

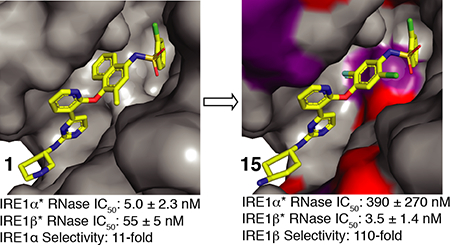
Abstract
The dual kinase endoribonuclease IRE1 is a master regulator of cell fate decisions in cells experiencing endoplasmic reticulum (ER) stress. In mammalian cells there are two paralogs of IRE1: IRE1α and IRE1β. While IRE1α has been extensively studied, much less is understood about IRE1β and its role in signaling. In addition, whether the regulation of IRE1β’s enzymatic activities vary compared to IRE1α is not known. Here, we show that the RNase domain of IRE1β is enzymatically active and capable of cleaving an XBP1 RNA mini-substrate in vitro. Using ATP-competitive inhibitors, we find that, like IRE1α, there is an allosteric relationship between the kinase and RNase domains of IRE1β. This allowed us to develop a novel toolset of both paralog specific and dual-IRE1α/β kinase inhibitors that attenuate RNase activity (KIRAs). Using sequence alignments of IRE1α and IRE1β we propose a model for paralog-selective inhibition through interactions with non-conserved residues that differentiate the ATP-binding pockets of IRE1α and IRE1β.
Graphical Abstract
Introduction
Various perturbations within the cell can cause endoplasmic reticulum (ER) stress, resulting in unfaithful folding and the accumulation of proteins within the lumen of the ER.1,2 In mammalian cells, these perturbations are sensed by the unfolded protein response (UPR), which consists of three transmembrane sensor proteins: IRE1, PERK, and ATF6.3–7 The initial aim of the UPR is to restore ER homeostasis, where all three UPR sensor initiate signals to upregulate genes that aid in ER stress adaptation.8 Additionally, PERK mediates a global translation block to help decrease the ER’s protein folding burden.5 If these initial adaptive responses fail, the UPR switches to terminal outputs that result in apoptosis.9 IRE1α is the most ancient and conserved component of the UPR, and is thought to be one of the dominant factors in cell fate decision making in cell experiencing ER stress.4,10 IRE1α contains a stress sensing N-terminal lumenal domain that is connected to cytosolic protein kinase and RNase domains through a transmembrane linker.11 Upon ER stress, IRE1α becomes active through lumenal domain dimerization and kinase trans-autophosphorylation. Phosphorylation of the kinase promotes IRE1α dimerization on the cytosolic face of the ER, an event required for RNase domain activity.12,13 Under an adaptive UPR, the RNase domain of IRE1α facilitates the cleavage of XBP1 mRNA, which results in the subsequent generation of a mature form of XBP1 that encodes for an active transcription factor that upregulates targets that aid in ER adaptation.14–16 Conversely, prolonged ER stress leads to the non-specific cleavage of ER-localized mRNA by IRE1α’s RNase in an event called regulated IRE1 dependent decay, or RIDD.17–19 RIDD increases ER stress and promotes apoptosis through the cleavage of mRNA encoding for adaptive ER proteins and anti-apoptotic proteins, as well as cleaving miRNA functioning to suppress pro-apoptotic and proinflammatory proteins.20
In mammalian cells, IRE1α has a highly homologous paralog called IRE1β. While IRE1α is ubiquitously expressed, IRE1β’s expression is mainly isolated to epithelial cells in the gastrointestinal tract and bronchia.21 IRE1β’s domain architecture is the same as IRE1α’s but much less is known about its RNase activity or its functional role in the cell. While it has been shown that IRE1β’s kinase retains phosphotransferase function, there have been conflicting reports about the ability of its RNase domain to cleave XBP1 and the efficiency of this cleavage event compared to IRE1α.16,22 It has been suggested that under ER stress, rather than prioritizing XBP1 cleavage, IRE1β’s RNase primarily cleaves ER-localized mRNA encoding secretory proteins.23 Furthermore, there is evidence that IRE1β might have a distinct set of mRNA substrates from IRE1α.23–25 These differences in enzymatic activity have been highlighted in vivo, where IRE1β but not IRE1α has been shown to be functionally required for mucin production.25 This relationship suggests that selective IRE1β inhibition may be an attractive target in diseases characterized by the overproduction of mucin, like cystic fibrosis, chronic obstructive pulmonary disease, and asthma.26 Additionally, mice that lack IRE1β develop colitis at an advanced rate and have more pronounced hyperlipidemia, revealing that IRE1β may play an essential role in lipid metabolism and inflammation responses in gut and bronchial tissues.21,27
Here, we demonstrate that IRE1β’s RNase domain is similarly efficient as IRE1α in cleaving an XBP1 mini-substrate in vitro. Using different ATP-competitive inhibitors that have been shown to either activate or inhibit IRE1α’s RNase activity, we investigated the allostery between IRE1β’s kinase and RNase domains. We find that an ATP-competitive inhibitor that is a potent activator of IRE1α’s RNase activity has a similar effect on IRE1β. Furthermore, we demonstrate that, like IRE1α, IRE1β’s RNase activity can be allosterically inhibited with ATP-competitive ligands called kinase inhibitor RNase attenuators (KIRAs). This result motivated us to determine whether we could develop KIRAs that demonstrate paralog selectivity. By performing medicinal chemistry around the scaffold of a KIRA developed to target IRE1α, we were able to develop both dual IRE1α/β and paralog-selective KIRAs. Using a kinobead-based profiling method, we demonstrate that these inhibitors are selective for IRE1 on the kinome level. Together, we expect our IRE1-targeting toolset will be valuable reagents for defining paralog-specific function in cells and disease models.
