

Abstract
The Siamese crocodile (Crocodylus siamensis) is a freshwater, endangered crocodile with high economic value in the farming industry. Gut microflora plays an essential role in host physiological activity, and it contributes significantly to both the health and diseased states of animals. However, thus far, no study has focused on the correlation between diseases and intestinal bacterial communities in crocodilians. Here, we first compared the composition and function of gut microbial communities in captive juvenile C. siamensis suffering from anorexia and healthy crocodile controls using deep amplicon sequencing. The gut microbial diversity of anorexic crocodiles was much lower than the healthy individuals. Obvious changes in gut microbial composition were observed between sick and healthy crocodiles, except for Cetobacterium somerae of phylum Fusobacteria. In particular, the abundance of Bacteroides luti, Clostridium disporicum, Plesiomonas shigelloides, and Odoribacter sp. in the gut flora of healthy crocodiles was distinctly higher than the diseased group. Conversely, the species Edwardsiella tarda was overrepresented in the gut of anorexic crocodiles compared to the healthy group. Furthermore, in anorexic crocodiles, the predicted microbial functions that were related to amino acid metabolism, biosynthesis of other secondary metabolites, nucleotide metabolism, replication and repair, and translation were significantly reduced, while signal transduction was significantly enriched. These findings of the present study provide a reference to enrich the field of gut microorganism studies in crocodilians and suggest that alterations in the composition and function of gut bacteria in C. siamensis juveniles may be associated with anorexia in crocodiles.
Keywords: anorexia, bacterial diversity, Crocodylus siamensis, gut microbes, microbial function
In this study, we demonstrate for the first time, the diversity of intestinal bacterial communities in captive juvenile Crocodylus siamensis suffering from anorexia and healthy crocodile controls using deep amplicon sequencing. Our findings provide a reference to enrich the field of gut microorganism studies in crocodilians and suggest that alterations in the composition and function of gut bacteria in C. siamensis may be associated with anorexia in crocodilians

1. INTRODUCTION
The Siamese crocodile (Crocodylus siamensis) is an endangered freshwater crocodilian that is native to most countries in Southeast Asia, including Cambodia, Laos, Indonesia, Thailand, and Vietnam (Bezuijen et al., 2013). As far back as the mid‐1980s, wild crocodiles were exported from Cambodia to China as farm animals (Guo et al., 2018). In China, successful breeding of C. siamensis only occurs in a few southern provinces, such as Fujian and Hainan, because wild crocodiles have particular climate and temperature requirements for a suitable habitat (Guo et al., 2018). In addition to the requirements of the environmental conditions, bacterial diseases have also severely restricted the development of the crocodile farming industry (Camus & Hawke, 2002; Kim, Lee, & Kwak, 2016; Roh et al., 2011). The captive Siamese crocodile is universally acknowledged as “soft gold in aquatics” because it has significant economic benefits. Their skin is used in the leather industry, and their blood has potential effects in antibiotic therapy (Leelawongtawon, Siruntawineti, Chaeychomsri, & Sattaponpan, 2010). Their oil is used for medical treatment, and crocodiles are beneficial to the tourism industry (Li et al., 2012; Ryan, 1998).
The intestinal tract is an indispensable digestive organ that plays a key role in the defense of animals' immune system, and it is where considerable amounts of microbial flora colonize (Eckburg, 2005). Normally, the gut microflora is interdependent and interactive, which maintains the homeostasis of the internal environment, and it greatly influences the physiological activities of the host (Hooper, 2001). The composition and structure of the vertebrates' gut microbial communities are influenced by multiple factors (such as diet and environmental conditions) that also contribute to disease (Feng, Chen, & Wang, 2018; Scott, Gratz, Sheridan, Flint, & Duncan, 2013; Sharpton, 2018).
Prior studies on the gut bacterial communities of nonmammalian vertebrates have performed on birds, fish, amphibians, and reptiles (Colston & Jackson, 2016; Waite & Taylor, 2015). However, so far, the study on crocodilian's gut microbiome is still scarce, there was only one crocodilian species, American alligator (Alligator mississippiensis), which has been reported (Keenan, Engel, & Elsey, 2013). In those alligators, Fusobacteria was a unique and core flora of the gut microbiome, which is distinguished from other reptiles' gut microbiome (mainly consisted of Firmicutes and Bacteroidetes; Colston & Jackson, 2016; Keenan & Elsey, 2015). To expand the understanding of gut microflora of crocodilians, here we perform the 16S rRNA gene amplicon sequencing to compare the diversity of gut bacteria in healthy and anorexic Siamese crocodiles. The data observed from this work will elucidate the basic composition and function of the gut microbial communities in farmed crocodiles, and it will identify key bacteria that may influence the healthy growth of crocodilian.
2. MATERIALS AND METHODS
2.1. Sample collection
In May 2016, a sudden disease occurred in captive Crocodylus siamensis juveniles (1‐year‐old), which was observed at the Xiamen Lonsun crocodile zoo in Fujian Province. The clinical symptom of the sick Siamese crocodiles was anorexia (apparent decrease in daily feeding activity) with no trauma. The experimental crocodiles (0.65–0.78 m, 1.04–1.37 kg) were divided into two groups: the healthy group (labeled as H, n = 3) and the diseased group (labeled as D, n = 3). Six crocodiles were fed the same diet and reared in individual feeding areas before sampling. Cloacal swabs were used for sampling the crocodile gut flora which were an acceptable source for nondestructive sampling the reptiles' intestinal microbiota (Colston, Noonan, & Jackson, 2015; Jiang et al., 2017; Johnston, Porter, Scott, Rhodes, & Webster, 2010). The cloacal samples of the H group were labeled as H1–H3, while the cloacal samples of the D group were labeled as D1–D3. Specimens were stored in liquid nitrogen and immediately transported to the laboratory for DNA extraction.
2.2. DNA extraction
The total bacterial genomic DNA was extracted directly from each frozen sample (220 mg) using the PowerFecal® DNA Isolation Kit (Qiagen), following the manufacturer's protocol. The quality and integrity of each DNA extraction was determined using 1% agarose gel electrophoresis before deep sequencing.
