

Abstract
The Phosphoprotein Phosphatase Calcineurin (CN, PP2B, PP3) recognizes and binds to two short linear motifs (SLiMs), PxIxIT and LxVP, in its regulators and substrates. These interactions enable CN function in many key biological processes. The identification of SLiMs is difficult because of their short, degenerate sequence and often low binding affinity. Here we combine Structure Based Shape Complementarity (SBSC) analysis and proteome-wide affinity purification-mass spectrometry to identify PxIxIT and LxVP containing CN interactors to expand and thereby redefine the LxVP motif. We find that the new πφ-LxVx primary sequence defines an ensemble of binding competent confirmations and thus the binding on-rate, making it difficult to predict the LxVP binding strength from its sequence. Our analysis confirms existing and, more importantly, identifies novel CN interactors, substrates, and thus biological functions of CN.
Graphical Abstract
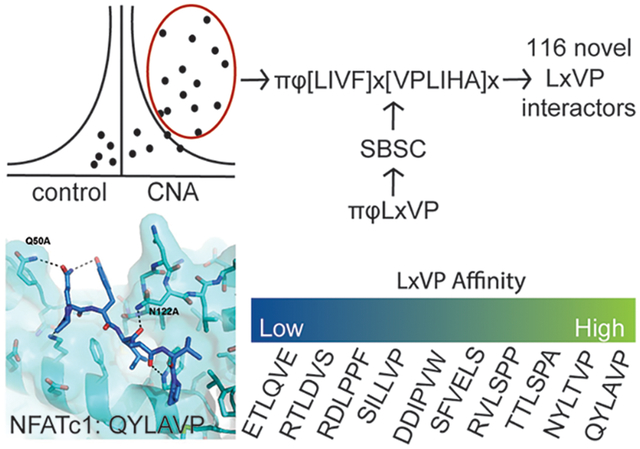
Cellular signaling is driven by protein:protein interactions, which result in the establishment of signaling platforms and the recruitment of substrates. While many of these interactions are between folded proteins, others are driven by the interaction of folded, globular proteins with short linear motifs (SLiMs) located within intrinsically disordered regions (IDRs) of proteins.1 These SLiM interactions provide several advantages:2 (1) small sequence variations can alter SLiM binding affinities; (2) hydrophobic contacts, which often form the core of SLiM interactions, can be modulated by charge:charge interactions; (3) avidity of multiple SLiMs can strengthen binding; and (4) posttranslational modifications including phosphorylation modulate binding affinity. In particular, SLiM-based interactions are critical for kinase and phosphatase signaling.3–5
Calcineurin (PP2B, PP3, CN)6 is an essential enzyme that is highly conserved from yeast to humans. It is ubiquitously expressed and crucial for diverse biological functions, including development, cardiac function, and the immune response.7 CN is a member of the family of Phosphoprotein Phosphatases (PPPs).3 It is a constitutive heterodimer consisting of a catalytic subunit CNA/calcineurin A and CNB that binds Ca2+, which is required for CN activation.8 Although CN has been studied for decades, the mechanism(s) by which CN identifies and dephosphorylates specific substrates is still only poorly understood.
Ser/Thr kinases phosphorylate phosphoacceptor residues surrounded by a specific recognition motif, making kinase-substrate relationships largely predictable.9 In contrast, PPP active sites do not recognize specific, distinct motif sequences surrounding the phosphorylated residues.3 However, PPPs still exhibit exquisite specificity for their substrates.10 The emerging view is that PPPs use PPP-specific SLiMs for substrate recruitment, which has been shown for PP1,11 PP2A,12 PP4,13 and CN.14,15 These SLiMs bind complementary SLiM-binding pockets in the cognate PPP. The two known CN SLiMs are the PxIxIT14 and LxVP15 motifs, which bind the PxIxIT binding pocket on CNA and the LxVP binding pocket at the CNA/CNB interface (Figure 1A,B), respectively. Both binding pockets are >15 Å away from the CN active site. The LxVP binding pocket has also been identified as the binding site of cyclosporin A (CSA) and FK-506, two critical immunosuppressant drugs that are necessary to suppress organ rejection after transplantations.15,16
Figure 1.

