

Abstract
Raman spectroscopy is the most commonly used method to investigate structures of materials. Recently, few-layered IV-VI 2D materials (SnS, SnSe, GeS, and GeSe) have been found and ignited significant interest in electronic and optical applications. However, unlike few-layer graphene, in which its interlayer structures such as the number of its layers are confirmed through measurement of the Raman scattering, few-layer IV-VI 2D materials have not yet been developed to the point of understanding their interlayer structure. Here we performed first-principles calculations on Raman spectroscopy for few-layer IV-VI 2D materials. In addition to achieving consistent results with measurements of bulk structures, we revealed significant red and blue shifts of characteristic Raman modes up to 100 cm−1 associated with the layer number. These shifts of lattice vibrational modes originate from the change of the bond lengths between the metal atoms and chalcogen atoms through the change of the interlayer interactions. Particularly, our study shows weak covalent bonding between interlayers, making the evolution of Raman signals according to the thickness different from other vdW materials. Our results suggest a new way for obtaining information of layer structure of few-layer IV-VI 2D materials through Raman spectroscopy.
Subject terms: Magnetic properties and materials, Structural properties, Two-dimensional materials
Introduction
Recently, van der Waals (vdW) materials such as graphene and transition metal dichalcogenides have attracted substantial attention from nanoscience and technology fields because they have exotic electronic properties and potential applications for energy storage and harvest. In addition, their properties have been shown to be dependent on the number of layers they have1–6. For example, for graphene, it has a Dirac cone in a single layer while it is gone more than one layer, leading to different physical phenomena such as massless and mass Fermions, respectively7,8. Bilayer graphene shows an exotic superconducting phase as the twisting angle between the layers9. By contrast, single-layer MoS2 is semiconducting with a direct bandgap of 1.57 eV, and the properties of few-layer MoS2 are significantly tuned to be indirect-bandgap semiconductors by the number of layers, because of interlayer interaction10–12. Although the technology for controlling the thickness of vdW materials has been developed by exfoliation and atomic layer deposition methods13–18, it is highly appreciated to conveniently verify the number of layers or thicknesses of vdW materials for device applications.
As one of the most fundamental tools to study structures, Raman spectroscopy can be employed to determine the thickness of few-layer vdW materials such as graphene19 and transition metal dichalcogenides20 because the lattice vibration modes are dependent on the number of layers of the vdW materials. For instance, for graphene, the number of layers can be determined based on the results of Raman spectroscopy: the change of the position of G peak or the position and shape of the 2D band21. In the case of MoS2, the positions of the E12g and A1g peaks provide clues for the thickness22–24. For black phosphorus, the intensities of Ag1 and B2g peaks play the same roles25.
More recently, in few-layer metal monochalcogenides (MX, M = Sn, Ge; X = S, Se, etc.), members of layered IV-VI compounds have been discovered through exfoliation from bulk structures26–28. The structures are the same as that of black phosphorus, which is called the puckered structure, where M and X are bonded alternatively (Fig. 1). These novel materials show intriguing electric polarization properties26,27,29,30. In addition, they have properties of vertical dielectric screening, resulting in changes to the electronic and optical properties27,31,32. On the other hand, due to their high absorption coefficient, they can be used for photovoltaic cells3,33–38 and show high performance. They can also be used for lithium-ion battery anodes39,40, because they have layered structures where Li are intercalated to space between layers, exhibiting high Li capacity. In addition, MX has attracted much attention due to the ~1 eV electronic bandgap close to bulk silicon. Thus MX has a huge potential for use in electric devices41. Similar to almost layer stacking structures, monolayer or few-layer MX could have a distinctive property or better performance for certain devices, like thin-film devices42. Recently, there have been many attempts to synthesize few-layer MX, including through the CVD method43–48, chemical bath deposition38,49,50, atomic layer deposition18,51, and spray pyrolysis52. Heterostructures with MX have also been studied as field-effect transistor device materials48.
Figure 1.

(a) Schematic diagram of the crystal structure of MX. It is a puckered structure like that of black phosphorus. d denotes the interlayer distance. (b) L1 and L2 represent bonding lengths of the vertical and horizontal directions of the plane, respectively. (c) Top view of the crystal structure.
However, like graphene and transition metal dichalcogenides, verifying the number of layers in few-layer MX structures is crucial when they are used as device materials. In stacking a layer, we average the length of all similar direction bonding. While MX has attracted great interest, verifying the number of layers of MX sample has not yet been sufficiently studied. For example, there has only been one case verifying the thickness of a sample using an electronic bandgap10; that is a good method for figuring out the thickness. However, measuring a bandgap has too many more steps than Raman and can ruin an exfoliated crystal sample. Therefore, nondestructive Raman method might be the best method, but there are no relevant reference data. In this paper, we performed Raman simulation for MX as the number of layers varied from a monolayer to bulk in order to provide standard Raman spectroscopy data. We focus on SnS, SnSe, GeS, and GeSe as representative examples for MX. Our calculations show that the Raman spectroscopy for metal monochalcogenides depends on the number of layers. We found that the red shift and blue shift occur continuously as the number of layers increases by ~100 cm−1. This can be explained by the fact that the shifts come from the change of the bond lengths between the metal atoms and chalcogen atoms by the change of the interlayer interaction, leading to the slight modification of a vibration mode. These results provide benchmarks for determining the number of layers of metal monochalcogenides by Raman spectroscopy analysis.