Results and Discussion
Activity and Allosteric Regulation of IRE1β’s RNase Domain
We first performed biochemical characterization of IRE1β with a construct that contains just the cytosolic kinase and RNase domains, which we refer to as IRE1β*. Previous studies have demonstrated that a similar construct of IRE1α, IRE1α*, possesses both kinase and RNase activity.28,29 For in vitro studies of IRE1, IRE1α* and IRE1β* were expressed in baculovirus-infected insect cells and purified in the activation loop phosphorylated form. Activation loop phosphorylation of IRE1 promotes the formation of an RNase active dimer, commonly referred to as the back-to-back dimer.13 In contrast, dephosphorylated versions of IRE1 are monomeric and RNase inactive.28,29 To assess the RNase activities of IRE1α* and IRE1β*, we used a fluorescence-quenched XBP1 mini-substrate labeled with a 5’-AlexaFluor647 and a 3’-IowaBlack fluorescence quencher (Figure 1A). We tested the activity of IRE1β*’s RNase by monitoring the real-time cleavage of a XBP1 mini-substrate in vitro. Despite previous reports that IRE1β’s RNase is less capable or incapable of cleaving XBP1,22 a comparison of equal concentrations of IRE1α* and IRE1β* reveal that both are able to efficiently cleave the XBP1 mini-substrate (Figure 1B). Further inspection of the XBP1 mini-substrate product revealed that the RNase domains of IRE1α* and IRE1β* generate fragments of similar size, supporting the notion that these paralogs cleave XBP1 at the same site (Supp. Figure 1). Next, we measured the rate of IRE1α* and IRE1β* cleavage for varying concentrations of XBP1 mini-substrate to determine their Michaelis constant (KM) values for this RNA substrate. The KM values for the XBP1 mini-substrate between the two paralogs were nearly identical (Figure 1C). This result reveals that IRE1β* can form back-to-back dimers, which are necessary for RNase activity. Additionally, when dimerized, the RNase domains of IRE1α* and IRE1β* are similarly able to bind and cleave XBP1.
Figure 1. In Vitro Activity of IRE1β’s RNase Domain.

A. Schematic of the XBP1 cleavage assay with IRE1α* and IRE1β*. An increase in fluorescence is observed upon cleavage of a FRET-quenched XBP1 mini-substrate. B. Real time fluorescence curves for 75 nM IRE1α* and 75 nM IRE1β* with 250 nM of the XBP1 mini-substrate. C. Michaelis-Menten curves of IRE1α* and IRE1β* for the XBP1 mini-substrate. Data shown are mean ± SEM, n=3.
Previously, it has been established that the kinase and RNase domains of IRE1α are allosterically coupled.28 The basis of this allostery relies on the conformation of a structural element in the ATP-binding site called helix-αC. When IRE1α is unphosphorylated and inactive, helix-αC adopts a conformation that is incompatible with the back-to-back dimer required for RNase activity. Phosphorylation of IRE1α’s activation loop stabilizes the active conformation of helix-αC, which promotes back-to-back dimerization and RNase activity.29 It has been shown that ATP-competitive inhibitors can have divergent effects on IRE1α’s RNase activity by preventing or promoting formation of the back-to-back dimer through modulation of helix-αC’s conformation.13,28 For example, AT9283 potently activates IRE1α’s RNase activity by promoting IRE1α dimerization, presumably though stabilization of the active conformation of helix-αC.29 In contrast, inhibitor 1 (KIRA8) inactivates IRE1α’s RNase domain by stabilizing an inactive helix-αC conformation and preventing IRE1α dimerization.31 We refer to ATP-competitive inhibitors that dually inhibit kinase and RNase activity, like 1, as kinase inhibitor RNase attenuators (KIRAs). To examine the allostery between the kinase and RNase domain of IRE1β, we tested the effects of AT9283 and 1 on IRE1β’s RNase activity. To allow us to measure both activation and inhibition of RNase activity, we tested these inhibitors under two different IRE1β* concentration regimes. We used a concentration of IRE1β* (Low [IRE1β*]) that is mainly monomeric and has low RNase activity to measure activation of IRE1β’s RNase activity by AT9283 (Figure 2A). A higher concentration of IRE1β* (High [IRE1β*]), which contains a substantial amount of the RNase active dimer, was used to test the inhibitory properties of KIRA 1. We found that Low [IRE1β*] treated with AT9283 demonstrated a 12-fold higher rate of XBP1 mini-substrate cleavage than the apo form (Figure 2B). In contrast, treatment of High [IRE1β*] with 1 led to an almost complete loss in its ability to cleave the XBP1 mini-substrate (Figure 2C). The ability of these two classes of ATP-competitive inhibitors to divergently modulate IRE1β*’s RNase activity suggests that the allosteric communication between its kinase and RNase domains is similar to IRE1α.
Figure 2. Divergent Modulation of IRE1β’s RNase Domain with ATP-Competitive Inhibitors.

A. Concentration regimes for testing the allosteric modulation of IRE1β*’s RNase domain. (top) Schematic of the oligomeric states of Low [IRE1β*] and High [IRE1β*]. (bottom) Real time fluorescence curves and initial rates for Low [IRE1β*] and High [IRE1β*]. Data shown are mean ± SEM, n=3. B. Activation of IRE1β*’s RNase activity. (top) Structure of the ATP-competitive RNase activator AT9283. (bottom) Real time fluorescence curves and rates of XBP1 mini-substrate cleavage for Low [IRE1β*] treated with DMSO (light gray) or AT9283 (purple) in the in vitro RNase assay. Data shown are mean ± SEM, n=3. C. Inhibition of IRE1β*’s RNase activity. (top) Structure of KIRA 1. (bottom) Real time fluorescence curves and rates of XBP1 mini-substrate cleavage for High [IRE1β*] treated with DMSO (dark gray) or 1 (coral) in the in vitro RNase assay. Data shown are mean ± SEM, n=3.
