

Abstract
Potential inhibitors of a target biomolecule, NAD-dependent deacetylase Sirtuin 1, were identified by a contest-based approach, in which participants were asked to propose a prioritized list of 400 compounds from a designated compound library containing 2.5 million compounds using in silico methods and scoring. Our aim was to identify target enzyme inhibitors and to benchmark computer-aided drug discovery methods under the same experimental conditions. Collecting compound lists derived from various methods is advantageous for aggregating compounds with structurally diversified properties compared with the use of a single method. The inhibitory action on Sirtuin 1 of approximately half of the proposed compounds was experimentally accessed. Ultimately, seven structurally diverse compounds were identified.
Subject terms: Virtual drug screening, Information technology
Introduction
In the early stages of the drug discovery process, active compounds, inhibitors and/or activators, for a target biomolecule are sought. The high-throughput screening (HTS) campaign can be experimentally used to identify as many active compounds as possible from a compound library. However, the odds of finding active compounds using HTS is low. A previous study concluded that the success rate ranges from 0.05% to 0.14% depending on the class of target biomolecule (kinase, GPCR, protein-protein interaction, etc.)1. To improve odds, computer-aided drug discovery or virtual screening (VS) methods have been employed2,3.
VS methods utilize structural information of a target biomolecule and/or experimentally known actives/inactives. The former is known as structure-based VS (SBVS), and the latter, as ligand-based VS (LBVS). The representative of SBVS is docking study, which docks test compounds to a modeled target biomolecule. There are many parameters that are used in docking studies to determine the ranking of test compounds, e.g., how the target biomolecule is modeled, whether the target biomolecules are treated as a rigid body or a flexible body, how the compounds of interest are oriented in relation to the target molecule, and how the score of the placed compound is defined4,5. LBVS is based on a hypothesis that test compounds with similar properties of known actives would have similar activities. To rank the test compounds, a simple comparison of the similarity of the chemical structure or structure-activity relationship (SAR) using actives and inactives is performed6. Combining LBVS and SBVS is also common2.
Performance comparisons of various VS methods have been addressed using known active/inactive compounds; in other words, benchmarks have been conducted retrospectively7. Such benchmark results have led to obscure outcomes when VS methods are used in a prospective way. In addition, the performance of VS methods differs under various conditions, and no set standard exists8. Hence, currently we are not supplied with enough knowledge for choosing a suitable method for a designated compound library and target biomolecule before experimental validation.
Utilizing various VS methods for a certain problem is an alternative approach for relying on a single VS approach. For the various methods, we conducted two compound screening contests, in which participants were asked to propose potential active compounds of a target biomolecule from commercially available 2.2- and 2.4-million compound lists. The inhibitory functions of the proposed compounds were experimentally assessed. In the first and second contests held in 20149 and 201510, respectively, we identified several hit compounds with inhibitory activity of the target molecule, i.e., the tyrosine-protein kinase Yes. Participants employed various methods. In the second contest, we found that iterating the contest with the same target would give improved hit rates and enable identification of the statistically warranted method.
To verify robustness of the concept of the contest-based approach, we conducted another contest in 2016 employing a new target biomolecule, nicotinamide adenine dinucleotide (NAD)-dependent deacetylase Sirtuin 1, hereafter referred to as Sirtuin 1. Sirtuin 1 is a member of the sirtuin family (Sirtuin 1 to Sirtuin 7). The structure of Sirtuin 1 changes from an open state to a closed state upon the addition of cofactor (NAD+) and a substrate, and both open and closed structures have been reported (PDBID: 4IG9, 4KXQ)11. Other relevant structural information is also available (see Table 1). Information of actives and inactives has been deposited in open databases, such as BindingDB12,13 and ChEMBL14. Histone deacetylase (HDAC) inhibitors and decoys were previously compiled in the MUBD-HDACs database, in which information of Sirtuin 1 is included15. Hence, SBVS and LBVS can be employed for the target.
Table 1.
Summary of the methods used by participating groups.
| Group | Modeling of Sirtuin 1 structure | Ligand preparation | Processing method of compound library | |||
|---|---|---|---|---|---|---|
| 3D structure modeling/prediction methods/tools | PDB ID used | Filter class | Actives | Decoys | ||
| 1 | — | 4KXQ11 | OMEGA61 | LB→SB | Cambinol, HR73, salermide, sirtinol, suramin, and tenovin | |
| 2 | HM (FAMS)21 | 4BN520 | Open babel62 | LB→SB | Cocrystalized ligands in PDB and ChEMBL (IC50 < 1 μM) | — |
| 3 | — | — | PaDEL-Descriptor23 | LB | MUBD-HDACs63 | |
| 4 | — | 4KXQ | CORINA64 | Hybrid (LB&SB) | PubChem (CID703333, CID71459392) | — |
| 5 | — | 4ZZJ65 | Open babel. Dock27 | Hybrid (LB&SB) | Known Sirtuin inhibitors | — |
| 6 | — | — | RDKit66 | LB | ChEMBL (CHEMBL4506, CHEMBL4462, CHEMBL4461), PubChem (AID 652115), BindingDB (Target = NAD-Dependent Deacetylase Sirtuin 1) | |
| 7 | 4IG9,11 4IF6, 4ZZI,65 4I5I,33 4ZZJ | Ligprep67 | Hybrid (LB&SB) | Cocrystalized ligands | ||
| 8 | MD (myPresto, cosgene37) | 4ZZI | myPresto (AM1, AM1BCC) | Hybrid (LB → SB) | Selisistat (EX-527), Compound 2835 | — |
| 9 | HM and minimization (SWISS model, Foldit) | 4ZZI | Open babel, Discovery Studio visualizer | SB | ||
| 10 | HM (Prime)41,42 | 4ZZI (selected from 4I5I, 4IF6, 4IG9, 4KXQ, 4ZZI, 4ZZJ, 5BTR68) | LigPrep | Hybrid (LB&SB) | ChEMBL (IC50 < 20 μM) | ChEMBL (IC50 > 100 μM) |
| 11 | myPresto (tplgeneX)37,38 | 4I5I | myPresto (create3D) | SB | EX-527 analogue33 | DUD-E8 |
| 12 | — | — | RDKit | LB | BindingDB | ZINC47 |
| 13 | MD | 4I5I | OMEGA | Hybrid (LB, SB&visual inspection) | Sun et al.49 | — |
| 14 | — | 4ZZI | LigandBOX52 | Hybrid (LB & SB) | 8 compounds including Splitomicin, Cambinol, Salerminde | — |
| 15 | — | 4I5I | OMEGA | Hybrid (LB&SB) | Cocrystalized ligands in PDB (4I5I, 4ZZI, 4IF6) | — |
| 16 | 4ZZI | Hybrid (LB → SB) | ||||
Software names are given in italic.
