nrf2 target genes (Hyun et al. 2006; Bruns et al. 2015) and naked mole rats that exhibit a remarkable extension in lifespan have increased Nrf2 signaling (Lewis et al. 2015). Similarly, administration of a known activator of Nrf2, conjugated linoleic acid, to older adults is reported to reduce systemic oxidative stress and skeletal muscle oxidative damage (Konopka et al. 2017). Thus, age-associated reduction in Nrf2 activity with age might contribute to age-related increase in necroptosis through induction of oxidative stress.
Necroptosis has also been reported to be induced by mTOR activation. Activation of mTOR has been observed to increase with age in various tissues (Baar et al. 2016) and increased mTORC1 signaling is associated with various age-related diseases such as Alzheimer’s disease (An et al. 2003, Caccamo et al. 2010), diabetes (Inoki et al.2011; Völkers et al.2014), and cancer (Bar-Peled et al.2013; Grabiner et al.2014). Dietary interventions (such as calorie restriction and protein restriction) and genetic manipulation (e.g., Ames Dwarf mice) that extend lifespan are associated with reduced mTORC1 signaling (Solon-Biet et al.2014; Lamming et al.2015; Sharp and Bartke 2005) and inhibition of mTOR by rapamycin extends the lifespan of yeast (Powers et al.2006), Drosophila melanogaster (Bjedov et al. 2010), C. elegans (Robida-Stubbs et al. 2012), and mice (Miller et al. 2011). Several studies suggest that mTOR pathway plays a role in activating necroptosis. In the hippocampal neuronal cell line HT22, induction of necroptosis was blocked by the combined treatment of Akt and mTOR inhibitors, suggesting a potential role of Akt-mTOR pathway in necroptosis (Liu et al. 2014). In the mouse fibroblast cell line L929, activation of the PI3K-Akt-mTOR signaling pathway by insulin promotes necrotic cell death via suppression of autophagy (Wu et al. 2009). Similarly, treatment of schwannoma cells with lithium chloride (a chemical that reduces cancer risk) induces necroptosis through activation of the Akt-mTOR pathway. Aberrant activation of mTOR by genetic deletion of TSC1 in intestinal epithelial cells resulted in the overexpression of RIPK3, epithelial necrosis, and subsequent colitis (Xiao 2018). A strong association between mTOR pathway and neuroinflammation is also reported. In a mouse model of cerebral palsy, mTOR inhibitor rapamycin has been shown to prevent neuroinflammation and neuronal cell death (Srivastava et al. 2016). Rapamycin treatment has also been shown to block lipopolysaccharide-induced neuroinflammation in rats (Mengke et al. 2016). Rapamycin injection after focal ischemic stroke in rats reduced production of proinflammatory cytokines and chemokines by macrophages and microglia, and blocked brain macrophage polarization towards the M1 type (Xie et al. 2014). A dual inhibitor of mTORC1 and mTORC2, KU0063794, is shown to reduce neuroinflammation associated with spinal cord injury in mice (Cordaro et al. 2017). These findings show a strong association between mTOR inhibition and reduced neuroinflammation suggesting that mTOR-mediated activation of necroptosis might play a role in neuroinflammation. However, this possibility needs to be tested in the future.
Another pathway that could contribute to necroptosis is cellular senescence. Cell senescence is a cellular response to persistent DNA damage, which initiates several signaling cascades, resulting in an irreversible growth arrest. Senescent cells exhibit a secretory phenotype, characterized by the secretion of numerous proinflammatory cytokines (termed the senescence-associated secretory phenotype or SASP), which can alter the function of nearby normal cells (Coppé et al. 2008, 2010). Senescent cells accumulate with age in many tissues and are resistant to cell death/apoptosis. They are proposed to be an important factor in many age-related pathologies and inflammaging (Coppé et al. 2010; Baker et al. 2011). The proinflammatory cytokine TNF-α is one of the SASPs secreted by the senescent cells, and the circulating levels of TNF-α have been reported to be increased with age in humans (Bruunsgaard et al. 2003; Kirwan et al. 2001). Importantly, TNF-α is one of the well-characterized inducers of necroptosis (Laster et al. 1988), e.g., binding of TNF-α to its receptor TNFR1 induces necroptosis in various cell lines in the presence of a caspase inhibitor. Therefore, it is possible that age-associated increase in cell senescence, which results in increased proinflammatory cytokines, could trigger the increase in necroptosis in neighboring cells with age. Removal of senescent cells using senolytics reduced neuroinflammation and improved cognition in a mouse model of AD (Zhang et al. 2019b). Similarly, in a mouse model of tau-dependent neurodegenerative disease model, clearance of senescent cells using INK-ATTAC transgenic mice reduced neuroinflammation and improved cognitive function (Bussian et al. 2018). These studies support a role of senescence in neuroinflammation. It will be interesting to see whether removal of senescent cells could affect necroptosis markers in these mouse models.