Paralog-Selective KIRAs
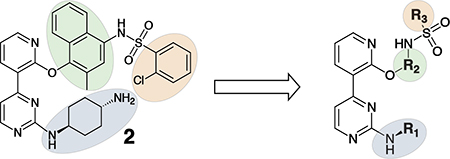
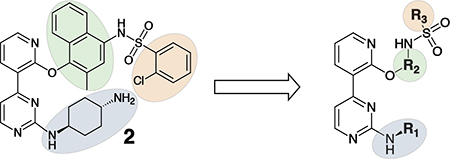
Having demonstrated that the RNase activity of IRE1β can modulated through its ATP-binding site, we next determined whether it would be possible to develop KIRAs with enhanced paralog selectivity. To do this, we performed a structure-activity-relationship (SAR) study around the pyrimidine-pyridine scaffold of KIRA 1 and a close analog (KIRA 2) (Figure 3A,B). Although KIRAs 1 and 2 were optimized for IRE1α, they were selected as a starting points for derivatization because of the demonstrated marked selectivity of KIRA 1 on the kinome level.31,35 To visualize potential differences in KIRA-binding residues between IRE1α and IRE1β, we used a co-crystal structure of KIRA 2 bound to IRE1α to generate a model of potential inhibitor contact residues with IRE1β (Figure 3B,C). The basic trans-1,4-cyclohexandiamine moiety of 2 makes several hydrophobic contacts with residues lining the ATP-binding site of IRE1α and forms a salt bridge with Glu651 located in IRE1α’s helix αH. While IRE1β contains a Glu at the same position (Glu600), an alanine residue (Ala646) in IRE1α that makes hydrophobic contacts with the piperidine ring is substituted with an Arg (Arg595) in IRE1β (Figure 3B,C). Therefore, we introduced various R1 alicyclic groups that contain a basic amine in our SAR to potentially exploit these differences (Figure 3A). The naphthyl group of 2 bridges the core pyrimidine-pyridine scaffold and the sulfonamide group. While most of the residues in the ATP-binding site of IRE1α that form contacts with the bridging naphthyl group of 2 are conserved in IRE1β, the identity of the gatekeeper residue–Ile in IRE1α (Ile642) and Leu in IRE1β (Leu591)–is a clear difference between the two paralogs in this region of the binding pocket. We thus varied the naphthyl group of 2 with various naphthyl and substituted phenyl R2 groups to see if inhibitors could differentiate between IRE1α and IRE1β in this region of the kinase active site. The arylsulfonamide of 2 occupies a pocket created by movement of helix-αC to an inactive conformation. A notable difference in this pocket between IRE1α and IRE1β includes a change from an Ala (Ala609) in the helix-αC of IRE1α to a Val residue in IRE1β (Val558). Therefore, we also generated analogs that contain various sulfonamides at the R3 position to see if the pocket formed by the movement of helix-αC could be used to gain selectivity between IRE1β and IRE1α.
Figure 3. SAR of ATP-Competitive KIRAs.

A. Chemical structure of KIRA 1 (top). Structural elements of 1 that are varied in our study are colored. RNase IC50 values for 1 against both IRE1α* and IRE1β* and the fold selectivity for IRE1α (bottom). Inhibitor data are shown as the mean ± SEM, n=3. B. LigPlot map detailing the binding interactions (yellow sticks) between inhibitor 2, a close analog of 1, and the ATP-binding site of IRE1α (PDB: 4U6R). Residues involved in hydrogen-bond interactions are shown as sticks. Residues involved in hydrophobic interactions are shown as gray eyelashes. C. LigPlot map detailing the hypothesized binding pocket of IRE1β and its interactions with pyridine-pyrimidine based ligands. Residues that are conserved between IRE1β and IRE1α are shown in gray. Conservative mutations are shown in purple and non-conservative mutations are shown in red.
To facilitate the rapid generation of inhibitors, we introduced diverse R1-R3 groups into the conserved pyrimidine-pyridine scaffold of 1 (Scheme 1). A four-step synthetic route was used to diversify the commercially available 2-chloro-4-(2-fluoro-3-pyridinyl)pyrimidine scaffold. R1 groups were introduced through the nucleophilic aromatic substitution (SNAr) of 2-chloro-4-(2-fluoro-3-pyridinyl) pyrimidine with mono boc-protected diaminocycloalkanes. A subsequent SNAr with commercially available 4-amino-phenols or amino-naphthols was used to generate R2 group diversity. Diverse R3 groups were introduced by sulfonylation with various sulfonyl chlorides. Following sulfonylation, final inhibitors were generated by boc-deprotection with TFA (Scheme 1).
Scheme 1.

a Reagents and conditions: (i) R1-amine, TEA, DMSO, 80 °C, 18 hr, (ii) 4-aminophenols, 5-aminonaphthols, or 4-aminonaphthols, K2CO3, DMF, 155 °C, μW, 2 hr (iii) sulfonyl chloride, pyridine, DCM, RT, 18 hr (iv) TFA:DCM (1:1), RT, 2 hr
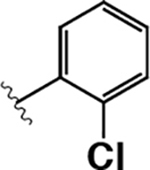
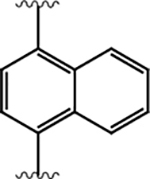
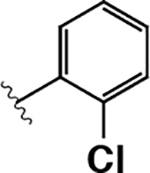
We screened KIRAs for the ability to inhibit the RNase activities of IRE1α* and IRE1β* with our fluorescence-quenched XBP1 mini-substrate assay (Figure 1A). We first generated and tested an analog of 2 (inhibitor 3) that contains identical trans-1,4-cyclohexandiamine R1 and 2-chlorophenyl R3 groups but a slightly different 1,4-substituted 4-amino-1-naphthol R2 group (Table 1). Inhibitor 3 is almost equipotent against IRE1α* and IRE1β*. Replacement of the trans-1,4-cyclohexandiamine R1 group with a 6-amino-2-aza-spiro[3.3] heptane moiety (inhibitor 4), while maintaining the same R1 and R3 groups as inhibitor 3, led to decreased inhibition of both IRE1β*’s and IRE1α*’s RNase domains, albeit disproportionately, resulting in 4.2-fold selectivity for IRE1β*. Next, we explored whether R3 sulfonamide groups affect paralog selectivity. Varying the R3 group of 3 from a 2-chlorophenyl to a 2-chlorobenzyl group (inhibitor 5) led to a ~30-fold reduction in potency against IRE1β*’s RNase activity with minimal influence on IRE1α* potency. We speculated that the Ala to Val substitution present in the helix-αC of IRE1β would create a more restricted binding pocket that would favorably accommodate smaller R3 groups. However, we found that inhibitor 6, which contains identical R1 and R2 substituents as inhibitor 3 but a smaller cyclobutyl R3 group, is remarkably selective (>2000-fold) for IRE1α*. Finally, we determined how changing the display of the R3 sulfonamide from the R2 naphthyl group would affect paralog potency and selectivity. We observed that Inhibitor 7, which contains the same R1 and R3 groups as 6 but a 1,5-naphthyl substitution instead of a 1,4, was also highly selective (>300-fold) for IRE1α*.
Table 1.
