

Abstract
Accumulating evidence demonstrates that the intestinal microbiota enhance mammalian enteric virus infections. For example, we and others have previously reported that commensal bacteria stimulate acute and persistent murine norovirus infections. In apparent contradiction to these results however, the virulence of murine norovirus infection was unaffected by antibiotic treatment. This prompted us to perform a detailed investigation of murine norovirus infection in microbially deplete mice, revealing a more complex picture whereby commensal bacteria inhibit viral infection of the proximal small intestine while simultaneously stimulating infection of distal regions of the gut. Thus, commensal bacteria can regulate viral regionalization along the intestinal tract. We further show that the mechanism underlying bacteria-dependent inhibition of norovirus infection in the proximal gut is bile acid priming of type III interferon. Finally, the regional effects of the microbiota on norovirus infection may result from distinct regional expression profiles of key bile acid receptors which regulate the type III interferon response. Overall, these findings reveal that biotransformation of host metabolites by the intestinal microbiota directly and regionally impacts infection by a pathogenic enteric virus.
Accumulating evidence has emerged in recent years highlighting the influence of the intestinal microbiota on enteric virus infections (1,2). For all mammalian enteric viruses studied to date, the intestinal microbiota has been reported to stimulate viral infection (3–8). Mechanisms of stimulation include direct facilitation of cellular infection (6,9), virion stabilization (9), and indirect regulation of the mucosal immune response in a manner promoting viral infection (4,7,10). In contrast to these examples of mammalian enteric viruses, the intestinal microbiota of insects provide protection from oral viral infections by priming an antiviral immune pathway or degrading mucin (11–13). Similarly, commensal bacteria have been shown to limit infection of mammalian viral infections outside of the intestinal tract, due at least in part to commensal bacteria-mediated priming of interferon (IFN) pathways (14–17). For respiratory influenza virus infection, recent findings implicate the bacterial metabolite desaminotyrosine in priming type I IFN responses (18), uncovering a chain of events whereby commensal microbes contribute to host metabolism, the products of which regulate host immunity and pathogen susceptibility. Here we show that the intestinal microbiota have opposing regional effects on murine norovirus (MNV) infection, inhibiting viral infection of the proximal gut while promoting viral infection of the distal gut. We further show that inhibition of proximal gut infection is due to priming of a type III IFN response by bacteria-biotransformed bile acids. This antiviral response fails to block infection in the distal gut, possibly because of regional differences in expression of a key bile acid receptor. Overall, these studies highlight the complex and varied effects of the intestinal microbiota on viral infections along the intestinal tract.
We and others have demonstrated reduced MNV infectious titers and genome copy number in antibiotic (Abx)-treated and germ-free mice (5–7). Although prior studies of the role of the intestinal microbiota on MNV infection did not explore the effect of the intestinal microbiota on MNV morbidity or mortality, we logically predicted that infection would be attenuated under conditions of microbial depletion as is the case for poliovirus, reovirus, and rotavirus (3,8). To test this, highly susceptible type I interferon receptor-deficient (Ifnar−/−) mice were pretreated with PBS or Abx, infected with 105 TCID50 units MNV-1, and monitored for weight loss and survival kinetics. Importantly, type I IFN has been demonstrated to be dispensable for the effect of commensal bacteria on MNV-CR6 infection (7). As expected based on our previous work with Ifnar−/− mice (19), PBS-treated control mice rapidly lost weight (Fig. 1a) and succumbed to lethal infection by 9 dpi (Fig. 1b). To our surprise, Abx-treated mice were equally susceptible to MNV-1 infection. The lower dose of 104 TCID50 units MNV-1 caused only modest and comparable weight loss (Fig. 1a) and was nonlethal in both groups (Fig. 1b).
Figure 1. The intestinal microbiota play opposing roles in regulating proximal gut versus distal gut MNV infection.
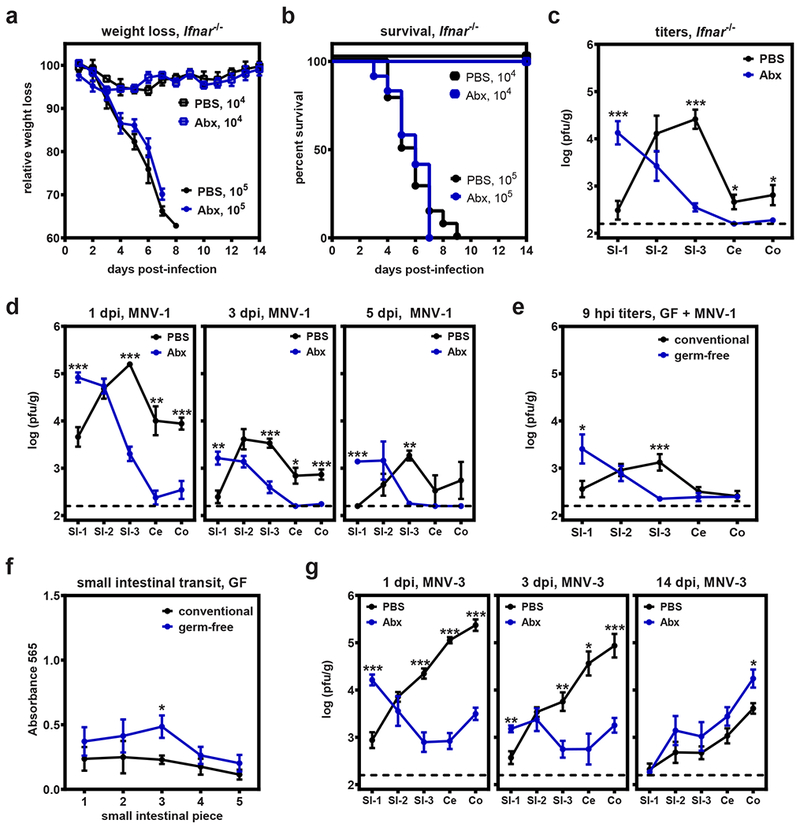
a-b) Groups of Ifnar−/− mice were treated with PBS or Abx and then infected with 105 pfu MNV-1 (n = 19 for PBS, n= 17 for Abx) or 104 TCID50 units MNV-1 (n = 4 for both groups) and followed for weight loss (a) and survival (b). c) Groups of Ifnar−/− mice (n = 7) were treated with PBS or Abx and then infected with 105TCID50 units MNV-1. At 1 day post-infection (dpi), virus titers were determined in the indicated segments of the intestinal tract. d) Groups of PBS- and Abx-treated wild-type B6 mice were inoculated with 107TCID50 units MNV-1 and virus titers determined at 1 (n = 10 for PBS, n = 8 for Abx), 3 (n = 8 for PBS, n = 6 for Abx), and 5 dpi (n = 3 per group). Groups were also tested at 14 dpi but no virus was detected. e) Conventional (n = 13) or germ-free mice (n = 15) were inoculated with 106 pfu neutral red-labeled MNV-1. At 9 hours post-infection (hpi), tissues were collected and plaque assays were used to measure newly synthesized virus. f) Conventional (n = 5) or germ-free (n = 4) mice were administered Evan’s blue dye and intestinal transit times were determined 15 min later. g) Groups of PBS- and Abx-treated wild-type B6 mice were inoculated with 107TCID50 units MNV-3 and virus titers determined at 1 (n = 7 for PBS, n = 8 for Abx), 3 (n = 6 per group), and 14 (n = 6 per group) dpi. Error bars indicate the mean of all data points. Unpaired two-tailed Student’s t-tests were used for statistical purposes. *P < 0.05, **P < 0.01, ***P < 0.001. See Supplementary Table 1 for detailed statistical information.
Because commensal bacteria did not influence MNV-1 virulence in spite of reported reductions in virus titers (5–7), we speculated that there are regional differences in bacterial regulation of viral infection that were inadvertently missed in earlier studies. Previous studies in microbially deplete mice assessed MNV infection by quantifying virus titers or viral genome copy number in only the distal regions of the intestine (ileum, colon, or stool) (5–7). Therefore, we measured MNV-1 titers along the entire gastrointestinal (GI) tract of Ifnar−/− mice, including three longitudinal segments of the small intestine (SI-1, SI-2, and SI-3), the cecum (Ce), and the colon (Co) (Fig. 1c). Consistent with previous studies, SI-3 and Co titers were reduced 117-fold and 5-fold, respectively, in Abx-treated mice compared to PBS-treated controls. There was also a 4-fold reduction in Ce titers. However, Abx treatment had no significant effect on virus titers in SI-2 and remarkably it resulted in a 53-fold increase in SI-1 titers compared to controls. The cumulative intestinal titer in bacteria-replete and bacteria-deplete mice was not significantly different (Extended Data Fig. 1), providing an explanation for similar virulence under these conditions despite the difference in regionalization of viral infection. These data reveal that commensal bacteria direct the intestinal regionalization of MNV-1 infection, in contrast to the previous conclusion that they globally stimulate viral infection along the GI tract.