PDB = Protein Data Bank; LB = ligand-based; SB = structure-based; HM = homology modeling; MD = molecular dynamics simulation.
The third compound screening contest was organized by the Initiative for Parallel Bioinformatics (IPAB). The submission period of compound proposals started in January 20, 2016, and ended in May 20, 2016. Sixteen groups participated in the contest. The participants were asked to propose a prioritized set of 400 compounds. We selected approximately 200 compounds from each group and, in total, 3,192 unique compounds were assayed. Seven potent compounds with half-maximal inhibitory concentrations (IC50) less than 20 μM were identified. The benefits of collecting proposed compounds via various methods are discussed in the following.
Methods
Preparation of the compound library
To create a compound library for the contest, we obtained a compound library from Enamine Ltd., which listed 2,459,912 available compounds in their inventory. We searched this inventory for compounds that were reported to interact or not interact with Sirtuin 1 to 7 in BindingDB12,13 and ChEMBL version 2016. We found 1,443 unique compounds, among which 44 compounds in the compound library were eliminated. Finally, the compound library used in the contest contained 2,459,868 compounds, which was distributed to the participants of the contest.
Methods used by the participants
Sixteen groups participated in the contest, proposing various methods as shown in Table 1. Proposed compounds in canonical SMILES format with prioritized ranking are given in the supporting materials. Here, we briefly describe each method.
Group 1 (G1)
Compounds in the library were first filtered by two-dimensional (2D) similarity with six known drugs for the target using SIMCOMP17. The top 20% of similar compounds were selected and used for structure-based virtual screening. The human Sirtuin 1 crystal structure (PDBID: 4KXQ) was used as a receptor. From the prescreened compound library, PL-PatchSurfer218 was used to select the top 2,000 molecules. These molecule were docked to Sirtuin 1 by AutoDock Vina19. The compounds were ranked by a sum of Z-scores from each program.
Group 2 (G2)
Compounds in the library were filtered by the Tanimoto coefficient of 0.8 for the active chemicals detected in ChEMBL in relation to the target protein (query: SIR1_HUMAN). The protein structure (PDBID: 4BN520), containing cocrystalized coenzyme carba-nicotinamide-adenine-denucleotide (CNA) and ligand N-[2-[3-(piperazin-1-ylmethyl)imidazo[2,1-B][1,3]thiazol-6-yl]phenyl]quinoxaline-2-carboxamide (SR7), was used to model a template protein with the FAMS program21. An in silico screening approach, ChooseLD22, was applied to the model. Both the original version and the hydrophobic interaction, including a version of ChooseLD, were used in the presence or absence of Coenzyme CNA. Then, four kinds of ChooseLD patterns were determined, among which highly ranked compounds were equally selected.
Group 3 (G3)
An SAR model was trained by utilizing Sirtuin 1 and HDAC inhibitors and decoys from the MUBD-HDACs database.15 In detail, 770 1D and 2D descriptors and 881 PubChem fingerprints through the PaDEL descriptor23 for compounds were generated. The extremely random tree algorithm24 was employed for learning SAR models. Next, a trained model was applied to the compound library to predict the compound activity. Finally, the top-ranked 400 compounds were proposed for further validation based on the predicted score.
Group 4 (G4)
A series of rational methods was applied to screen the compound library against the target protein. The methods included SAR analysis, docking simulation, database mining, substructure searching and empirical inspection. For structure-based studies, a post refined protein model originally retrieved from PDB (PDBID: 4KXQ) was applied as a target structure. Multiple docking tools were used in the screening. Machine learning methods25,26 were then applied to assess the binding potentials and to identify the most predictive binding mode that originated from those docking tools.
Group 5 (G5)
In total, 110,000 compounds were chosen from the compound library based on their structural similarity to known Sirtuin 1 inhibitors and Lipinski’s rule of five. To further screen the compounds, the complex structures of Sirtuin 1 (PDBID: 4ZZJ) with the selected compounds were modeled, and the grid scores were obtained by docking calculations with Dock6 program code27. A new measure named the Ligand Triangle (LT) score was also introduced, and the candidate compounds were ranked according to the square sum of the two scores.
Group 6 (G6)
Assay data of not only Sirtuin 1 but also its paralogous enzymes were utilized to train a regression model using a transfer learning method. Here, activity values of the training data were standardized to inhibition rates at 20 μM using Hill’s equation28 under the assumption that the Hill coefficient is 1. Compounds with similar physicochemical properties to those of known inhibitors were extracted from the library using a modified method of QED29,30. Inhibition rates of these compounds were predicted with the trained model24. Then, compounds that were predicted to possess the highest inhibition rates were proposed.
Group 7 (G7)
Structure- and pharmacophore-based approaches were used to identify Sirtuin 1 inhibitors. In the structure-based method, open (PDBID: 4IG9), closed (PDBID: 4IF6), and liganded (PDBID: 4ZZI, 4I5I, 4ZZJ) conformations were used, and 500 compounds were shortlisted from the compound library. Clustering yielded 300 compounds with chemical diversity. In the pharmacophore-based approach,31,32 the cocrystallized structures of Sirtuin 1 with an inhibitor and a substrate were used to derive a pharmacophore with the features and the geometry suitable to bind the active site of Sirtuin 1. Hit-libraries that resulted from structure-based screenings were screened again using the pharmacophore to obtain 100 compounds with the best fitness. In total, 400 compounds were submitted.
Group 8 (G8)
Among the compound library, compounds that possess an amide or thioamide group bound to a ring, lactams and thiolactams were first extracted, considering previous SAR studies33–35. Then, nondrug-like compounds and smaller or larger compounds were excluded. A molecular dynamics (MD) simulation of the structure of Sirtuin 1 (PDB ID: 4ZZI) was performed, and multiple coordinates were obtained from the MD trajectory. myPresto36–38 and a cloud computing environment was utilized for the docking simulation. The results were scored by the multiple target screening (MTS) method.36 Finally, inappropriate structures were eliminated by visual inspection.