Summary
Necroptosis most likely evolved as an alternative form of cell death to kill cells infected by viral pathogens and to promote inflammatory and immune responses to limit the spread of the viruses (Dondelinger et al. 2016). As shown in Fig. 5, viral RNA transcribed by the host genome following viral infection is sensed by Z-DNA binding protein 1 (ZBP1, also known as DAI or DLM-1), which dimerizes with and activates RIPK3. Activated RIPK3 then triggers necroptotic cell death through phosphorylation and membrane translocation of MLKL. Importantly, viral induced, ZBP1-dependent necroptosis depends on RIPK3 recruitment and does not require RIPK1 (Upton et al. 2012; Maelfait et al. 2017).
Fig. 5.
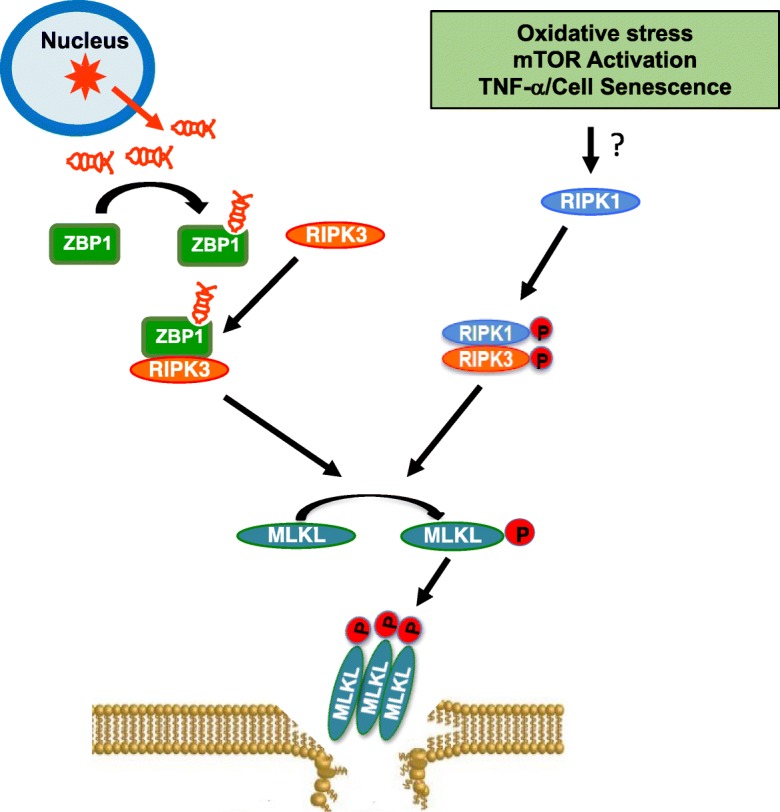
Necroptosis is an example of antagonistic pleiotropy. Following viral infection, viral RNA (shown in red) transcribed by the host genome is sensed by ZBP1, which dimerizes with RIPK3 through its RHIM domain to activate RIPK3. Activated RIPK3 triggers necroptotic cell death through phosphorylation and membrane translocation of MLKL and this process of independent of RIPK1. Necroptosis is also induced by the age-related activation of pathways such as oxidative stress, mTOR signaling, and cell senescence, possibly through RIPK1-RIPK3-MLKL pathway. ZBP1: Z-DNA binding protein, also known as DAI or DLM-1; RHIM, RIP homotypic interaction motifs.