All of the calculations were carried out using density functional theory (DFT) and density functional perturbation theory (DFPT)53, as implemented in the PWSCF package of the QUANTUM-ESPRESSO54. The DFT is employed for the geometry optimization of the IV-VI layers, and the DFPT is employed for calculations of the Raman spectra, phonons, and dynamical matrices. We used norm conserved local density approximation (LDA) for pseudopotentials55,56, which gives consistent results with PBE-D3. The kinetic energy cutoff was taken to be 30 Ry. We use 9 × 9 × 3 and 9 × 9 × 1 k-point for the bulk and layered structures, respectively. Geometrical optimizations are performed for every single structure until the Hellman-Feynman force is below 10−8 Ry/Bohr. All of our calculations were converged to ~10−4 Ry/atom. Detail information for the optimization of the lattice vectors and the lattice constants is described in supporting information Figure S1, Figure S2, and Table S1.
We first performed calculations on the geometry optimizations for AB staked multilayered MXs (Fig. 1a) because it has been confirmed that, for MX bulks, AB stacked MX layers in bulks are the energetically most favorable configuration57,58. Figure 1 shows the optimized atomics structures of AB-stacked multilayer MX structures, where L1 and L2 denote the bonding lengths for out of plane and in-plane, respectively. The calculated values of the L1 and L2 and the interlayer distances for AB stacked MX multilayers are presented in Table 1, which are consistent with the values in the literature for a multi-layer59. For instance, L1 and L2 were calculated to be 2.56 and 2.70 Å for bilayer SnS, respectively. The bond lengths are increased as the atomic numbers of M and X increase. However, the interlayer distance, d is 2.64 Å (Supporting information Figure S3), slightly dependent on the type of MX layers, which corresponds to the equilibrium distance of ~3.4 Å via vdW interaction. Furthermore, the calculated values of L1, L2, and d as the number of layers varied from 1 to 3 are also presented in Table 1. We also carried out the calculations on the interlayer binding energy of bilayer and trilayer MXs in Table 2. For comparison, the interlayer binding energy and the interlayer distance were also calculated with other functional, i.e., meta-GGA60. The binding energy and the interlayer distance obtained with the LDA is slightly larger and smaller than those with the meta-GGA, respectively. We found that there are small changes in L1 and L2 as well as interlayer distance, similar to other 2D materials12,61.
Table 1.
Calculated local geometry information for AB stacked MX layers as the number of layers.
| Materials | # of layers | L1 | L2 | Δ1 | Δ2 | d |
|---|---|---|---|---|---|---|
| SnS | 1 | 2.53 | 2.73 | 0 | 0 | — |
| 2 | 2.56 | 2.70 | 0.03 | −0.03 | 2.64 | |
| 3 | 2.58 | 2.69 | 0.05 | −0.04 | 2.64 | |
| ∞ | 2.61 | 2.64 | 0.08 | −0.09 | 2.57 | |
| SnSe | 1 | 2.66 | 2.91 | 0 | 0 | — |
| 2 | 2.69 | 2.84 | 0.03 | −0.07 | 2.79 | |
| 3 | 2.70 | 2.79 | 0.04 | −0.12 | 2.71 | |
| ∞ | 2.75 | 2.77 | 0.09 | −0.14 | 2.66 | |
| GeS | 1 | 2.36 | 2.46 | 0 | 0 | — |
| 2 | 2.39 | 2.45 | 0.03 | −0.01 | 2.49 | |
| 3 | 2.39 | 2.44 | 0.03 | −0.02 | 2.46 | |
| ∞ | 2.43 | 2.42 | 0.07 | −0.04 | 2.36 | |
| GeSe | 1 | 2.49 | 2.59 | 0 | 0 | — |
| 2 | 2.51 | 2.59 | 0.02 | 0 | 2.80 | |
| 3 | 2.52 | 2.56 | 0.03 | −0.03 | 2.58 | |
| ∞ | 2.55 | 2.56 | 0.06 | −0.03 | 2.51 |
Δ1(Δ2) denotes L1 (L2) for a given layer minus L1 (L2) of monolayer presented in Fig. 1(b). The unit of the values below is angstroms. ∞ means multilayer of MX. d indicates the interlayer distance.
Table 2.