2.3. Deep amplicon sequencing
The genomic DNA was sequenced by the Majorbio Bio‐technology Company using the Illumina MiSeq PE300 platform (Illumina). PCR primers 338F (5′‐ACTCCTACGGGAGGCAGCAG‐3′) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′) with dual barcode sequences were used to amplify the V3–V4 region of the 16S rRNA gene for all DNA samples. Each 20 μl of PCR mixture included 5× FastPfu buffer (4 μl), FastPfu Polymerase (0.4 μl), 2.5 mM dNTPs (2 μl), 5 μM forward primer (0.8 μl), 5 μM reverse primer (0.8 μl), and template DNA (10 ng). The PCR protocol was amplified using the conditions as following: 95°C for 3 min (initial denaturation); 25 cycles of 95°C for 30 s (denaturation), 55°C for 30 s (annealing), 72°C for 45 s (elongation), and 72°C for 10 min (final elongation). Then, the PCR products were detected by gel electrophoresis using 2% agarose and Tris–acetate–EDTA buffer, and finally, the amplicons (reads with an average length of 468 bp) were used for paired‐end sequencing analysis.
2.4. Bioinformatic and statistical analysis
Raw amplicon sequences obtained by deep sequencing were demultiplexed, quality‐filtered, and analyzed by using the software Mothur v1.35.1 (Schloss et al., 2009). All unqualified sequences, such as joint pollution, primer mismatches, low complexity, incorrect barcodes, and ambiguous bases, were discarded. Reads with a Q (base quality score) <20 and tags with less than 80% of the total base number were also removed. After the filtering and trimming procedures, all unique tags observed from each group were clustered into operational taxonomic units (OTUs) with a 3% distance level using Usearch v7.0 software (Edgar, 2010). Finally, all OTUs were classified taxonomically through the Ribosomal Database Project (RDP) Classifier, which is based on Naive Bayesian, with an 80% confidence threshold (Wang, Garrity, Tiedje, & Cole, 2007). As a result, the ACE, Chao1, Shannon, and Simpson indexes were calculated using Mothur v.1.35.1 software. The principal coordinates analysis (PCoA, weighted UniFrac distances) and bacterial taxa analysis were calculated and drawn in R v3.5.2 software. The alpha diversity indexes and relative abundance of gut microbial communities (phylum and genus level) that were identified from healthy (n = 3) and diseased (n = 3) groups were comparatively analyzed by Student's t test, and p < .05 was considered significant. Linear discriminant analysis (LDA) effect size (LEfSe) was employed to determine the key contributors of gut bacteria in healthy and anorexic crocodiles, and the LDA score threshold was 3.5 (Segata et al., 2011). Furthermore, PICRUSt analysis via the Kyoto Encyclopedia of Gene and Genomes (KEGG) database was used to predict functional profiles of gut bacteriome in healthy and diseased crocodiles (Langille et al., 2013). STAMP v2.1.3 software was used to statistically analyze the gene functions using Student's t test with Bonferroni correction (Parks, Tyson, Hugenholtz, & Beiko, 2014). A q‐value (adjusted p) < .05 with an effect size >0.2 was considered significant. A brief description of the total bioinformatic analyses of crocodile gut genomic DNA is shown in Figure 1.
Figure 1.
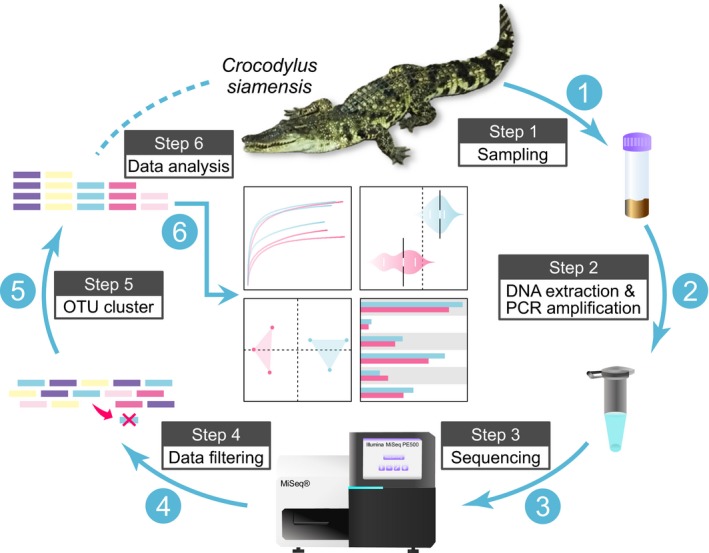
Brief description of the analysis approach for the gut microbial diversity of healthy and anorexic captive Crocodylus siamensis
3. RESULTS
3.1. Sequence survey
A total of 258,748 valid reads were gathered from cloacal samples from 6 individual crocodiles. The number of effective unique tags ranged from 38,125 to 48,216 per sample, resulting in 168 OTUs with same sequence similarity values of 97% (Table 1). The number of OTUs obtained from each sample ranged from 51 to 88. In this study, 99.98%–99.99% coverage of species was obtained in all samples, which demonstrates that the majority of the bacterial phylotypes present in the specimens were identified.
Table 1.
Summary of species richness estimators, including observed sequence reads, OTUs, estimated OTU richness (ACE and Chao1), diversity index (Shannon and Simpson), and estimated sample coverage between different cloacal samples
| Sample | Reads | OTUs | ACE | Chao1 | Shannon* | Simpson* | Coverage |
|---|---|---|---|---|---|---|---|
| H1 | 43,776 | 88 | 92.22 | 91.50 | 2.41 | 0.16 | 99.98% |
| H2 | 41,681 | 84 | 87.91 | 85.88 | 2.52 | 0.13 | 99.99% |
| H3 | 38,125 | 66 | 75.11 | 72.43 | 2.16 | 0.20 | 99.98% |
| D1 | 48,216 | 51 | 52.29 | 52.20 | 1.01 | 0.44 | 99.99% |
| D2 | 39,584 | 58 | 63.33 | 60.15 | 1.49 | 0.33 | 99.98% |
| D3 | 47,366 | 88 | 93.58 | 93.14 | 1.79 | 0.28 | 99.98% |
H1 to H3 represent the healthy crocodile cloacal samples. D1 to D3 represent the anorexic crocodile cloacal samples. OTUs clustered at 97% sequence identity. * indicates a significant difference between the healthy group (contained H1, H2, and H3) and the diseased group (contained D1, D2, and D3), as determined by Student's t test. p < .05 was considered significant.
3.2. Alpha and beta diversity
The rarefaction curve of the D group quickly reached the saturation plateau under 97% similarity values, which indicates lower species richness compared to the H group (Figure 2a). The Shannon indexes of the gut communities (Table 1; Figure 2b) in the H group were significantly higher (p = .021) than in the D group, and the Simpson indexes (Table 1) were significantly lower (p = .022) than the D group. These results indicate that the H group had richer microbial diversity than the D group. The PCoA score plot (Figure 3) revealed that the PC1, PC2, and PC3 axes included almost all variations (98.3%) of principal components found among the cloacal samples from 6 individual crocodiles. However, the H group samples were separated from the D group samples along the vast major component PC1 axis, which accounted for 86.3% of total variations.
