

Abstract
Prexasertib (LY2606368) is a potent and selective small molecule inhibitor of cell-cycle checkpoint CHK1 and CHK2 protein kinases and is currently under clinical evaluation for treatment of pediatric malignancies. As a candidate therapy for pediatric Group 3 medulloblastoma (G3MB), prexasertib CNS penetration was evaluated in mice using cerebral microdialysis and pharmacokinetic modeling. A plasma pharmacokinetic study with a population-based design was performed in CD1 nude mice bearing G3MB orthotopically implanted in the brain and receiving a single dose of prexasertib (10 mg/kg, subcutaneously) to characterize prexasertib disposition and to establish a limited plasma sampling model for the microdialysis studies. The microdialysis studies were performed in both non-tumor bearing mice and in mice bearing G3MB receiving 10 mg/kg prexasertib subcutaneously, for up to 24 hours post-dose. Plasma and extracellular fluid (ECF) concentrations were quantified using validated LC MS/MS methods, and analyzed using a population pharmacokinetic model. Model-derived prexasertib tumor/ECF to plasma partition coefficient Kp,uu (ratio of tumor/brain ECF to unbound plasma AUC0–24h) was significantly greater in G3MB tumor-bearing mice (0.17 ± 0.08) compared to non-tumor bearing mice (0.09 ± 0.04, p=0.04). A pharmacodynamic study was then performed in mice bearing G3MB (20 mg/kg, IV) to evaluate prexasertib-induced target engagement after a single dose. Phosphorylated CHK1 serine 345 (pCHK1 S345), phosphorylated Histone 2A variant (γ-H2AX), and cleaved caspase-3 were quantified in mouse G3MB tumor tissues by immunohistochemistry at different time points up to 24 hours post-dose. The induction of pCHK1 S345 and γ-H2AX peaked at 2 hours after the dose and was elevated above baseline for at least 6 hours, reflecting relevant CHK1 inhibition and DNA damage. Cleaved caspase-3 levels increased at 24 hours suggesting initiation of cell apoptosis. Adequate unbound prexasertib exposure reached the brain tumor site relative to target engagement in G3MB tumor bearing mice at a clinically relevant dosage. These results support further preclinical and clinical development of prexasertib to treat children with medulloblastoma.
Keywords: prexasertib, cerebral microdialysis, pharmacokinetic modeling, non-specific binding, pediatric brain tumors, phosphorylated CHK1
Graphical Abstract
1. Introduction:
Medulloblastomas are the most common malignant brain cancer in children (Cronin et al., 2018). These brain tumors are highly heterogeneous, and divide molecularly into different subgroups, with distinctive transcriptional and epigenetic signatures, and prognostic implications (Gajjar and Robinson, 2014; Northcott et al., 2011; Taylor et al., 2012). Contemporary therapy for medulloblastoma includes surgerical resection followed by craniospinal irradiation and high-dose chemotherapy. While this therapy led to a cure rate exceeding 80% (Gajjar et al., 2015), 30% of patients with high risk tumors and those with recurrent tumors have a poor prognosis The successful therapeutic improvement mainly relies on the refined use of conventional chemotherapy rather than the emergence of new treatments. However, we are now reaching a plateau in terms of survival rate with still severe acute and long-term complications (Armstrong et al., 2009; Leary and Olson, 2012). Thus, new therapeutic strategies are needed to improve the progression-free survival rate and reduce the acute and long-term sequalae associated with current therapy.
Checkpoint kinase protein (CHK) inhibitors have attracted considerable interest as therapeutic agents. After DNA damage or replication stress, cells stop proliferating at cell cycle checkpoints upon activation of critical effectors CHK1 and 2, resulting in either DNA repair or induction of apoptosis (McNeely et al., 2014). CHK1 activates and maintains the S and G2/M cell-cycle checkpoints, and is crucial for homologous recombination repair of double-stranded DNA breaks (Dai and Grant, 2010). Inhibiting CHK1 leads to the loss of DNA damage checkpoints, increases replication stress and cell death (Lin et al., 2017). As such, CHK1 inhibitors represent a promising target to drive tumor cell death in different types of cancers, including medulloblastoma.
Prexasertib (LY2606368) is a novel, second generation, selective small molecule inhibitor of cell-cycle checkpoints CHK1, and of CHK2 to a lesser extent, that demonstrated a potent activity in non-clinical studies (King et al., 2015; Lowery et al., 2017; Sen et al., 2017). In vitro, in a panel of established cancer cell lines, prexasertib treatment showed target engagement through the activation of different markers of CHK1 inhibition and DNA damage (King et al., 2015; Lowery et al., 2017). In vivo, prexasertib inhibits tumor growth at low concentrations in cancer xenografts: small and non-small cell lung cancer, and neuroblastoma, as single agent and in combination DNA damaging agents, with cisplatin and olaparib (King et al., 2015; Lowery et al., 2017; Sen et al., 2017). The potential anticancer activity of prexasertib is currently under evaluation in adults with advanced solid tumors (Hong et al., 2016; Lee et al., 2018). The results of the first clinical studies indicate that prexasertib is well tolerated, albeit with manageable hematological toxicity (Hong et al., 2016; Lee et al., 2018).
As a candidate therapy for pediatric medulloblastoma, it is essential to understand the central nervous system (CNS) penetration of prexasertib and its ability to attain adequate exposure at the target site, which is limited by selective permeability of the blood-brain-barrier. Cerebral microdialysis allows the sampling of brain extracellular fluid (ECF) samples and the measurement of unbound drug concentrations in ECF over a set amount of time. This technique has been combined with pharmacokinetic modeling and simulation in preclinical studies with tumor subgroup-specific models of CNS tumors to further characterize drug brain penetration (Jacus et al., 2014). Herein we applied this preclinical approach for prexasertib in both mice orthotopically implanted with Group 3 medulloblastoma (G3MB), a major subgroup of medulloblastoma, and in non-tumor bearing (NTB) mice to evaluate its CNS penetration.
The objectives of the current study were to characterize the plasma disposition of prexasertib in mice bearing G3MB, to derive a limited plasma sampling model for the microdialysis study, to characterize the tumor and brain ECF disposition of prexasertib in G3MB and NTB mice, and to perform preliminary pharmacodynamic studies in tumor tissue from G3MB-bearing mice.