The SBSC-MS analysis of CN. (A) Structure of CN (A: catalytic domain, black; B: regulatory domain, cyan) bound to the AKAP PxIxIT SLiM (green, PDBID: 3LL8) within the PxIxIT binding pocket (dark blue) and the NFATc1 LxVP SLiM (magenta, PDBID: 5SVE) bound within the LxVP binding pocket (light blue). CN active site metals Zn2+ and Fe3+: yellow and orange spheres, respectively. CNB Ca2+ ions: red spheres. (B) Close-up of the NFATc1 LxVP bound to the CN LxVP binding pocket (blue). Binding pocket core (dashed outline) with highlighted areas (yellow) shows the interaction surfaces for the i, i+2, and i+3 residues in the LxVP motif. (C) Experimental setup to identify CNA interactors. (D) Volcano plot of label-free AP-MS identification of CN interactors. Red: known CN interactors; blue: new interactors selected for validation. (E) FLAG-IP schematic for validation of CNA interactions. (F) Western blot analysis of immunoprecipitations from nontransfected control cells and cells expressing FLAG-tagged protein and their interaction with CNA.
Identifying functional SLiMs in interactors and substrates is difficult because their sequence features are short and degenerate, making bioinformatics prediction challenging. One way to overcome this is to use proteomic phage display to directly define SLiM sequences.2,17 However, as this technique is less sensitive to the low-to-moderate affinity interactions (5–1000 μM) typical for SLiMs, alternative approaches for identifying SLiMs in novel regulators and substrates are needed. Recently, we developed a Structure Based Shape Complementarity (SBSC) approach. The SBSC uses the shape of the preformed, rigid SLiM binding pocket to identify amino acids with binding complementarity. This approach has been applied both to PP2A:B56 (LxxIxE motif)12 and to the LxVP site of CN.18
In recent years, identification of novel CN regulators has focused on only the PxIxIT motif19–21 due in part to the greater predictive power of the PxIxIT SLiM, which spans 6 to 7 amino acids (the motif can either be PxIxIT14 or PxxIxIT,22 which both fit into the same binding pocket on CN). The PxIxIT binding pocket is used by regulators, substrates, and scaffolding proteins such as AKAP79, which targets CN to its cellular location.23 In contrast, the smaller LxVP motif is more challenging to predict but equally important for CN substrate recruitment.
Here we combine proteome-wide affinity mass spectrometry (MS) with SBSC to further expand the CN interactome. Using surface plasmon resonance (SPR), we show that LxVP SLiM binding to CN is on-rate-dependent, confirming that SBSC is an ideal approach to identify and expand SLiM sequences. Applying a new, expanded definition for the LxVP and previously established PxIxIT definition, we identify 119 CN regulators and substrates containing one or both of these SLiMs, leading to the redefinition of the πφ-LxVx motif and the identification of a novel role for CN in the regulation of nuclear-cytoplasmic transport. This analysis expands both the technical approach to identifying SLiM based interactions on a proteome-wide scale and significantly expands our understanding of the biological function of CN.
RESULTS AND DISCUSSION
Unbiased Proteome-Wide Interaction Screen Using Affinity Mass Spectrometry (MS).