Figure 2.
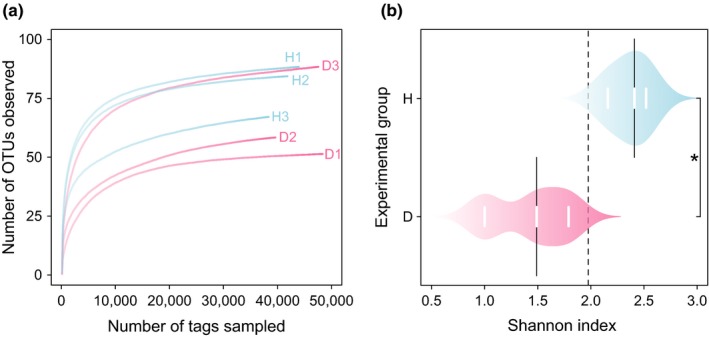
Alpha diversity of the gut microflora of captive Siamese crocodiles. (a) Rarefaction curve sequences show the species richness in the healthy group (H1, H2, and H3) and the diseased group (D1, D2, and D3) at the 3% distance cutoff. (b) The Shannon indexes of the cloacal samples from 6 individual crocodiles. H (n = 3) indicates the healthy group. D (n = 3) indicates the diseased group. * indicates a significant difference between the H group and the D group, as determined by Student's t test. p < .05 was considered significant
Figure 3.
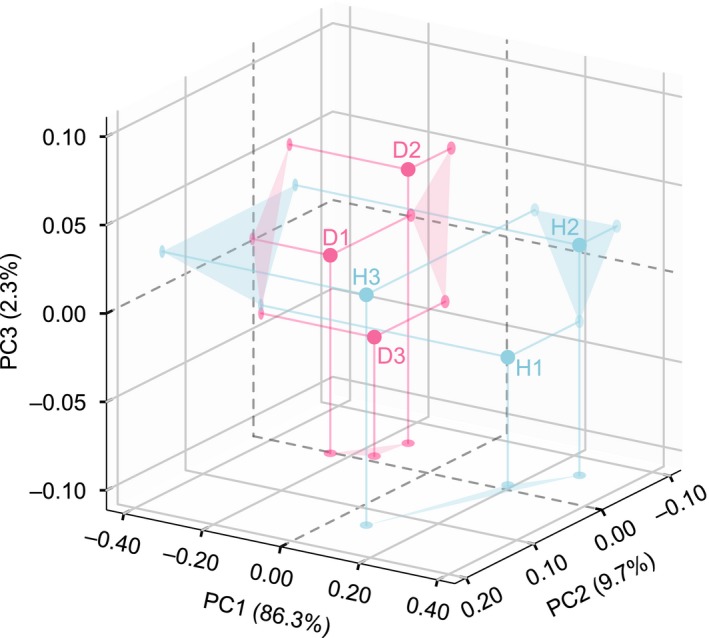
Principal coordinate analysis (PCoA) of differences in gut microbial communities based on the weighted UniFrac distances observed from 6 individual crocodiles. H indicates the healthy crocodile group (containing H1, H2, and H3). D indicates the diseased crocodile group (containing D1, D2, and D3)
3.3. Taxonomic composition and comparison
At the phylum level (Figure 4), the core microbes in the H (H1, H2, and H3) and D (D1, D2, and D3) libraries were Fusobacteria (H: 43.30%; D: 45.57%, p = .82), Bacteroidetes (H: 33.14%; D: 8.06%, p = .03, significantly enriched in the H group), Firmicutes (H: 12.03%; D: 1.10%, p = .82), Tenericutes (H: 7.79%; D: <0.01%, p = .18), and Proteobacteria (H: 3.63%; D: 44.96%, p < .001, significantly enriched in the D group).
Figure 4.
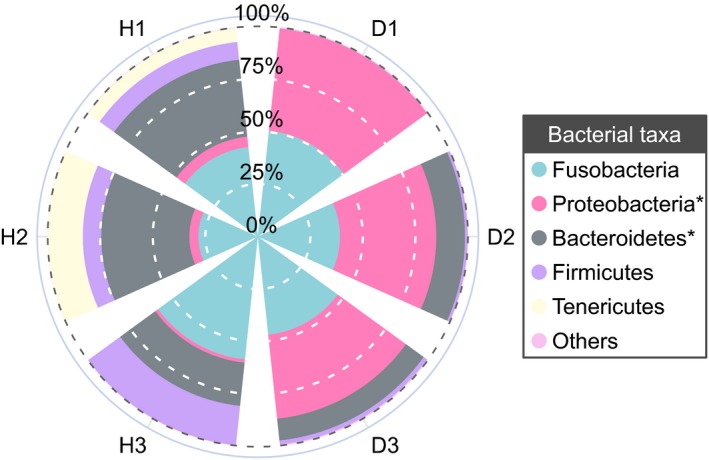
Relative abundance of gut bacterial composition of 6 individual crocodiles, organized at the phylum level. H1, H2, and H3 represent the healthy group; D1, D2, and D3 represent the diseased group. Genera with an observed relative abundance less than 1% and unclassified bacteria in both groups were assigned as “Others.” * indicates a significant difference between the healthy group and the diseased group, as determined by Student's t test. p < .05 was considered significant
At the genus level (Figure 5), the H libraries displayed a distinct structure of bacterial composition (mean relative abundance >1%) from the D libraries, except for Cetobacterium (p = .82). This shared genus was also the most dominant genus identified in all samples; it accounted for 43.15% in the H group and 45.40% in the D group. The common gut microbial communities presented in both the H and D groups also included Bacteroides (H: 17.10%; D: 1.26%, p = .09) and Macellibacteroides (H: 2.14%; D: 1.45%, p = .64). Moreover, the genera Clostridium (7.33%, p = .23), Parabacteroides (5.33%, p = .10), Plesiomonas (2.29%, p = .05, significantly enriched), Odoribacter (2.09%, p = .03, significantly enriched), and Terrisporobacter (1.17%, p = .07) were the dominant bacteria in the H group. The major components of the D group were Edwardsiella (39.28%, p = .02, significantly enriched), Aeromonas (3.01%, p = .27), Porphyromonas (2.19%, p = .39), and Raoultella (1.18%, p = .04, significantly enriched).
Figure 5.