While necroptosis evolved to protect organisms from viral infection, the age-related activation of pathways such as oxidative stress, mTOR signaling, and cell senescence could make the cells in old animals prone to undergo necroptosis and release DAMPs as shown in Fig. 5. We propose that the increase in necroptosis contributes to chronic inflammation that increases with age. Thus, necroptosis is an example of antagonistic pleiotropy, i.e., a process beneficial early in life that suppresses viral infection but is detrimental later in life when necroptosis is induced by oxidative stress, mTOR activation, or cell senescence resulting in inflammaging that in turn results in an age-related increase in pathology, disease and reduced physiological functions. Because recent studies show that blocking/reducing necroptosis either genetically or pharmaceutically can reduce inflammation, it is possible that similar treatments might prevent, reduce, or retard inflammaging and lead to improved lifespan/healthspan and a reduction in age-related diseases.
Funding information
This work was supported by NIH/NIA R01 AG059718, Oklahoma Center for the Advancement of Science and Technology research grant (HR18-053) and Presbyterian Health Foundation (OUHSC) Seed grant to Dr. Sathyaseelan S Deepa. The research was also supported by grants awarded to Dr. Arlan Richardson from the National Institute on Aging (P01AG020591, R01AG045693).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, Castro RE, Rodrigues CM. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) 2015;129:721–739. doi: 10.1042/CS20140732. [DOI] [PubMed] [Google Scholar]
- Ahmadi-Abhari S, Luben RN, Wareham NJ, Khaw KT. Seventeen year risk of all-cause and cause-specific mortality associated with C-reactive protein, fibrinogen and leukocyte count in men and women: the EPIC-Norfolk study. Eur J Epidemiol. 2013;28:541–550. doi: 10.1007/s10654-013-9819-6. [DOI] [PubMed] [Google Scholar]
- Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med. 2007;261:511–228. doi: 10.1111/j.1365-2796.2007.01821.x. [DOI] [PubMed] [Google Scholar]
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pe J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–166. doi: 10.1111/acel.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi E, Candela M, Franceschi C, Brigidi P. The aging gut microbiota: new perspectives. Ageing Res Rev. 2011;10:428–429. doi: 10.1016/j.arr.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Bian P, Zheng X, Wei L, Ye C, Fan H, Cai Y, Zhang Y, Zhang F, Jia Z, Lei Y. MLKL mediated necroptosis accelerates JEV-induced neuroinflammation in mice. Front Microbiol. 2017;8:303. doi: 10.3389/fmicb.2017.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. 2004;125:811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Brubaker AL, Palmer JL, Kovacs EJ. Age-related dysregulation of inflammation and innate immunity: lessons learned from rodent models. Aging Dis. 2011;2:346–360. [PMC free article] [PubMed] [Google Scholar]
- Bruns DR, Drake JC, Biela LM, Peelor FF, 3rd, Miller BF, Hamilton KL. Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxidative Med Cell Longev. 2015;2015:732596. doi: 10.1155/2015/732596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruunsgaard H, Ladelund S, Pedersen AN, Schroll M, Jørgensen T, Pedersen BK. Predicting death from tumour necrosis factor-alpha and interleukin-6 in 80-year-old people. Clin Exp Immunol. 2003;132:24–31. doi: 10.1046/j.1365-2249.2003.02137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–582. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between ammalian target of rapamycin (mTOR), amyloid-beta, and tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S. Necroptosis activation in Alzheimer’s disease. Nat Neurosci. 2017;20:1236–1246. doi: 10.1038/nn.4608. [DOI] [PubMed] [Google Scholar]
- Calvo A, Moglia C, Balma M, Chiò A. Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: a therapeutic opportunity? CNS Neurol Disord Drug Targets. 2010;9(3):325–330. doi: 10.2174/187152710791292657. [DOI] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5(2):e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaro M, Paterniti I, Siracusa R, Impellizzeri D, Esposito E, Cuzzocrea S. KU0063794, a Dual mTORC1 and mTORC2 inhibitor, reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. Mol Neurobiol. 2017;54:2415–2427. doi: 10.1007/s12035-016-9827-0. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613–629. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martinis M, Franceschi C, Monti D, Ginaldi L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005;579:2035–2039. doi: 10.1016/j.febslet.2005.02.055. [DOI] [PubMed] [Google Scholar]
- Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. 2011;62:141–155. doi: 10.1146/annurev-med-042909-093756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepa SS, Unnikrishnan A, Matyi S, Hadad N, Richardson A. Necroptosis increases with age and is reduced by dietary restriction. Aging Cell. 2018;17:e12770. doi: 10.1111/acel.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepa SS, Van Remmen H, Brooks SV, Faulkner JA, Larkin L, McArdle A, Jackson MJ, Vasilaki A, Richardson A. Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice. Free Radic Biol Med. 2019;132:19–23. doi: 10.1016/j.freeradbiomed.2018.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Gasser T. The role of inflammation in sporadic and familial Parkinson's disease. Cell Mol Life Sci. 2013;70:4259–4273. doi: 10.1007/s00018-013-1352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didier ES, Sugimoto C, Bowers LC, Khan IA, Kuroda MJ. Immune correlates of aging in outdoor-housed captive rhesus macaques (Macaca mulatta) Immun Ageing. 2012;9(1):25. doi: 10.1186/1742-4933-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016;26:721–732. doi: 10.1016/j.tcb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- Duan S, Wang X, Chen G, Quan C, Qu S, Tong J. Inhibiting RIPK1 limits neuroinflammation and alleviates postoperative cognitive impairments in D-galactose-induced aged mice. Front Behav Neurosci. 2018;12:138. doi: 10.3389/fnbeh.2018.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Dvoriantchikova G, Degterev A, Ivanov D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp Eye Res. 2014;123:1–7. doi: 10.1016/j.exer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Feldman N, Rotter-Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev. 2015;24:29–39. doi: 10.1016/j.arr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL. The origins of age-related proinflammatory state. Blood. 2005;105:2294–2299. doi: 10.1182/blood-2004-07-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S. Human immunosenescence: the prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine. 2000;18:1717–1720. doi: 10.1016/s0264-410x(99)00513-7. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, Savelkoul HFJ, De Jonge MI, Boekschoten MV, Smidt H, Faas MM, de Vos P. Aged gut microbiota contributes to systemical inflammaging after transfer to germ-free mice. Front Immunol. 2017;8:1385. doi: 10.3389/fimmu.2017.01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012;13:322–330. doi: 10.1038/embor.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev. 2015;265:63–74. doi: 10.1111/imr.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M. Inflammaging (inflammation + aging): a driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci Trends. 2008;2:218–230. [PubMed] [Google Scholar]
- Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager K, Machein U, Krieger S, Platt D, Seefried G, Bauer J. Interleukin-6 and selected plasma proteins in healthy persons of different ages. Neurobiol Aging. 1994;15:771–772. doi: 10.1016/0197-4580(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Han CH, Guan ZB, Zhang PX, Fang HL, Li L, Zhang HM, Zhou FJ, Mao YF, Liu WW. Oxidative stress induced necroptosis activation is involved in the pathogenesis of hyperoxic acute lung injury. Biochem Biophys Res Commun. 2017;495:2178–2183. doi: 10.1016/j.bbrc.2017.12.100. [DOI] [PubMed] [Google Scholar]
- Hanus J, Anderson C, Wang S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res Rev. 2015;24:286–298. doi: 10.1016/j.arr.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Möller T, Orre M, Kamphuis W, Hol EM, Boddeke EW, Eggen BJ. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zhou T, Sun X, Zheng Y, Cheng B, Li M, Liu X, He C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018;25:180–189. doi: 10.1038/cdd.2017.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A. 2006;103:19908–19912. doi: 10.1073/pnas.0608008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, Massimino L, Rubio A, Morabito G, Caporali L, Tagliavini F, Musumeci O, Gregato G, Bezard E, Carelli V, Tiranti V, Broccoli V. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018;22:2066–2079. doi: 10.1016/j.celrep.2018.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iantorno M, Campia U, Di Daniele N, Nistico S, Forleo GB, Cardillo C, Tesauro M. Obesity, inflammation and endothelial dysfunction. J Biol Regul Homeost Agents. 2014;28:169–176. [PubMed] [Google Scholar]
- Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Katsumoto A, Takeuchi H, Takahashi K, Tanaka F. Microglia in Alzheimer’s disease: risk factors and inflammation. Front Neurol. 2018;9:978. doi: 10.3389/fneur.2018.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan JP, Krishnan RK, Weaver JA, Del Aguila LF, Evans WJ. Human aging is associated with altered TNF-alpha production during hyperglycemia and hyperinsulinemia. Am J Physiol Endocrinol Metab. 2001;281:E1137–E1143. doi: 10.1152/ajpendo.2001.281.6.E1137. [DOI] [PubMed] [Google Scholar]
- Konopka AR, Laurin JL, Musci RV, Wolff CA, Reid JJ, Biela LM, Zhang Q, Peelor FF, 3rd, Melby CL, Hamilton KL, Miller BF. Influence of Nrf2 activators on subcellular skeletal muscle protein and DNA synthesis rates after 6 weeks of milk protein feeding in older adults. Geroscience. 2017;39:175–186. doi: 10.1007/s11357-017-9968-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Cummings NE, Rastelli AL, Gao F, Cava E, Bertozzi B, Spelta F, Pili R, Fontana L. Restriction of dietary protein decreases mTORC1 in tumors and somatic tissues of a tumor-bearing mouse xenograft model. Oncotarget. 2015;6:31233–31240. doi: 10.18632/oncotarget.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land WG. The role of damage-associated molecular patterns in human diseases: Part I - Promoting inflammation and immunity. Sultan Qaboos Univ Med J. 2015;15:e9–e21. [PMC free article] [PubMed] [Google Scholar]
- Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91:142–149. doi: 10.1177/0022034511421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi GC, Accardi G, Monastero R, Nicoletti F, Libra M. Ageing: from inflammation to cancer. Immun Ageing. 2018;15:1. doi: 10.1186/s12979-017-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, Buffenstein R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc Natl Acad Sci U S A. 2015;112:3722–3727. doi: 10.1073/pnas.1417566112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Meng L, Xu T, Su Y, Liu X, Zhang Z, Wang X. RIPK1-RIPK3-MLKL-dependent necrosis promotes the aging of mouse male reproductive system. Elife. 2017;6:e27692. doi: 10.7554/eLife.27692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Qiu J, Liang M, Golinski J, van Leyen K, Jung JE, You Z, Lo EH, Degterev A, Whalen MJ. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 2014;5:e1084. doi: 10.1038/cddis.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu BQ, Li JH, Jing L, Jiang JL, Tang J, Chen ZN. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5:3174–3185. [PMC free article] [PubMed] [Google Scholar]
- Liu ZM, Chen QX, Chen ZB, Tian DF, Li MC, Wang JM, Wang L, Liu BH, Zhang SQ, Li F, Ye H, Zhou L. RIP3 deficiency protects against traumatic brain injury (TBI) through suppressing oxidative stress, inflammation and apoptosis: Dependent on AMPK pathway. Biochem Biophys Res Commun. 2018;499:112–119. doi: 10.1016/j.bbrc.2018.02.150. [DOI] [PubMed] [Google Scholar]
- Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017;36:2529–2543. doi: 10.15252/embj.201796476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masternak Michal M., Bartke Andrzej. Growth hormone, inflammation and aging. Pathobiology of Aging & Age-related Diseases. 2012;2(1):17293. doi: 10.3402/pba.v2i0.17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhaney Janet E, Effros Rita B. Immunosenescence: what does it mean to health outcomes in older adults? Current Opinion in Immunology. 2009;21(4):418–424. doi: 10.1016/j.coi.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- Meng L, Jin W, Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112:11007–11012. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengke NS, Hu B, Han QP, Deng YY, Fang M, Xie D, Li A, Zeng HK. Rapamycin inhibits lipopolysaccharide-induced neuroinflammation in vitro and in vivo. Mol Med Rep. 2016;14:4957–4966. doi: 10.3892/mmr.2016.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Strong R. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Moreno-Gonzalez G, Vandenabeele P, Krysko DV. Necroptosis: a novel cell death modality and its potential relevance for critical care medicine. Am J Respir Crit Care Med. 2016;194:415–428. doi: 10.1164/rccm.201510-2106CI. [DOI] [PubMed] [Google Scholar]