Group 9 (G9)
An initial 3D structure of Sirtuin 1 was prepared by the SWISS model using the structure of Sirtuin 1 (PDBID: 4ZZI) as a modeling template39. To clean up structural error in the prepared model, sidechain structures were optimized by Foldit standalone version40. To reduce calculation costs, the compounds in the library were filtered by human inspection considering the Sirtuin 1 binding site condition. The filtered compounds were evaluated by docking simulation (AutoDock Vina19 with PyRx 0.8.). Finally, possibly favorable compounds were selected by visual inspection and scored by AutoDock Vina.
Group 10 (G10)
Homology modeling by Prime41,42 was used to obtain the target protein structure. Seven homology models were evaluated by decoy docking of 122 known active compounds and 200 inactive compounds obtained from the ChEMBL database. Compounds in the library with a molecular weight less than 500 were screened by Glide SP mode docking43,44. Finally, the top 50,000 compounds were reranked by the SIEVE-Score,45 and the similarity of the interaction energy between a compound and each amino acid residue compared with the known active and decoy compounds was evaluated.
Group 11 (G11)
A Sirtuin 1 structure (PDBID: 4I5I) was chosen to carry out the docking simulation. 4I5I contains a ligand that inhibits Sirtuin 1 activity (EX-527).33 Therefore, the place where the ligand was bound attracted our attention, and our group attempted to find molecules that could bind at that location. Ligand efficiencies of the compounds in the library were calculated using the myPresto system37,38. Then, after eliminating optical isomers, the top 400 compounds that had the highest ligand efficiency were chosen as molecules with inhibitory activity.
Group 12 (G12)
RankSVM,46 a machine learning method for ranking prediction, was used to construct the prediction model. Training data consisted of two parts. One part comprised samples of compounds with an IC50 report of Sirtuin 1, 2 and 3 in BindingDB. The other part comprised random samples from ZINC47 as pseudo inactive compound data. ECFP448 fingerprint was used as a feature vector. The cost parameter of RankSVM was chosen from 2−17,2−15, …, 215 with 5-fold cross validation. The evaluation was based on the top-k version of normalized discounted cumulative gain (NDCG) with a k of 100.
Group 13 (G13)
First, LBVS was performed for the compound library using known actives described by Sun et al.49 Then, the output compounds from LBVS were applied to ensemble-docking using Sirtuin 1 pockets prepared by MD simulation with the initial coordinate of PDBID: 4I5I. Top-ranked compounds in the ensemble docking were collected, and their docking poses were produced. The docking poses were visually presented to volunteer voters. The voters conducted visual inspection, and selected “good,” “bad,” or “no idea” for each of the docking poses. Finally, the compounds were reranked by the vote result.
Group 14 (G14)
Three methods were employed: MTS (structure-based),36 machine-learning MTS (ML-MTS) (hybrid of structure-based and ligand-based),50 and docking score index (DSI) (ligand-based)51 methods using myPresto. A target protein structure (PDB ID: 4ZZI) was used in MTS and ML-MTS methods. Eight known inhibitors including Splitomicin and Cambinol were used in the ML-MTS and DSI methods. Calculations were performed for the compound library included in a ready-to-dock compound database, LigandBOX52.
Group 15 (G15)
In the process of seeking inhibitors of the target protein, an LBSV method, VS-APPLE53,54, was used. While conventional ligand-based VS methods adopt single ligand as a template, VS-APPLE adopts the multiple-ligand template, which is composed of multiple ligands bound to the same pocket, as a template. Three ligands of the target protein were chosen and then each of them was used as a single template for independent VS. This procedure was performed, because only a few known protein-ligand complexes were available for the target in PDB and these ligands bind to different part of the binding pocket.
Group 16 (G16)
ECFP1248 fingerprints of 514 Sirtuin 1 binders downloaded from BindingDB were generated and used to build lasso, elastic net, ridge and random forest machine learning models relating Ki. A protein-structure-based pharmacophore was generated as an alternative approach. The machine learning and pharmacophore models were applied to ECFP12 fingerprints of the compound library to predict Ki values. Prioritized 200-300 compounds from each of the methods above, together with hits from the similarity search using known active Sirtuin 1 binders were prepared. This protocol resulted in approximately 2,000 compounds. Finally, rescoring and clustering based on the docking of the compounds to the target protein (PDBID: 4ZZI) using GOLD were used to prioritize 400 representative compounds.
Screening of Compounds
Screening of potential inhibitors
Selection of compounds for assaying from the proposed lists and the flow of the assays are described in Fig. 1a,b. The experimental procedures are described in the supporting information in detail. Here, we briefly describe the screening flow and results.
Figure 1.

(a) A flowchart of the contest. Each group (G1-G16) proposed 400 compounds (cmpds) with a prioritized rank from compound library using their own methods. The proposed compounds that were not stocked-out were selected until the number of compounds reached 200 for each group. If there was duplication in the proposed compounds among different groups, these groups attained additional compounds to be assayed. For this reason, there are differences among the number of selected compounds of each group. Finally, the selected compounds were assayed. (b) The screening flow of the compounds in the experimental assay. The filtering criteria are shown in a trapezium. The number of compounds applied to each screening is shown in parenthesis. This flow was conducted twice with NAD+ (the first number in parenthesis) and without NAD+ (the second number in parenthesis). (c) IC50 hits found based on TSA screening without (w/o) and with (w/) NAD+ (see Screening of potential inhibitors in the main text).
A total of 3,192 compounds extracted from the proposed lists were first screened by thermal shift assay (TSA), which determined a melting temperature shift (ΔTm) of Sirtuin 1 upon the addition of a test compound at a concentration of 10 µM (n = 1) with primary hit criteria of ΔTm > 0.45 K or ΔTm < −1.5 K. Primary hits (48 compounds) were retested in the same assay at three concentrations (5, 10, 20 µM, n = 4) for hit confirmation and for dose-dependency check. Secondary hits (41 compounds) were checked their selectivity for Sirtuin 1 based on counterscreening on unrelated protein targets (bovine carbonic anhydrase (CA) and recombinant SH2 domain of hABL1 kinase (ABL1)) at three different concentrations (5, 10, 20 µM, n = 4), which yielded 38 hit compounds, hereafter referred to as TSA hits. The TSA screening above was conducted in the absence of NAD+. As noted, the structure of Sirtuin 1 is different in the presence of NAD+ and substrate. The chemical profile of TSA hits found from the different conditions could be different. Hence, we applied the same TSA in the presence of NAD+ to the 3,192 compounds. As a result, 39 TSA hits were found. Regardless of the presence of NAD+, 29 compounds were shared in both TSA hits.