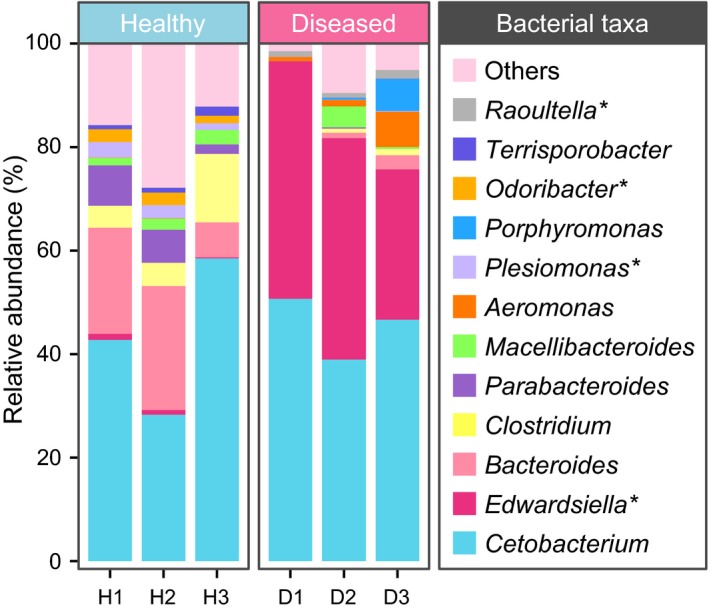
Relative abundance and composition of gut bacteria from 6 individual crocodiles, organized at the genus level. H1, H2, and H3 indicate the healthy group; D1, D2, and D3 indicate the diseased group. Genera with an observed relative abundance less than 1%, and unclassified bacteria in both groups were assigned as “Others”. * indicates a significant difference between the healthy group and the diseased group, as determined by Student's t test. p < .05 was considered significant
Specifically, all sequences recognized in the two sample groups that were within the genera Cetobacterium, Edwardsiella, Aeromonas, Plesiomonas, Terrisporobacter, and Raoultella belonged to Cetobacterium somerae, Edwardsiella tarda, Aeromonas hydrophila, Plesiomonas shigelloides, Terrisporobacter petrolearius, and Raoultella planticola, respectively. Furthermore, Bacteroides luti, Clostridium disporicum, and Porphyromonas pogonae were the major sequence contributors (69.82%, 39.71%, and 99.7%, respectively) from the genera Bacteroides, Clostridium, and Porphyromonas, respectively.
3.4. Significant alterations of the gut microbial community in healthy and diseased Siamese crocodiles
In this study, LEfSe analysis was employed to identify any key contributors that have a statistically significant role in the H and D groups. These data were calculated and analyzed by the nonparametric factorial Kruskal–Wallis test and pairwise Wilcoxon test with the same p value of .05. An LDA score value > 3.5 was considered to have reached statistical significance. The cladogram plot (Figure 6a) demonstrated that the two groups could be separated at the phylum level of the significant bacteria. It indicated that the H group contained all Firmicutes and Bacteroidetes, whereas the D group contained mostly Proteobacteria. Compared to the D group, reads from the H group indicated that the species B. luti (LDA score = 4.34), C. disporicum (LDA score = 3.65), P. shigelloides (LDA score = 3.62), and Odoribacter sp. (LDA score = 3.61) may have a highly significant effect on the healthy growth of crocodiles based on their LDA score (LDA score > 3.5). In addition, the species E. tarda (LDA score = 4.83) was significantly enriched in the D group, and it may have a negative effect on growth (Figure 6b).
Figure 6.
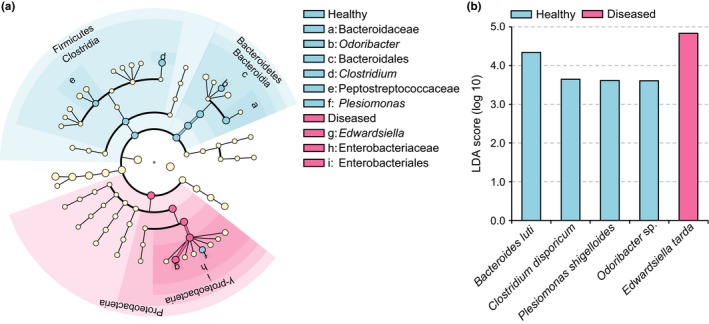
The linear discriminant analysis (LDA) effect size (LEfSe) analysis that shows the significant differences in gut flora between healthy and diseased groups. (a) Cladogram plot demonstrating the significant gut bacteria in both healthy and diseased groups. (b) Highly significant bacterial species with an LDA score >3.5
3.5. Comparison of the functional profiles of gut flora from healthy and diseased Siamese crocodiles
The present study used PICRUSt analysis to predict the major gene functions to determine the functional profiles of the gut microflora in the H and D groups. The identified genes that were predicted in all specimens, which were primarily involved in the KEGG level 1 pathways of metabolism (H: 72.68%; D: 68.79%), genetic information processing (H: 13.00%; D: 10.68%), environmental information processing (H: 9.32%; D: 15.45%), and cellular processes (H: 0.98%; D: 1.89%). These were further assigned as dominant predicted genes of 18 functional categories of KEGG level 2 pathways (Figure 7a). When compared to the H group (Figure 7b), several functional pathways (at level 2) of metabolism and genetic information processing were significantly reduced (q‐value <.05, effect size >.2). The pathways of metabolism included amino acid metabolism, biosynthesis of other secondary metabolites, and nucleotide metabolism. The pathways of genetic information processing included replication, repair, and translation. Moreover, the environmental information processing function that is related to signal transduction was significantly enriched (q‐value <.05, effect size >.2) in the D group. This was revealed by STAMP analysis using Student's t test coupled with Bonferroni correction.
Figure 7.