2. Materials and Methods
2.1. Drugs and chemicals
5-(5-[2-(3-Aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl}amino)pyrazine-2-carbonitri le methanesulfonate hydrate [prexasertib methanesulfonate] and 5-({5-[2-(3-aminopropoxy-1,1,3,3-d4)-6-methoxyphenyl]-1H-pyrazol-3-yl}amino)pyrazine-2-carbonitrile hydrochloride [prexasertib hydrochloride-d4, internal standard (ISTD)] were supplied by Eli Lilly (Indianapolis, IN). Ammonium formate (for mass spectrometry, ≥99.0%) and albumin from bovine serum (lyophilized powder, ≥98% purity) were purchased from Millipore Sigma (St. Louis, MO). Formic acid (LC-MS/MS grade, 99.5% purity), acetonitrile (HPLC grade), and tert-butyl methyl ether (99% purity) were purchased from Fisher Scientific (Waltham, MA). Ringer’s Solution was purchased from Frey Scientific (Nashua, NH). CD-1 mouse plasma with sodium heparin as anticoagulant was purchased from BioreclamationIVT (Baltimore, MD). All water was prepared using Millipore Q-advantage water purification system (Temecula, CA). Captisol® used for dosing solution was purchased from Ligand (San Diego, CA).
2.2. Animals and cell lines
Female CD-1 nude mice were purchased from Charles River Laboratories. Experimental cohorts of mice were cortically implanted with 1×105 mouse G3MB purified tumor cells retrovirally transduced with luciferase (Morfouace et al., 2014). Tumor growth was measured weekly by bioluminescence imaging using a Xenogen system (Caliper Life Science, Waltham, MA). Animals were kept under controlled environment where temperature, humidity, and 12 hours day and night cycles were maintained artificially. All animal studies performed were approved by the St. Jude Children’s Research Hospital Institutional Animal Care and Usage Committee (IACUC) and met the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC).
2.3. Plasma protein binding study
The extent of prexasertib plasma protein binding was determined using equilibrium dialysis. Prexasertib was added to CD-1 mouse plasma to make final concentrations of 2, 10, 100, 500, 1000, 1250, 1500, and 2000 ng/ml. Blank Ringer’s solution (200 μl) was transferred to the buffer side of a 96-well Equilibrium Dialyzer™ (Cat #74–2330, Harvard Apparatus). Plasma samples (200 μl each) were transferred to the sample side of a 96-well Equilibrium Dialyzer™ in triplicate. The dialysis plate was inserted in to a plate rotator and kept in the incubator set to 37°C for 24 hours. After 24 hours, 100 μl samples from each side of 96-well Equilibrium Dialyzer™ were collected in separate tubes and stored at −80°C until further analysis. Fraction unbound of prexasertib in plasma (FU) was calculated as ratio of unbound prexasertib concentration in buffer compartment to total prexasertib concentration in sample compartment of 96-well Equilibrium Dialyzer™.
2.4. Plasma pharmacokinetic studies
Two plasma pharmacokinetic studies were performed: one to characterize the mouse equivalent prexasertib clinical dosage and the limited sampling model for the cerebral microdialysis study, and a subsequent one to support the pharmacodynamic study. In both studies, nine female CD-1 nude mice implanted with mouse G3MB cells were enrolled once their bioluminescence signals reached ~1×107 photons/sec. Population-based study designs were used with 2–3 samples collected per mouse, up to 24 hours post-dose. In the first pharmacokinetic study, mice were administered 10 mg/kg prexasertib subcutaneously (SQ). In the second study, mice received 20 mg/kg prexasertib mesylate intravenously (IV). Each dosing formulation was prepared by diluting an appropriate amount of prexasertib and 20% w/v Captisol®. The first blood samples (75 μl) were collected via retro-orbital eye bleed and the final blood collection (1 ml) for each group was collected via cardiac stick. The blood was immediately spun to plasma and stored at −80°C until analysis using a published LC MS/MS method (Zhong et al., 2018).
2.5. In vitro probe recovery studies
The cerebral microdialysis technique allows for dynamic sampling of ECF by implantation of a microdialysis probe (Cat # MD2211, Bioanalytical Systems, Inc.) in the target tissue. For this study, microdialysis recovery for each probe was determined using in vitro retrodialysis recovery. Each probe was placed in a stirred blank Ringer’s solution maintained at 37°C. The probe was perfused with 100 ng/ml prexasertib in 0.5% bovine serum albumin (BSA) in Ringer’s solution at a flow rate of 0.5 μl/min. After equilibration for 1-hour, three consecutive fractions of 1-hour interval each were collected. Dialysate samples were stored at −80°C until further analysis. Probe recovery was calculated using the ratio of prexasertib concentration in the dialysate sample to that in the stock solution. Unbound concentration in tumor or brain ECF was calculated by dividing concentration observed in dialysate samples collected during microdialysis study with the recovery ratio.
2.6. Studies to assess the in vitro non-specific binding of prexasertib
The results of an initial microdialysis study strongly suggested prexasertib was binding to components of the microdialysis system (data not shown). Thus, several experiments were performed to evaluate and reduce prexasertib non-specific binding to the microdialysis system: syringes, tubing, probe or collection vials. The objective of the first study was to determine the extent of the binding of prexasertib and to determine if the use of plastic or glass tubes were appropriate for sample collection. Thus, recovery studies were done using 4 separate probes. For two of them, all samples were collected in plastic vials, and for the other two probes, samples were collected in glass vials.
In vitro retrodialysis recovery studies were repeated with and without the addition of 0.5%BSA to the perfusate (Ringer’s solution) to assess the effect of BSA on the non-specific binding of prexasertib.
Lastly, a rinsing solution (i.e., 50% ACN/50% ddH2O/0.1% formic acid) was evaluated to eliminate residual prexasertib from the microdialysis syringes. Four syringes were filled with 100 ng/ml prexasertib in 0.5% BSA in Ringer’s solution. The drug solution remained in the syringes for 0.5 hour before being washed. The first two syringes were washed twice using the organic solution followed with ddH2O twice. The second two syringes were washed four times using ddH2O. All syringes were filled with 0.5% BSA in Ringer’s solution and samples were collected from each syringe after 5 min in to glass collection tubes.
2.7. Development of pharmacokinetic limited sampling modeling for prexasertib
Due to limited blood volume that could be withdrawn, and the minimum plasma volume required for bioanalysis, we were limited to only three plasma samples per mouse during the microdialysis study. To determine the three most informative time points for plasma sample collection, the pharmacokinetic parameters obtained from the first plasma disposition study were used to develop a limited plasma sampling model (LSM) using the D-optimality method implemented in ADAPT 5 (BMSR, Los Angeles, CA, USA).