The 48 ( = 38 + 39 −29) unique TSA hits were evaluated by an inhibitory assay, which determined inhibition rates of enzymatic activity of Sirtuin 1 of the test compounds at a concentration of 10 µM (n = 4) with a hit criterion of an inhibitory rate >15%. Note that the inhibitory assay was conducted with NAD+. As a result, 20 hit compounds, 12 from TSA without NAD+ and 16 from TSA with NAD+, were found. An IC50 determination of the 20 hits was conducted for dilutions of 100 μM to 0.05 μM (n = 4). We identified seven compounds satisfying the hit criteria of IC50 < 20 μM with clear dose-response relationship as shown in Figure S6 of the supporting information. The chemical structures of the seven hits are given in Table 2.
Table 2.
IC50 values of hit compoundsa and their similar known inhibitors.
| Compound ID | Chemical Structureb | IC50 μM | 95% CI μM | Group | Similar compound ID & Chemical structure | Similarityc & Inhibition | |
|---|---|---|---|---|---|---|---|
| lower | upper | ||||||
| Z56773446 | 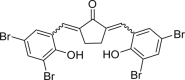 |
4.1 | 1.9 | 8.9 | 6 (LB) |
 CHEMBL260149 (Compound 4b)57 CHEMBL260149 (Compound 4b)57
|
0.95 (Inh. rate@25 μM: 50%)57 |
| Z62466600* | 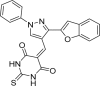 |
7.1 | 3.0 | 17 | 6,12,13 (LB) |
 CHEMBL1714005 (Compound 18),58 CHEMBL1483080 (Compound 3)59 CHEMBL1714005 (Compound 18),58 CHEMBL1483080 (Compound 3)59
|
0.78 (IC50: 6 μM)58 (IC50: 5.9 μM)59 |
| Z235344735 | 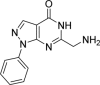 |
7.6 | 6.4 | 9.1 | 15 (Hybrid) |
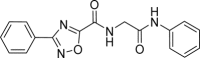 CHEMBL449117 CHEMBL449117 |
0.64 (Inhibition rate@200 μM: 28%)69 |
| Z21813138 | 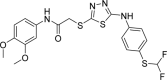 |
7.7 | 6.4 | 9.3 | 3 (LB) |
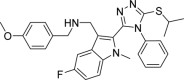 CHEMBL2436120 CHEMBL2436120 |
0.65 (Inh. rate@40 μM: 70%)70 |
| Z165049452 | 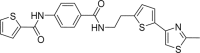 |
12 | 9.3 | 14 | 13 (Hybrid) |
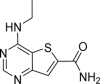 CHEMBL2332048 CHEMBL2332048 |
0.68 (IC50> 50 μM d)35 |
| Z165047618* | 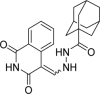 |
13 | 7.6 | 23 | 9 (SB) |
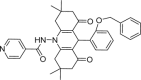 CHEMBL1762050 CHEMBL1762050 |
0.60 (Inh. rate@50 μM = 70%)71 |
| Z605844126 | 15 | 10 | 22 | 1 LB->SB |
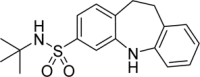 CHEMBL3222149 CHEMBL3222149 |
0.65 (Inh. rate@100 μM = 10%)72 | |
a) The melting temperature shift (ΔTm) of Sirtuin 1 upon addition of a test compound and the inhibition rates are shown in the Supporting Information along with the canonical SMILES. The final Sirtuin 1 concentrations used for the IC50 determination were 30 nM for compounds marked with * and 20 nM for the others. b) The stereochemistry of compounds was not determined. c) Similarity scores were calculated using the Tanimoto coefficients of the MACCS descriptor. d) This compound was included as a known inhibitor despite its weak potency, because the series of its analogues are inhibitors.35 IC50 = inhibitory concentrations; CI = confidence interval.
We first noticed that Z56773446 and Z21813138 were found only through primary screening without NAD+; on the other hand, Z165047618 and Z62466600 were found only through primary screening with NAD+. To confirm whether the results of primary screenings of Z56773446 and Z21813138 with NAD+ and Z165047618 and Z62466600 without NAD+ were false negatives or not, we conducted additional experiments of TSA on Z165047618 and Z21813138 with NAD+ (n = 4) and Z56773446 and Z62466600 without NAD+ (n = 4). We confirmed that the ΔTm values of Z165047618, Z21813138, and Z56773446 were not as large as the hit criterion, i.e., −0.03, 0.32, and −0.05, respectively. However, the ΔTm of Z62466600 was 0.87, and its hit confirmation and dose-dependence of the three concentrations were also confirmed. Hence, we decided that the first primary screening of Z62466600 was a false negative and that it should be classified as an IC50 hit (w/ & w/o NAD+). It is not clear why results that were shown in Fig. 1c were obtained, based on the limited information of the present study.
The seven hit compounds were compared to the pan-assay interference compounds (PAINS) filters,55 which collects substructures of frequent hitters that appeared in many biochemical high-throughput screenings. As a result, Z21813138, Z235344735, Z605844126, Z165049452, and Z165047618 were confirmed not to have the potential problematic substructures. This result indicates that these compounds are promising candidates for further investigation. On the other hand, Z56773446 and Z62466600 were found to have potentially problematic substructures, i.e., Z56773446 has a divinylketone (PAINS filter entry: ene_one_ene_A), and Z62466600 contains alkylidene barbiturate (PAINS filter entry: ene_six_het_A). Despite of these potentially problematic substructures, we regarded these compounds as hit compounds, because there are known inhibitors similar to them, as shown in Table 2, and these compounds passed the counterscreening on unrelated protein targets. Note that the ΔTm values of Z56773446 and Z62466600 at 5, 10, and 20 μM (determined without NAD+) were as follows: For Z56773446, Sirtuin 1: 0.4, 0.6, 1.0; CA: 0.2, 0.5, 0.5; and ABL1: 0.0, 0.1, 0.2; and for Z62466600, Sirtuin 1: 0.2, 0.3, 0.5; CA: 0.0, 0.0, 0.0; and ABL1: −0.2, 0.1, 0, respectively.