Calculated binding energy (Eb) between interlayers for few-layer MXs and interlayer distance (d). The exchange-correlation functional is treated using LDA and strongly constrained and appropriately normed (SCAN) meta-generalized gradient approximation (meta-GGA).
| Functionals | # of layers | SnS | SnSe | GeS | GeSe |
|---|---|---|---|---|---|
| LDA | 2 | Eb = 0.50 eV | 0.50 eV | 0.49 eV | 0.38 eV |
| d = 2.64 Å | 2.79 Å | 2.49 Å | 2.80 Å | ||
| 3 | Eb = 0.46 eV | 0.51 eV | 0.49 eV | 0.39 eV | |
| d = 2.64 Å | 2.71 Å | 2.46 Å | 2.58 Å | ||
| Meta-GGA | 2 | Eb = 0.26 eV | 0.26 eV | 0.28 eV | 0.28 eV |
| d = 2.97 Å | 3.15 Å | 2.73 Å | 3.04 Å |
We performed calculations on the Raman spectra of multilayer MX using DFPT calculation. In order to verify the computational accuracy of the DFPT, the calculated results were compared to the experimental data for AB stacked multilayer SnS18. We focused on the four characteristic peaks in the spectra of 53, 76, 104, and 244 cm−1 for monolayer SnS, because their Raman signals are remarkable and have been observed in experiments18,62. Remarkably, the calculated Raman spectra for SnS is strongly consistent with the experimental data (Fig. 2)18. Thus, we believe that our calculations on the Raman spectra for other MX layers are reliable for predicting their properties. Moreover, it was verified that there is high accuracy in Raman simulations for monolayer using the ab initio method63.
Figure 2.
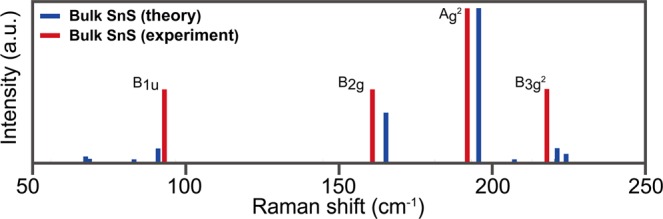
Calculated and experimental Raman spectroscopy for AB stacked multilayer SnS. The experimental data was in ref. 62.
We further analyzed the lattice vibrational modes of AB-stacked multilayer SnS corresponding to the characteristic Raman peaks in order to determine how the peaks are generated. We found that the phonon modes of the four B1u, Ag2, B2g, and B3g2 shown in Fig. 3(e–h), respectively, are associated with the peaks for 91, 166, 196, and 221 cm−1 for multilayer SnS, respectively. Hereafter, we refer to the four peaks in the Raman spectra as B1u, Ag2, B2g, and B3g2, respectively. Monolayer SnS has the same phonon modes in Fig. 3(a–d). For the B1u phonon mode, Sn atoms move out of plane and S atoms move in the oblique plane (Fig. 3(a,e)). For the Ag2 phonon mode, Sn and S atoms move in the oblique plane (Fig. 3(c,g)). For the B2g, and B3g2 phonon modes, the Sn and S atoms move in the same plane in the opposite direction (Fig. 3(b,d,f,h)). These modes can be approximately classified by atomic moving direction into three kinds: vertical of the MX plane, horizontal of the MX plane, and hybrid. B2g is horizontal while B3g2 is vertical of the SnS plane. B1u looks like a hybrid of the two. In Ag2 mode, atoms have a vibration in both directions, out of plane and in-plane. However, moving in out of plane appears to be negligible. Therefore, we can classify Ag2 as the horizontal mode.
Figure 3.

Phonon modes of monolayer ((a–d)) and multilayer (e–h) SnS: B1u, B2g, Ag2, and B3g2.
In order to investigate how the Raman spectra of few-layer MX materials depend on the number of layers, we performed calculations for few-layers MX as the number of layers varied. An attractive feature is that the Raman spectrum of SnS is dependent on the number of layers. For SnS, we found that B1u, B2g, and Ag2 are a blue shift by ~40 cm−1, ~90 cm−1, and ~92 cm−1, respectively, while B3g2 are red shift by ~23 cm−1.(Fig. 4(a)) Since Raman peaks are naturally split by stacking layers, we selected the most significant peak among several split peaks. If certain peaks are degenerate in the same position, the measured intensity is higher than that of the theoretical prediction. Increasing the number of layers yields a red shift for B3g2 and a blue shift for B2g and Ag2. These trends appear in all four MX except for B1u. For the SnS and GeS layers, the B1u mode shows a blue shift. For GeSe, the B1u mode has a red shift trend, while it does not show any shift trend for SnSe. For other peaks, the changes appear to be sufficient to distinguish the number of layers of samples. On the other hand, the interaction addition as the number of layers is significant in a vertical direction compared with a horizontal direction because The thickness with respect to the vertical to the plane of MXs is increased as the number of their layers increases because L1 increases as the number of layers increases (see Table 1). Therefore, the frequency shift in a complete vertical mode, i.e., B3g2 is more significant than that other modes as shown in Fig. 4.