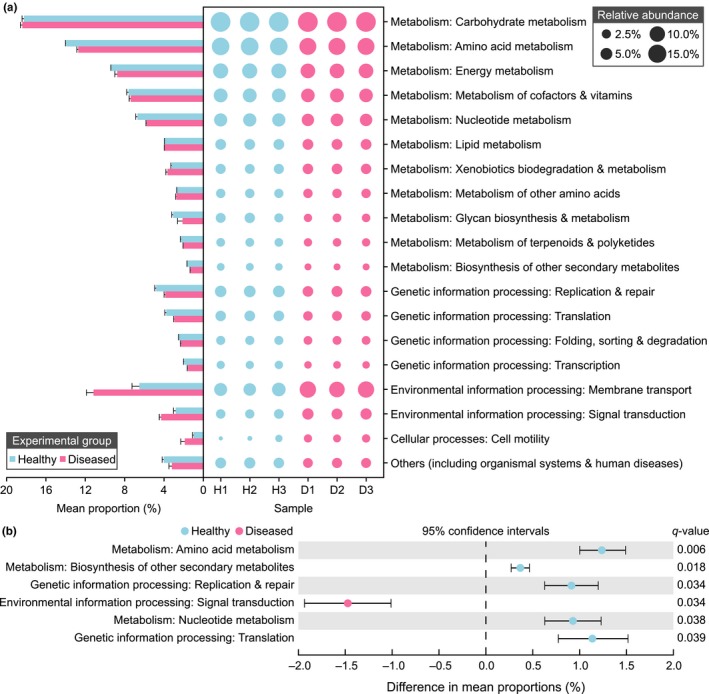
Predicted gut functional composition and the differences between the healthy and diseased groups. These were predicted by PICRUSt against the KEGG pathway database (at level 1 and level 2). (a) Functional families of healthy (H1, H2, and H3) and diseased (D1, D2, and D3) cloacal samples. (b) Extended error bar plot showing the significant differences in gene functions against KEGG database pathways (at level 2). The q‐value (adjusted p) was tested by Student's t test and multiple‐corrected using the Bonferroni method. A q‐value <.05 and effect size >.2 were considered significant
4. DISCUSSION
Gut microbiota is generally recognized as an indivisible “organ” of the host that is closely related to various diseases in animals (Dou et al., 2017; Roy et al., 2013; Li et al., 2017; Nicholson et al., 2012; Stanley, Hughes, & Moore, 2014). In the entire dataset, the gut microbes (mean relative abundance >10%) identified in healthy crocodiles were dominated by the phyla Fusobacteria, which was followed by Bacteroidetes and Firmicutes. According to previous studies that sequenced 16S rRNA genes, Bacteroidetes and Firmicutes were found to be the core gut communities of most amphibians and reptiles (Bletz et al., 2016; Colston et al., 2015; Jiang et al., 2017; Kohl et al., 2016). However, the Fusobacteria were dominated in the gut microbiota of freshwater American alligators, which is consistent with the current study (Keenan et al., 2013). As a result, there was a substantial alteration in the gut microbial composition between anorexic crocodiles and healthy crocodiles. An exception was the species C. somerae because it was the most dominant bacterium identified in both the healthy and diseased groups. C. somerae was first isolated from human feces, and it has been universally identified in the gut of freshwater fish (Bledsoe, Peterson, Swanson, & Small, 2016; Larsen, Mohammed, & Arias, 2014; Lin et al., 2019). It has been found to produce vitamin B12 and acetic acid, which are beneficial for the host (Finegold et al., 2003; Tsuchiya, Sakata, & Sugita, 2008). However, C. somerae do not seem to be strongly associated with healthy Siamese crocodiles.
The proportion of Bacteroidetes and Firmicutes in the healthy group was higher than it was in the diseased group. Among the Bacteroidetes, Bacteroides was the most abundant genera in the healthy group, and the species B. luti was significantly enriched compared to the diseased group. Bacteroides sp. is a component in the gut flora of various vertebrates, including carps, cottonmouth snakes, and crocodile lizards, and B. luti was first isolated from methanogenic sludge (Colston et al., 2015; Hatamoto, Kaneshige, Nakamura, & Yamaguchi, 2014; Jiang et al., 2017; Li et al., 2015). However, the effect of B. luti on the host‐bacteria ecosystem is vastly underexplored. Odoribacter was also a core genus of the Bacteroidetes phylum observed in this study, and it demonstrated a positive correlation with crocodile health. This was closely related to the human gut microflora and may improve host metabolism (Kulagina et al., 2012; Lim et al., 2017). Firmicutes was the third‐most dominant phylum identified in healthy crocodile cloacal samples, which mainly consisted of the genus Clostridium. According to previous studies, most Clostridium sp. can produce a type of fatty acid called butyrate, which provides many benefits to the health of the host gut (Hamer et al., 2009; Pryde, Duncan, Hold, Stewart, & Flint, 2002). The Clostridium organisms observed in the healthy group had the highest bacterial diversity, which included 9 different kinds of Clostridium species (all mean relative abundance >0.1%). However, only C. disporicum reached statistical significance when compared to the diseased group. C. disporicum is an uncommon, fermentative, and anaerobic gut bacterium, and it is primarily found in mammal feces, such as rats and pigs (Horn, 1987; Su, Yao, Perez‐Gutierrez, Smidt, & Zhu, 2008). It has been found to be related to the degradation of complex organic macromolecules (Vilajeliu‐Pons et al., 2015). Moreover, we found that the rarely known T. petrolearius was also a major component in the gut of healthy crocodiles’ group. T. petrolearius was first isolated from oilfields, and this is the first report that suggests this microbe is associated with animal gut flora (Deng et al., 2015). Interestingly, the present study found that the human pathogen P. shigelloides in the phyla Proteobacteria was also significantly enriched in the cloacal samples of healthy crocodiles (Chen et al., 2013). This appears to be a normal component of the gut flora of aquatic animals (Johnston et al., 2010; Larsen et al., 2014; Lin et al., 2019; Silva, Brito, Farias, & Nicoli, 2005). In contrast, the intestinal microflora of diseased Siamese crocodiles was significantly enriched in E. tarda, which belongs to the phyla Proteobacteria. E. tarda is widely known as a zoonotic pathogen, and it is generally found in animals of an aquatic environment, such as bullfrogs, alligators, and eels (Johnston et al., 2010; Lin et al., 2019; Mauel, Miller, & Frazier, 2002). In addition, E. tarda can infect a broad range of hosts and cause various diseases, which most frequently present as gastroenteritis and septicemia (Leung, Siame, Tenkink, Noort, & Mok, 2012; Miyazawa et al., 2018; Wang et al., 2005).
Furthermore, the result of the functional prediction indicated that the diseased group had a reduction in pathways related to amino acid metabolism, biosynthesis of other secondary metabolites, nucleotide metabolism, replication and repair, and translation, and there was a significant enrichment in the signal transduction pathway in the diseased group. Metabolism is a basic requirement for maintaining the normal growth of hosts, and this is commonly related to the function of gut bacteria in animals, such as snakes, mice, birds, and goats (Mclaughlin, Cochran, & Dowd, 2015; Suzuki & Nachman, 2016; Wang, Jin, Xue, Wang, & Peng, 2019; Wang et al., 2018). Healthy crocodiles demonstrated distinctly higher rates for some metabolic pathways. This may be related to higher energy consumption, which is required to fulfill the normal growth of the host. When some of the diseased crocodiles suffered from anorexia, there was a significant decrease in several metabolic functions of the gut microbes which were compared to the healthy controls. Besides, some functional pathways of cellular processes (including replication, repair, and translation) were also significantly reduced in the gut of anorexic crocodiles. Based on the prior work, it has been shown that the signal transduction system contributed to antibiotic resistance, biofilm formation, environmental persistence, virulence protein, and pathogenicity in E. tarda (Lv et al., 2012). Therefore, a high abundance of E. tarda may be strongly associated with an increase in the signal transduction pathway, and this may contribute to the negative effect observed in sick crocodiles. Thus, we hypothesized that during the growth stage of the captive Siamese crocodiles, some juvenile individuals might have been infected with some pathogens such as E. tarda, and then abnormal alterations of composition and function of healthy crocodiles' gut bacteria might be related to the disease by causing anorexia.