2.8. Prexasertib cerebral microdialysis studies
Cerebral microdialysis experiments were conducted in both mice G3MB-bearing (N=8) and NTB mice (N=7) to sample prexasertib in the tumor and normal brain ECF after a single dose of prexasertib (10 mg/kg, SQ). G3MB tumor cells and/or microdialysis guide cannula (MD-2255, BASi) were implanted into the cerebral cortex of mice using a stereotactic instrument as described previously (Atkinson et al., 2011; Carcaboso et al., 2010). Once the tumor reached a bioluminescence of ~1 ×107 photons/sec, tumor-bearing mice were enrolled in the study. On the day of the experiment, a 1 mm microdialysis probe with 38 KDa MWCO membrane (MD-2211, BASi) was inserted into the guide cannula, and infused with Ringer’s solution at a flow-rate of 0.5 μl/min for 1 hour to equilibrate with the in vivo environment. After equilibration, all the mice were dosed with 10 mg/kg prexasertib SQ. Dialysate samples were collected over 1-hour intervals for up to 24 hours. Blood samples from each mouse were collected at the LSM derived time points. Immediately after collection, blood samples were centrifuged, and plasma was extracted. Plasma and dialysate samples were stored at −80°C until further analysis by LC MS/MS. After the experiment, each animal was euthanized and the whole head was fixed in 10% neutral buffered formalin for at least 72 hours and embedded in paraffin. Hematoxylin and eosin (H&E)-stained sections (5 μm) were examined microscopically to inspect the brain tissue surrounding the cannula and probe for probe placement, inflammation, and hemorrhage.
2.9. Pharmacokinetic modeling of prexasertib plasma and ECF data
Population compartmental modeling of prexasertib in plasma and ECF was performed in NONMEM 7.3 (ICON Development Solutions, Ellicott City, Maryland). GraphPad Prism (version 7 for Windows, www.graphpad.com) were used for graphical presentations. The first conditional estimation method with interaction was used to derive the population pharmacokinetic parameters and the inter-individual variability (IIV). IIV terms were assumed to follow a log-normal distribution. Additive and/or proportional residual variability models were applied for plasma and ECF prexasertib concentrations. The Laplacian Estimation method and the Beal’s M3 censoring method were used to handle the ECF concentrations below the limit of quantification (Beal, 2001). Model selection was based upon the goodness-of fit plots (Nguyen et al., 2017), precision of the parameter estimates (relative standard errors RSE%) and change in the objective function value. The final pharmacokinetic models were evaluated using visual predictive checks based on 500 replicates of the analysis dataset (Holford, 2005).
The prexasertib plasma concentration-time data obtained from the first pharmacokinetic study were modeled, and the LSM was derived. The prexasertib plasma data obtained during the microdialysis studies were analyzed using a Bayesian forecasting approach (Sheiner and Beal, 1982). The final parameter estimates from the previous model were used as “prior” information and computed with the limited plasma data (MAXEVAL=0 approach). Prexasertib plasma disposition was assumed similar in tumor-bearing and NTB mice. The individual model parameters (Maximum A Posteriori Bayesian estimates) were used to derive full plasma pharmacokinetic profiles for each mouse which were linked to the tumor and brain ECF model to avoid over-parameterization. Only free drug was assumed to penetrate the CNS, so that prexasertib amount distributed from the plasma into the tumor or brain ECF compartment was multiplied by the plasma FU (Di et al., 2013). During the microdialysis studies, the dialysate was collected in fractions and not as a single time-point. This was accommodated in the model by incorporating a separate compartment for each of the new collected fractions and using the model-change-time function MTIME (Beal et al., 1989–2013). The microdialysate flow rate, tubing lag time, and the total volume of each fraction were fixed to experimental values. All the tumor and brain ECF concentration-time data obtained in G3MB and NTB mice were simultaneously analyzed. Potential differences between G3MB and NTB mice were characterized using the categorical covariate: “G3MB group” versus “NTB group”, modeled using a fractional change (θ) on a model parameter. The covariate was selected if it showed a significant decrease of at least 3.84 points (χ2-test, p-value <0.05) in the objective function value.
The area under total plasma, and unbound tumor or brain ECF concentration-time curve (AUCplasma, AUCECF) were derived for each mouse using the pharmacokinetic model. To assess the CNS penetration of unbound prexasertib in G3MB and NTB mice, the unbound tumor or brain to plasma partition coefficient (Kp,uu) was calculated for each mouse using the following equation:
The model-derived prexasertib AUCplasma and AUCECF, as well as the Kp,uu values were statistically compared between the G3MB and NTB mice using two-tailed unpaired t-tests. A p-value of ≤ 0.05 was considered significant.
2.10. Pharmacodynamic study
A pharmacodynamic study was performed in mice bearing G3MB (N=21) to characterize the dynamics of several biomarkers in tumor tissue after treatment with prexasertib and evaluate target engagement. Tumor-bearing mice were enrolled in the study once their bioluminescence signal reached ~ 1×107 photons/sec and were randomized in one control group receiving vehicle and one treatment group receiving single IV bolus of 20 mg/kg prexasertib. Mice were sacrificed, and their brain tissues were harvested and fixed with paraformaldehyde at different time-points (i.e, pre-dose, 1, 2, 6, and 24 hours post-dose). The tissues were processed for immunohistochemistry analysis to quantify protein levels of the CHK1 inhibition marker phosphorylated CHK1 serine 345 (pCHK1 S345), double-stranded DNA breaks through the phosphorylation of histone H2A variant X (γ-H2AX), and programmed cell-death through the cleaved caspase-3. Stained slides were scanned into the Aperio ScanScope system for image analysis and quantification of stained cells (Aperio ImageScope software, Leica Biosystems, Nussloch, Germany).
A pharmacokinetic-pharmacodynamic (PK/PD) model was developed to characterize the exposure-response relationship of prexasertib in mice bearing G3MB. Prexasertib plasma data collected in the second pharmacokinetic study were implemented in the previous plasma model, and the model parameters were re-estimated. Using this model, the typical tumor ECF profile after 20 mg/kg prexasertib IV bolus was derived and used as an input to model pCHK1 S345 and γ-H2AX. The pharmacodynamic model was developed using a naïve-pooled approach in ADAPT 5. Direct effect and indirect response models were tested to link prexasertib ECF concentrations, pCHK1, and γ-H2AX dynamics (Felmlee et al., 2012). Model selection was based on the Akaike Information Criterion value, the precision of the parameter estimates, and the model fits.
3. Results
3.1. Prexasertib protein binding in mouse plasma
Prexasertib was negligibly bound to the dialysis membrane contained in the Equilibrium Dialyzer™ dialysis plate, suggesting that equilibrium dialysis was a suitable technique for determining prexasertib protein binding. The median (range) FU of prexasertib in CD1 mouse plasma was 0.11 (0.09–0.13). No significant differences were observed among the different tested concentrations, suggesting a linear binding process within the range 2–2000 ng/ml (p=0.0755).
3.2. In vitro nonspecific binding studies
The initial non-specific binding studies were designed to assess the extent of binding to plastic and glass collection tubes in Ringer’s solution with no additive. However, the results were highly variable and generally uninterpretable. These studies were repeated with the use of an additive (i.e., 0.5% BSA), which reduced the non-specific binding (data not shown).