SAR of KIRAs Containing Naphthyl R2 Groupsa
 | ||||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | IRE1β RNase IC50 (nM) | IRE1α RNase IC50 (nM) | IRE1β RNase Selectivity (Fold) |
| 1 |  |
 |
 |
55 ± 5 | 5.0 ± 2.3 | 0.09 |
| 3 | 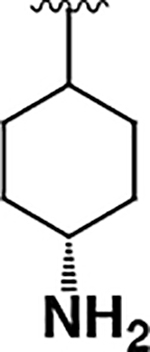 |
 |
 |
15 ± 2 | 22 ± 9 | 1.4 |
| 4 | 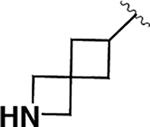 |
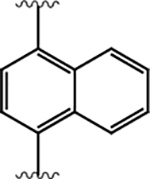 |
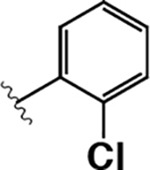 |
100 ± 10 | 430 ± 230 | 4.2 |
| 5 | 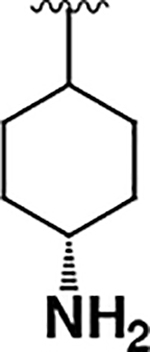 |
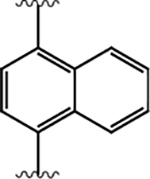 |
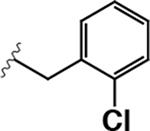 |
490 ± 20 | 9.4 ± 1.0 | 0.02 |
| 6 | 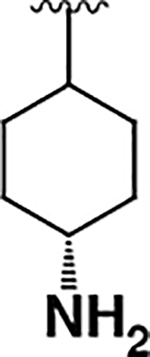 |
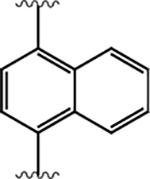 |
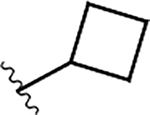 |
>20000 | 10 ± 1 | <0.0005 |
| 7 | 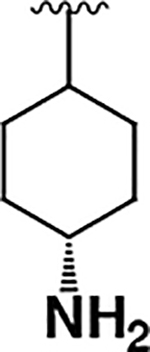 |
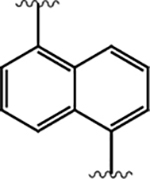 |
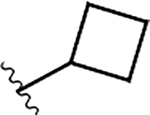 |
14000 ± 4000 | 42 ± 5 | 0.003 |
RNase IC50 data are shown as mean ± SEM, n=3. RNase selectivity was determined by dividing IRE1α* RNase IC50 by IRE1β* RNase IC50 value for each inhibitor (individual IC50 curves provided in Supp. Figure 2).
We found that a 4-amino-1-naphthol R2 group provided inhibitors that are IRE1α selective or equipotent for IRE1α and IRE1β while maintaining reasonably potent RNase inhibition (RNase IC50 < 20 nM). To develop KIRAs that are potent and selective for IRE1β, we next focused on optimizing the R2 position for this paralog (Table 2). A notable difference between IRE1α and IRE1β is the identity of their gatekeeper residues. IRE1α has an isoleucine residue (Ile642), while IRE1β has a leucine (Leu591) at the gatekeeper position. This led us to reason that it may be possible to selectively target IRE1β over IRE1α by using smaller R2 substituents, specifically substituted 4-aminophenol groups. First, we generated inhibitor 8, which contains a trans1,4-cyclohexanediamine R1 group, a 2-chlorophenyl R3 group, and a 4-amino-3-fluorophenol at the R2 position. Inhibitor 8, which contains the same R1 and R3 groups as IRE1α/IRE1β equipotent inhibitor 3, demonstrated single-digit nanomolar potency against IRE1β* and is 5-fold selective for this paralog over IRE1α*. Thus, replacing the 4-amino-1-naphthol R2 group of 3 with a 4-amino-3-fluorophenol increased IRE1β* selectivity. Replacing the trans-1,4-cyclohexanediamine R1 group of 8 with a (S)-3-aminopiperidine (inhibitor 9) resulted in less potent inhibition of IRE1α* and IRE1β*. An analog of 9 that contains a 2-chlorobenzylsulfonamide (10) was found to be almost 10-fold selective for IRE1α* over IRE1β*.
Table 2.
KIRAs Containing 4-aminophenol R2 Groupsa
 | ||||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | IRE1β RNase IC50 (nM) | IRE1α RNase IC50 (nM) | IRE1β RNase Selectivity (Fold) |
| 8 | 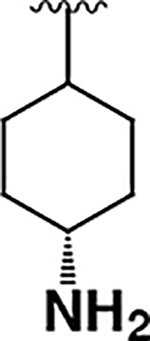 |
 |
 |
4.4 ± 0.7 | 22 ± 9 | 5.0 |
| 9 | 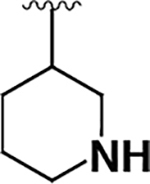 |
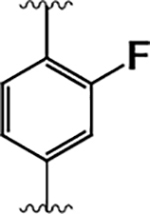 |
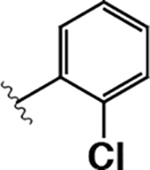 |
120 ± 40 | 120 ± 10 | 1.0 |
| 10 | 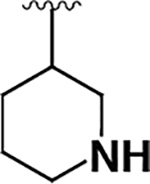 |
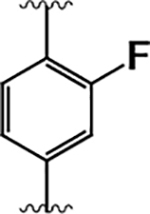 |
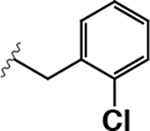 |
50 ± 2 | 6.0 ± 1.7 | 0.12 |
| 11 | 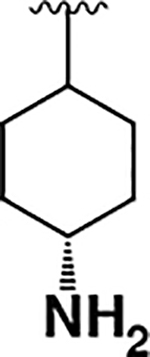 |
 |
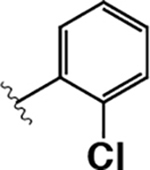 |
99 ± 56 | 57 ± 11 | 0.57 |
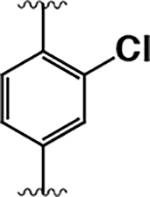
| 12 | 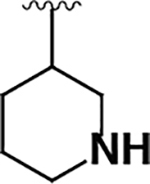 |
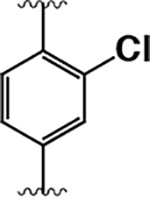 |
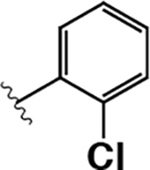 |
150 ± 30 | 130 ± 70 | 0.87 |
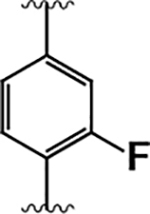
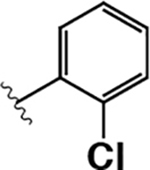
| 13 | 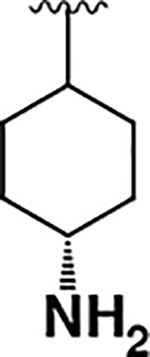 |
 |
 |
3.9 ± 1.1 | 240 ± 50 | 60 |
| 14 | 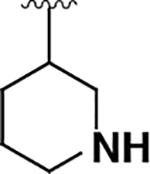 |
 |
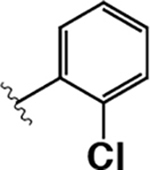 |
13 ± 2 | 110 ± 10 | 8.1 |
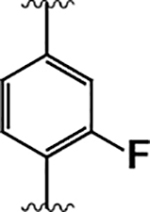
| 15 | 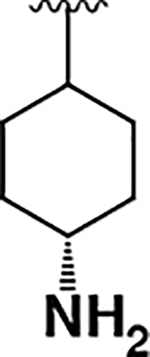 |
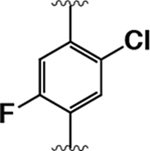 |
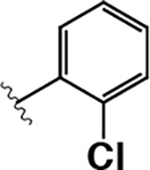 |
3.5 ± 1.4 | 390 ± 270 | 110 |
| 16 | 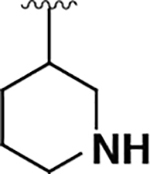 |
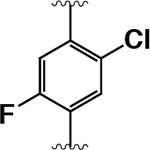 |