To identify calcineurin (CN) interacting proteins (regulators and substrates), 3xFLAG-tagged Calcineurin (CNA; A-subunit) was stably expressed in HEK293T cells, immunoprecipitated using an anti-FLAG antibody, and analyzed using label-free quantitative mass spectrometry (Figure 1C). To distinguish specific CN interactors from nonspecifically bound proteins, we compared protein abundances in CNA and control (vector only) immunoprecipitations. We identified 176 (CN176) proteins that interact specifically with CN (increase in abundance of at least 5-fold in CN versus control immunoprecipitations, p-value < 0.05) (Table S1). These proteins include the indispensable CN B-subunit (PPP3R1/CANB1), established CN-interacting proteins such as the RCAN family of CN inhibitors (RCAN1, RCAN2, and RCAN3),24 and CN-substrates like the family of nuclear factor of activated T-cells transcription factors (NFATC1 and NFATC4),25 among others (Figure 1D). We also identified more recently established CN interactors, including the CREB-regulated transcription coactivator 2 (CRTC2),26 the ER chaperone Calnexin (CALX),27 and the CN inhibitor Cabin 1 (CABIN).28 Notably, we found several proteins previously predicted to interact directly with CN via an LxVP sequence identified using a structure-based analysis of the CN:NFAT1c LxVP interaction.18 These proteins included the HEAT repeat-containing protein 3 (HEAT3), the serine/threonine-protein kinase SIK3, the histone deacetylase complex subunit SAP130 (SP130), the neuronal protein FAM126A/hyccin (HYCCI), the microtubule stabilizer Nucleolar and spindle-associated protein 1 (NUSAP), the kinase GSK3β (GSK3B), and the chromatin remodeling protein AT-rich interactive domain-containing protein 1A (ARI1A). Together, these data show that the experimental conditions used allow for the identification of new and known CN regulators and substrates.
To further validate that some of the newly identified proteins bind CN, we conducted reciprocal immunoprecipitation experiments. For this, we selected MCM2, NUP53, RANBP3, and XPO1, which exhibit 11- to 230-fold enrichments in CN-binding versus control. 3xFLAG-tagged versions of these proteins were expressed in HEK293FT cells, immunoprecipitated using anti-FLAG antibody, and the immune complexes were analyzed by Western blot for their ability to bind endogenous CN (Figure 1E). The data show that all four proteins immunoprecipitated endogenous CN, confirming that the MS workflow successfully identifies new CN interactors (Figure 1F).
Using the SBSC-Derived LxVP SLiM To Identify Direct CN Interactors.
Previously, we used the crystal structure of the substrate NFATC1 LxVP peptide bound to CN, coupled with a conservative SBSC study, to define an expanded CN LxVP motif ([NQDESRTH][YRTDFILV]Lx[VPL][PK] → πϕLx[VPL]-[PK]). We employed this approach to identify potential LxVP motifs in proteins in the CN176 data set. First, we applied an IDR filter (IUPRED ≥ 0.4) to exclude SLiM sequences that are present in structured domains. We then required the presence of experimentally observed phospho-Ser/Thr residues (as determined using the PhosphoSitePlus database29). Lastly, we searched for PxIxIT SLiMs in the same targets using established PxIxIT SLiM motifs.14 This resulted in the identification of 11 proteins from the CN176 data set (6.25%) that contain a πϕLx[VPL][PK] motif (Table S1).
The limited number of CN interactors containing a πϕLx[VPL][PK] motif was not surprising because of the conservative approach originally used to expand the LxVP SLiM.18 Thus, we used SBSC to establish a more degenerate LxVP SLiM: πϕ[LIVF]x[VPLIHA]x. In this degenerate SLiM, the “L” of the LxVP motif, also referred to as the “i” position, was expanded from L → [LIVF], the “V” or “i+2” residue expanded from [VPL] → [VPLIHA], and the “P” or “i+3” residue expanded from [PK] to “x”. The key assumptions used to establish this degenerate SLiM were: (1) the LxVP binding pocket of CN is rigid (all crystal structures of CNA/B determined thus far show that the LxVP binding pocket is conformationally invariant8,14,15,18,22) and (2) that both pocket shape and chemistry determine the amino acids that effectively bind in the pocket (Figure 1B). First, the very deep “L” binding pocket (i position) was determined to readily accommodate F, I, and V residues; consistent with this determination, an “F” residue was recently experimentally confirmed to bind this pocket.30 Second, the shallow “V” (i+2) binding pocket was determined to accommodate a diverse set of hydrophobic residues, including I, A, and H in addition to V, P, and L. Third, the “P” (i+3) interaction site is not a pocket, but rather a surface. In our original in silico search, the i+3 residue was restricted to be either a “P” or “K” residue, as these residues had been previously experimentally determined to bind at this site.15,18 Here, our SBSC analysis revealed that any amino acid could interact at this position, and thus, the i+3 position can accommodate all 20 amino acids. Using this new, degenerate LxVP motif, π-ϕ-[LIVF]-x-[VPLIHA]-x (πφ-LxVx), 119 proteins in the CN176 data set were identified to contain either an πφ-LxVx or a PxIxIT motif in IDPs that contain experimentally confirmed phospho-Ser/Thr residues. Ninety-one of these are predicted to contain both an πφ-LxVx and a PxIxIT SLiM, while 25 and 3 are predicted to contain only an πφ-LxVx or a PxIxIT SLiM, respectively (Figure S1).