The stocks containing no additive collected in glass tubes during in vitro dialysis studies were within 2% of the expected concentration (Table 1). All measured stocks containing 0.5% BSA in the in vitro retrodialysis study were within 3% of the expected concentration (Table 2). In vitro retrodialysis recovery values were also much less variable than in the previous study in which 0.5% BSA was not used. Because of these results, future microdialysis studies would use glass collection tubes and 0.5% BSA in Ringer’s solution as the perfusate.
Table 1.
In vitro dialysis results comparing plastic and glass collection vials using 0.5% BSA in the perfusate.
| Plastic | Glass | |||
|---|---|---|---|---|
| Probe E | Probe F | Probe G | Probe H | |
| In vitro- 1h (ng/ml) | 26.66 | 27.19 | 28.27 | 21.46 |
| In vitro- 2h (ng/ml) | 27.91 | 24.43 | 25.97 | 21.55 |
| In vitro- 3h (ng/ml) | 26.99 | 27.83 | 28.96 | 22.59 |
| Stock average (ng/ml)† | 69.68 | 69.68 | 98.95 | 98.95 |
| In vitro recovery rate (%) | 39.02 | 37.97 | 28.03 | 22.10 |
Expected concentration of 100 ng/ml
Table 2.
In vitro retrodialysis (RD) results comparing plastic and glass collection vials using 0.5% BSA in the perfusate.
| Plastic | Glass | |||
|---|---|---|---|---|
| Probe E | Probe F | Probe G | Probe H | |
| In vitro RD- 1h (ng/ml) | 83.75 | 81.04 | 51.15 | 79.20 |
| In vitro RD- 2h (ng/ml) | 81.36 | 73.87 | 74.48 | 82.82 |
| In vitro RD- 3h (ng/ml) | 78.34 | 71.51 | 105.77 | 83.74 |
| Stock average (ng/ml)† | 102.98 | 101.55 | 102.95 | 102.51 |
| In vitro RD recovery rate (%) | 21.20 | 25.68 | 25.08 | 20.09 |
Expected concentration of 100 ng/ml
During the early microdialysis studies, we observed residual amounts of prexasertib bound to the glass microdialysis syringe after rinsing with ddH2O. Thus, a different rinsing solution was tested to eliminate this binding. In this experiment a rinsing solution of 50% ACN/50% ddH2O/0.1% formic acid was evaluated to eliminate residual prexasertib from the microdialysis syringe. After 100 ng/ml prexasertib in 0.5% BSA in Ringer’s solution was perfused through the microdialysis system, rinsing with an organic solution (i.e., 50% ACN/50% ddH2O/0.1% formic acid) decreased the residual drug in the syringe by 4.60-fold compared to the control approach of rinsing with water (data not shown). This approach was used in the future studies as an approach to reduce the non-specific binding of prexasertib in the microdialysis system.
3.3. Prexasertib plasma disposition after SQ administration
Total prexasertib plasma concentration-time data obtained after SQ dosing were best fitted using a two-compartment population model with linear absorption and elimination (Fig. 1A). The raw data and the visual predictive checks depicted in Fig. 1B show a good overlay between the observed and model predictions. All population and random pharmacokinetic parameters were well estimated (RSE <30%) and are summarized in Table 3. Due to the limited number of animals and time-points per animal, both residual error and inter-individual variabilities couldn’t be estimated properly. The residual error was described by a proportional error model fixed to 5%, while between-animal variabilities were implemented on pharmacokinetic parameters. The mean elimination half-life of prexasertib was estimated at 4.5 hours. The model-derived prexasertib AUCplasma, 0−∞ was estimated at 1819 ± 462 ng·h/ml.
Figure 1. Prexasertib plasma pharmacokinetic model and visual predictive checks.

(A) Prexasertib plasma pharmacokinetic model structure: a two-compartment linear model parameterized with apparent central clearance (CL) and volume (Vc), peripheral clearance (Q) and volume (Vp), and an absorption rate constant (ka) to describe the subcutaneous administration. (B) Prexasertib plasma concentration-time profile in mice bearing G3MB: observations (open-circles) vs model predictions. The solid line represents the median of model predictions, and the shaded area depicts the 90th prediction interval.
Table 3.
Parameter estimates for prexasertib plasma pharmacokinetic model
| Parameters† | Units | Estimate (RSE%) | IIV% (RSE%) | |
|---|---|---|---|---|
| ka | Absorption rate constant | h−1 | 1.07 (12%) | 20.0 (21%) |
| CL | Plasma (central) clearance | l/h/kg | 5.64 (4.3%) | 21.5 (23%) |
| Vc | Plasma (central) volume | l/kg | 1.81 (18%) | - |
| Q | Tissue (peripheral) clearance | l/h/kg | 0.58 (13%) | 16.3 (23%) |
| Vp | Tissue (peripheral) volume | l/kg | 3.42 (8.0%) | - |
RSE% relative standard error, IIV% inter-individual variability reported as coefficient of variation The residual proportional error was fixed to 5%.
Estimated prexasertib clearance and volume were apparent parameters in the absence of data collected after IV in this model.
The mean plasma pharmacokinetic parameters were used to derive an LSM for use in the microdialysis study with a maximum of three samples. The most informative time-points were 0.25, 6.5, and 24 hours post-dose.
3.4. Prexasertib ECF disposition and CNS penetration
After optimizing the conditions, microdialysis studies were conducted in NTB mice and mice bearing G3MB. The in vitro retrodialysis recovery coefficients were on average ± SD 21.5% ± 7.9% (range: 16.0 to 29.8%) for the tumor ECF concentrations, and 20.3% ± 5.0% (range: 12.5 to 26.1%) for the normal brain ECF concentrations.
All the prexasertib plasma concentrations obtained at the LSM time-points were in the same range compared to those obtained during the previous plasma pharmacokinetic study. In mice bearing G3MB, the maximum tumor ECF concentrations were observed after 1-hour post-dose (range: 6.7 to 24.4 ng/ml), and about 47% of the data were below the limit of quantification all after 8 hours post-dose (Fig. 2B). In NTB mice, the maximum brain ECF concentrations were also observed after 1-hour post-dose, but at a lower range (1.5 to 9.6 ng/ml), and about 66% of the data were below the limit of quantification all after 6 hours post-dose (Fig. 2C).
Figure 2. Prexasertib ECF pharmacokinetic model and visual predictive checks.

Prexasertib ECF model structure (A): a two-compartment linear model parameterized with influx and efflux clearances (CLin and CLeff), and peripheral rate constants (kb45 and kb54). The amount of drug moving from plasma to ECF was multiplied by the fraction unbound FU. A separate compartment was used to model the microdialysis fractions. Model predictions of prexasertib ECF concentrations in mice bearing G3MB (B) and in non-tumor bearing mice (C). In (B) and (C), the open-circles are the observed ECF concentrations, the crosses are the simulated data below the limit of quantification, the solid line represents the median of model predictions, the shaded area depicts the 90th prediction interval.