Figure 4.

Raman shifts for four peaks in (a) SnS, (b) SnSe, (c) GeS, and (d) GeSe as the number of layers. With an increasing number of layers, Raman shifts increase or decrease depending on the type of phonon mode.
Moreover, we observe that the tendency depends on the direction of phonon mode, horizontal or vertical. In Fig. 5, the horizontal modes (such as B2g), in-plane modes, appear as the blue shift tendency, while the vertical modes (such as B3g2), out of plane modes, appear as the red shift trend when increasing the number of layers for SnS. This relation is consistent across the four kinds of MX.
Figure 5.

Calculated Raman spectroscopy of AB stacked few-layered SnS as the number of layers: (a) monolayer, (b) bilayer, (c) trilayer, and (d) multilayer SnS.
We investigate what causes the difference between red and blue shift trends. In order to determine the blue and red shifts of the Raman spectroscopy, we investigated the local geometry of them with varying numbers of layers. The calculated values of the bond lengths (L1, L2) between L1, L2, and interlayered distance as the number of layers varies are presented in Table 1. Importantly, we found that there is a small change of length for unit cell vectors, as in other 2D materials12,61. Further, there are certain changes of atomic bonding length from interlayer interactions. This can change the lattice vibration motion, which might be one of the causes of the shift tendency. We can understand that the change of bond lengths can be interpreted by the change of the spring constant using a simple harmonic approximation. Hence, when a bonding length is decreasing, a frequency is increased. This is the reason for why Raman shift frequency, a peak position, is changed according to the number of layers. According to the number of layers, the changes for L1 and L2 are shown in Table 1. With increasing the number of layers, L1, an average bond length of the vertical direction, is increasing, while L2, an average bond length of the horizontal direction, is decreasing. In other words, with increasing the number of layers, the vertical mode has a red shift and the horizontal mode has a blue shift. This relation holds for all our studied MXs.
In order to understand the origin of the bond lengths of few-layer MXs resulting in the shift of the phonon energy, we investigate the bond between layers. We found that this change of the lattice parameter via the interaction between layers stems from the weakly covalent bonding between interlayers, as shown in Fig. 6, where the charges transfer between interlayers. This differs from the bond between interlayer of few-layer vdW materials like graphene or MoS2. In addition, the binding energy of the layers is calculated to range from 0.2 eV to 0.5 eV, slightly dependent on the type of sheets (Table 2), which is greater than the value of a few ten meV in graphite and MoS264–66. This result is consistent with those of previous studies67,68. Therefore, we believe that, because of the stronger interaction between layers than other vdW materials, the Raman shifts as the number of layers occur.
Figure 6.

Calculated charge density difference between interlayers: (a) Oblique view (b) Side view. The red and blue colors indicate the accumulation and depletion of electrons, respectively. (c) In-plane projected charge density along with z direction.
The relation between Raman shifts (Δv (cm−1)) and the number of layers for MXs is shown in Fig. 4. We confirmed that this relationship remains very consistent for Ag2, B3g2, and B2g. The data could almost be fitted linearly by a reciprocal number of layers as follows:
| 1 |
where the subscript i indicates the index of phonon modes, ai and bi are fitting coefficients for a given i and N indicates the number of MX layers. The calculated coefficients for MX layers were calculated by a linear fitting (Supporting information Figure S1), which are presented in Table 3. The negative and positive values of ai mean red and blue shifts as N increases, respectively. This will be very beneficial in experiments to know the number of layers for a sample. Even though some shifts are of a small value per layer, like GeSe, modern Raman equipment has sufficient resolution (<1 cm−1) in room temperature to identify them69. In addition, for all examples, there are more than two rapid shift peaks, enough to identify thickness. The blue shift peaks, the horizontal modes, might be useful indicators for verifying the number of layers for almost MX. Another important point is that we propose a method of distinguishing the number of layers by comparing predicted Raman shift using Eq. (1) with experimental results of the Raman spectrum.
Table 3.
Coefficients (ai, bi) to Eq. (1) for each phonon mode. The unit of the values below is cm−1.
| B1u | B2g | Ag2 | B3g2 | |
|---|---|---|---|---|
| SnS | (−37.7, 92.5) | (−88.9, 160.2) | (−93.3, 194.3) | (22.6, 220.3) |
| SnSe | (−1.2, 58.0) | (−88.3, 115.6) | (−99.4, 128.0) | (23.4, 149.6) |
| GeS | (−21.4, 106.8) | (−52.2, 215.6) | (−59.9, 269.8) | (21.9, 248.3) |
| GeSe | (11.2, 69.1) | (−28.8, 148.0) | (−26.8, 184.3) | (23.0, 178.4) |
In conclusion, we have done the first-principles calculations for Raman spectroscopy based on the density functional theory. We found important things for a shift of the Raman peaks of metal monochalcogenide (MX), as its the number of layers. (1) Raman spectroscopy is considerably dependent upon the number of layers. (2) We have discovered the shifting or red trend of Raman peaks as the number of layers due to the change in atomic bonding length along with the direction of phonon. (3) We believe that our graphs for Raman peaks are useful benchmarks for identifying the number of layers of an arbitrary MX sample. Our results propose a method for distinguishing the number of layers in recently-reported few-layer MXs.