The SBSC Approach Identifies πφ-LxVx Sequences That Bind Directly to CN.
To test the validity of the SBSC-based πφ-LxVx predictions, we used cellular binding assays. We selected eight proteins predicted to contain an πφ-LxVx motif: SKP1, GCFC2, RANBP3, NUP53, NUP160, MCM2, IPO7, and XPO1, that are involved in diverse biological functions, including DNA binding/transcription, cell cycle regulation, and nuclear pore complex assembly.31–34 To determine if these proteins require their πφ-LxVx motifs to bind CN, we synthesized nine πφ-LxVx -containing peptides (SKP1 has two predicted πφ-LxVx sequences) and nine πφ-LxVx controls (AAAA replacing the LxVx sequence). The peptides were immobilized on sepharose resin, incubated with HEK293T lysates, and analyzed for CN binding by Western blot (Figure 2A). Eight of the nine πφ-LxVx peptides interacted specifically with CN, an interaction that was abolished or strongly reduced with the control peptides (LxVx →AAAA). For the MCM2 peptide, we could not consistently detect an interaction with CNA, despite detecting binding of CN to full-length MCM2 (Figure 1F). This indicates that the LxVx motif was either incorrectly predicted or that the interaction affinity was too weak to be detected using this method. Taken together, these results showed that the πφ-LxVx sequences are correctly predicted using the SBSC-analyses, increasing the confidence that the 116 proteins identified in the CN176 data sets are novel CN regulators or substrates.
Figure 2.

LxVP SLiM binding to CN. (A) Western Blot analysis of endogenous CNA binding to immobilized WT LxVP and mutant AAAA peptides. (B) kon and koff rates for denoted LxVP SLiM peptides binding to CN as detected by SPR. (C) Model of CN bound to the πφLxVP motif (TTLSPA) of Nup53 (using PDBID: 5SVE) to highlight CNBN122A. (D) SPR traces for Nup53 binding with CN and CNN122A.
The Affinities of πφ-LxVx Motifs for CN Are Driven by Their On-Rates.
To understand how πφ-LxVx amino acid composition contributes to CN affinity, we used surface plasmon resonance (SPR). Specifically, we measured the binding kinetics of 10 πφ-LxVx peptides derived from either our newly identified CN interactors (7 distinct LxVx peptides), NFATc2, Na+/H+ Exchanger 1 (NHE1, SLC9A1), or NFATc1 (Table 1). The πφ-LxVx sequences from NFATc2 and NHE1 were established previously,16,35 while the πφ-LxVx peptide from NFATc1 was used as a binding control (equilibrium and kinetic SPR experiments showed that the affinity of the NFATc1 LxVx for CN, KD = 6 μM, was, as expected, similar that determined previously using ITC18). The sequences of these 10 πφ-LxVx peptides differed in the core LxVP residues (positions i to i+3) as well as residues in the i-1 and i-2 positions (the location of the πΦ-motif), a region recently recognized to contribute to CN binding.18
Table 1.