To test whether bacteria-directed regionalization of MNV-1 infection is influenced by type I IFN signaling, we next repeated these experiments in wild-type B6 mice (Fig. 1d). Mirroring results in Ifnar−/− mice at 1 dpi, Abx treatment stimulated viral infection of SI-1 (9-fold increase); had no effect on infection of SI-2 titers; and suppressed infection of SI-3, Ce, and Co (44-fold, 93-fold, and 11-fold, respectively). These phenotypes were temporally maintained from 1 to 5 dpi (Fig. 1d). As observed in Ifnar−/− mice, the cumulative intestinal titers in B6 mice treated with PBS or Abx were not significantly different from one another at any time point tested (Extended Data Fig. 1). Thus, bacteria direct the regionalization of MNV-1 in a type I IFN-independent manner.
To exclude the possibility that these patterns were influenced by residual Abx-resistant commensal bacteria in our Abx treatment group, germ-free mice were infected with neutral red-labeled MNV-1. Consistent with results in Abx-treated mice, MNV-1 titers in germ-free mice were 25-fold higher in SI-1, unaffected in SI-2, and 10-fold lower in SI-3 at 9 hpi as compared to conventional mice (Fig. 1e) while cumulative titers were comparable (Extended Data Fig. 1). Large intestinal titers were negligible in this experiment likely because of the early time point. Because these titers represent only newly synthesized virus, these data also exclude the possibility that our findings can be explained by slower transit time in microbially deplete mice. Further supporting this conclusion, regional phenotypes were maintained in Abx-treated mice through 5 d of infection (Fig. 1d) and we observed only a modest difference in intestinal transit of Evans blue dye in conventional and germ-free mice (Fig. 1f).
There are multiple pathogenically distinct MNV strains that display inherent differences in their intestinal regional preference under conditions of microbial colonization. For example, MNV-1 reaches peak titers in SI-3 of B6 mice while a highly genetically related strain called MNV-3 reaches peak titers in Co (20). Considering that peak MNV-1 infection shifted from SI-3 to SI-1 in microbially deplete hosts, it was of interest to test whether this phenotype is generalizable to other MNV strains. Indeed, MNV-3 titers were 16-fold higher in SI-1, unaffected in SI-2, and reduced in SI-3 (17-fold), Ce (110-fold), and Co (68-fold) of Abx-treated mice compared to controls at 1 dpi (Fig. 1g), phenocopying MNV-1 results. While MNV-1 is considered an acute strain, other strains including MNV-3 establish a persistent infection in the large intestine (21–23). Although persistence of another MNV strain, MNV-CR6, was shown to require commensal bacteria (7), we observed minimal effect of Abx treatment on MNV-3 persistence at 14 dpi (Fig. 1g), suggesting virus strain-specific differences in the role of the intestinal microbiota on persistent MNV infection. In sum, commensal bacteria suppress acute MNV infection of the most proximal segment of the small intestine while enhancing acute infection of distal segments.
One mechanism underlying bacteria-driven regulation of enteric virus infections is skewing of an antiviral immune response (1,4,7,11). Having excluded a role for type I IFN in regulating MNV-1 intestinal regionalization (Fig. 1c), we examined a panel of other immune mediators that play important roles in intestinal immune responses, namely IL-10, lymphocytes, and type III IFN (Fig. 2a). The absence of IL-10 had no effect on overall MNV-1 titers or MNV-1 regionalization. Although titers in Rag1−/− mice were slightly lower compared to B6 controls in most regions of the gut (which was expected based on our previous findings (6,24)), the regional pattern of infection was comparable in B6 and Rag1−/− mice under bacteria-replete and bacteria-deplete conditions. Thus, we conclude that type I IFN, IL-10, and lymphocytes are dispensable for driving MNV-1 regionalization in the gut. In contrast, MNV-1 SI-1 titers in conventional Ifnlr1−/− mice were elevated 37-fold compared to B6 mice (Fig. 2a, left graph), demonstrating that type III IFN plays a key role in inhibiting MNV-1 infection of the proximal intestine. Titers in all other tissues were comparable in Ifnlr1−/− and B6 mice. If commensal bacteria and type III IFN suppress MNV-1 infection in distinct manners, then we reasoned there would be a combinatorial effect reflected in increased SI-1 titers in Ifnlr1−/− Abx-treated mice compared to Abx-treated control B6 mice. However, SI-1 titers in Abx-treated Ifnlr1−/− and B6 mice were comparable (Fig. 2a, right graph). We conclude that type III IFN is necessary for bacteria-driven inhibition of MNV-1 proximal gut infection. None of the immune factors analyzed in this study influenced bacteria-driven enhancement of MNV-1 distal gut infection, highlighting that these regional phenotypes are driven by distinct mechanisms.
Figure 2. Bacteria-dependent type III IFN signaling in the proximal gut protects intestinal immune cells from MNV-1 infection.
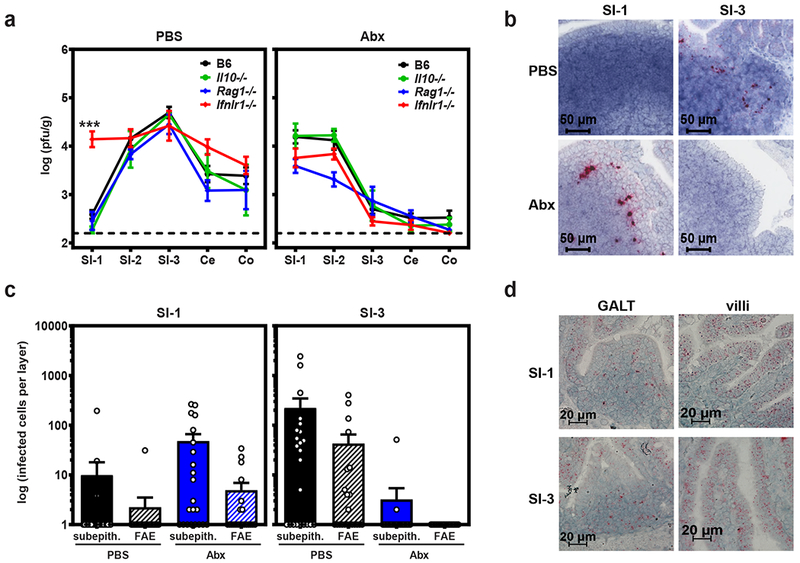
a) Groups of wild-type B6 mice (n = 18) or mice deficient in IL-10 (n = 4), Rag1 (n = 6 for PBS, n = 7 for Abx), or Ifnlr1 (n = 9 for PBS, n = 11 for Abx) were treated with PBS or Abx and then infected with 107 TCID50 units MNV-1. At 1 dpi, virus titers were determined in the indicated segments of the intestinal tract. P <0.0001 for SI-1 titers in B6 versus Ifnlr1−/− mice; titers in all other intestinal segments in knockout mice were statistically similar to titers in B6 mice in both PBS and Abx groups. Unpaired two-tailed Student’s t-tests were used for statistical purposes. ***P < 0.001. b) Groups of B6 mice were treated with PBS or Abx, infected with mock inoculum (n = 4 per condition) or 107 TCID50 units MNV-1 (n = 6 per condition), and intestinal segments harvested at 1 dpi for the purpose of RNAscope-based ISH. Serial sections were hybridized with a probe specific to the minus-strand MNV-1 antigenome indicative of productive infection. Representative images are shown. c) The relative contribution of subepithelial cell (subepith.) versus follicle-associated epithelium (FAE) infection, as indicated by minus-strand viral RNA signal, was quantified as described in the Methods. d) Serial sections from SI-1 and SI-3 of mock-inoculated PBS-treated mice (n = 5 per condition) were hybridized with a probe specific to the Ifnlr1 mRNA and analyzed by RNAscope-based ISH. Representative images from intestinal villi and areas of GALT are shown for each condition. For panels a and c, error bars indicate the mean of all data points.