Supplementary information
Acknowledgements
This paper was supported by Konkuk University in 2019.
Author contributions
L.Y. and H.L. conceived and designed the study. M.P. performed the calculations. M.P., J.C., L.Y. and H.L. interpreted the data. All authors wrote the manuscript and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Li Yang, Email: lyang@physics.wustl.edu.
Hoonkyung Lee, Email: hkiee3@konkuk.ac.kr.
Supplementary information
is available for this paper at 10.1038/s41598-019-55577-x.
References
- 1.Peng Q, De S. Outstanding mechanical properties of monolayer MoS2 and its application in elastic energy storage. Phys. Chem. Chem. Phys. 2013;15:19427–37. doi: 10.1039/c3cp52879k. [DOI] [PubMed] [Google Scholar]
- 2.Baugher BW, Churchill HO, Yang Y, Jarillo-Herrero P. Intrinsic Electronic Transport Properties of High-Quality Monolayer and Bilayer MoS2. Nano Lett. 2013;13:4212–4216. doi: 10.1021/nl401916s. [DOI] [PubMed] [Google Scholar]
- 3.Xia C, et al. A type-II GeSe/SnS heterobilayer with a suitable direct gap, superior optical absorption and broad spectrum for photovoltaic applications. J. Mater. Chem. A Mater. 2017;5:13400–13410. doi: 10.1039/C7TA02109G. [DOI] [Google Scholar]
- 4.Chen X, Xia F. Black phosphorous optoelectronic devices. Proc.of SPIE. 2017;10194:101940E. doi: 10.1117/12.2263244. [DOI] [Google Scholar]
- 5.Miranda HPC, et al. Quantum Interference Effects in Resonant Raman Spectroscopy of Single- and Triple-Layer MoTe2from First-Principles. Nano Lett. 2017;17:2381–2388. doi: 10.1021/acs.nanolett.6b05345. [DOI] [PubMed] [Google Scholar]
- 6.Favron A, et al. Photooxidation and quantum confinement effects in exfoliated black phosphorus. Nat. Mater. 2015;14:826–832. doi: 10.1038/nmat4299. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Direct observation of a widely tunable bandgap in bilayer graphene. Nature. 2009;459:820–823. doi: 10.1038/nature08105. [DOI] [PubMed] [Google Scholar]
- 8.Mucha-Kruczyński M, et al. Characterization of graphene through anisotropy of constant-energy maps in angle-resolved photoemission. Phys. Rev. B. 2008;77:195403–12. doi: 10.1103/PhysRevB.77.195403. [DOI] [Google Scholar]
- 9.Cao Y, et al. Unconventional superconductivity in magic-angle graphene superlattices. Nature. 2018;556:43–50. doi: 10.1038/nature26160. [DOI] [PubMed] [Google Scholar]
- 10.Brent JR, et al. Tin(II) Sulfide (SnS) Nanosheets by Liquid-Phase Exfoliation of Herzenbergite: IV–VI Main Group Two-Dimensional Atomic Crystals. J. Am. Chem. Soc. 2015;137:12689–12696. doi: 10.1021/jacs.5b08236. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, et al. Two-dimensional GeS with tunable electronic properties via external electric field and strain. Nanotechnology. 2016;27:274001. doi: 10.1088/0957-4484/27/27/274001. [DOI] [PubMed] [Google Scholar]
- 12.Ahmad S, Mukherjee S. A Comparative Study of Electronic Properties of Bulk MoS2 and Its Monolayer Using DFT Technique: Application of Mechanical Strain on MoS2 Monolayer. Graphene. 2014;3:52–59. doi: 10.4236/graphene.2014.34008. [DOI] [Google Scholar]
- 13.Johnson RW, Hultqvist A, Bent SF. A brief review of atomic layer deposition: from fundamentals to applications. Mater. Today. 2014;17:236–246. doi: 10.1016/j.mattod.2014.04.026. [DOI] [Google Scholar]
- 14.Miikkulainen V, Leskelä M, Ritala M, Puurunen RL. Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends. J. Appl. Phys. 2013;113:021301–102. doi: 10.1063/1.4757907. [DOI] [Google Scholar]
- 15.Song L, et al. Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers. Nano Lett. 2010;10:3209–3215. doi: 10.1021/nl1022139. [DOI] [PubMed] [Google Scholar]
- 16.Geim AK, Grigorieva IV. Van der Waals heterostructures. Nature. 2013;499:419–425. doi: 10.1038/nature12385. [DOI] [PubMed] [Google Scholar]