The prexasertib unbound tumor and brain ECF concentration-time data were best fitted using a two-compartment model with influx and efflux linear clearance terms (CLin and CLef) (Fig. 2A). The volume of the tumor ECF central compartment was fixed to 0.001 l/kg based on published data (Daryani et al., 2016). The ECF central and peripheral compartments were linked using rate constants (kb45 and kb54). Significant differences were found between mice bearing G3MB and the NTB mice for each parameter (p<0.05). NTB mice exhibited a lower CLin, but higher CLef, kb45, and kb54 compared to G3MB mice. All population and random pharmacokinetic parameters were reasonably well estimated, except for one parameter (kb45 θNTB RSE 77%), and are summarized in Table 4. The central tendency and the variability observed in both tumor ECF and brain ECF concentration-time data were well captured by the model as shown by the visual predictive checks in Fig. 2B–C.
Table 4.
Parameter estimates for prexasertib tumor and brain ECF pharmacokinetic model.
| Parameters | Units | Estimate (RSE%) | IIV% (RSE%) | |
|---|---|---|---|---|
| CLin | Plasma to tumor influx clearance | l/h/kg | 1.12–10−4 (18%) | 43.2 (34%) |
| θNTB | Proportional shift for NTB mice | - | 0.93 (45%) | - |
| CLef | Plasma to tumor efflux clearance | l/h/kg | 6.1610−4 (28%) | 46.1 (26%) |
| θNTB | Proportional shift for NTB mice | - | 2.20 (38%) | - |
| kb45 | Peripheral rate constant | h−1 | 0.39 (24%) | - |
| θNTB | Proportional shift for NTB mice | - | 5.90 (77%) | - |
| kb54 | Peripheral rate constant | h−1 | 0.05 (41%) | - |
| θNTB | Proportional shift for NTB mice | - | 17.9 (32%) | - |
RSE% relative standard error, IIV% inter-individual variability reported as coefficient of variation, NTB non-tumor bearing mice. The residual errors for prexasertib tumor ECF and brain ECF models were 21.9% and 25.1%, respectively.
Mean ± SD plasma AUCplasma of unbound prexasertib derived from the individual total drug exposures and prexasertib FU were 291 ± 59 ng·h/mL for mice bearing G3MB, and 271 ± 23 ng·h/ml for NTB mice (p=0.42) at 24 hours post-dose. Mean ± SD unbound prexasertib AUCECF was estimated at 47.3 ± 17 ng·h/ml in tumor-bearing mice, and 24.8 ± 11.8 ng·h/ml in NTB mice (p=0.013) at 24 hours post-dose. Model-derived Kp,uu values (mean ± SD) were estimated at 0.17 ± 0.08 in mice bearing orthotopic G3MB, and 0.09 ± 0.04 in NTB mice (p=0.04).
3.5. Prexasertib pharmacodynamics and exposure-response relationships
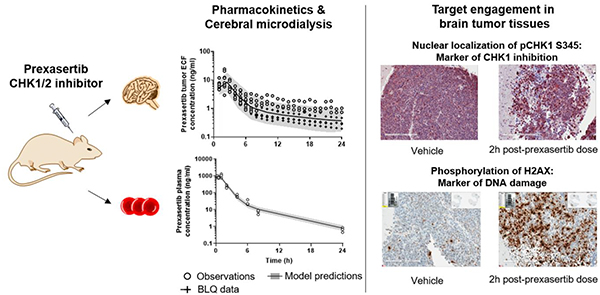
After a single dose of 20 mg/kg prexasertib (IV), we observed a 2.2-fold peak in the pCHK1 S345 nuclear staining at 2 hours, which stayed above the baseline for at least 6 hours after the prexasertib dose (Fig. 3A and 3B). At 24 h post-dose, the levels of pCHK1 S345 nucleus staining were similar to those at baseline (data not shown). Similar results were observed with γ-H2AX marker (Fig. 3A and 3C). The apoptotic marker cleaved caspase-3 didn’t statistically increase during the study, although a modest 1.4-fold increase was seen at 24 hours post-prexasertib dose (Fig. 3A and D).
Figure 3. Prexasertib-induced changes in dynamics of efficacy markers in mouse G3MB tumors.

(A) Representative immunohistochemistry images from tumors of mice treated with vehicle (control) or single dose prexasertib (20 mg/kg, IV) stained with antibodies against pCHK1 S345, γ-H2AX and cleaved caspase-3, at 1, 2, 6, and/or 24 h post-dose. Quantification of the percentage of cells staining positive for nuclear pCHK1 S345 (B), γ-H2AX (C), and cleaved caspase-3 (D) in control and treated animals at 1, 2, 6, and/or 24 h post-dose.
An indirect-response PK/PD model was used to describe the dynamics of both pCHK1 S345 and γ-H2AX (Fig. 4A). Prexasertib plasma disposition after IV bolus was similar to that observed after SQ administration, with a bioavailability close to 1 (Fig. 4B). To describe the dynamics of pCHK1 S345 and γ-H2AX levels, the production rate kin of each marker was stimulated by the model-simulated prexasertib ECF concentrations according to an Emax model. The data were adequately predicted by the pharmacodynamic models (Fig. 4C–D), and the associated EC50 parameters were estimated at 10.2 ng/ml and 19.4 ng/ml for pCHK1 S345 and γ-H2AX, respectively. As shown in Fig. 4B, the prexasertib ECF concentrations were sustained above these EC50 values approximately 2 hours post-dose. All the estimated pharmacokinetic-pharmacodynamic parameters are reported in Table 5.
Figure 4. Prexasertib pharmacokinetic-pharmacodynamic model and predictions.

(A) Indirect response model used to predict pCHK1 S345 and γ-H2AX dynamics. kin and kout represent the production and degradation rates. Prexasertib ECF concentrations were used to stimulate kin with an Emax/EC50 model. Emax is the maximum extent of simulation, and EC50 is the prexasertib ECF concentration for 50% stimulation of maximum function of kin. (B) Prexasertib observed concentrations (open-circles), mean predicted prexasertib plasma concentration-time profile (solid line), and mean predicted ECF concentration-time profile (dashed line) after 20 mg/kg IV. (C) pCHK1 S345 observations (open-circles) and model predictions (solid line). (D) γ-H2AX observations (open-circles) and model predictions (solid line).
Table 5.