Because bacterial inhibition of a mammalian enteric virus has not been demonstrated previously, we further probed the underlying mechanism in the proximal gut. We first explored the cellular basis of the bacteria-dependent type III IFN response in the proximal gut. Although it is generally well-accepted that epithelial cells are the major Ifnlr1-expressing cell type (25), we discovered that intestinal immune cells in the gut-associated lymphoid tissue (GALT) are the main target of acute MNV-1 infection in bacteria-replete mice (24). Consistent with immune cell tropism of MNV-1, Wilen et al. recently reported that, although tuft cells serve as the persistent reservoir for the MNV-CR6 strain, acute MNV-1 requires the viral receptor CD300lf on radiation-sensitive immune cells but not on radiation-resistant epithelial cells for normal levels of gut infection (26). Moreover, MNV-1 titers were comparable in Pou2f3−/− mice that lack tuft cells and wild-type controls (Extended Data Fig. 2). Thus, we predicted that bacteria-dependent antiviral signaling prevents MNV-1 infection of intestinal immune cells in the proximal gut. To test this, we performed RNAscope-based in situ hybridization (ISH) with a probe specific to the viral minus-strand RNA on SI-1 and SI-3 sections prepared from PBS- and Abx-treated mice. Consistent with our previous results, the predominant target of MNV-1 infection in bacteria-replete PBS-treated mice was subepithelial cells in the GALT in SI-3 whereas very few infected cells were detected in SI-1 of these mice (Fig. 2b–c). In bacteria-deplete Abx-treated mice, the predominant target was also subepithelial cells in the GALT but these were only observed in SI-1 and not SI-3 (Fig. 2b–c), supporting our hypothesis that the bacteria-dependent antiviral response in the proximal gut normally protects intestinal immune cells from MNV infection.
Although the Ifnlr1 subunit is generally described to be expressed on epithelial cells, there are multiple reports of its expression on immune cells in certain contexts (27). Considering that the IFN-λ response protects immune cells from MNV-1 infection, we next examined the expression profile of the Ifnlr1 subunit using RNAscope ISH. Along the intestinal villi, it was expressed nearly exclusively by epithelial cells in both SI-1 and SI-3 (Fig. 2d, villi). A distinct expression pattern was observed in the GALT where few follicle-associated epithelial cells expressed Ifnlr1 but substantial numbers of cells in the underlying subepithelial dome were Ifnlr1-positive (Fig. 2d, GALT). Based on the tropism of MNV-1 for GALT immune cells (24), it is intriguing to speculate that these subepithelial Ifnlr1-positive cells play a key role in protecting from MNV-1 infection, a hypothesis to be tested in future studies. Relevant to our studies, the overall expression pattern was similar in SI-1 and SI-3 excluding a regional difference in the expression pattern of the IFN-λ receptor as underlying the regional bacterial effect on MNV infection.
We next examined the nature of the bacteria-dependent signal required for viral inhibition in the proximal gut. Bacterial metabolites have significant effects on host homeostasis, immune function, and even response to infections (28,29). Considering that the microbial load in the proximal small intestine is relatively low yet there is a profound bacteria-dependent inhibition of MNV-1 infection, it seemed plausible that this phenotype is driven by a bacterial metabolite. We hypothesized that bacterially regulated bile acids (i.e. unconjugated 1° and 2° bile acids) could be key players since they interact with bile acid receptors on many cell types to regulate metabolic and immune functions (30,31) and they have established proviral and antiviral functions (32,33). In fact, they have been shown to regulate norovirus infections in vitro and in vivo (34,35). Based on the prior observation that clindamycin treatment of mice causes a major disruption of bile acid composition while nalidixic acid treatment causes minimal change (36,37), we tested the effect of these single antibiotics on MNV-1 infection in the proximal small intestine. As expected, there were substantial reductions in the overall levels of unconjugated 1° bile acids and all 2° bile acids in the SI-1 and SI-3 lumenal contents of Abx- and clindamycin-treated naive mice compared to PBS- and nalidixic acid-treated mice (Fig. 3a). This pattern was also reflected when measuring individual bile acids (Extended Data Fig. 3). Next, we infected clindamycin-treated and nalidixic acid-treated mice. Remarkably, MNV-1 titers in SI-1 and SI-3 of clindamycin-treated mice phenocopied results observed following treatment with the full Abx cocktail while nalidixic acid had no significant effect (Fig. 3b). If bile acids biotransformed by commensal bacteria in turn regulate MNV infection, then commensal bacteria known to biotransform bile acids should be sufficient to rescue viral inhibition in the proximal gut when reconstituted into Abx-treated mice. While many bacteria possess enzymes capable of deconjugating 1° bile acids, the bacteria encoding dehydroxylases which convert 1° bile acids to 2° bile acids are restricted. The most well-established bacteria responsible for this process is Clostridium scindens (36,38). Indeed, reconstitution of Abx-treated B6 mice with C. scindens was sufficient to restore viral inhibition in SI-1 although it was not sufficient to restore viral enhancement in SI-3 (Fig. 3c).
Figure 3. Bile acids biotransformed by the intestinal microbiota prime type III IFN induction to suppress MNV infection of the proximal gut.
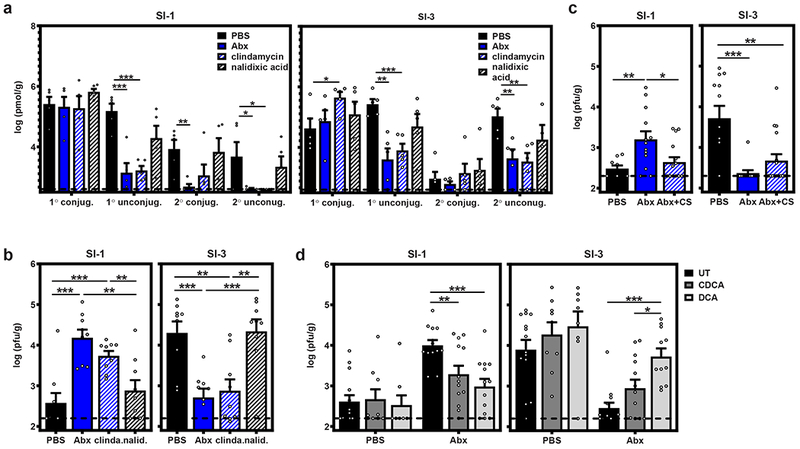
a) Groups of B6 mice (n = 5) were administered PBS, Abx, clindamycin, or nalidixic acid. Lumenal flushes from SI-1 (left graph) and SI-3 (right graph) were analyzed for conjugated 1°, unconjugated 1°, conjugated 2°, or unconjugated 2° bile acids. b) Groups of B6 mice (n = 9) were administered PBS, Abx, clindamycin, or nalidixic acid and then infected with 107 TCID50 units MNV-1. At 1 dpi, SI-1 and SI-3 titers were determined by plaque assay. c) Groups of B6 mice were administered PBS (n = 9) or Abx (n = 13 for Abx; n = 15 for Abx + CS). At day 6, Abx were removed from one group of Abx-treated mice and they were gavaged with 108 cfu C. scindens on days 7 and 8. On day 14, mice in all three groups were orally inoculated with MNV-1. At 1 dpi, SI-1 and SI-3 titers were determined by plaque assay. d) Groups of B6 mice were administered PBS (n = 14 for UT, n = 9 for CDCA, and n = 8 for DCA) or Abx(n = 13 for each condition) and then fed normal chow (UT) or chow supplemented with 0.6%CDCA or DCA for 14 d. Mice were then infected with 107 TCID50 units MNV-1. At 1 dpi, SI-1 (left graphs) and SI-3 (right graphs) titers were determined by plaque assay. Error bars indicate the mean of all data points. Unpaired two-tailed Student’s t-tests were used for statistical purposes. *P < 0.05, **P < 0.01, ***P < 0.001. See Supplementary Table 1 for detailed statistical information.
To directly test whether bacteria-regulated bile acids affect MNV infection in a regional manner, we supplemented Abx-treated mice with chenodeoxycholic acid (CDCA) or deoxycholic acid (DCA), representative unconjugated 1° and 2° bile acids that were significantly reduced in Abx- and clindamycin-treated mice. Both bile acids partially restored viral inhibition of Abx-treated mice in SI-1 (Fig. 3d), demonstrating that individual bile acids can rescue viral inhibition in the proximal gut in the absence of the intestinal microbiota. The 2° bile acid DCA partially rescued enhancement of viral infection of the distal gut while the 1° bile acid CDCA had no significant effect in this region (Fig. 3d). This latter phenotype is consistent with recent work demonstrating that certain bile acids can promote MNV binding to its host receptor CD300lf (39). Collectively, these data support a model whereby commensal bacteria modulate the bile acid pool along the intestinal tract and specific bile acids in turn regulate enteric virus infection in regionally distinct manners.