In conclusion, the present study is the first to report the composition and function of the gut microflora of captive juvenile Siamese crocodiles in both healthy and diseased conditions. The presence of B. luti, C. disporicum, P. shigelloides, and Odoribacter sp. may have beneficial contributions to the healthy growth of crocodiles, but E. tarda may negatively influence the health of the host. Our findings revealed that alterations in the composition and function of the intestinal bacteria of sick and healthy crocodiles might be associated with anorexia. However, it is still unclear how gut microbes interact with each other, which should be explored in further research. Besides, future studies should seek to increase the sample size of the host in order to enhance the statistical power for detailed bioinformatic analyses.
CONFLICT OF INTERESTS
None declared.
AUTHOR CONTRIBUTIONS
Conceptualization: ML (lead) and CXZ (supporting); Data curation: CXZ; Formal analysis: CXZ (lead), ML (equal), ZQL (equal), YM (supporting); Funding acquisition: ML; Investigation: CXZ (lead), ZQL (equal), YM (equal), and XQJ (supporting); Methodology: CXZ (lead), ZQL (equal), YM (equal), and XQJ (supporting); Project administration: ML; Supervision: ML; Software: CXZ (lead) and ZQL (supporting); Validation: CXZ (lead), ZQL (equal), YM (equal); Visualization: CXZ; Writing–original draft: CXZ (equal) and ML (equal). All authors reviewed and edited the manuscript, and gave the final approval for publication.
ETHICS STATEMENT
This study complied to the guidelines for the kindly care and use of experimental animals established by the Ministry of Science and Technology of the People's Republic of China (Approval No. 2006–398). Besides, the research protocol was reviewed and approved by the Animal Ethics Committee of Jimei University (Approval No. JMULAC201603). No hunting and destructive sampling involved.
ACKNOWLEDGMENTS
This study was financially supported by Regional Demonstration of Marine Economy Innovative Development Project (No. 16PZY002SF18), Natural Science Foundation of Fujian Province (No. 2019J01695), and National Natural Science Foundation of China (No. 31202030 and No. 31272669).
Lin M, Zeng C, Li Z, Ma Y, Jia X. Comparative analysis of the composition and function of fecal‐gut bacteria in captive juvenile Crocodylus siamensis between healthy and anorexic individuals. MicrobiologyOpen. 2019;8:e929 10.1002/mbo3.929
Mao Lin and Chenxi Zeng are contributed equally to this study and share the first authorship.
DATA AVAILABILITY STATEMENT
All raw sequencing data obtained in this study were deposited in the Sequence Read Archive of the National Center for Biotechnology Information (NCBI), and the SRA submission data are SRX4396839‐SRX4396844.
REFERENCES
- Bezuijen, M. R. , Cox, J. H. , Thorbjarnarson, J. B. , Phothitay, C. , Hedemark, M. , & Rasphone, A. (2013). Status of Siamese crocodile (Crocodylus siamensis) schneider, 1801 (reptilia: Crocodylia) in Laos. Journal of Herpetology, 47, 41–65. 10.1670/11-157 [DOI] [Google Scholar]
- Bledsoe, J. W. , Peterson, B. C. , Swanson, K. S. , & Small, B. C. (2016). Ontogenetic characterization of the intestinal microbiota of channel catfish through 16S rRNA gene sequencing reveals insights on temporal shifts and the influence of environmental microbes. PLoS ONE, 11, e0166379 10.1371/journal.pone.0166379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bletz, M. C. , Goedbloed, D. J. , Sanchez, E. , Reinhardt, T. , Tebbe, C. C. , Bhuju, S. , … Steinfartz, S. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitat‐specific predicted functions. Nature Communications, 7, 13699 10.1038/ncomms13699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus, A. C. , & Hawke, J. P. (2002). Providencia rettgeri‐associated septicemia and meningoencephalitis in juvenile farmed American alligators Alligator mississippiensis . Journal of Aquatic Animal Health, 14, 149–153. 10.1577/1548-8667(2002)014<0149:PRASAM>2.0.CO;2 [DOI] [Google Scholar]
- Chen, X. , Chen, Y. U. , Yang, Q. , Kong, H. , Yu, F. , Han, D. , … Li, L. (2013). Plesiomonas shigelloides infection in southeast China. PLoS ONE, 8, e77877 10.1371/journal.pone.0077877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colston, T. J. , & Jackson, C. R. (2016). Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Molecular Ecology, 25, 3776–3800. 10.1111/mec.13730 [DOI] [PubMed] [Google Scholar]
- Colston, T. J. , Noonan, B. P. , & Jackson, C. R. (2015). Phylogenetic analysis of bacterial communities in different regions of the gastrointestinal tract of Agkistrodon piscivorus, the cottonmouth snake. PLoS ONE, 10, e0128793 10.1371/journal.pone.0128793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, S. , Gadonna‐Widehem, P. , Rome, V. , Hamoudi, D. , Rhazi, L. , Lakhal, L. , … Abdennebi‐Najar, L. (2017). Characterisation of early‐life fecal microbiota in susceptible and healthy pigs to post‐weaning diarrhoea. PLoS ONE, 12, e0169851 10.1371/journal.pone.0169851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg, P. B. (2005). Diversity of the human intestinal microbial flora. Science, 308, 1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than blast. Bioinformatics, 26, 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Feng, Q. , Chen, W. D. , & Wang, Y. D. (2018). Gut microbiota: An integral moderator in health and disease. Frontiers in Microbiology, 9, 151 10.3389/fmicb.2018.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegold, S. M. , Vaisanen, M.‐L. , Molitoris, D. R. , Tomzynski, T. J. , Song, Y. , Liu, C. , … Lawson, P. A. (2003). Cetobacterium somerae, sp. nov. from human feces and emended description of the genus Cetobacterium . Systematic and Applied Microbiology, 26, 177–181. 10.1078/072320203322346010 [DOI] [PubMed] [Google Scholar]