Parameter estimates for prexasertib pharmacokinetic and pharmacodynamic model.
| Parameters | Units | Estimate (RSE%) | |
|---|---|---|---|
| Pharmacokinetic plasma parameters | |||
| ka | Absorption rate constant | h−1 | 1.16 (10%) |
| CL | Plasma clearance | l/h/kg | 6.03 (8.0%) |
| Vc | Plasma volume | l/kg | 2.67 (13%) |
| Q | Tissue (peripheral) clearance | l/h/kg | 0.807 (10%) |
| Vp | Tissue (peripheral) volume | l/kg | 3.79 (6.0%) |
| pCHK1 S345 pharmacodynamic parameters | |||
| Baseline | Initial value | % | 18.5 (7.7%) |
| kin | Zero-order production rate constant | h−1 | 17.4 (25.1%) |
| Emax | Maximum extent of stimulation | - | 2.0 (-) |
| EC50 | Prexasertib ECF concentration for 50% stimulation of maximum function of kin | ng/ml | 10.15 (26.5%) |
| pH2Ax pharmacodynamic parameters | |||
| Baseline | Initial value | % | 21.4 (16.5%) |
| kin | Zero-order production rate constant | h−1 | 8.9 (36.2%) |
| Emax | Maximum extent of stimulation | - | 5.0 (-) |
| EC50 | Prexasertib ECF concentration for 50% stimulation of maximum function of kin | ng/ml | 19.4 (34.1%) |
For both pCHK1 S345 and pH2Ax, the Emax parameter had to be fixed based upon a sensitivity analysis, due to the small sampling size and the absence of data collected at different prexasertib doses.
4. Discussion
In this work, plasma, tumor and brain ECF disposition of prexasertib were described in mice bearing G3MB and NTB mice receiving a single dose of prexasertib (10 mg/kq, SQ) using cerebral microdialysis and pharmacokinetic modeling. The CNS penetration of prexasertib in tumor-bearing mice was characterized by a mean Kp,uu = 0.17, and was statistically greater than that observed in NTB mice (Kp.uu = 0.09, p-value = 0.04). Pharmacodynamic studies were further performed in tumor-bearing mice to characterize the dynamics of markers of prexasertib efficacy. The increases of nuclear pCHK1 S345 and pH2Ax levels in tumor tissues after a single dose of prexasertib (20 mg/kg, IV) confirmed a treatment-induced target engagement, and thus, an adequate CNS penetration of the CHK1 inhibitor. These results support further preclinical and clinical investigation in treating adult and pediatric CNS malignancies.
The ability to cross the blood-tumor/brain-barrier and reach an adequate unbound exposure at the target sites was the major obstacle for brain therapy drug candidates. Therefore, preclinical studies evaluating CNS penetration along with drug efficacy were warranted. The methods used involved cerebral microdialysis with pharmacokinetic modeling and simulation techniques, as previously described (Jacus et al., 2014). This approach has been applied to several anticancer drugs in different murine CNS tumor models and has been critical to support or condemn further development in the context of new therapies for pediatric brain tumors (Daryani et al., 2016; Patel et al., 2015). The CNS penetration studies were usually performed with intracranial tumor-bearing animal models in which the blood-brain-barrier was compromised (Deeken and Löscher, 2007). However, it is also of interest to perform these studies in non-tumor bearing animals, as the integrity of the blood-brain-barrier may remain intact in some children with medulloblastoma (Barai et al., 2005). Indeed, the pediatric brain tumors such as medulloblastoma or glioma are very heterogenous tumors with different epigenetic alterations and immunosuppressive environments between different tumor subgroups, but also heterogenous in their composition with some solid or more infiltrative components (Barai et al., 2005; Vogelbaum, 2018). Therefore, to get a complete evaluation of the CNS penetration of a drug and determine its ability to cross the blood-brain-barrier whether the barrier is disrupted or intact, it is valuable to conduct experiments in both tumor-bearing and heatlhy animals.
To translate our preclinical findings to the clinical setting, it was important to use clinically relevant dosages of drugs in our cerebral microdialysis experiments, which would lead to drug exposures in target tissues comparable to those obtained in clinic. Large interpatient pharmacokinetic variability was observed in adult patients across dosages, and at the recommended phase II dosage of 105 mg/m2, mean ±SD prexasertib plasma exposure (AUCplasma, 0−∞) was 2410 ± 1229 ng·h/ml (Hong et al., 2016). A prexasertib dosage of 10 mg/kg SQ was selected for our pharmacokinetic studies in mice bearing orthotopic G3MB. The model-derived prexasertib AUCplasma, 0−∞ ranged from 1461 to 2880 ng·h/ml with a mean of 1819 ng·h/ml. These values were in the lower range of that reported for adult patients receiving a tolerable prexasertib dosage of 105 mg/m2 (Hong et al., 2016). Thus, our dosage was considered as relevant in terms of systemic drug exposure and was used for the subsequent microdialysis studies.
Our first attempt to perform cerebral microdialysis for prexasertib was challenged by non-specific binding issues, which required modification of the experimental study methodology for the microdialysis studies. Non-specific binding to the microdialysis tubing and the probe membrane were critical issues to evaluate and address prior to performing any microdialysis experiments to avoid underestimation of target concentrations (Nirogi et al., 2012).
Our initial hypothesis was that prexasertib would exhibit greater CNS penetration in mice bearing G3MB compared to that in NTB mice. In the presence of brain primary and metastatic malignancies, the integrity of the blood-brain barrier may be altered (Deeken and Löscher, 2007). Thus, the blood-tumor-barrier is less intact which allows for increased permeability and access of chemotherapy treatment to reach the tumor. The simple visual inspection of the tumor and brain ECF concentration-time profiles showed differences between tumor-bearing and NTB mice, with lower ECF concentrations falling more rapidly below the limit of quantification in NTB mice. The modeling analysis confirmed our hypothesis of a greater CNS penetration of prexasertib in tumor-bearing mice. Model-derived unbound prexasertib exposures showed a two-fold difference between mice bearing G3MB (mean AUCtECF = 47.6 ng·h/mL) and NTB mice (mean AUCECF = 24.8 ng·h/mL, p-value = 0.013), while similar prexasertib plasma exposures were observed between the two groups of animals. The Kp,uu values derived in both tumor-bearing and NTB mice (0.17 versus 0.09, p-value = 0.04) were both below 1, suggesting efflux transport of prexasertib at the blood-brain-barrier and blood-tumor-barrier. The different model parameters estimated in tumor-bearing and NTB mice would suggest a slower CNS penetration and more rapid brain clearance of prexasertib in NTB mice. However, caution should be used with regards to the interpretation of the model parameters, as our pharmacokinetic model was empirical and not physiologically-based.