Having determined that type III IFN signaling and bacteria-biotransformed bile acids are both involved in inhibiting MNV infection in the proximal gut, we next questioned whether bile acids directly prime type III IFN induction. To this end, we tested two 1° bile acids, CDCA and cholic acid (CA), and two 2° bile acids, DCA and lithocholic acid (LCA), for their ability to modulate IFN-λ induction in vitro. CMT-93 cells (a murine intestinal epithelial cell line) pretreated with CDCA or DCA for 12 h expressed significantly more IFN-λ upon transfection with the viral analog poly(I:C) than unprimed cells whereas CA and LCA failed to enhance IFN-λ expression (Fig. 4a). Importantly, bile acid treatment in the absence of poly(I:C) activation failed to induce IFN-λ, supporting a two-signal activation pathway. In order to test whether bile acids enhance IFN-λ induction in the context of MNV-1 infection, we took advantage of an intestinal epithelial cell line engineered to express the MNV receptor CD300lf referred to as M2C-CD300lf (40). Pretreatment with DCA resulted in significantly more IFN-λ induction when these cells were infected with MNV-1, and there was a similar trend for CDCA although not statistically significant (Fig. 4b).
Figure 4. Bile acids prime IFN-λ induction in vitro.
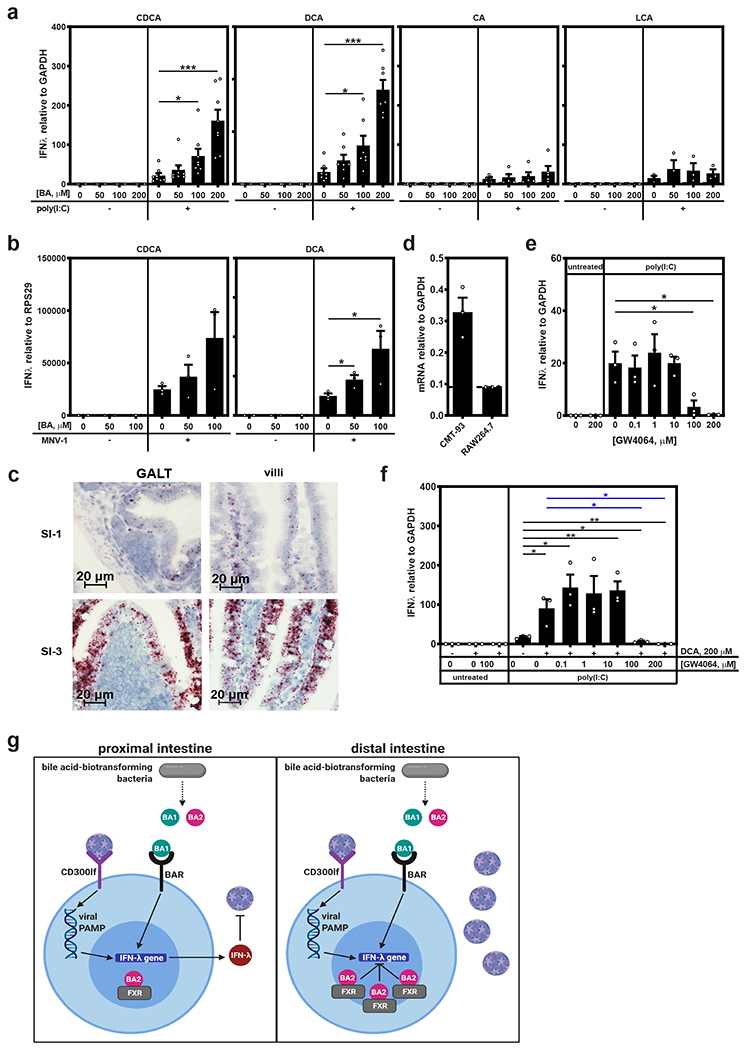
a) Duplicate wells of CMT-93 cells were treated with the indicated concentration of CDCA (n = 8 experimental repeats), DCA (n = 7 experimental repeats), CA (n = 5 experimental repeats) or LCA (n = 3 experimental repeats) for 12 h and then transfected with 5 μg poly(I:C) or untreated. At 6 h post-transfection, IFN-λ mRNA levels were determined. Data are reported as relative IFN-λ levels normalized to GAPDH as a housekeeping control. All untransfected controls (−) had undetectable levels of IFN-λ. Cells transfected with a control plasmid in place of poly(I:C) also failed to express IFN-λ irrespective of bile acid pretreatment. b) Duplicate wells of M2C-CD300lf cells were pretreated with the indicated concentration of CDCA or DCA for 12 h and then infected with MNV-1 at MOI 1. At 24 hpi, IFN-λ mRNA levels were determined. Data are reported as relative IFN-λ levels normalized to RPS29 as a housekeeping control (n = 3 experimental repeats). All uninfected controls had undetectable levels of IFN-λ. c) Serial sections from SI-1 and SI-3 of mock-inoculated PBS-treated mice (n = 5 mice per condition) were hybridized with a probe specific to the Fxr mRNA and analyzed by RNAscope-based ISH. Representative images from intestinal villi and areas of GALT are shown for each condition. d) Total RNA was extracted from duplicate wells of CMT-93 and RAW 264.7 cells and analyzed for Fxr mRNA levels. Data are reported as relative Fxr levels normalized to GAPDH housekeeping control (n = 3 experimental repeats). e) Duplicate wells of CMT-93 cells were treated with the indicated concentration of GW4064 for 12 h and then transfected with 5 μg poly(I:C) or untreated. At 6 h post-transfection, IFN-λ mRNA levels were determined. Data are reported as relative IFN-λ levels normalized to GAPDH as a housekeeping control (n = 3 experiments). All untransfected controls (−) had undetectable levels of IFN-λ. f) Duplicate wells of CMT-93 cells were treated with 200 μM DCA and the indicated concentration of GW4064 for 12 h, transfected with 5 μg poly(I:C) for 6 h, and then analyzed for IFN-λ gene expression (n = 3 experiments). Each poly(I:C)-transfected condition was compared to no treatment (black asterisks) and DCA-treated (blue asterisks) samples for statistical purposes. g) A model of the regional nature of bacteria-mediated effects on MNV infection. In both the proximal and distal regions of the intestinal tract, commensal bacteria regulate the pool of bile acids in the gut lumen; and a key bacteria-biotransformed bile acid primes IFN-λ gene expression when cells recognize viral pattern associated molecular patterns (PAMPs) during MNV-1 infection. In the proximal gut, this antiviral response inhibits MNV-1 infection. In the distal gut, high levels of FXR interacting with its cognate ligand suppress this process and MNV-1 replicates successfully. This model was created with BioRender.com. In all experiments, values for technical repeats were averaged and error bars indicate the mean of experimental repeats. Unpaired two-tailed Student’s t-tests were used for statistical purposes. *P < 0.05, **P < 0.01, ***P < 0.001. See Supplementary Table 1 for detailed statistical information.
While these collective data uncover an antiviral process whereby specific bile acids prime IFN-λ induction to inhibit MNV infection of intestinal immune cells, they do not provide an explanation for the regional nature of this process in vivo. Our results exclude regional differences in the expression pattern of the IFN-λ receptor (Fig. 2d), and argue against regional differences in bile acid profiles (Fig. 3a), as key factors. An alternative explanation could be regional differences in expression of a key bile acid receptor (BAR). The BAR farnesoid X receptor (FXR) has been reported to be expressed more abundantly in the distal gut than the proximal gut (41–44). We confirmed this regional difference using RNAscope-based ISH (Fig. 4c). FXR is known to play an immunoregulatory role (30), having the capacity to be both proviral (45) and antiviral (33). Its regional expression pattern led us to speculate that high levels of FXR activation suppress bile acid-mediated enhancement of IFN-λ expression, thereby providing an explanation for the lack of IFN-λ priming in the distal gut during MNV infection. To test this possibility, we first confirmed that CMT-93 cells express FXR (Fig. 4d). Next, CMT-93 cells were pretreated with the FXR agonist GW4064 and transfected with poly(I:C). GW4064 inhibited IFN-λ expression in a dose-dependent manner (Fig. 4e). Finally, to determine whether FXR activation could suppress the ability of a bile acid to enhance IFN-λ expression, cells were treated with a constant dose of 200 μM DCA and a range of GW4064 doses for 12 h prior to poly(I:C) transfection. Higher doses of GW4064 completely blocked DCA-mediated enhancement of IFN-λ expression (Fig. 4f). Overall, our findings support a model in which bacteria-biotransformed bile acids prime IFN-λ expression which in turn blocks MNV-1 replication; but this antiviral activity can be suppressed by sufficient FXR engagement (Fig. 4g). Although we have not yet discovered the BAR responsible for enhancing antiviral activity on our system, the BAR Takeda G protein-coupled receptor 5 (TGR5) was recently reported to amplify type I IFN production (46) so this represents a strong candidate to test in future studies.