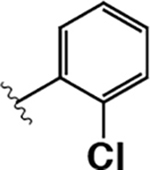 |
430 ± 70 | 2000 ± 100 | 4.5 |
| 17 | 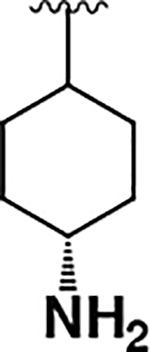 |
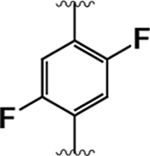 |
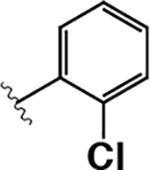 |
74 ± 14 | 270 ± 70 | 3.6 |
| 18 | 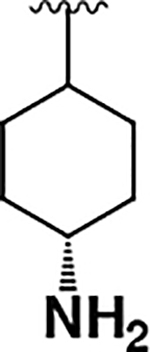 |
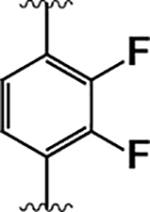 |
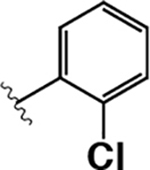 |
17 ± 2 | 83 ± 8 | 5.8 |
RNase IC50 data are shown as mean ± SEM, n=3. RNase selectivity was determined by dividing the IRE1α RNase IC50 by the IRE1β RNase IC50 value for each inhibitor (individual IC50 curves provided in Supp. Figure 2).
For all inhibitors tested, we found that varying away from a 2-chlorophenylsulfonamide R3 group was detrimental to IRE1β* potency. Thus, for the remainder of inhibitors generated in this SAR study, a 2-chlorophenylsulfonamide group was maintained at the R3 position. Because the introduction of a 4-amino-3-fluorophenol R2 group modestly improved selectivity for IRE1β* (compare inhibitor 3 to inhibitor 8), we next determined whether increasing the size of the halogen at this position could enhance the ability to discriminate between the two paralogs. Unfortunately, inhibitors containing a 4-amino-3-chlorophenol (11 and 12) were markedly less potent against IRE1β*. We next determined the effect of changing the position of the halogen from the 3-position to the 2-position on the 4-aminophenol R2 group by generating inhibitors 13 and 14, which contain 4-amino-2-fluorophenols. Both inhibitors demonstrated promising selectivity for IRE1β*, with inhibitor 13, which contains a trans-1,4-cyclohexanediamine R1 group, exhibiting single-digit nanomolar potency against IRE1β* and 60-fold selectivity over IRE1α*. Finally, we looked at how adding an additional halogen to the 4-amino-2-fluorophenol R2 group affected the potency and paralog selectivity of inhibitors. Inhibitor 15, which contains a 4-amino-5-chloro-2-fluorophenol R2 group and a trans-1,4-cyclohexanediamine R1 group demonstrated potent inhibition of IRE1β* and >100-fold selectivity over IRE1α*. Replacing the trans-1,4-cyclohexanediamine R1 group of 15 with an (S)-3-aminopiperidine (16) led to reduced potency against IRE1β* and diminished selectivity. Addition of a fluorine to the 3- or 5-positions (inhibitor 17 and 18, respectively) of the 4-amino-2-fluorophenol R2 group was found to be generally detrimental to IRE1β* inhibition. From this SAR, it is clear that employing 4-amino-2-halophenol R2 groups and a trans-1,4-cyclohexanediamine R1 substituent can impart high IRE1β RNase selectivity.
KIRAs Inhibit the Kinase Activity of IRE1α/β
KIRA 1 was previously confirmed to be an ATP-competitive inhibitor of IRE1α and we assumed that all of the analogs generated in this study inhibit IRE1 RNase activity through the ATP-binding site.31 To verify that our inhibitors also inhibit the kinase activities of IRE1α* and IRE1β*, we measured the propensity of a representative set of compounds to block IRE1-mediated phosphorylation of an exogenous substrate–myelin basic protein (MBP) (Figure 4A). As expected, we found that all compounds tested also inhibited the kinase activity of IRE1α* and IRE1β* (Figure 4B). While we were not able to accurately determine kinase IC50 values for a number of our more potent inhibitors (3, 11, 13, 14, 15, and 18) against IRE1β* due to constraints of the assay, we found that kinase IC50 values were, in general, several-fold lower than RNase IC50 values for this paralog. This is in contrast to IRE1α*, where inhibitors generally exhibited similar IC50 values against kinase and RNase activity. The reason for this discrepancy between the two paralogs is unknown but may reflect differences in the energetic penalties required to disrupt the dimeric states of IRE1α* and IRE1β*, and, in turn, RNase inhibition. Thus, inhibitors typically demonstrate slightly greater selectivity for IRE1β* in the kinase assay than in the RNase assay. Collectively, these results show that trends in inhibitor selectivity are consistent in both kinase and RNase assays.
Figure 4. Kinase Inhibition by Exemplary KIRAs.