- Guo, X. , Wang, Y. , Yang, Z. , Kong, D. , Chen, X. , Wang, H. , … He, M. (2015). Terrisporobacter petrolearius sp. nov. isolated from an oilfield petroleum reservoir. International Journal of Systematic and Evolutionary Microbiology, 65, 3522–3526. 10.1099/ijsem.0.000450 [DOI] [PubMed] [Google Scholar]
- Hamer, H. M. , Jonkers, D. M. A. E. , Bast, A. , Vanhoutvin, S. A. L. W. , Fischer, M. A. J. G. , Kodde, A. , … Brummer, R.‐J. (2009). Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clinical Nutrition, 28, 88–93. 10.1016/j.clnu.2008.11.002 [DOI] [PubMed] [Google Scholar]
- Hatamoto, M. , Kaneshige, M. , Nakamura, A. , & Yamaguchi, T. (2014). Bacteroides luti sp. nov. an anaerobic, cellulolytic and xylanolytic bacterium isolated from methanogenic sludge. International Journal of Systematic and Evolutionary Microbiology, 64, 1770–1774. 10.1099/ijs.0.056630-0 [DOI] [PubMed] [Google Scholar]
- Hooper, L. V. (2001). Commensal host‐bacterial relationships in the gut. Science, 292, 1115–1118. 10.1126/science.1058709 [DOI] [PubMed] [Google Scholar]
- Horn, N. (1987). Clostridium disporicum sp. nov. a saccharolytic species able to form two spores per cell, isolated from a rat cecum. International Journal of Systematic Bacteriology, 37, 398–401. 10.1099/00207713-37-4-398 [DOI] [Google Scholar]
- Jiang, H.‐Y. , Ma, J.‐E. , Li, J. , Zhang, X.‐J. , Li, L.‐M. , He, N. , … Chen, J.‐P. (2017). Diets alter the gut microbiome of crocodile lizards. Frontiers in Microbiology, 8, 2073 10.3389/fmicb.2017.02073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, M. A. , Porter, D. E. , Scott, G. I. , Rhodes, W. E. , & Webster, L. F. (2010). Isolation of faecal coliform bacteria from the American alligator (Alligator mississippiensis). Journal of Applied Microbiology, 108, 965–973. 10.1111/j.1365-2672.2009.04498.x [DOI] [PubMed] [Google Scholar]
- Keenan, S. W. , & Elsey, R. M. (2015). The good, the bad, and the unknown: Microbial symbioses of the American alligator. Integrative and Comparative Biology, 55, 972–985. 10.1093/icb/icv006 [DOI] [PubMed] [Google Scholar]
- Keenan, S. W. , Engel, A. S. , & Elsey, R. M. (2013). The alligator gut microbiome and implications for archosaur symbioses. Scientific Reports, 3, 2877 10.1038/srep02877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. T. , Lee, S. H. , & Kwak, D. (2016). Sudden death of a Siamese crocodile (Crocodylus siamensis) due to systemic aspergillosis. Journal of Veterinary Medical Science, 78, 1723–1726. 10.1292/jvms.16-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, K. D. , Brun, A. , Magallanes, M. , Brinkerhoff, J. , Laspiur, A. , Acosta, J. C. , … Bordenstein, S. R. (2016). Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions, and transmission. Molecular Ecology, 26, 1175–1189. 10.1111/mec.13921 [DOI] [PubMed] [Google Scholar]
- Kulagina, E. V. , Efimov, B. A. , Maximov, P. Y. , Kafarskaia, L. I. , Chaplin, A. V. , & Shkoporov, A. N. (2012). Species composition of bacteroidales order bacteria in the feces of healthy people of various ages. Bioscience, Biotechnology, and Biochemistry, 76, 169–171. 10.1271/bbb.110434 [DOI] [PubMed] [Google Scholar]
- Langille, M. G. I. , Zaneveld, J. , Caporaso, J. G. , McDonald, D. , Knights, D. , Reyes, J. A. , … Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, A. M. , Mohammed, H. H. , & Arias, C. R. (2014). Characterization of the gut microbiota of three commercially valuable warmwater fish species. Journal of Applied Microbiology, 116, 1396–1404. 10.1111/jam.12475 [DOI] [PubMed] [Google Scholar]
- Le Roy, T. , Llopis, M. , Lepage, P. , Bruneau, A. , Rabot, S. , Bevilacqua, C. , … Gérard, P. (2013). Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut, 62, 1787–1794. 10.1136/gutjnl-2012-303816 [DOI] [PubMed] [Google Scholar]
- Leelawongtawon, R. , Siruntawineti, J. , Chaeychomsri, W. , & Sattaponpan, C. (2010). Antibacterial and antifungal activities from Siamese crocodile blood. Journal of the Medical Association of Thailand, 93, S58–64. [PubMed] [Google Scholar]
- Leung, K. Y. , Siame, B. A. , Tenkink, B. J. , Noort, R. J. , & Mok, Y. K. (2012). Edwardsiella tarda ‐ virulence mechanisms of an emerging gastroenteritis pathogen. Microbes and Infection, 14, 26–34. 10.1016/j.micinf.2011.08.005 [DOI] [PubMed] [Google Scholar]
- Li, H. L. , Chen, L. P. , Hu, Y. H. , Qin, Y. , Liang, G. , Xiong, Y. X. , & Chen, Q. X. (2012). Crocodile oil enhances cutaneous burn wound healing and reduces scar formation in rats. Academic Emergency Medicine, 19, 265–273. 10.1111/j.1553-2712.2012.01300.x [DOI] [PubMed] [Google Scholar]
- Li, T. , Li, H. , Gatesoupe, F.‐J. , She, R. , Lin, Q. , Yan, X. , … Li, X. (2017). Bacterial signatures of “red‐operculum” disease in the gut of crucian carp (Carassius auratus). Microbial Ecology, 74, 510–521. 10.1007/s00248-017-0967-1 [DOI] [PubMed] [Google Scholar]
- Li, T. T. , Long, M. , Gatesoupe, F. J. , Zhang, Q. Q. , Li, A. H. , & Gong, X. N. (2015). Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microbial Ecology, 69, 25–36. 10.1007/s00248-014-0480-8 [DOI] [PubMed] [Google Scholar]
- Lim, M. Y. , You, H. J. , Yoon, H. S. , Kwon, B. , Lee, J. Y. , Lee, S. , … Ko, G. P. (2017). The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut, 66, 1031–1038. 10.1136/gutjnl-2015-311326 [DOI] [PubMed] [Google Scholar]
- Lin, M. , Zeng, C. X. , Jia, X. Q. , Zhai, S. W. , Li, Z. Q. , & Ma, Y. (2019). The composition and structure of the intestinal microflora of Anguilla marmorata at different growth rates: A deep sequencing study. Journal of Applied Microbiology, 126, 1340–1352. 10.1111/jam.14174 [DOI] [PubMed] [Google Scholar]
- Lv, Y. Z. , Xiao, J. F. , Liu, Q. , Wu, H. Z. , Zhang, Y. X. , & Wang, Q. Y. (2012). Systematic mutation analysis of two‐component signal transduction systems reveals EsrA‐EsrB and PhoP‐PhoQ as the major virulence regulators in Edwardsiella tarda . Veterinary Microbiology, 157, 190–199. 10.1016/j.vetmic.2011.12.018 [DOI] [PubMed] [Google Scholar]