In summary, our findings reveal a more complex picture of commensal bacterial regulation of norovirus infection than previously appreciated and they provide mechanistic insight into this regulation. First, the intestinal microbiota play opposing roles in the proximal small intestine and more distal regions of the gut. Throughout the entire course of acute MNV-1 infection, commensal microbes inhibit proximal gut infection while simultaneously enhancing distal gut infection. The mechanisms underlying these phenotypes are distinct from one another, as highlighted by the finding that proximal gut inhibition is dependent on type III IFN signaling while distal gut enhancement occurs in a type III IFN-independent manner. It is fascinating to consider the consequences of bacteria-directed regionalization of enteric virus infections. For example, the mucosal immune system along the GI tract is highly compartmentalized (47) so preferential viral infection of the distal gut may result in a fundamentally distinct host immune response compared to infection of the proximal gut. More generally, our observations represent an example of the intestinal microbiota inhibiting a mammalian enteric virus. Understanding natural host defense mechanisms against pathogens can inform the development of therapeutic strategies. We demonstrated that bile acids, host metabolites that are biotransformed in the gut lumen by commensal bacterial enzymes, prime type III IFN induction in the proximal gut which in turn inhibits MNV-1 infection of intestinal immune cells.
METHODS
Cells and viruses.
The RAW 264.7 (ATCC), 293T (provided by Fanxiu Zhu, Florida State University), CMT-93 (ATCC), and M2C-CD300lf (40,48) cell lines tested negative for mycoplasma contamination. Cell lines were not authenticated. All lines were maintained in Dulbecco’s modified Eagle medium (DMEM; Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Omega), 100 U penicillin/mL, and 100 μg/mL streptomycin. Stocks of recombinant MNV-1.CW3 (GenBank accession number KC782764, referred to as MNV-1) and MNV-3 (GenBank accession number KC792553) were generated as described previously (20). Briefly, 106293T cells were transfected with 5 μg infectious clone using Lipofectamine 2000 (Life Technologies), cells were freeze-thawed after 1 d, lysates titrated with a standard TCID50 assay (49), and RAW264.7 cells infected at MOI 0.05. RAW 264.7 lysates were freeze-thawed when cultures displayed 90% cytopathic effect and supernatants were clarified by low-speed centrifugation followed by purification through a 25% sucrose cushion. The viral genomes of stocks were sequenced completely to confirm no mutations arose during stock generation. A mock inoculum stock was prepared using RAW 264.7 lysate from uninfected cultures. A neutral red-labeled MNV-1 stock was generated as previously described (50). Briefly, RAW 264.7 cells were infected at MOI 0.05 with MNV-1 in the presence of 10 μg/ml neutral red (Sigma), a light sensitive dye. After 2 d, supernatant was collected in a darkened room using a red photolight (Premier OMNI). Stocks were freeze-thawed twice and stored in a light safe box at −80°C. Viral titer was determined by neutral red virus plaque assay without white light exposure.
Mice.
Specific-pathogen-free (SPF) mice used in this study were bred and housed in animal facilities at the University of Florida, Washington University School of Medicine and Yale University. Germ-free mice were bred and housed at University of Michigan Germ-Free & Gnotobiotic Mouse Facility; germ-free status was verified regularly through fecal analysis and control necropsies. All animal experiments were performed in strict accordance with federal and university guidelines. The animal protocols were approved by the Institutional Animal Care and Use Committees at the University of Florida (study number 201408668), Washington University School of Medicine (protocol number 20190162), University of Michigan (protocol number 00006658), and Yale University (study number 201820198). In all experiments, age- and sex-matched groups were used. Mice were 6 to 12 weeks of age, and comparable numbers of male and female mice were used in each experiment. Sample sizes were determined using power calculations based on our prior experience with the MNV system. Blinding was not used in our study as it does not have any clinical data or field sample collection. Wild-type B6 mice and the following knockout strains on the B6 background were used in these studies: Ifnar−/−, Il10−/−, Rag1−/−, and Ifnlr1−/−, Ifnl2/3−/−, Fxr−/−. Pou2f3−/− mice were newly generated for this study: Fertilized zygotes from C57BL/6J mice (Jackson Laboratories) were injected with Cas9 mRNA and sgRNA (5’ AGGCCATGCCACCTGAGCCANGG 3’) targeting Pou2f3. Founder and F1 mice were genotyped by deep sequencing of the target locus. Additional generations were genotyped from tail biopsies by qPCR (Transnetyx, Inc). All experimental mice were derived from breeding Pou2f3 heterozygotes. The CRISPR targeting resulted in an eight base pair deletion and three base pair substitution deletion Pou2f3 exon 4 as shown below:
| WT | TGGGTGACTCTCTACAGCAGACCCTCTCCCACAGGCCATGCCACCTGAGCCAAGGACCTA |
| KO | TGGGTGACTCTCTACAGCAGACCCTCTCCCACAGGCCT--------CTCCCAAGGACCTA |
| ************************************* *********** |
Antibiotic treatments and microbial reconstitutions.
The intestinal microbiota were depleted from mice as described previously (6). Briefly, mice were administered daily a cocktail of 10 mg each of ampicillin (Sigma), neomycin (Sigma), metronidazole (Acros Organics), and vancomycin (Fisher Scientific) via oral gavage for five days. After day five, antibiotics (Abx) were added to the drinking water at a concentration of 1 g/L for ampicillin, metronidazole, and neomycin and 500 mg/L for vancomycin. To confirm efficient depletions, fecal samples were collected from mice at the fifth day of gavage, homogenized, plated on brain-heart infusion agar with 10% sheep blood, and cultured under anaerobic or aerobic conditions at 37°C for 2 d. Abx were maintained in the drinking water throughout the entire experiment and the infections were only carried out after the Abx-treated animals were verified to be free of detectable intestinal bacteria. Alternatively, mice were administered 2 mg clindamycin or 6 mg nalidixic acid (Sigma) daily via oral gavage for 7 days, based on previous work (37). Bacteria-replete controls received phosphate buffered saline (PBS) in place of antibiotics in all experiments. For C. scindens colonization experiments, mice were administered Abx as described above, Abx were discontinued on day 6, and mice were gavaged with 108 colony forming units (cfu) of ATCC strain 35704 on days 7 and 8. On day 14, mice were orally inoculated with MNV-1.
Mouse infections and other in vivo treatments.
For all acute MNV infection experiments, six- to ten-week old, sex-matched mice were inoculated perorally (p.o.) with the indicated virus strain in 25 μl inoculum. For virulence assays, Ifnar−/− mice were observed for weight loss and survival daily for 14 days. The daily weights were compared to day 0 weights to calculate the relative weight loss. For MNV titer determination, tissue samples were harvested at the indicated time points and titrated by plaque assay, as previously described (51,52). For certain experiments, neutral red-labeled virus was used in order to differentiate between inoculum and newly synthesized virus along the GI tract (50,53). When using tissues from mice infected with neutral red virus, samples were exposed to white light for 30 min prior to titering in order to inactive input virus (50). To measure intestinal transit time using a previously published method (54), mice were fasted for 18 h, gavaged orally with 100 μl 50 mg/ml Evans Blue solution prepared with 1% methylcellulose, and euthanized 15 min later. The small intestine was divided into five consecutive pieces, each piece was minced in 0.1 M sodium hydroxide, 6 mM N-acetylcysteine, sonicated, and the absorbance at 565 nm determined. For bile acid supplementations, mice were treated with PBS or Abx for one week and then fed either standard chow or chow supplemented with 0.6% CDCA or DCA (Sigma) for 14 d by grinding the bile acid powder into the chow. Chow was replaced daily. PBS or Abx was delivered in the drinking water throughout the bile acid supplementation. All animal experiments were repeated 2-4 times and data from all experimental replicates are included in the figures. For studies in the newly generated tuft cell-deficient mice, Pou2f3−/− or WT littermate controls were infected at 6-8 weeks of age with 106 plaque forming units (PFU) MNV-1. Seven days post-infection spleen, MLN, distal ileum, and proximal colon were harvested and immediately flash frozen. Total RNA was extracted with TRIzol and viral genomes were quantified relative to host actin mRNA by qPCR as described previously (26).
RNAScope staining.