Information of the assayed compounds is given in the supporting information, including the results of ΔTm, dose-dependence check, selectivity, inhibition rates, and IC50 values.
Discussion
Hit rate of assayed compounds
Among 3,192 assayed compounds, 7 and 4 compounds showed IC50 values less than 20 μM and 10 μM, respectively, as shown in Table 2. To evaluate the success odds of the contest, we compared this value (4/3192) with a hit rate obtained from a previous HTS study on inhibitors against recombinant Sirtuin 1. Therein, 58 inhibitors with an IC50 less than 10 μM were identified from among 147,000 different compounds56. Thus, the success odds of the contest (0.13% = 4/3192) under the same hit definition was higher than that reported previously (0.04%). To determine whether this improvement is statistically significant, we performed an exact binomial test with a significance level of 0.05. The p-value of the test was 0.04 and less than the defined significance level. Although the previously applied experimental conditions and compound library were not identical to the contest, the results indicated that a contest-based approach would be successful in comparison with a simple HTS even in the hit rate. The total hit rate in this study, 7/3192, was lower than that of the previous contest, 11/1991, which targeted the tyrosine-protein kinase Yes when IC50 < 20 μM. This reduction in the hit rate may have resulted from a greater difficulty in identifying an inhibitor of the target molecule, i.e., the hit rate for HTS of Sirtuin 1 (0.04%)56 was lower than that for kinase (0.06%)1.
The most successful methods in terms of the hit rate have been proposed by G6 and G13, both of which identified two hits. The performance of these methods, relative to that of other methods, was evaluated using the binomial test. Provided that the average hit rate was 7/3192, the p-values of G6 and G13 were both 0.07; hence, they may not be considered promising methods relative to the other methods. Assuming that the success odds of the HTS study, 0.04%, can be considered the baseline for random screening, a hit rate of 2/181 of G6 under the hit criterion of IC50 < 10 μM yielded a p-value of 0.0025. Even with Bonferroni correction, this p-value indicated that the method of G6 is statistically promising (the p-value < 0.05/16) than the HTS.
In this study, we do not review each method in detail, but rather, we present an overview of the contest-based approach for the identification of potential inhibitors for the target molecule.
Diversity of proposed compounds
We have repeatedly emphasized that the contest-based approach realized diversification in the screening results in terms of the chemical diversity of the assayed compounds.9,10 This observation was applicable to the present results, as shown in Fig. 2. The average similarity of the different compounds in each group, 0.44 ± 0.09 (average and standard deviation of 16 methods, diagonal elements of Fig. 2b, of which values are summarized in Table S2), was higher than the average similarity of the different compounds in all groups, 0.36 ± 0.08 (average and standard deviation of 120 + 16 combinations, off-diagonal and diagonal elements of Fig. 2b), i.e., the p-value of Welch’s t-test with the null hypothesis that the two averages are equal was 0.005. In addition, as seen in Tables S2 and S3, we did not observe that a specific filter class mainly contribute to diversifying screened compounds.
Figure 2.

(a) Similarity of the compounds proposed from each group. The similarity scores are defined with the Tanimoto coefficient of the MACCS descriptor60. The number of identical compounds proposed from different two groups is shown in Figure S7, which indicates that identical compounds were rarely proposed from different groups, except for the combinations of G6 and G12 (19 compounds) and G6 and G13 (5 compounds), which used ligand information. (b) Averaged similarity scores in each cell of (a), in which identical compounds on the diagonal are not included for averaging. (c) Assayed compounds from each group are projected to the first and second principal components (PC1: x-axis, PC2: y-axis). Hit compounds are projected to PC1 and PC2 as well. Principal component analysis was applied to the compound library using the MACCS descriptor. The cumulative variance of the PC1 and PC2 are 26% and 50%, respectively. A randomly chosen 3% of the compounds in the library are projected (gray points).
There were two groups that showed higher diversity than the average of all groups, i.e., G3 (LB approach) and G11 (SB approach), of which average diversities of assayed compounds were 0.30 and 0.33, respectively. Even though the two methods’ average diversities were higher than the average of all groups, each method typically yielded compounds from different places in the chemical space of the compound library, as visualized in Fig. 2c, indicating that the contest-based approach can realize diversified screening.
The final hit compounds were also chemically diverse, as seen in Table 2. The maximum similarity among the hit compounds was 0.54, which was the score between Z56773446 and Z62466600.
Novelty of the assayed and hit compounds
Finding chemically novel compounds in the early stage of drug discovery process is crucially important. To evaluate the performance of the contest-based approach to find such compounds, we compared the hits to the known inhibitors of Sirtuin 1. The known inhibitors in this study were collected from ChEMBL version 20 with queries of Standard type including IC, EC, or inhibition and were then filtered by the criteria of an inhibition rate less than 0% or an IC50 greater than 100 μM. Removing duplicates, 892 unique inhibitors were identified. The inhibitor set included Sirtuin 1 ligands defined in the MUBD-HDACs database15 except a compound (ChEMBL ID CHEMBL99779). In this study, the compound was not defined as an inhibitor, because relevant information for the compound regarding inhibition of Sirtuin 1 was not found in ChEMBL and BindingDB.
First, we evaluated the average similarity of assayed compounds to the known inhibitors. The average similarities were 0.33 ± 0.02, 0.24 ± 0.06, and 0.34 ± 0.01 for LB, SB, or hybrid methods, respectively. These values were low, and each filter class proposed novel compounds on average.
Thereafter, we investigated similarities among hit compounds with the known inhibitors. We regard that Z235344735, Z21813138, Z165049452, Z165047618, and Z605844126 are novel, because their maximum similarity to known compounds was 0.68, and comparison of the chemical structures between a hit and the corresponding inhibitor were not similar as shown in Table 2. Among them, four compounds were proposed from methods that use ligand information (LB, hybrid, LB- > SB methods) (G1, G3, G13, G15), and one compound, from the SBVS method (G9). This means that the use of ligand information does not necessarily yield similar compounds to known inhibitors. The most novel compound (Z165047618) was proposed from SBVS. However, the potency of the hit was classified as weak among the seven hits, as shown in Fig. 3. In the previous study, novel but weak hits were also found from SBVS10. Hence, novelty and potency tend to display a tradeoff association.