- Muller Florian L., Song Wook, Liu Yuhong, Chaudhuri Asish, Pieke-Dahl Sandra, Strong Randy, Huang Ting-Ting, Epstein Charles J., Roberts L. Jackson, Csete Marie, Faulkner John A., Van Remmen Holly. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radical Biology and Medicine. 2006;40(11):1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y, Kayama M, Thanos A, Nakatake S, Notomi S, Hisatomi T, Ikeda Y, Ishibashi T, Connor KM, Miller JW, Vavvas DG. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014;21:270–277. doi: 10.1038/cdd.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem. 2016;85:743–763. doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Ni HM, Chao X, Kaseff J, Deng F, Wang S, Shi YH, Li T, Ding WX, Jaeschke H. Receptor-interacting serine/threonine-protein kinase 3 (RIPK3)-mixed lineage kinase domain-like protein (MLKL)-mediated necroptosis contributes to ischemia-reperfusion injury of steatotic livers. Am J Pathol. 2019;189:1363–1374. doi: 10.1016/j.ajpath.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. 2011;69:743–758. doi: 10.1002/ana.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, Das S, Adiconis X, Chen H, Zhu H, Kelliher MA, Levin JZ, Yuan J. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:E8788–E8797. doi: 10.1073/pnas.1714175114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Bruunsgaard H, Weis N, Hendel HW, Andreassen BU, Eldrup E, Dela F, Pedersen BK. Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech Ageing Dev. 2003;124:495–502. doi: 10.1016/s0047-6374(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, Benatti S, Gibellini L, Cotichini R, Stazi MA, Trenti T, Franceschi C, Cossarizza A. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur J Immunol. 2014;44:1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FKM, Pasparakis M, Kelliher MA. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193:1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R, Ten Hacken NH, Krysko DV, Vandenabeele P, de Vries M, van Oosterhout AJ, Heijink IH, Nawijn MC. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Phys Lung Cell Mol Phys. 2016;310:L377–L386. doi: 10.1152/ajplung.00174.2015. [DOI] [PubMed] [Google Scholar]
- Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzianowska-Kuźnicka M, Owczarz M, Wieczorowska-Tobis K, Nadrowski P, Chudek J, Slusarczyk P, Skalska A, Jonas M, Franek E, Mossakowska M. Interleukin-6 and C-reactive protein, successful aging, and mortality: the PolSenior study. Immun Ageing. 2016;13:21. doi: 10.1186/s12979-016-0076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Galvan V, Lin AL, Oddo S. How longevity research can lead to therapies for Alzheimer’s disease: the rapamycin story. Exp Gerontol. 2015;68:51–58. doi: 10.1016/j.exger.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubenoff R, Harris TB, Abad LW, Wilson PW, Dallal GE, Dinarello CA. Monocyte cytokine production in an elderly population: effect of age and inflammation. J Gerontol A Biol Sci Med Sci. 1998;53:M20–M26. doi: 10.1093/gerona/53a.1.m20. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed WK, Jun DW, Jang K, Oh JH, Chae YJ, Lee JS, Koh DH, Kang HT (2019) Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J Gastroenterol Hepatol. 10.1111/jgh.14740 [DOI] [PubMed]
- Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127:3240–3249. doi: 10.1172/JCI90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289:35237–32245. doi: 10.1074/jbc.R114.619304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin CJ, Dodson MB, Madhavan L, Zhang DD. Redox regulation by NRF2 in aging and disease. Free Radic Biol Med. 2019;134:702–707. doi: 10.1016/j.freeradbiomed.2019.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux PF, Hoenicke L, Dang H, Klotz S, Robinson L, Doré G, Rozenblum N, Kang TW, Chawla R, Buch T, Vucur M, Roth M, Zuber J, Luedde T, Sipos B, Longerich T, Heikenwälder M, Wang XW, Bischof O, Zender L. Author correction: Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;564:E9. doi: 10.1038/s41586-018-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, Torres-Hernandez A, Pergamo M, Ochi A, Zambirinis CP, Pansari M, Rendon M, Tippens D, Hundeyin M, Mani VR, Hajdu C, Engle D, Miller G. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–249. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Sharp ZD, Bartke A. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2005;60:293–300. doi: 10.1093/gerona/60.3.293. [DOI] [PubMed] [Google Scholar]
- Shobin E, Bowley MP, Estrada LI, Heyworth NC, Orczykowski ME, Eldridge SA, Calderazzo SM, Mortazavi F, Moore TL, Rosene DL. Microglia activation and phagocytosis: relationship with aging and cognitive impairment in the rhesus monkey. Geroscience. 2017;39:199–220. doi: 10.1007/s11357-017-9965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon-Biet SM, McMahon AC, Ballard JW, Ruohonen K, Wu LE, Cogger VC, Warren A, Huang X, Pichaud N, Melvin RG, Gokarn R, Khalil M, Turner N, Cooney GJ, Sinclair DA, Raubenheimer D, Le Couteur DG, Simpson SJ. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014;19:418–430. doi: 10.1016/j.cmet.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding CC, Walford RL, Effros RB. Calorie restriction inhibits the age-related dysregulation of the cytokines TNF-alpha and IL-6 in C3B10RF1 mice. Mech Ageing Dev. 1997;93:87–94. doi: 10.1016/s0047-6374(96)01824-6. [DOI] [PubMed] [Google Scholar]
- Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis. 2016;85:144–154. doi: 10.1016/j.nbd.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, Pasparakis M, Offermanns S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–218. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma N, Ohmae S, Toriguchi K, Tanabe K, Tanaka H, Seo S, Taura K, Machida K, Takeda N, Saji S, Uemoto S, Asagiri M. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Völkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S, Ornelas L, Thuerauf DJ, Gude N, Friedrich K, Herzig S, Glembotski CC, Sussman MA. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 2014;6:57–65. doi: 10.1002/emmm.201303183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen F, Ye L, Zirkin B, Chen H. Steroidogenesis in Leydig cells: effects of aging and environmental factors. Reproduction. 2017;154:R111–R122. doi: 10.1530/REP-17-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkeler A, Boisgard R, Martin A, Tavitian B. Radioisotopic imaging of neuroinflammation. J Nucl Med. 2010;51:1–4. doi: 10.2967/jnumed.109.065680. [DOI] [PubMed] [Google Scholar]
- Wu YT, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle. 2009;8:3267–3273. doi: 10.4161/cc.8.20.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YT, Tan HL, Huang Q, Ong CN, Shen HM. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 2009;5:824–834. doi: 10.4161/auto.9099. [DOI] [PubMed] [Google Scholar]
- Xiao H (2018) J Immunol May 1 200(1 Supplement)53.12
- Xiao X, Du C, Yan Z, Shi Y, Duan H, Ren Y. Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am J Nephrol. 2017;46:131–138. doi: 10.1159/000478746. [DOI] [PubMed] [Google Scholar]
- Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J, Shi Y. Structural basis of RIP1 inhibition by necrostatins. Structure. 2013;21:493–499. doi: 10.1016/j.str.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, Su DM, Simpkins JW, Greenberg DA, Jin K. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. 2014;192:6009–6019. doi: 10.4049/jimmunol.1303492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Zhang L, Fan H, Qi C, Zhang K, Liu X, Fei L, Chen S, Wang M, Kuang F, Wang Y, Wu S. RIPK3/MLKL-mediated neuronal necroptosis modulates the M1/M2 polarization of microglia/macrophages in the ischemic cortex. Cereb Cortex. 2018;28:2622–2635. doi: 10.1093/cercor/bhy089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, Dixit VD. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology. 2011;152:4039–4045. doi: 10.1210/en.2011-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Münzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013;497:211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ikeno Y, Bokov A, Gelfond J, Jaramillo C, Zhang HM, Liu Y, Qi W, Hubbard G, Richardson A, Van Remmen H. Dietary restriction attenuates the accelerated aging phenotype of Sod1(-/-) mice. Free Radic Biol Med. 2013;60:300–306. doi: 10.1016/j.freeradbiomed.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Davies KJA, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Radic Biol Med. 2015;88:314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang Y, Li D, Wu J, Si W, Wu Y. Necrostatin-1 attenuates inflammatory response and improves cognitive function in chronic ischemic stroke mice. Medicines (Basel) 2016;3(3):E16. doi: 10.3390/medicines3030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Feng Q, Wang T. Necrostatin-1 protects against paraquat-induced cardiac contractile dysfunction via RIP1-RIP3-MLKL-dependent necroptosis pathway. Cardiovasc Toxicol. 2018;18:346–355. doi: 10.1007/s12012-017-9441-z. [DOI] [PubMed] [Google Scholar]
- Zhang J, Qin D, Yang YJ, Hu GQ, Qin XX, Du CT, Chen W. MLKL deficiency inhibits DSS-induced colitis independent of intestinal microbiota. Mol Immunol. 2019;107:132–141. doi: 10.1016/j.molimm.2019.01.018. [DOI] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M, Mattson MP. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22:719–728. doi: 10.1038/s41593-019-0372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkin BR, Tenover JL. Aging and declining testosterone: past, present, and hopes for the future. J Androl. 2012;33:1111–1118. doi: 10.2164/jandrol.112.017160. [DOI] [PMC free article] [PubMed] [Google Scholar]