- 17.Jaramillo, R. et al. Making Record-efficiency SnS Solar Cells by Thermal Evaporation and Atomic Layer Deposition. J. Vis. Exp. e52705 (2015).10.3791/52705 [DOI] [PMC free article] [PubMed]
- 18.Sinsermsuksakul P, Heo J, Noh W, Hock AS, Gordon RG. Atomic Layer Deposition of Tin Monosulfide Thin Films. Adv. Energy Mater. 2011;1:1116–1125. doi: 10.1002/aenm.201100330. [DOI] [Google Scholar]
- 19.Tan PH, et al. The shear mode of multilayer graphene. Nature Mater. 2012;11:294–300. doi: 10.1038/nmat3245. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, et al. Raman spectroscopy of shear and layer breathing modes in multilayer MoS2. Phys. Rev. B. 2013;87:115413. doi: 10.1103/PhysRevB.87.115413. [DOI] [Google Scholar]
- 21.Ferrari AC. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007;143:47–57. doi: 10.1016/j.ssc.2007.03.052. [DOI] [Google Scholar]
- 22.Li H, et al. From Bulk to Monolayer MoS2: Evolution of Raman Scattering. Adv. Funct. Mater. 2012;22:1385–1390. doi: 10.1002/adfm.201102111. [DOI] [Google Scholar]
- 23.Lee C, et al. Anomalous Lattice Vibrations of Single- and Few-Layer MoS2. ACS Nano. 2010;4:2695–2700. doi: 10.1021/nn1003937. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, et al. Phonon and Raman scattering of two-dimensional transition metal dichalcogenides from monolayer, multilayer to bulk material. Chem. Soc. Rev. 2015;44:2757–2785. doi: 10.1039/C4CS00282B. [DOI] [PubMed] [Google Scholar]
- 25.Castellanos-Gomez A, et al. Isolation and characterization of few-layer black phosphorus. 2D Mater. 2014;1:025001. doi: 10.1088/2053-1583/1/2/025001. [DOI] [Google Scholar]
- 26.Mehboudi M, et al. Structural Phase Transition and Material Properties of Few-Layer Monochalcogenides. Phys. Rev. Lett. 2016;117:246802–5. doi: 10.1103/PhysRevLett.117.246802. [DOI] [PubMed] [Google Scholar]
- 27.Koteeswara Reddy N, Devika M, Gopal ESR. Review on Tin (II) Sulfide (SnS) Material: Synthesis, Properties, and Applications. Crit. Rev. Solid State Mater. Sci. 2015;40:359–398. doi: 10.1080/10408436.2015.1053601. [DOI] [Google Scholar]
- 28.Wu L, et al. Few-Layer Tin Sulfide: A Promising Black-Phosphorus-Analogue 2D Material with Exceptionally Large Nonlinear Optical Response, High Stability, and Applications in All-Optical Switching and Wavelength Conversion. Adv. Opt. Mater. 2017;6:1700985–10. doi: 10.1002/adom.201700985. [DOI] [Google Scholar]
- 29.Fei R, Li W, Li J, Yang L. Giant piezoelectricity of monolayer group IV monochalcogenides: SnSe, SnS, GeSe, and GeS. Appl. Phys. Lett. 2015;107:173104–6. doi: 10.1063/1.4934750. [DOI] [Google Scholar]
- 30.Fei R, Kang W, Yang L. Ferroelectricity and Phase Transitions in Monolayer Group-IV Monochalcogenides. Phys. Rev. Lett. 2016;117:097601–6. doi: 10.1103/PhysRevLett.117.097601. [DOI] [PubMed] [Google Scholar]
- 31.Gomes LC, Carvalho A, Castro Neto AH. Enhanced piezoelectricity and modified dielectric screening of two-dimensional group-IV monochalcogenides. Phys. Rev. B. 2015;92:214103. doi: 10.1103/PhysRevB.92.214103. [DOI] [Google Scholar]
- 32.Chowdhury C, Karmakar S, Datta A. Monolayer Group IV–VI Monochalcogenides: Low-Dimensional Materials for Photocatalytic Water Splitting. J. Phys. Chem. C. 2017;121:7615–7624. doi: 10.1021/acs.jpcc.6b12080. [DOI] [Google Scholar]
- 33.Devika M, Reddy NK, Gunasekhar KR. Structural, electrical, and optical properties of as-grown and heat treated ultra-thin SnS films. Thin Solid Films. 2011;520:628–632. doi: 10.1016/j.tsf.2011.07.074. [DOI] [Google Scholar]
- 34.Franzman MA, Schlenker CW, Thompson ME, Brutchey RL. Solution-Phase Synthesis of SnSe Nanocrystals for Use in Solar Cells. J. Am. Chem. Soc. 2010;132:4060–4061. doi: 10.1021/ja100249m. [DOI] [PubMed] [Google Scholar]