A. Schematic of the in vitro IRE1 kinase assay. Phosphotransferase activity of IRE1 was measured by monitoring the IRE1α*/IRE1β*-mediated phosphorylation of the exogenous substrate myelin basic protein (MBP) by ATP[γP32/33]. B. Ki values for IRE1α* and IRE1β* kinase activity. All Ki values were calculated using the Cheng-Prusoff equation. For IRE1α*, the Ki values shown are mean ± SEM, n=3. For IRE1β, the Ki values shown are the mean ± SEM, n=3 (denoted by a *) or the average values from two measurements (individual inhibitory values provided in Supp. Table 1). Inhibitors with Ki values lower than the concentration of IRE1α* or IRE1β* used in the assay are denoted with a ‡. Individual IC50 curves provided in Supp. Figure 2
Kinome Selectivity of Paralog-Selective KIRAs
From our optimization efforts we were able to generate inhibitors with three different profiles: IRE1α-selective inhibitors, dual-IRE1 inhibitors that are equipotent against IRE1α and IRE1β, and IRE1β-selective inhibitors. Inhibitors with these profiles would be excellent tools for examining the contributions of IRE1α and/or IRE1β activity in cells if they possess sufficient general kinase selectivity. Therefore, we used a lysate profiling method to determine if representative inhibitors from these three classes are selective on the kinome level. Specifically, we measured the abilities of representative inhibitors to compete for binding of lysate kinases to an affinity matrix containing seven different nonselective ATP-competitive inhibitors (kinobeads).32–34 Kinobeads enrich kinases through their ATP-binding sites and allow inhibitors to be quantitatively profiled against a large percentage of the human kinome. Binding of an inhibitor prevents enrichment with the kinobead matrix and kinome selectivity can be determined by comparing the relative enrichment of a kinase target between DMSO and inhibitor-treated lysates using quantitative mass spectrometry (Supp. Figure 3). In total, we profiled five inhibitors (1 (IRE1α-selective), 4 and 9 (dual-IRE1), and 13 and 15 (IRE1β-selective)) with an HCT116/HEK293 cell lysate mixture spiked with exogenous recombinant IRE1β* (or IRE1α* for profiling of 1). Addition of exogenous IRE1 was required because we were not able to reproducibly quantify the endogenous protein.
The kinome selectivity of 1 was previously assessed with in vitro activity assays against a large panel of recombinant kinases, where it was shown to be highly selective across the kinome.31,35 These results were verified in our kinobead assay, where IRE1α was the only kinase identified as a target of 1 (Supp. Figure 4). Out of ~150 kinases quantified in the assay, IRE1β was the primary target of 3, 9, 13, and 15, with only one off-target identified for each respective inhibitor (Figure 5). Inhibitor 3, which contains a naphthol R2 group, prevented the enrichment of PKD2 in addition to IRE1β. Compounds 9, 13, and 15, which all contain 4-amino-halophenol R2 groups, share the same off-target, CDK2. To determine whether the level of competition observed in our kinobead assay would lead to potent inhibition of CDK2 activity, we tested the ability of 9, 13, and 15 to inhibit the CDK2/cyclin A complex in vitro. Despite the ability of 9, 13, and 15 to moderately prevent CDK2 binding to the kinobead matrix in our lysate profiling experiments (Supp. Figure 5A), all three KIRAs showed weak inhibition of CDK2/cyclin A kinase activity (IC50 values >5 μM; Supp. Figure 5B). For 9, 13, and 15, the discrepancy between the activity assay with CDK2/cyclin A and our kinobead profiling experiment could possibly be due to the high concentrations (10 μM) of these inhibitors tested in the profiling experiment, which allows sufficient occupation of CDK2’s ATP-binding site despite their weak affinities for this kinase. Another possibility is that these inhibitors can only interact with inactive CDK2, but not active CDK2-cyclin complexes. Previously, it has been shown that cyclin binding causes a conformational change in CDK2’s ATP-binding site that makes it inaccessible to certain types of ATP-competitive inhibitors.36
Figure 5. Kinome Profiling of KIRAs 3, 9, 13, and 15.

The kinome selectivity of 3, 9, 13, and 15 were determined with a previously described kinobead lysate profiling method. Kinases that were quantified in the experiment are shown as circular nodes, where node size and color has been scaled to the log2 difference (difference in LFQ intensity between DMSO treated and inhibitor treated lysates) between DMSO and treatment with 10 μM of KIRA 3 (A), 9 (B), 13 (C), or 15 (D). Gray nodes represent kinases that were quantified but no competition with an inhibitor was observed. Values shown are the mean of four replicates.
Structural Model of Inhibitor Paralog Selectivity
While extensive structural characterization has been performed with IRE1α, equivalent information is not available for IRE1β.29,31,37–42 To build a structural model of IRE1β inhibition and selectivity, we used a sequence alignment of the kinase and RNase domains of IRE1α and IRE1β. The kinase domains of IRE1α and IRE1β share 81% sequence identity, while the RNase domains are 61% identical (Figure 6A).24 Due to the high degree of sequence identity between the kinase domains of the two paralogs, we predicted that KIRAs would have similar binding modes in their ATP-binding sites. From this sequence alignment, we mapped sequence differences between IRE1α and IRE1β onto a co-crystal structure of IRE1α bound to the pyrimidine-pyridine KIRA 2 (PDB: 4U6R).31 KIRA 2 is nearly identical to potent dual-IRE1 inhibitor 3, differing only by the presence of a methyl group at the 2-position of the R2 naphthyl ring. Using the mutagenesis tool in PyMol, residue side chain differences between IRE1α and IRE1β were visualized for the co-crystal structure of IRE1α bound to 2. We next looked at residues within 5 Å of inhibitor 2. Of the 23 residues identified, only four residues are not identical in IRE1α and IRE1β: Ala609 (Val558 in IRE1β), Ile642 (Leu591 in IRE1β), Ala646 (Arg595 in IRE1β), Thr648 (Ser597 in IRE1β) (Figure 6B,C). Although the specific interactions that each paralog makes with inhibitors cannot be known in the absence of high-resolution structural information, these sequence differences likely play a major role in paralog selective inhibition. The IRE1α residues Ala646 (Arg595 in IRE1β) and Thr648 (Ser597 in IRE1β) are directed towards the alicyclic portion of 2’s trans-1,4-cyclohexandiamine R1 group, which forms a salt bridge with Glu651 in IRE1α and most likely Glu600 in IRE1β. While we were unsuccessful in identifying R1 groups that were optimal for IRE1β’s Arg595 and Ser597 residues, our SAR data show that IRE1α is more permissive of variability at this position. The stabilization of an inactive helix-αC “out” conformation by KIRAs leads to IRE1 monomerization and RNase inhibition. The 2-chlorophenyl aryl sulfonamide R3 group of 2 occupies the pocket created by movement of helix-αC. In this inactive conformation, Ala609 in IRE1α (Val559 in IRE1β) from helix-αC is projected towards inhibitor R3 groups. Our SAR data demonstrate that the smaller Ala residue of IRE1α allows it to favorably accommodate a greater diversity of R3 substituents than IRE1β. Finally, the sidechain of IRE1α’s gatekeeper residue, Ile642, points directly towards the methyl naphthyl R2 group of 2 (Figure 6B). Interestingly, selectivity for IRE1β, which possesses a Leu gatekeeper, over IRE1α was achieved most effectively by introducing 4-amino-2-fluorophenol R2 groups. Thus, we hypothesize that 4-amino-2-fluorophenol R2 groups are able to form more favorable interactions with the Leu residue of IRE1β than with the Ile residue of IRE1α and that paralogs selectivity can be achieved by optimizing gatekeeper/R2 group interactions.