RNAscope in situ hybridization (ISH) was performed as recently described by our group (24). In brief, formalin-fixed paraffin embedded (FFPE) intestinal swiss rolls were deparaffinized, dehydrated, and stained using the RNAscope 2.5 HD Assay-RED kit according to manufacturer’s instructions (Advanced Cell Diagnostics). Tissue sections were hybridized with a probes targeting the minus-strand MNV-1 RNA (custom probe), Ifnlr1 (RNAscope Probe-Mm-Ifnlr1 catalog # 512981), or Fxr (RNAscope Probe-Mm-Nr1h4 catalog # 484491) followed by hematoxylin staining to visualize the cellular architecture. Commercial control probes specific to the PPIB housekeeping gene transcript and the bacterial dapB gene transcript were also used as positive and negative controls, respectively, in each experiment. Virus minus-strand images were acquired using an Aperio ScanScope XT slide scanner and analyzed using Aperio ImageScope software. Ifnlr1 and Fxr images were acquired using a Nikon Ti-E Live Cell Widefield Microscope and analyzed using NIS Elements BR 3.22.11 software. Swiss rolls from mock inoculated PBS- and Abx-treated mice were included in all experiments to evaluate nonspecific signal. Increased background signal was observed in SI-1 sections hybridized with the minus-strand MNV probe in both mock inoculated- and virus-inoculated animals, regardless of Abx treatment. However this nonspecific signal was blurry and faint, and clearly distinguishable from the expected punctate positive signal. Infection of subepithelial cells and cells within the follicle-associated epithelium (FAE) was quantified as previously described (24). In brief, individual dots corresponding to viral minus strand RNA were counted throughout entire sections of SI-1 or SI-3. For each mouse, 3-5 layers of a given swiss roll were counted in this manner and data from all layers of all six mice per condition were averaged. The hematoxylin staining enabled the differentiation of subepithelial cells and cells within the FAE in most instances. In cases where the location of the dot was questionable, we did not include those in our quantification since the main objective of this experiment was to determine the predominant cell type targeted by MNV-1. No dots were detected in any of the 4 PBS-treated or 4 Abx-treated animals in SI-1 or SI-3.
Bile acid analyses.
Bile acids in intestinal lumenal contents were quantified by the University of Pennsylvania Microbial Culture and Metabolomics Core associated with the PennCHOP Microbiome Program using a Waters Acquity uPLC System with a QDa single quadrupole mass detector and an autosampler (192 sample capacity). Samples were analyzed on an Acquity uPLC with a Cortecs UPLC C-18+ 1.6 mm 2.1 x 50 mm column using a flow rate of 0.8 ml/min, injection volume of 4 μl, column temperature of 30°C, sample temperature of 4°C, and run time of 4 min per sample. 0.1% formic acid in water was used as eluent A and 0.1% formic acid in acetonitrile was used as eluent B. The weak needle wash was 0.1% formic acid in water, the strong needle wash was 0.1% formic acid in acetonitrile, and the seal wash was 10% acetonitrile in water. The gradient was 70% eluent A for 2.5 minutes, gradient to 100% eluent B for 0.6 minutes, and then 70% eluent A for 0.9 minutes. The mass detection channels were: +357.35 for chenodeoxycholic acid (CDCA) and deoxycholic acid (DCA); +359.25 for lithocholic acid (LCA); −407.5 for cholic acid (CA), alphamuricholic acid (αMCA), betamuricholic acid (βMCA), gamma muricholic acid (γMCA), and omegamuricholic acid (ωMCA); −432.5 for glycolithocholic acid (GLCA); −448.5 for glycochenodeoxycholic acid (GCDCA) and glycodeoxycholic acid (GDCA); −464.5 for glycocholic acid (GCA); −482.5 for taurolithocholic acid (TLCA); −498.5 for taurochenodeoxycholic acid (TCDCA) and taurodeoxycholic acid (TDCA); and −514.4 for taurocholic acid (TCA). Samples were quantified against standard curves of at least five points run in triplicate. Standard curves were run at the beginning and end of each metabolomics run. Quality control checks (blanks and standards) were run every eight samples. The range of the assay was at least 50 nM – 10,000 nM; the limit of detection was set at 100 nM and the limit of quantitation was >10,000 nM. Data for combined conjugated 1° bile acids (TCDCA, TCA, GCDCA, and GCA), unconjugated 1° bile acids (CDCA, CA, αMCA, and βMCA), conjugated 2° bile acids (TDCA, GDCA, TLCA, and GLCA), and unconjugated 2° bile acids (γMCA, ωMCA, DCA, and LCA) are presented in the main text (Fig. 3a). Levels of each individual bile acids are presented in Extended Data Fig. 3 and source data is available in Supplementary Table 2.
In vitro IFN-λ assays.
5.0 x 105CMT-93 cells were grown in six-well plates and allowed to adhere overnight. Cells were treated with CDCA, DCA, CA, LCA, the FXR agonist GW4064, or vehicle control (Sigma) at predetermined nontoxic doses. At 12 hours post-treatment, supernatants were replaced with antibiotic-free DMEM plus 10% fetal bovine serum and cells were transfected with 5 μg poly(l:C) (Invivogen) using Lipofectamine 2000 (Invitrogen). Cells were then harvested at 6 h post-transfection and total RNA was extracted using the RNeasy Mini kit (Qiagen). Untransfected cells and cells transfected with a pLJM1-EGFP control plasmid treated with bile acids were tested in every experiment, and duplicate wells were tested for all conditions. To quantify IFN-λ levels, total RNA was subjected to DNase treatment using the Turbo DNA-free kit (Ambion) and cDNA was synthesized using the ImProm-II Reverse Transcription system (Promega). Quantitative real-time PCR was performed using a CFX96 Real-Time System C1000 Touch Thermal iCycler (Bio-Rad) and SYBR Green Master Mix (Thermo Scientific). Primer sequences were as follows: GAPDH sense: 5’-CATGGCCTTCCGTGTTCCTA-3’ antisense: 5’-CCTGCTTCACCACCTTCTTGAT-3’; IFN-λ sense: 5’-CCGCAGTGCTGACAAGAACC-3’ antisense: 5’-ACTGGCCACACACTTGAGG-3’. 5.0 x 104 M2C-CD300lf cells were grown in six- well plates and allowed to adhere overnight. Cells were then treated with the indicated concentrations of bile acids for 12 hours, and infected with MNV-1 at an MOI of 1.0. At 24 hours post-infection, cells were harvested and total RNA was extracted using Direct-zol kits (Zymo Research). Taqman assays were used to detect IFN-λ (Mm04204156_gH) (Thermo Fisher Scientific) and RPS29 (RPS29 sense: 5’-CAAATACGGGCTGAACATG-3’ antisense 5’-GTCCAACTTAATGAAGCCTATGTC-3’ probe 5’-HEX-CCTTCGCGTACTGCCGGAAGC-3’). All samples were run in triplicate for each primer pair and normalized cytokine levels were calculated using the comparative Ct method. The entire experiment was repeated four times.
Statistical Analyses.
All data were analyzed with GraphPad Prism software. Error bars denote standard errors of mean in all figures and P values were determined using two-tailed Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001).
DATA AVAILABILITY
All data that support the findings of this study are available from the corresponding authors on request. Source data for bile acid analyses performed in this study are included in Supplementary Table 2.
Extended Data
Extended Data Fig. 1. Cumulative intestinal titers.
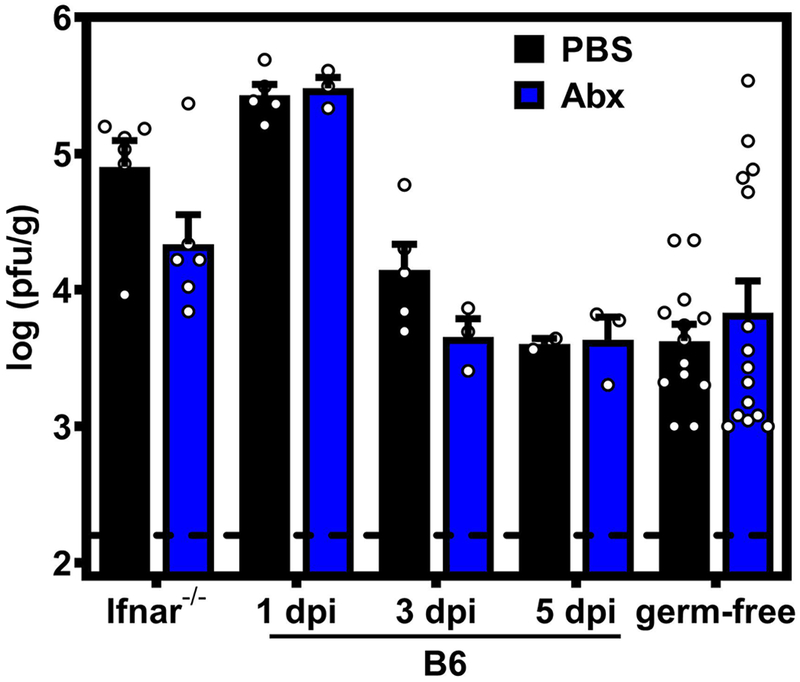
Intestinal titers from SI-1, SI-2, SI-3 cecum and colon were combined for each mouse, and data for all mice per group averaged. Data for individual intestinal regions for Ifnar−/− mice are shown in Fig. 1c, for B6 mice are shown in Fig. 1d, and for germ-free mice are shown in Fig. 1e.