- 35.Shi G, Kioupakis E. Anisotropic Spin Transport and Strong Visible-Light Absorbance in Few-Layer SnSe and GeSe. Nano Lett. 2015;15:6926–6931. doi: 10.1021/acs.nanolett.5b02861. [DOI] [PubMed] [Google Scholar]
- 36.Loferski JJ. Theoretical Considerations Governing the Choice of the Optimum Semiconductor for Photovoltaic Solar Energy Conversion. J. Appl. Phys. 1956;27:777–784. doi: 10.1063/1.1722483. [DOI] [Google Scholar]
- 37.Sinsermsuksakul P, et al. Overcoming Efficiency Limitations of SnS-Based Solar Cells. Adv. Energy Mater. 2014;4:1400496–7. doi: 10.1002/aenm.201400496. [DOI] [Google Scholar]
- 38.Reghima M, Akkari A, Guasch C, Castagné M, Kamoun-Turki N. Synthesis and characterization of Fe-doped SnS thin films by chemical bath deposition technique for solar cells applications. J. Renew. Sustain. Energy. 2013;5:063109–11. doi: 10.1063/1.4830256. [DOI] [Google Scholar]
- 39.Im HS, et al. Germanium and Tin Selenide Nanocrystals for High-Capacity Lithium Ion Batteries: Comparative Phase Conversion of Germanium and Tin. J. Phys. Chem. C. 2014;118:21884–21888. doi: 10.1021/jp507337c. [DOI] [Google Scholar]
- 40.Kim Y, et al. SnSe alloy as a promising anode material for Na-ion batteries. Chem. Commun. 2014;51:50–53. doi: 10.1039/C4CC06106C. [DOI] [PubMed] [Google Scholar]
- 41.Chandrasekhar HR, Humphreys RG, Zwick U, Cardona M. Infrared and Raman spectra of the IV-VI compounds SnS and SnSe. Phys. Rev. B. 1977;15:2177. doi: 10.1103/PhysRevB.15.2177. [DOI] [Google Scholar]
- 42.Zhang C, et al. Two-Dimensional Tin Selenide Nanostructures for Flexible All-Solid-State Supercapacitors. ACS Nano. 2014;8:3761–3770. doi: 10.1021/nn5004315. [DOI] [PubMed] [Google Scholar]
- 43.Kevin P, Lewis DJ, Raftery J, Malik MA, O’Brien P. Thin films of tin(II) sulphide (SnS) by aerosol-assisted chemical vapour deposition (AACVD) using tin(II) dithiocarbamates as single-source precursors. J. Cryst. Growth. 2015;415:93–99. doi: 10.1016/j.jcrysgro.2014.07.019. [DOI] [Google Scholar]
- 44.Kana AT, et al. Organotin unsymmetric dithiocarbamates: synthesis, formation and characterisation of tin(II) sulfide films by atmospheric pressure chemical vapour deposition. Polyhedron. 2001;20:2989–2995. doi: 10.1016/S0277-5387(01)00908-1. [DOI] [Google Scholar]
- 45.Ramasamy K, et al. Organotin Dithiocarbamates: Single-Source Precursors for Tin Sulfide Thin Films by Aerosol-Assisted Chemical Vapor Deposition (AACVD) Chem. Mater. 2013;25:266–276. doi: 10.1021/cm301660n. [DOI] [Google Scholar]
- 46.Hibbert TG, Mahon MF, Molloy KC, Price LS, Parkin IP. Deposition of tin sulfide thin films from novel, volatile (fluoroalkythiolato)tin(iv) precursors. J. Mater. Chem. 2001;11:469–473. doi: 10.1039/b005863g. [DOI] [Google Scholar]
- 47.Bade BP, Garje SS, Niwate YS, Afzaal M, O’Brien P. Tribenzyltin(IV)chloride Thiosemicarbazones: Novel Single Source Precursors for Growth of SnS Thin Films. Chem. Vap. Deposition. 2008;14:292–295. doi: 10.1002/cvde.200806687. [DOI] [Google Scholar]
- 48.Sánchez-Juárez A, Tiburcio-Silver A, Ortiz A. Fabrication of SnS2/SnS heterojunction thin film diodes by plasma-enhanced chemical vapor deposition. Thin Solid Films. 2005;480–481:452–456. doi: 10.1016/j.tsf.2004.11.012. [DOI] [Google Scholar]
- 49.Ray SC, karanjai MK, DasGupta D. Structure and photoconductive properties of dip-deposited SnS and SnS2 thin films and their conversion to tin dioxide by annealing in air. Thin Solid Films. 1999;350:72–78. doi: 10.1016/S0040-6090(99)00276-X. [DOI] [Google Scholar]
- 50.Gao C, Shen H. Influence of the deposition parameters on the properties of orthorhombic SnS films by chemical bath deposition. Thin Solid Films. 2012;520:3523–3527. doi: 10.1016/j.tsf.2011.12.077. [DOI] [Google Scholar]