Figure 3.
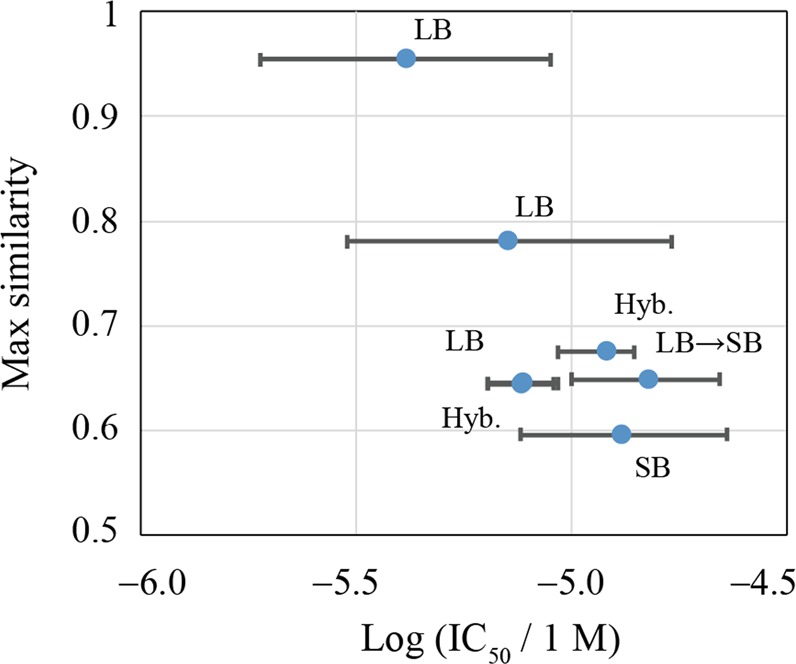
The similarity of each hit compound to known Sirtuin 1 inhibitors (see Novelty of the assayed and hit compounds Section) is plotted against the experimental inhibition activity. The error bar represents 95% confidence intervals estimated from IC50 assays. For each point, the category of method used is presented (see Table 1). The similarity in the figure was calculated with the Tanimoto coefficient of the MACCS descriptor60.
The remaining two compounds were similar to the known inhibitors, as shown in Table 2. The similar known compound of Z56773446 was reported as compound 4b57 (Tanimito coefficient of the MACCS descriptor was 0.95). In the literature, the authors evaluated the effect of 4b on apoptosis induction and granulocytic differentiation in the human leukemia U937 cells and found high differentiating activity. Hence, cell-based assay of Z56773446 is considered interesting. Similar known compounds of Z62466600 were reported as compound 1858 and compound 359 in two studies (both Tanimoto coefficients were 0.78). Compound 18 was derived from optimizing Cambinol, a sirtuin inhibitor that shows antitumor activity in preclinical models. Compound 18 showed selectivity for Sirtuin 1 (Sirtuin 1 IC50 = 6 μM, and Sirtuin 2 IC50 = 20 μM). Compound 3 showed selectivity for Sirtuin 1 (Sirtuin 1 IC50 = 5.9 μM, Sirtuin 2 IC50 = 20.3 μM, Sirtuin 3 inhibition rate = 14% at 50 μM, Sirtuin 5 IC50 = 46.5 μM). Hence, further investigation of Z62466600 would be valuable.
Conclusion
A compound screening contest was conducted to identify inhibitors of Sirtuin 1. Seven inhibitors were identified with highly diverse structures. Assuming that the hit rate of a reported HTS study56 is potentially applicable as a baseline for random screening, the contest-based approach could be considered significantly better than the HTS. The diversity could be attained by the collection of various methods in this approach. This observation was consistent with the previous contests. We speculated that prospective benchmarking of various methods based on the identical conditions (the compound library, experimental conditions, time period of proposal) would enable identifying a promising method for finding inhibitors of the target molecule. However, none of the proposed methods were better than the others on comparing them on the basis of the average hit rate determined from all methods, probably because of increased difficulty in identifying a novel inhibitor for Sirtuin 1 rather than the target of the previous contest, wherein a promising method was reported. Under these circumstances, an increase in the number of compounds assayed from a group would help identify a promising method. Furthermore, on comparing the proposed methods with low hit rates, consideration of not only hit rates but also potency of hits could be useful.
Supplementary information
Acknowledgements
We gratefully acknowledge the financial support of The Japan Biological Informatics Consortium (JBIC), Schrödinger K.K., Namiki Shoji Co., Ltd., Microsoft Japan Co., Ltd., DataDirect Networks Japan, Inc., Fast Track Initiative, Inc., NEC Corporation, HPCTECH Corporation, IMSBIO Co., Ltd., DiscoveResource, Inc., Biomodeling Research Co., Ltd., Lisit, Co., Ltd., Leave a Nest Co., Ltd., Level Five Co., Ltd., Research Organization for Information Science and Technology (RIST), Teijin Pharma Limited, DELL, NVIDIA Corporation, and Innovations and Future Creation Inc. (MIRAI SOUZOU) which made it possible to complete our contest. We are deeply grateful to Tokyo Institute of Technology, Ministry of Economy, Trade and Industry (METI), Japan Pharmaceutical Manufacturers Association (JPMA), Information Processing Society of Japan (IPSJ), Japanese Society of Bioinformatics (JSBi), Chem-Bio Informatics (CBI) Society, The Japan DataScientist Society, The Science News Ltd., and Nikkan Kogyo Shimbun Ltd. We would like to offer our special thanks to Mr. Toshiaki Miyaki and Ms. Kanako Ozeki. This work is partially supported by the Research Complex Program “Wellbeing Research Campus: Creating new values through technological and social innovation” from Japan Science and Technology Agency (JST) and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP19am0101112j0003.