Extended Data Fig. 2. Tuft cells are not required for MNV-1 infection.
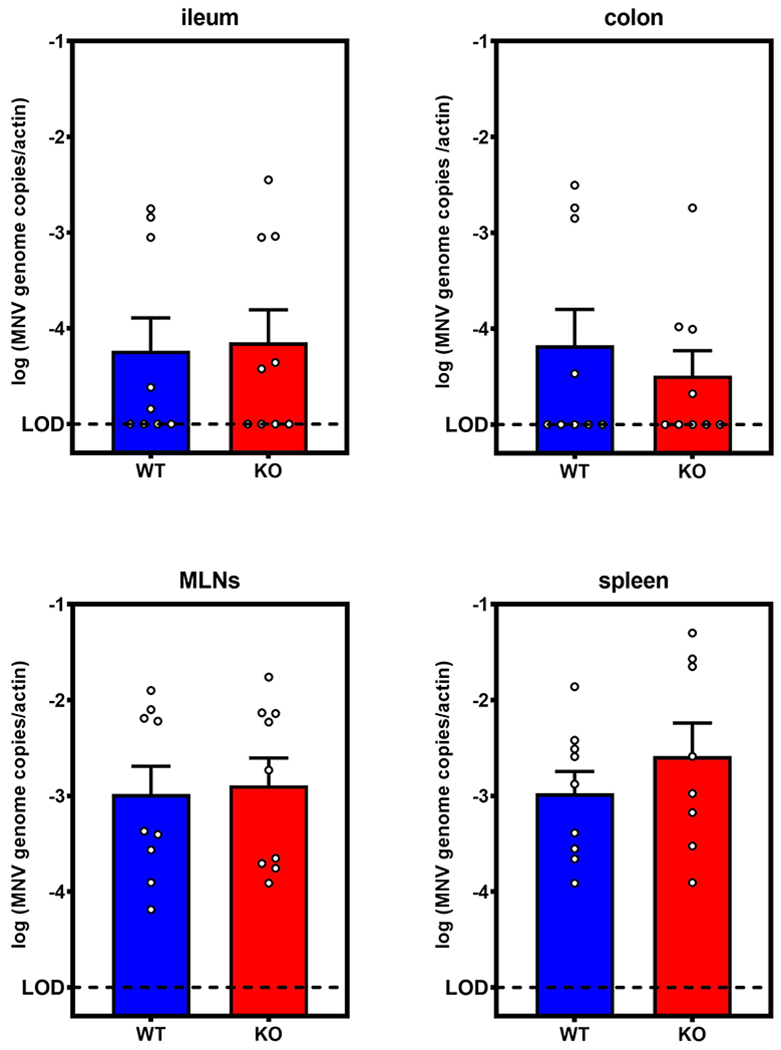
Tuft cell-deficient Pou2f3−/− or wild-type littermates (n = 9 mice per group) were challenged with 106 plaque-forming units (PFU) MNV-1 and viral genomes were quantified at 7 dpi. Pou2f3−/− (KO) and WT mice had statistically similar viral genomes in ileum, colon. MLNs, and spleen. Data is pooled from three independent experiments. Error bars indicate the mean of all data points. LOD, limit of detection.
Extended Data Fig. 3. Measurements of individual bile acids.
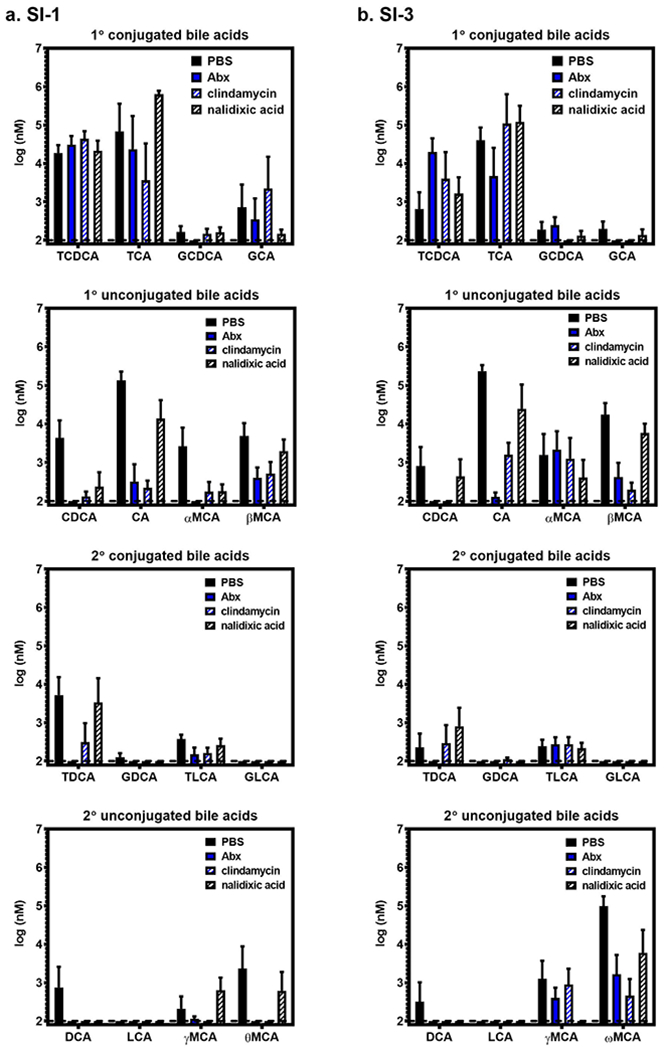
Lumenal contents were collected from SI-1 (a) and SI-3 (b) of groups of B6 mice (n = 5) treated with PBS, Abx, clindamycin, or nalidixic acid. The indicated bile acids were measured at the University of Pennsylvania Microbial Culture and Metabolomics Core using a Waters Acquity vPLC System. Source data are provided in Supplementary Table 2.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge the Washington University Genome Engineering and iPSC center, Michael White and Darren Kreamalmeyer. S.M.K. was funded by NIH R01AI116892, NIH R01AI081921, and NIH R01AI141478. M.T.B. was supported by NIH R01AI141478, NIH K22 AI127846-01, DDRCC grant P30 DK052574, and the Global Probiotics Council’s Young Investigator Grant for Probiotics Research. C.E.W. was funded in part by NIH R21 AI103961 and the University of Michigan Host-Microbiome Initiative. E.W. and M.P. were supported by T90DE021990. H.T. was supported by T32DK094775. C.B.W was supported by NIH grant K08 AI28043 and the Burroughs Wellcome Fund.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCEES
- 1.Karst SM. The influence of commensal bacteria on infection with enteric viruses. Nat Rev Microbiol. 2016. April; 14(4): 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfeiffer JK, Virgin HW. Transkingdom control of viral infection and immunity in the mammalian intestine. Science. 2016. January 15;351(6270):aad5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, et al. Intestinal Microbiota Promote Enteric Virus Replication and Systemic Pathogenesis. Science. 2011. October 14; 334(6053):249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, et al. Successful Transmission of a Retrovirus Depends on the Commensal Microbiota. Science. 2011. October 14;334(6053):245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kernbauer E, Ding Y, Cadwell K. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014. December 4;516(7529):94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, et al. Enteric Bacteria Promote Human and Murine Norovirus Infection of B cells. Science. 2014. November 7; 346(6210): 755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, et al. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science. 2015;347(6219):266–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchiyama R, Chassaing B, Zhang B, Gewirtz AT. Antibiotic Treatment Suppresses Rotavirus Infection and Enhances Specific Humoral Immunity. J Infect Dis. 2014. July 15;210(2): 171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson CM, Jesudhasan PR, Pfeiffer JK. Bacterial Lipopolysaccharide Binding Enhances Virion Stability and Promotes Environmental Fitness of an Enteric Virus. Cell Host Microbe. 2014. January 15;15(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilks J, Lien E, Jacobson AN, Fischbach MA, Qureshi N, Chervonsky AV, et al. Mammalian Lipopolysaccharide Receptors Incorporated into the Retroviral Envelope Augment Virus Transmission. Cell Host Microbe. 2015. October 14;18(4):456–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sansone CL, Cohen J, Yasunaga A, Xu J, Osborn G, Subramanian H, et al. Microbiota-Dependent Priming of Antiviral Intestinal Immunity in Drosophila. Cell Host Microbe. 2015. November 11;18(5): 571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez JL, Souza-Neto J, Cosme RT, Rovira J, Ortiz A, Pascale JM, et al. Reciprocal Tripartite Interactions between the Aedes aegypti Midgut Microbiota, Innate Immune System and Dengue Virus Influences Vector Competence. PLOS Negl Trop Dis. 2012. March 6;6(3):e1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu P, Sun P, Nie K, Zhu Y, Shi M, Xiao C, et al. A Gut Commensal Bacterium Promotes Mosquito Permissiveness to Arboviruses. Cell Host Microbe. 2019. January 9;25(1):101–112.e5. [DOI] [PubMed] [Google Scholar]
- 14.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci. 2011. March 29;108(13):5354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity. 2012. July 27;37(1): 158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McFarlane AJ, McSorley HJ, Davidson DJ, Fitch PM, Errington C, Mackenzie KJ, et al. Enteric helminth-induced type I interferon signaling protects against pulmonary virus infection through interaction with the microbiota. J Allergy Clin Immunol. 2017. October;140(4):1068–1078.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thackray LB, Handley SA, Gorman MJ, Poddar S, Bagadia P, Briseño CG, et al. Oral Antibiotic Treatment of Mice Exacerbates the Disease Severity of Multiple Flavivirus Infections. Cell Rep. 2018. March 27;22(13):3440–3453.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science. 2017. August 4;357(6350):498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu S, Watanabe M, Kirkpatrick E, Murray AB, Sok R, Karst SM. Regulation of norovirus virulence by the VP1 protruding domain correlates with B cell infection efficiency. J Virol. 2015. December 30;JVI.02880-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu S, Regev D, Watanabe M, Hickman D, Moussatche N, Jesus DM, et al. Identification of Immune and Viral Correlates of Norovirus Protective Immunity through Comparative Study of Intra-Cluster Norovirus Strains. PLoS Pathog. 2013. September 5;9(9):e1003592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, et al. Murine Noroviruses Comprising a Single Genogroup Exhibit Biological Diversity despite Limited Sequence Divergence. J Virol. 2007. October 1;81(19):10460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu CC, Riley LK, Wills HM, Livingston RS. Persistent Infection with and Serologic Crossreactivity of Three Novel Murine Noroviruses. Comp Med. 2006. August;56:247–51. [PubMed] [Google Scholar]