After characterizing the CNS exposure of prexasertib, we established if whether this exposure was adequate for target engagement. We first compared the tumor ECF concentration-time profiles to the EC50 values (< 1nM) reported from a cell proliferation assay in neuroblastoma cell lines since that data in medulloblastoma cell lines were not available (Lowery et al., 2017). The majority of the G3MB bearing mice exhibited prexasertib tumor concentrations above the in vitro EC50 for at least 15 hours post-dose. Even in NTB mice, prexasertib brain ECF concentrations were sustained above the EC50 for approximately 6–12 hours post-dose. These findings were encouraging, suggesting that prexasertib may exert its pharmacological activity against G3MB in vivo.
For in vivo pharmacodynamic studies, the prexasertib dosing regimen was redefined at 20 mg/kg, IV bolus to match the clinical route of administration of the drug, and to reach the upper range of clinical exposures that were observed in adult patients receiving 105 mg/m2 prexasertib (Hong et al., 2016). After 20 mg/kg, IV, the model-derived prexasertib AUCplasma, 0−∞ ranged from 1793 to 4192 ng·h/ml with a mean of 2985 ng·h/ml. We selected three different biomarkers: pCHK1 S345, γ-H2AX, and cleaved caspase-3 to evaluate prexasertib target engagement. The induction of pCHK1 S345 was chosen as a marker of CHK1 inhibition (Parsels et al., 2011). Briefly, in presence of a CHK1 inhibitor, the autophosphorylation of CHK1 on serine S296 was blocked, and as such, the activity of CHK1 kinase resulted in double-strand DNA breaks. This leads to the activation of the DNA damage response, which can be observed by the nuclear localization of different DNA damage sensors and increased phosphorylation of CHK1 S345 by different mechanisms (Parsels et al., 2011). The two other markers, γ-H2AX and cleaved caspase-3, are commonly used as pharmacodynamic biomarkers of DNA damage and apoptosis, respectively (Gown and Willingham, 2002; Redon et al., 2010). Prexasertib-induced target engagement in mice bearing G3MB after a single prexasertib dose was successfully observed with consistent increases in levels of pCHK1 S345 and γ-H2AX reflecting CHK1 inhibition and a DNA damage response in brain tumor tissues. Regarding the cleaved caspase-3 marker, an increased staining was only observed at 24 hours post-dose. This suggested that the cells initiated apoptosis after 24 hours. Thus, a delay was observed between the peak activity at 2 hours post-dose for pCHK1 S345 and γ-H2AX markers. This might be due to the time required to activate the other mechanisms responsible for apoptosis in tumor cells. The prexasertib ECF exposure-response relationships for the induction of pCHK1 S345 and γ-H2AX were further characterized using pharmacokinetic/pharmacodynamic modeling. The EC50 values that were estimated may be considered as preliminary parameters, as data were collected after only one single prexasertib dose. Data obtained from different dosages would be necessary to accurately estimate the pharmacodynamic parameters. Nonetheless, our estimated EC50 values were consistent with the observed ECF concentration-time profiles.
These results suggested that adequate unbound prexasertib reached the brain tumor relative to target engagement after single dose (20 mg/kg, IV) and as a single agent. These findings have led to further preclinical pharmacodynamic studies in mouse models of pediatric medulloblastoma evaluating the actual tumor growth inhibition to assess prexasertib efficacy as a single agent, and also in combination with chemotherapeutic agents such as cyclophosphamide and gemcitabine. Combination therapy with prexasertib and chemotherapy was shown to lead to significant antitumor activity in multiple preclinicals of pediatric cancer (Lowery et al., 2019). Thus, it may represent a promising strategy to treat pediatric medulloblastoma.
Developing a translational model to further predict the pharmacokinetic and pharmacodynamic profiles expected in pediatrics based on the results obtained in mice was beyond the scope of this analysis. To do so, a physiological-based pharmacokinetic (PBPK) model describing the different CNS compartments will have to be used (Yamamoto et al., 2017; Yamamoto et al., 2018). The developement of a CNS PBPK model will allow to take into account the critical physiological differences between mice and adults, and between adults and pediatrics that control the CNS penetration. Those physiological parameters include the blood flows, the organ volumes, the expression and activity of the enzymes and transporters implicated in the pharmacokinetics of prexasertib, and the extent of plasma protein binding. It will be interesting to develop this PBPK model including a disrupted blood-brain-barrier as well as an intact barrier as the higher blood-brain-barrier permeability typically assumed can be modulated by the tumor size and location.
To conclude, our results show that prexasertib can penetrate both the mouse CNS and G3MB tumor at an adequate exposure for target engagement after a clinically relevant and tolerable dosage. This work paved the way to further preclinical studies investigating prexasertib in combination with chemotherapeutic agents cyclophosphamide and gemcitabine to treat pediatric CNS malignancies. These studies were the rationale for a recently opened international clinical trial SJELIOT ().
Acknowledgments:
We thank the St. Jude Children’s Research Hospital Veterinary Pathology Core for their assistance in the microdialysis studies, Sarah Robinson for her expert technical assistance in the pharmacodynamic studies, Drs Nick Gottardo and Raelene Endsberry for the γ-H2AX staining protocol, and Dr. Aimee Bence of Eli Lilly for her helpful comments during the conduct of this study.
Funding: This work was supported in part by grants from Eli Lilly, the National Cancer Institute (CA21765 (MFR; CFS) and CA096832 (MFR)), and the American Lebanese Syrian Associated Charities (ALSAC) at St. Jude Children’s Research Hospital.
Footnotes
Declaration of interests: none.
References
- Armstrong GT, Liu Q, Yasui Y, Huang S, Ness KK, Leisenring W, Hudson MM, Donaldson SS, King AA, Stovall M, Krull KR, Robison LL, Packer RJ, 2009. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst 101, 946–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson JM, Shelat AA, Carcaboso AM, Kranenburg TA, Arnold LA, Boulos N, Wright K, Johnson RA, Poppleton H, Mohankumar KM, Feau C, Phoenix T, Gibson P, Zhu L, Tong Y, Eden C, Ellison DW, Priebe W, Koul D, Yung WK, Gajjar A, Stewart CF, Guy RK, Gilbertson RJ, 2011. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell 20, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barai S, Bandopadhayaya GP, Julka PK, Kale SS, Kumar R, Malhotra A, Haloi AK, Seith A, Naik KK, Dhanapathi H, 2005. Evaluation of Tc99m-glucoheptonate for SPECT functional imaging of medulloblastoma. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 12, 36–38. [DOI] [PubMed] [Google Scholar]
- Beal SL, 2001. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28, 481–504. [DOI] [PubMed] [Google Scholar]
- Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ, 1989–2013. NONMEM 7.3.0 Users Guides. ICON Development Solutions, Hanover, MD. [Google Scholar]
- Carcaboso AM, Elmeliegy MA, Shen J, Juel SJ, Zhang ZM, Calabrese C, Tracey L, Waters CM, Stewart CF, 2010. Tyrosine Kinase Inhibitor Gefitinib Enhances Topotecan Penetration of Gliomas. Cancer Research 70, 4499–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, Kohler BA, Jemal A, 2018. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 124, 2785–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Grant S, 2010. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 16, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daryani VM, Patel YT, Tagen M, Turner DC, Carcaboso AM, Atkinson JM, Gajjar A, Gilbertson RJ, Wright KD, Stewart CF, 2016. Translational Pharmacokinetic-Pharmacodynamic Modeling and Simulation: Optimizing 5-Fluorouracil Dosing in Children With Pediatric Ependymoma. CPT: pharmacometrics & systems pharmacology 5, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeken JF, Löscher W, 2007. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res 13, 1663–1674. [DOI] [PubMed] [Google Scholar]
- Di L, Rong H, Feng B, 2013. Demystifying brain penetration in central nervous system drug discovery. Miniperspective. J Med Chem 56, 2–12. [DOI] [PubMed] [Google Scholar]
- Felmlee MA, Morris ME, Mager DE, 2012. Mechanism-based pharmacodynamic modeling. Methods Mol Biol 929, 583–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG, 2015. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 33, 2986–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar AJ, Robinson GW, 2014. Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol 11, 714–722. [DOI] [PubMed] [Google Scholar]
- Gown AM, Willingham MC, 2002. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem 50, 449–454. [DOI] [PubMed] [Google Scholar]
- Holford NH, 2005. The Visual Predictive Check — Superiority to Standard Diagnostic (Rorschach) Plots. PAGE 14, Abstr 738 [http://www.page-meeting.org/?abstract=972].
- Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J, 2016. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol 34, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacus MO, Throm SL, Turner DC, Patel YT, Freeman BB 3rd, Morfouace M, Boulos N, Stewart CF, 2014. Deriving therapies for children with primary CNS tumors using pharmacokinetic modeling and simulation of cerebral microdialysis data. Eur J Pharm Sci 57, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS, 2015. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther 14, 2004–2013. [DOI] [PubMed] [Google Scholar]
- Leary SE, Olson JM, 2012. The molecular classification of medulloblastoma: driving the next generation clinical trials. Current opinion in pediatrics 24, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, Bagheri MH, Botesteanu DA, Steinberg SM, Minasian L, Ekwede I, Kohn EC, 2018. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 19, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AB, McNeely SC, Beckmann RP, 2017. Achieving Precision Death with Cell-Cycle Inhibitors that Target DNA Replication and Repair. Clin Cancer Res 23, 3232–3240. [DOI] [PubMed] [Google Scholar]
- Lowery CD, Dowless M, Renschler M, Blosser W, VanWye AB, Stephens JR, Iversen PW, Lin AB, Beckmann RP, Krytska K, Cole KA, Maris JM, Hawkins DS, Rubin BP, Kurmasheva RT, Houghton PJ, Gorlick R, Kolb EA, Kang MH, Reynolds CP, Erickson SW, Teicher BA, Smith MA, Stancato LF, 2019. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin Cancer Res 25, 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF, 2017. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res 23, 4354–4363. [DOI] [PubMed] [Google Scholar]
- McNeely S, Beckmann R, Bence Lin AK, 2014. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther 142, 1–10. [DOI] [PubMed] [Google Scholar]
- Morfouace M, Shelat A, Jacus M, Freeman BB 3rd, Turner D, Robinson S, Zindy F, Wang YD, Finkelstein D, Ayrault O, Bihannic L, Puget S, Li XN, Olson JM, Robinson GW, Guy RK, Stewart CF, Gajjar A, Roussel MF, 2014. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 25, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Mouksassi MS, Holford N, Al-Huniti N, Freedman I, Hooker AC, John J, Karlsson MO, Mould DR, Perez Ruixo JJ, Plan EL, Savic R, van Hasselt JG, Weber B, Zhou C, Comets E, Mentre F, Model Evaluation Group of the International Society of Pharmacometrics Best Practice, C., 2017. Model Evaluation of Continuous Data Pharmacometric Models: Metrics and Graphics. CPT: pharmacometrics & systems pharmacology 6, 87–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirogi R, Kandikere V, Bhyrapuneni G, Benade V, Saralaya R, Irappanavar S, Muddana N, Ajjala DR, 2012. Approach to reduce the non-specific binding in microdialysis. J Neurosci Methods 209, 379–387. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD, 2011. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29, 1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsels LA, Qian Y, Tanska DM, Gross M, Zhao L, Hassan MC, Arumugarajah S, Parsels JD, Hylander-Gans L, Simeone DM, Morosini D, Brown JL, Zabludoff SD, Maybaum J, Lawrence TS, Morgan MA, 2011. Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res 17, 3706–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel YT, Jacus MO, Boulos N, Dapper JD, Davis AD, Vuppala PK, Freeman BB 3rd, Mohankumar KM, Throm SL, Gilbertson RJ, Stewart CF, 2015. Preclinical examination of clofarabine in pediatric ependymoma: intratumoral concentrations insufficient to warrant further study. Cancer Chemother Pharmacol 75, 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, Parchment RE, Doroshow JH, Pommier Y, 2010. Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res 16, 4532–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, Redwood AB, Fan YH, Li L, Glisson BS, Minna JD, Sage J, Gibbons DL, Piwnica-Worms H, Heymach JV, Wang J, Byers LA, 2017. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res 77, 3870–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheiner LB, Beal SL, 1982. Bayesian individualization of pharmacokinetics: simple implementation and comparison with non-Bayesian methods. J Pharm Sci 71, 1344–1348. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM, 2012. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelbaum MA, 2018. Targeted Therapies for Brain Tumors: Will They Ever Deliver? Clin Cancer Res 24, 3790–3791. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Valitalo PA, Huntjens DR, Proost JH, Vermeulen A, Krauwinkel W, Beukers MW, van den Berg DJ, Hartman R, Wong YC, Danhof M, van Hasselt JGC, de Lange ECM, 2017. Predicting Drug Concentration-Time Profiles in Multiple CNS Compartments Using a Comprehensive Physiologically-Based Pharmacokinetic Model. CPT: pharmacometrics & systems pharmacology 6, 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Valitalo PA, Wong YC, Huntjens DR, Proost JH, Vermeulen A, Krauwinkel W, Beukers MW, Kokki H, Kokki M, Danhof M, van Hasselt JGC, de Lange ECM, 2018. Prediction of human CNS pharmacokinetics using a physiologically-based pharmacokinetic modeling approach. Eur J Pharm Sci 112, 168–179. [DOI] [PubMed] [Google Scholar]
- Zhong B, Maharaj A, Davis A, Roussel MF, Stewart CF, 2018. Development and validation of a sensitive LC MS/MS method for the measurement of the checkpoint kinase 1 inhibitor prexasertib and its application in a cerebral microdialysis study. Journal of Pharmaceutical and Biomedical Analysis 156, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]