Author contributions
All authors made substantial contributions to this study and article. S.C., M.O., T.I., Y.A. and M.S. organized and operated the contest. S.Z., P.B. and A.G. carried out inhibitory assays. S.C., N.Y., R.Y., K.I., T. Honma, and T. Hirokawa evaluated data. N.Y., R.Y., W-H.S., D.K., M.I., H.U., T.I., R.T., K-Y.H., V.G., H.K., M.S., A.H., N.M., K.Y., M.M., C.R., A.M.T., D.V., M.M.G., I.N., N.U., H.H., M.T., H.K.N., S.D.S., T.I., M.K., K.K., S.T., K.Z.Y., Y.M., K.O., D.K., T.O., S.M., G.C., P.P., C.N., A.M., M.I. and K.M. participated in the contest and predicted hit compounds for the target protein by their method. S.C. and M.S. wrote the main manuscript text. All authors approved this version to be published.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-55069-y.
References
- 1.Bender A, et al. Which aspects of HTS are empirically correlated with downstream success? Curr Opin Drug Discov Devel. 2008;11:327–337. [PubMed] [Google Scholar]
- 2.Sliwoski G, Kothiwale S, Meiler J, Lowe EW., Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshino R, et al. In silico, in vitro, X-ray crystallography, and integrated strategies for discovering spermidine synthase inhibitors for Chagas disease. Sci Rep. 2017;7:6666. doi: 10.1038/s41598-017-06411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14:1923–1938. doi: 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acharya C, Coop A, Polli JE, Mackerell AD., Jr. Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Curr Comput Aided Drug Des. 2011;7:10–22. doi: 10.2174/157340911793743547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scior T, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model. 2012;52:867–881. doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 8.von Korff M, Freyss J, Sander T. Comparison of Ligand- and Structure-Based Virtual Screening on the DUD Data Set. J Chem Inf Model. 2009;49:209–231. doi: 10.1021/ci800303k. [DOI] [PubMed] [Google Scholar]
- 9.Chiba S, et al. Identification of potential inhibitors based on compound proposal contest: Tyrosine-protein kinase Yes as a target. Sci Rep. 2015;5:17209. doi: 10.1038/srep17209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiba S, et al. An iterative compound screening contest method for identifying target protein inhibitors using the tyrosine-protein kinase Yes. Sci Rep. 2017;7:12038. doi: 10.1038/s41598-017-10275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davenport AM, Huber FM, Hoelz A. Structural and functional analysis of human SIRT1. J Mol Biol. 2014;426:526–541. doi: 10.1016/j.jmb.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu TQ, Lin YM, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic. Acids. Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Lin Y, Liu M, Gilson MK. The Binding Database: data management and interface design. Bioinformatics. 2002;18:130–139. doi: 10.1093/bioinformatics/18.1.130. [DOI] [PubMed] [Google Scholar]
- 14.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic. Acids. Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia J, et al. Comparative Modeling and Benchmarking Data Sets for Human Histone Deacetylases and Sirtuin Families. J Chem Inf Model. 2015;55:374–388. doi: 10.1021/ci5005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bento AP, et al. The ChEMBL bioactivity database: an update. Nucleic. Acids. Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hattori M, Okuno Y, Goto S, Kanehisa M. Development of a Chemical Structure Comparison Method for Integrated Analysis of Chemical and Genomic Information in the Metabolic Pathways. J Am Chem Soc. 2003;125:11853–11865. doi: 10.1021/ja036030u. [DOI] [PubMed] [Google Scholar]
- 18.Shin WH, Christoffer CW, Wang J, Kihara D. PL-PatchSurfer2: Improved Local Surface Matching-Based Virtual Screening Method That Is Tolerant to Target and Ligand Structure Variation. J Chem Inf Model. 2016;56:1676–1691. doi: 10.1021/acs.jcim.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen GTT, Schaefer S, Gertz M, Weyand M, Steegborn C. Structures of human sirtuin 3 complexes with ADP-ribose and with carba-NAD+ and SRT1720: binding details and inhibition mechanism. Acta Crystallographica Section D. 2013;69:1423–1432. doi: 10.1107/S0907444913015448. [DOI] [PubMed] [Google Scholar]
- 21.Umeyama, H. & Iwadate, M. FAMS and FAMSBASE for protein structure. Current protocols in bioinformaticsChapter 5, Unit5 2, 10.1002/0471250953.bi0502s04 (2004). [DOI] [PubMed]
- 22.Takaya D, et al. Bioinformatics based ligand-docking and in-silico screening. Chem Pharm Bull. 2008;56:742–744. doi: 10.1248/cpb.56.742. [DOI] [PubMed] [Google Scholar]
- 23.Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011;32:1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 24.Geurts P, Ernst D, Wehenkel L. Extremely randomized trees. Mach Learn. 2006;63:3–42. doi: 10.1007/s10994-006-6226-1. [DOI] [Google Scholar]
- 25.Hsin Kun-Yi, Ghosh Samik, Kitano Hiroaki. Combining Machine Learning Systems and Multiple Docking Simulation Packages to Improve Docking Prediction Reliability for Network Pharmacology. PLoS ONE. 2013;8(12):e83922. doi: 10.1371/journal.pone.0083922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsin KY, et al. systemsDock: a web server for network pharmacology-based prediction and analysis. Nucleic Acids Res. 2016;44:W507–513. doi: 10.1093/nar/gkw335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen WJ, et al. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36:1132–1156. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubler H, Schopfer U, Jacoby E. Theoretical and Experimental Relationships between Percent Inhibition and IC50 Data Observed in High-Throughput Screening. J Biomol Screen. 2013;18:1–13. doi: 10.1177/1087057112455219. [DOI] [PubMed] [Google Scholar]