- Mauel, M. J. , Miller, D. L. , & Frazier, K. S. (2002). Bacterial pathogens isolated from cultured bullfrogs (Rana castesbeiana). Journal of Veterinary Diagnostic Investigation, 14, 431–433. 10.1177/104063870201400515 [DOI] [PubMed] [Google Scholar]
- Mclaughlin, R. W. , Cochran, P. A. , & Dowd, S. E. (2015). Metagenomic analysis of the gut microbiota of the timber rattlesnake, (Crotalus horridus). Molecular Biology Reports, 42(7), 1187–1195. 10.1007/s11033-015-3854-1 [DOI] [PubMed] [Google Scholar]
- Miyazawa, Y. , Murakami, K. , Kizaki, Y. , Itaya, Y. , Takai, Y. , & Seki, H. (2018). Maternal peripartum septic shock caused by intrauterine infection with Edwardsiella tarda: A case report and review of the literature. Journal of Obstetrics and Gynaecology Research, 44, 171–174. 10.1111/jog.13476 [DOI] [PubMed] [Google Scholar]
- Nicholson, J. K. , Holmes, E. , Kinross, J. , Burcelin, R. , Gibson, G. , Jia, W. , & Pettersson, S. (2012). Host‐gut microbiota metabolic interactions. Science, 336, 1262–1267. 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- Parks, D. H. , Tyson, G. W. , Hugenholtz, P. , & Beiko, R. G. (2014). STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics, 30, 3123–3124. 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde, S. E. , Duncan, S. H. , Hold, G. L. , Stewart, C. S. , & Flint, H. J. (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiology Letters, 217, 133–139. 10.1016/s0378-1097(02)01106-0 [DOI] [PubMed] [Google Scholar]
- Roh, Y. S. , Park, H. , Cho, H. U. , Cho, A. , Islam, M. R. , Cho, H. S. , … Kim, B. (2011). Aeromonas hydrophila‐associated septicemia in captive crocodiles (Crocodylus johnstoni and Crocodylus porosus). Journal of Zoo and Wildlife Medicine, 42, 738–742. 10.1638/2010-0234.1 [DOI] [PubMed] [Google Scholar]
- Ryan, C. (1998). Saltwater crocodiles as tourist attractions. Journal of Sustainable Tourism, 6, 314–327. 10.1080/09669589808667319 [DOI] [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , … Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, K. P. , Gratz, S. W. , Sheridan, P. O. , Flint, H. J. , & Duncan, S. H. (2013). The influence of diet on the gut microbiota. Pharmacological Research, 69, 52–60. 10.1016/j.phrs.2012.10.020 [DOI] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W. S. , & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology, 12, 1–18. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton, T. J. (2018). Role of the gut microbiome in vertebrate evolution. mSystems, 3(2), e00174‐17 10.1128/mSystems.00174-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, F. C. P. , Brito, M. F. G. , Farias, L. M. , & Nicoli, J. R. (2005). Composition and antagonistic activity of the indigenous intestinal microbiota of Prochilodus argenteus Agassiz. Journal of Fish Biology, 67, 1686–1698. 10.1111/j.1095-8649.2005.00877.x [DOI] [Google Scholar]
- Stanley, D. , Hughes, R. J. , & Moore, R. J. (2014). Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Applied Microbiology and Biotechnology, 98, 4301–4310. 10.1007/s00253-014-5646-2 [DOI] [PubMed] [Google Scholar]
- Su, Y. , Yao, W. , Perez‐Gutierrez, O. N. , Smidt, H. , & Zhu, W. Y. (2008). 16S ribosomal RNA‐based methods to monitor changes in the hindgut bacterial community of piglets after oral administration of Lactobacillus sobrius S1. Anaerobe, 14, 78–86. 10.1016/j.anaerobe.2007.12.004 [DOI] [PubMed] [Google Scholar]
- Suzuki, T. A. , & Nachman, M. W. (2016). Spatial heterogeneity of gut microbial composition along the gastrointestinal tract in natural populations of house mice. PLoS ONE, 11, e0163720 10.1371/journal.pone.0163720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya, C. , Sakata, T. , & Sugita, H. (2008). Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Letters in Applied Microbiology, 46, 43–48. 10.1111/j.1472-765X.2007.02258.x [DOI] [PubMed] [Google Scholar]
- Vilajeliu‐Pons, A. , Puig, S. , Pous, N. , Salcedo‐Dávila, I. , Bañeras, L. , Balaguer, M. D. , & Colprim, J. (2015). Microbiome characterization of MFCs used for the treatment of swine manure. Journal of Hazardous Materials, 288, 60–68. 10.1016/j.jhazmat.2015.02.014 [DOI] [PubMed] [Google Scholar]
- Waite, D. W. , & Taylor, M. W. (2015). Exploring the avian gut microbiota: Current trends and future directions. Frontiers in Microbiology, 6, 673 10.3389/fmicb.2015.00673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, I. K. , Kuo, H. L. , Chen, Y. M. , Lin, C. L. , Chang, H. Y. , Chuang, F. R. , & Lee, M. H. (2005). Extraintestinal manifestations of Edwardsiella tarda infection. International Journal of Clinical Practice, 59, 917–921. 10.1111/j.1742-1241.2005.00527.x [DOI] [PubMed] [Google Scholar]
- Wang, L. , Jin, L. , Xue, B. , Wang, Z. , & Peng, Q. (2019). Characterizing the bacterial community across the gastrointestinal tract of goats: Composition and potential function. MicrobiologyOpen, e820 10.1002/mbo3.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Zheng, S. S. , Li, L. X. , Yang, Y. S. , Liu, Y. B. , Wang, A. Z. , … Li, Y. (2018). Comparative metagenomics of the gut microbiota in wild greylag geese (Anser anser) and ruddy shelducks (Tadorna ferruginea). Microbiologyopen, e00725 10.1002/mbo3.725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J. , Zeng, J. , Guo, G. , Jiang, J. , Yang, N. , Wang, P. , … Wang, Y. (2018). An investigation of sudden death in farmed infant Siamese crocodiles during winter and spring in Hainan, China. Indian Journal of Animal Research, 52, 1058–1062. 10.18805/ijar.v0iOF.6999 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw sequencing data obtained in this study were deposited in the Sequence Read Archive of the National Center for Biotechnology Information (NCBI), and the SRA submission data are SRX4396839‐SRX4396844.