- 51.Kim JY, George SM. Tin Monosulfide Thin Films Grown by Atomic Layer Deposition Using Tin 2,4-Pentanedionate and Hydrogen Sulfide. J. Phys. Chem. C. 2010;114:17597–17603. doi: 10.1021/jp9120244. [DOI] [Google Scholar]
- 52.Koteswara Reddy N, Ramakrishna Reddy KT. Growth of polycrystalline SnS films by spray pyrolysis. Thin Solid Films. 1998;325:4–6. doi: 10.1016/S0040-6090(98)00431-3. [DOI] [Google Scholar]
- 53.Baroni S, de Gironcoli S, Dal Corso A, Giannozzi P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001;73:515–562. doi: 10.1103/RevModPhys.73.515. [DOI] [Google Scholar]
- 54.Giannozzi P, et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Condens. Matter Phys. 2009;21:395502–20. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 55.Hartwigsen C, Goedecker S, Hutter J. Relativistic separable dual-space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B. 1998;58:3641–3662. doi: 10.1103/PhysRevB.58.3641. [DOI] [PubMed] [Google Scholar]
- 56.Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B. 1996;54:1703–1710. doi: 10.1103/PhysRevB.54.1703. [DOI] [PubMed] [Google Scholar]
- 57.Taniguchi M, Johnson RL, Ghijsen J, Cardona M. Core excitons and conduction-band structures in orthorhombic GeS, GeSe, SnS, and SnSe single crystals. Phys. Rev. B. 1990;42:3634–3643. doi: 10.1103/PhysRevB.42.3634. [DOI] [PubMed] [Google Scholar]
- 58.Hu T, Dong J. Two new phases of monolayer group-IV monochalcogenides and their piezoelectric properties. Phys Chem Chem Phys. 2016;18:32514–32520. doi: 10.1039/C6CP06734D. [DOI] [PubMed] [Google Scholar]
- 59.Xin C, et al. Few-Layer Tin Sulfide: A New Black-Phosphorus-Analogue 2D Material with a Sizeable Band Gap, Odd–Even Quantum Confinement Effect, and High Carrier Mobility. J. Phys. Chem. C. 2016;120:22663–22669. doi: 10.1021/acs.jpcc.6b06673. [DOI] [Google Scholar]
- 60.Sun J, Ruzsinszky A, Perdew JP. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015;115:64–6. doi: 10.1103/PhysRevLett.115.036402. [DOI] [PubMed] [Google Scholar]
- 61.Shulenburger L, Baczewski AD, Zhu Z, Guan J, Tománek D. The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus. Nano Lett. 2015;15:8170–8175. doi: 10.1021/acs.nanolett.5b03615. [DOI] [PubMed] [Google Scholar]
- 62.Chao J, et al. Visible-light-driven photocatalytic and photoelectrochemical properties of porous SnSx(x = 1,2) architectures. CrystEngComm. 2012;14:3163–6. doi: 10.1039/c2ce06586j. [DOI] [Google Scholar]
- 63.Molina-Sánchez A, Wirtz L. Phonons in single-layer and few-layer MoS2 and WS2. Phys. Rev. B. 2011;84:155413. doi: 10.1103/PhysRevB.84.155413. [DOI] [Google Scholar]
- 64.Chen X, Tian F, Persson C, Duan W, Chen N-X. Interlayer interactions in graphites. Sci. Rep. 2013;3:693–695. doi: 10.1038/srep03046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Björkman T, Gulans A, Krasheninnikov AV, Nieminen RM. van der Waals Bonding in Layered Compounds from Advanced Density-Functional First-Principles Calculations. Phys. Rev. Lett. 2012;108:235502–5. doi: 10.1103/PhysRevLett.108.235502. [DOI] [PubMed] [Google Scholar]
- 66.Rydberg H, et al. Van der Waals Density Functional for Layered Structures. Phys. Rev. Lett. 2003;91:4978–4974. doi: 10.1103/PhysRevLett.91.126402. [DOI] [PubMed] [Google Scholar]
- 67.Jung JH, Park C-H, Ihm J. A Rigorous Method of Calculating Exfoliation Energies from First Principles. Nano Lett. 2018;18:2759–2765. doi: 10.1021/acs.nanolett.7b04201. [DOI] [PubMed] [Google Scholar]
- 68.Mounet N, et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018;13:246. doi: 10.1038/s41565-017-0035-5. [DOI] [PubMed] [Google Scholar]
- 69.Jin K, LIU D, Tian Y. Enhancing the interlayer adhesive force in twisted multilayer MoS2 by thermal annealing treatment. Nanotechnology. 2015;26:405708–405708. doi: 10.1088/0957-4484/26/40/405708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.