- 29.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mochizuki Masahiro, Suzuki Shogo D., Yanagisawa Keisuke, Ohue Masahito, Akiyama Yutaka. QEX: target-specific druglikeness filter enhances ligand-based virtual screening. Molecular Diversity. 2018;23(1):11–18. doi: 10.1007/s11030-018-9842-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salam NK, Nuti R, Sherman W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J Chem Inf Model. 2009;49:2356–2368. doi: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- 32.Loving K, Salam NK, Sherman W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided. Mol. Des. 2009;23:541–554. doi: 10.1007/s10822-009-9268-1. [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, et al. The 2.5 Å Crystal Structure of the SIRT1 Catalytic Domain Bound to Nicotinamide Adenine Dinucleotide (NAD+) and an Indole (EX527 Analogue) Reveals a Novel Mechanism of Histone Deacetylase Inhibition. J Med Chem. 2013;56:963–969. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 34.Napper AD, et al. Discovery of Indoles as Potent and Selective Inhibitors of the Deacetylase SIRT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 35.Disch JS, et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem. 2013;56:3666–3679. doi: 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- 36.Fukunishi Y, Mikami Y, Kubota S, Nakamura H. Multiple target screening method for robust and accurate in silico ligand screening. J Mol Graph Model. 2006;25:61–70. doi: 10.1016/j.jmgm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Fukunishi Y, Mikami Y, Nakamura H. The filling potential method: A method for estimating the free energy surface for protein-ligand docking. J. Phys. Chem. B. 2003;107:13201–13210. doi: 10.1021/jp035478e. [DOI] [Google Scholar]
- 38.Fukunishi Y, Mikami Y, Nakamura H. Similarities among receptor pockets and among compounds: analysis and application to in silico ligand screening. J Mol Graph Model. 2005;24:34–45. doi: 10.1016/j.jmgm.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleffner R, et al. Foldit Standalone: a video game-derived protein structure manipulation interface using Rosetta. Bioinformatics. 2017;33:2765–2767. doi: 10.1093/bioinformatics/btx283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobson MP, et al. A hierarchical approach to all‐atom protein loop prediction. Proteins: Struct., Funct., Bioinf. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 42.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002;320:597–608. doi: 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 43.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 44.Halgren TA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 45.Yasuo Nobuaki, Sekijima Masakazu. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. Journal of Chemical Information and Modeling. 2019;59(3):1050–1061. doi: 10.1021/acs.jcim.8b00673. [DOI] [PubMed] [Google Scholar]
- 46.Herbrich, R., Graepel, T. & Obermayer, K. Large margin rank boundaries for ordinal regression, Advances in Large Margin Classifiers (2000).
- 47.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers D, Hahn M. Extended-Connectivity Fingerprints. J Chem Inf Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 49.Sun Y, Zhou H, Zhu H, Leung SW. Ligand-based virtual screening and inductive learning for identification of SIRT1 inhibitors in natural products. Sci Rep. 2016;6:19312. doi: 10.1038/srep19312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukunishi Y, et al. Classification of Chemical Compounds by Protein−Compound Docking for Use in Designing a Focused Library. J Med Chem. 2006;49:523–533. doi: 10.1021/jm050480a. [DOI] [PubMed] [Google Scholar]
- 51.Fukunishi Y, Kubota S, Nakamura H. Noise Reduction Method for Molecular Interaction Energy: Application to in silico Drug Screening and in silico Target Protein Screening. J Chem Inf Model. 2006;46:2071–2084. doi: 10.1021/ci060152z. [DOI] [PubMed] [Google Scholar]
- 52.Fukunishi Y, et al. Advanced in-silico drug screening to achieve high hit ratio - Development of 3D-compound database. Synthesiology English edition. 2009;2:64–72. doi: 10.5571/syntheng.2.64. [DOI] [Google Scholar]
- 53.Okuno T, Kato K, Terada TP, Sasai M, Chikenji G. VS-APPLE: A Virtual Screening Algorithm Using Promiscuous Protein–Ligand Complexes. J Chem Inf Model. 2015;55:1108–1119. doi: 10.1021/acs.jcim.5b00134. [DOI] [PubMed] [Google Scholar]
- 54.Okuno T, et al. Importance of consensus region of multiple-ligand templates in a virtual screening method. Biophys Physicobiol. 2016;13:149–156. doi: 10.2142/biophysico.13.0_149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 56.Nayagam VM, et al. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J Biomol Screen. 2006;11:959–967. doi: 10.1177/1087057106294710. [DOI] [PubMed] [Google Scholar]
- 57.Mai A, et al. epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors. J Med Chem. 2008;51:2279–2290. doi: 10.1021/jm701595q. [DOI] [PubMed] [Google Scholar]
- 58.Blum CA, et al. SIRT1 Modulation as a Novel Approach to the Treatment of Diseases of Aging. J Med Chem. 2011;54:417–432. doi: 10.1021/jm100861p. [DOI] [PubMed] [Google Scholar]
- 59.Maurer B, et al. Inhibitors of the NAD+-Dependent Protein Desuccinylase and Demalonylase Sirt5. ACS Med Chem Lett. 2012;3:1050–1053. doi: 10.1021/ml3002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002;42:1273–1280. doi: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- 61.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Boyle NM, et al. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mendel DB, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 64.Sadowski J, Gasteiger J, Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Comput. Sci. 1994;34:1000–1008. doi: 10.1021/ci00020a039. [DOI] [Google Scholar]
- 65.Dai H, et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun. 2015;6:7645. doi: 10.1038/ncomms8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.RDKit: Open-source cheminformatics.
- 67.LigPrep v. 3.2 (Schrödinger, LLC, New York, NY, 2014).
- 68.Cao D, et al. Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015;29:1316–1325. doi: 10.1101/gad.265462.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huhtiniemi T, et al. Oxadiazole-carbonylaminothioureas as SIRT1 and SIRT2 inhibitors. J Med Chem. 2008;51:4377–4380. doi: 10.1021/jm800639h. [DOI] [PubMed] [Google Scholar]
- 70.Panathur N, et al. Identification and characterization of novel indole based small molecules as anticancer agents through SIRT1 inhibition. Eur J Med Chem. 2013;69:125–138. doi: 10.1016/j.ejmech.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 71.Alvala M, et al. Novel acridinedione derivatives: design, synthesis, SIRT1 enzyme and tumor cell growth inhibition studies. Bioorg Med Chem Lett. 2012;22:3256–3260. doi: 10.1016/j.bmcl.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 72.Di Fruscia Paolo, Ho Ka-Kei, Laohasinnarong Sasiwan, Khongkow Mattaka, Kroll Sebastian H. B., Islam Suhail A., Sternberg Michael J. E., Schmidtkunz Karin, Jung Manfred, Lam Eric W.-F., Fuchter Matthew J. The discovery of novel 10,11-dihydro-5H-dibenz[b,f]azepine SIRT2 inhibitors. MedChemComm. 2012;3(3):373. doi: 10.1039/c2md00290f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.