Figure 6. Sequence Alignment and Structural Comparison of IRE1α and IRE1β ATP-Binding Sites.

A. Sequence alignment of IRE1α and IRE1β (top) Sequence comparison between IRE1α and IRE1β mapped onto the crystal structure of IRE1α (PDB: 4U6R). Residues that are conserved are shown in gray, residues that have a conservative replacement are shown in purple, and residues with non-conservative replacements are shown in red. (bottom) Sequence alignment of IRE1α and IRE1β shows 80% sequence identity of the kinase domain and 61% sequence identity of the RNase domain. B. Interactions between 2, a close structural analog of KIRA 1, and the ATP-binding site of IRE1α. Compound 2 is shown as yellow sticks, key interacting residues are shown as gray sticks, and interactions are denoted with green dashed lines. C. Hypothesized binding mode of KIRA 2 by mapping of non-identical residues of IRE1β onto the structure of 4U6R. Conservative replacement non-identical residues are shown as purple sticks, non-conservative non-identical residues are shown as red sticks. Residue numbering is for IRE1β.
Conclusion
Pharmacological modulation of IRE1α’s RNase domain using ATP-competitive inhibitors has proven to be a useful method for examining the allosteric relationship between the kinase and RNase domains of IRE1α, and for better understanding the functional outputs of IRE1α’s RNase domain. While IRE1α’s close paralog IRE1β shares the same domain architecture, much less is known about the enzymatic activities of IRE1β and its function within the cell. Understanding IRE1β’s role in the cell has been particularly difficult to determine because while IRE1β expression is limited to epithelial cells of the gut and bronchia, all cells that express IRE1β also express IRE1α. Pharmacological tools that are able to discriminate between IRE1α and IRE1β will be useful reagents for defining paralog-specific function in cells.
There have been conflicting reports on how efficiently the RNase domain of IRE1β is able to cleave XBP1 mRNA.16,22 Here, we show that a cytosolic construct of IRE1β (IRE1β*) has RNase activity comparable to IRE1α* for an XBP1 mini-substrate. Therefore, IRE1β, like IRE1α, appears to form dimers that bind and cleave XBP1 mRNA in vitro. A reason for the observed discrepancies in IRE1β’s ability to cleave XBP1 may stem from differences in the phosphorylation state of the activation loop, which promotes formation of the RNase-active dimer, in the recombinant constructs used. The recombinant IRE1β* construct used in our study contains a glutathione S-transferase (GST) tag at its N-terminus. GST has been demonstrated to form dimers43, which likely enhances IRE1β* activation loop autophosphorylation when being expressed in insect cells. The N-terminal GST tag may also directly promote formation of RNase-active back-to-back dimers in the absence of activation loop phosphorylation. Regardless of the source of these discrepancies, our data clearly show that IRE1β can efficiently cleave an XBP1 mini-substrate when it is dimerized. The implications of our results for the ability of full-length IRE1β to cleave XBP1 mRNA in cells is unclear. In cells, IRE1β’s ability to undergo activation loop autophosphorylation appears to be hampered compared to its paralog IRE1α.44 Therefore, XBP1 mRNA may be a poor substrate for IRE1β in cells because the kinase domain does not undergo activation loop phosphorylation under ER stress. Differences in the ability of IRE1α and IRE1β to undergo autophosphorylation may also help explain previously reported disparities in the mRNA substrate preference between these two paralogs in cells. Under ER stress, XBP1 mRNA may be cleaved by activation loop phosphorylated IRE1α while IRE1β functions to aid in later stages of the UPR by cleaving ER-localized mRNAs as a part of RIDD.23–25
To examine the allosteric relationship between IRE1β’s kinase and RNase domains, we report the first ATP-competitive pharmacological modulators of IRE1β. We show that ATP-competitive inhibitors that activate IRE1α’s RNase domain also activate IRE1β’s RNase domain. Additionally, we find that ATP-competitive KIRAs are capable of inhibiting IRE1β’s RNase activity like IRE1α’s. The ability of ATP-competitive inhibitors to divergently modulate the RNase activity of IRE1β suggests that it shares all or most of the allosteric features of IRE1α.
By generating analogs of a KIRA that is moderately selective for IRE1α, we were able to identify inhibitors with two new paralog selectivity profiles: KIRAs that are equipotent against IRE1α and IRE1β and KIRAs that are highly selective for IRE1β. Lysate profiling experiments confirmed that all three classes of KIRAs are selective across the human kinome. Our SAR studies suggest that the key to discriminating between the very similar ATP-binding sites of IRE1α and IRE1β is optimizing the KIRA substituent that is in close proximity to their gatekeeper residues, which is Ile in IRE1α and Leu in IRE1β. Altogether, KIRAs with differing selectivity profiles (highly IRE1α-selective, dual-IRE1, and highly IRE1β-selective) will serve as useful tools for work characterizing the specific outputs of IRE1 in cells and, potentially, in animal models of gastrointestinal and pulmonary ER stress-related diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01DK116064 and R01DK100623 (F.R.P. and D.J.M.)).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supplemental figures S1–5, methods, protein amino acid sequence and DNA sequence, synthetic procedures and inhibitor characterization.