- 23.Arias A, Bailey D, Chaudhry Y, Goodfellow IG. Development of a Reverse Genetics System for Murine Norovirus 3; Long-Term Persistence Occurs in the Caecum and Colon. J Gen Virol. 2012. April 11;93(Pt 7):1432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grau KR, Roth AN, Zhu S, Hernandez A, Colliou N, DiVita BB, et al. The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nat Microbiol. 2017. December;2(12):1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-Lambda (IFN-λ) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLOS Pathog. 2008. March 14;4(3):e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL, et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science. 2018. 13;360(6385):204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015. July 21;43(1):15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008. August;7(8):678–93. [DOI] [PubMed] [Google Scholar]
- 29.Joyce SA, Gahan CGM. The gut microbiota and the metabolic health of the host: Curr Opin Gastroenterol. 2014. March;30(2): 120–7. [DOI] [PubMed] [Google Scholar]
- 30.Fiorucci S, Distrutti E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol Med. 2015. November;21(11):702–14. [DOI] [PubMed] [Google Scholar]
- 31.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J Immunol. 2009. November 15;183(10):6251–61. [DOI] [PubMed] [Google Scholar]
- 32.Schupp A-K, Trilling M, Rattay S, Le-Trilling VTK, Haselow K, Stindt J, et al. Bile Acids Act as Soluble Host Restriction Factors Limiting Cytomegalovirus Replication in Hepatocytes. J Virol. 2016. July 11;90(15):6686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Chang K-O. Inhibitory Effects of Bile Acids and Synthetic Farnesoid X Receptor Agonists on Rotavirus Replication∇. J Virol. 2011. December;85(23):12570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang K-O, Sosnovtsev SV, Belliot G, Kim Y, Saif LJ, Green KY. Bile acids are essential for porcine enteric calicivirus replication in association with down-regulation of signal transducer and activator of transcription 1. Proc Natl Acad Sci U S A. 2004. June 8;101(23):8733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, et al. Replication of human noroviruses in stem cell–derived human enteroids. Science. 2016. August 25;aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015. January 8;517(7533):205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun X, Winglee K, Gharaibeh RZ, Gauthier J, He Z, Tripathi P, et al. Microbiota-derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology [Internet], 2018. February 1 [cited 2018 Mar 13]; Available from: http://www.sciencedirect.com/science/article/pii/S0016508518301100 [DOI] [PMC free article] [PubMed]
- 38.Wells JE, Hylemon PB. Identification and Characterization of a Bile Acid 7α-Dehydroxylation Operon in Clostridium sp. Strain TO-931, a Highly Active 7α-Dehydroxylating Strain Isolated from Human Feces. Appl Environ Microbiol. 2000. March 1;66(3):1107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson CA, Wilen CB, Dai Y-N, Orchard RC, Kim AS, Stegeman RA, et al. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc Natl Acad Sci. 2018. September 25;115(39):E9201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Wilen CB, Orvedahl A, McCune BT, Kim K-W, Orchard RC, et al. Norovirus Cell Tropism Is Determined by Combinatorial Action of a Viral Non-structural Protein and Host Cytokine. Cell Host Microbe. 2017. October 11;22(4):449–459.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003. January 3;278(1):104–10. [DOI] [PubMed] [Google Scholar]
- 42.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006. August 25;126(4):789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higashiyama H, Kinoshita M, Asano S. Immunolocalization of farnesoid X receptor (FXR) in mouse tissues using tissue microarray. Acta Histochem. 2008. January 14;110(1):86–93. [DOI] [PubMed] [Google Scholar]
- 44.Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006. March 7;103(10):3920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang K-O, George DW. Bile Acids Promote the Expression of Hepatitis C Virus in Replicon-Harboring Cells. J Virol. 2007. September 15;81(18):9633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong Q, Huang H, Wang N, Chen R, Chen N, Han H, et al. Metabolite-Sensing G Protein Coupled Receptor TGR5 Protects Host From Viral Infection Through Amplifying Type I Interferon Responses. Front Immunol. 2018;9:2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014. October;14(10):667–85. [DOI] [PubMed] [Google Scholar]
- 48.Padilla-Nash HM, Hathcock K, McNeil NE, Mack D, Hoeppner D, Ravin R, et al. Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes Chromosomes Cancer. 2012. April;51(4):353–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, et al. Murine Noroviruses Comprising a Single Genogroup Exhibit Biological Diversity despite Limited Sequence Divergence. J Virol. 2007. October 1;81(19):10460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.González-Hernández MB, Perry JW, Wobus CE. Neutral Red Assay for Murine Norovirus Replication and Detection in a Mouse. Bio-Protoc. 2013. April 5;3(7). [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu S, Regev D, Watanabe M, Hickman D, Moussatche N, Jesus DM, et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of intra-cluster norovirus strains. PLoS Pathog. 2013;9(9):e1003592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kahan SM, Liu G, Reinhard MK, Hsu CC, Livingston RS, Karst SM. Comparative murine norovirus studies reveal a lack of correlation between intestinal virus titers and enteric pathology. Virology. 2011. December 20;421(2):202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald J, Ikizler M, Yagita H, et al. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol. 2014;88:6934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mária Bagyánszki, Mónika Krecsmarik, De Winter Benedicte Y, De Man Joris G, Fekete Éva, Pelckmans Paul A, et al. Chronic Alcohol Consumption Affects Gastrointestinal Motility and Reduces the Proportion of Neuronal NOS-Immunoreactive Myenteric Neurons in the Murine Jejunum. Anat Rec. 2010. August 20;293(9): 1536–42. [DOI] [PubMed] [Google Scholar]
- 55.Haug K, Salek RM, Conesa P, Hastings J, de Matos P, Rijnbeek M, et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013. January;41 (Database issue):D781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are available from the corresponding authors on request. Source data for bile acid analyses performed in this study are included in Supplementary Table 2.