References
- 1.Scheuner D, and Kaufman RJ (2008) The Unfolded Protein Response: A Pathway That Links Insulin Demand with β-Cell Failure and Diabetes. Endocr. Rev 29, 317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Anken E, Pena F, Hafkemeijer N, Christis C, Romijn EP, Grauschopf U, Oorschot VM, Pertel T, Engels S, Ora A, Lastun V, Glockshuber R, Klumperman J, Heck AJ, Luban J, and Braakman I (2009) Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc. Natl. Acad. Sci. U. S. A 106, 17019–17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter P, and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 4.Tirasophon W, Welihinda AA, and Kaufman RJ (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harding HP, Zhang Y, and Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. [DOI] [PubMed] [Google Scholar]
- 6.Haze K, Yoshida H, Yanagi H, Yura T, and Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a trans-membrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C, Chevet E, and Oakes SA (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, and Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258. [DOI] [PubMed] [Google Scholar]
- 9.Shore GC, Papa FR, and Oakes SA (2011) Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol 23, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, and Papa FR (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, and Ron D (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17, 5708–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, and Pearl LH (2011) Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 30, 894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KP, Dey M, Neculai D, Cao C, Dever TE, and Sicheri F (2008) Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Liang FX, and Wang X (2014) A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 55, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, and Hammarlund M (2014) The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 15, 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, and Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- 17.Hollien J, Lin JH, Li H, Stevens N, Walter P, and Weissman JS (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol 186, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollien J, and Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. [DOI] [PubMed] [Google Scholar]
- 19.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, and Papa FR (2012) IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, and Oakes SA (2012) IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, and Ron D (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 107, 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imagawa Y, Hosoda A, Sasaka S-I, Tsuru A and Kohno K (2008) RNase domains determine the functional difference between IRE1α and IRE1β. FEBS Lett. 582, 656–660. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura D, Tsuru A, Ikegami K, Imagawa Y, Fujimoto N, and Kohno K (2010) Mammalian ER stress sensor IRE1β specifically down-regulates the synthesis of secretory pathway proteins. FEBS Lett. 585, 133–138. [DOI] [PubMed] [Google Scholar]
- 24.Iwawaki T, Hosoda A, Okuda T, Kamigori Y, Nomura-Furuwatari C, Kimata Y, Tsuru A, and Kohno K (2001) Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Bio 3, 158–164. [DOI] [PubMed] [Google Scholar]
- 25.Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, Oneal WK, and Ribeiro CMP (2012) The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 6, 639–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hauber H-P, Foley SC and Hamid Q (2006) Mucin Overproduction in Chronic Inflammatory Lung Disease. Can. Respir. J 13, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iqbal J, Dai K, Seimon T, Jungreis R, Oyadomari M, Kuriakose G, Ron D, Tabas I, and Hussain MM (2008) IRE1β Inhibits Chylomicron Production by Selectively Degrading MTP mRNA. Cell Metab. 7, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Perera BG, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, Schurer SC, Oakes SA, Papa FR, and Maly DJ (2012) Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol 8, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feldman HC, Tong M, Wang L, Meza-Acevedo R, Gobillot TA, Lebedev I, Gliedt MJ, Hari SB, Mitra AK, Backes BJ, Papa FR, Seeliger MA, and Maly DJ (2016) Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands. ACS Chem. Biol 11, 2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papa FR, Zhang C, Shokat K, and Walter P (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302, 1533–1537. [DOI] [PubMed] [Google Scholar]
- 31.Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, Dellamaggiore K, Kendall R, Beckmann H, Jaeckel P, Materna-Reichelt S, Allen JR, and Lipford JR (2015) Unfolded Protein Response in Cancer: IRE1alpha Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med. Chem. Lett 6, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golkowski M, Vidadala RSR, Lombard CK, Suh HW, Maly DJ, and Ong S-E (2017) Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. J. Proteome Res 16, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, and Drewes G (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- 34.Golkowski M, Perera GK, Vidadala VN, Ojo KK, Van Voorhis WC, Maly DJ, and Ong SE (2018) Kinome chemoproteomics characterization of pyrrolo[3,4-c]pyrazoles as potent and selective inhibitors of glycogen synthase kinase 3. Mol. Omics 603, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, Mehdizadeh M, Ghosh R, Wang L, Colon-Negron K, Meza-Acevedo R, Backes BJ, Maly DJ, Bluestone JA, and Papa FR (2017) Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab. 25, 1207. [DOI] [PubMed] [Google Scholar]
- 36.Alexander LT, Möbitz H, Drueckes P, Savitsky P, Fedorov O, Elkins JM, Deane CM, Cowan-Jacob SW, and Knapp S (2015) Type II Inhibitors Targeting CDK2. ACS Chem. Biol 10, 2116–2125. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J, Sicheri F, and Ron D (2010) Flavonol Activation Defines an Unanticipated Ligand-Binding Site in the Kinase-RNase Domain of IRE1. Mol. Cell 38, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.A., Lucas JL, Vuga D, Lehmann L, Durocher D, Zeng Q, Patterson JB, and Sicheri F (2014) Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun 5, 4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, and Walter P (2009) The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi A, Newbatt Y, McAndrew PC, Stubbs M, Burke R, Richards MW, Bhatia C, Caldwell JJ, McHardy T, Collins I, and Bayliss R (2015) Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget 6, 13019–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colombano G, Caldwell JJ, Matthews TP, Bhatia C, Joshi A, McHardy T, Mok NY, Newbatt Y, Pickard L, Strover J, Hedayat S, Walton MI, Myers SM, Jones AM, Saville H, McAndrew C, Burke R, Eccles SA, Davies FE, Bayliss R, and Collins I (2019) Binding to an Unusual Inactive Kinase Conformation by Highly Selective Inhibitors of Inositol-Requiring Enzyme 1α Kinase-Endoribonuclease. J. Med. Chem 62, 2447–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Concha NO, Smallwood A, Bonnette W, Totoritis R, Zhang G, Federowicz K, Yang J, Qi H, Chen S, Campobasso N, Choudhry AE, Shuster LE, Evans KA, Ralph J, Sweitzer S, Heerding DA, Buser CA, Su D-S, and Deyoung MP (2015) Long-Range Inhibitor-Induced Conformational Regulation of Human IRE1 Endoribonuclease Activity. Mol. Pharmacol 88, 1011–1023. [DOI] [PubMed] [Google Scholar]
- 43.Fabrini R, Luca AD, Stella L, Mei G, Orioni B, Ciccone S, Federici G, Bello ML, and Ricci G (2009) Monomer−Dimer Equilibrium in Glutathione Transferases: A Critical Re-Examination. Biochemistry 48, 10473–10482. [DOI] [PubMed] [Google Scholar]
- 44.Grey MJ, Cloots E, Simpson MS, LeDuc N, Serebrenik YV, De Luca H, De Sutter D, Luong P, Thiagarajah JR, Paton AW, Paton JC, Seeliger MA, Eyckerman S, Janssens S, and Lencer WI IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. (2019) 586305 BioRxiv https://www.biorxiv.org/content/10.1101/586305v1, March 23, 2019. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.