Binding Kinetics for CN370 Association with LxVP Peptides Measured by SPRa
| protein | sequence | kon (M−1 s−1) | koff (s−1) | KD (μM) | χ2 | N |
|---|---|---|---|---|---|---|
| NFATc1 | DDYLAVPQHPYQWAKPK | 1.97 × 104 ± 3.23 × 103 | 0.113 ± 0.008 | 5.9 ± 1.3 | 0.79 ± 0.16 | 4 |
| NheI | QKINNYLTVPAHKLDSP | 9.45 × 103 ± 1.11 × 102 | 0.173 ± 0.010 | 18.3 ± 1.1 | 0.37 ± 0.04 | 3 |
| Nup53 | PRKTTLSPAQLDPFYTQ | 3.37 × 103 ± 7.97 × 102 | 0.117 ± 0.024 | 35.0 ± 3.5 | 0.73 ± 0.07 | 6 |
| Nup53b | 1.51 × 103 ± 4.43 × 102 | 0.218 ± 0.034 | 150 ± 30 | 0.29 ± 0.08 | 5 | |
| RANBP3 | GQNMSERVLSPPKLNEVS | 2.97 × 103 ± 5.32 × 102 | 0.248 ± 0.046 | 84.7 ± 17.2 | 0.49 ± 0.09 | 5 |
| NFATc2 | ESILLVPPTWPKPLVP | 1.20 × 103 ± 2.92 × 102 | 0.313 ± 0.059 | 265 ± 33 | 0.26 ± 0.08 | 5 |
| Nup160 | GALERSFVELSGGAERRERPR | 5.10 × 102 ± 7.72 × 101 | 0.227 ± 0.053 | 460 ± 179 | 0.35 ± 0.06 | 3 |
| SKP1.1 | ENKEKRTDDIPVWDQEFLK | 4.42 × 102 ± 5.38 × 101 | 0.268 ± 0.081 | 616 ± 208 | 0.33 ± 0.09 | 3 |
| GCFC2 | EGSESRTLDVSTDEEDKIH | 2.84 × 102 ± 3.65 × 101 | 0.211 ± 0.070 | 752 ± 251 | 0.25 ± 0.11 | 3 |
| IPO7 | RDVPNETLQVEEDDRPE | 2.17 × 102 ± 8.81 × 101 | 0.241 ± 0.026 | 1244 ± 442 | 0.29 ± 0.16 | 4 |
| MCM2 | DALTSSPGRDLPPFEDESE | 3.09 × 102 ± 5.76 × 101 | 0.267 ± 0.059 | 864 ± 147 | 0.25 ± 0.10 | 4 |
Values are represented as the mean ± standard deviation (SD).
CNBN122A
The SPR data showed that LxVx-containing peptides bind CN with a broad range of affinities, with KD values from 35 to >500 μM (Figure S2). The newly identified CN interactors with the tightest LxVx-CN affinities are Nup53 (KD, 35 μM, LsPA) and RANBP3 (KD, 78 μM, LsPP). In contrast, the LxVx sequence from NFATc2 (LlVP), a well-established CN substrate, had a much lower affinity (KD, 276 μM) (Table 1). Similarly, the Nup160 LxVx peptide (FvEL), which has phenylalanine in the “i” position, had an even lower affinity (KD, ~330 μM). Finally, the LxVx peptides from GCFC2 (LdVS), IPO7 (LqVE), SKP1.1 (IpVW), and MCM2 (LpPF) all had KDs for CN ≥ 500 μM (Table 1). Remarkably, there is no obvious correlation between the πφ-LxVx sequence and binding affinity. However, there is a strong correlation between binding affinity and the peptide on-rates (kon). Namely, while there is only a 2 to 3-fold difference between the πφ-LxVx peptide off-rates, there is ~100-fold difference in the corresponding on-rates and these correlate with the resulting KDs (Figure 2B). Taken together, these data show that while the sequence of the LxVx motif is important for binding (i.e., the AAAA peptides do not bind), the preexisting conformations of the πφ-LxVx motif that regulate (either allow for or inhibit) rapid binding to the preformed CN LxVP binding pocket determine the binding affinity. This demonstrates that predicting the binding strength of the LxVP motif using the amino acid sequence alone will be difficult.
CNB Asn122 Facilitates πφ-LxVx Peptide Binding.
To better understand the molecular basis for the distinct binding affinities between very closely related peptides (NFATc1 and Nup53 vs NFATc2 and RANBP3), we analyzed CN structures bound to πφ-LxVx motifs (CN:A238L, CN:NFATc1, CN:NHE1).15 We discovered that CNB residue Asn122 can hydrogen bond to the carbonyl of the residue in the “i” position of the LxVx motif. The CNB:Asn122 hydrogen bond can be further stabilized by polar residues in the i-1 position or disrupted by a bulky hydrophobic residue. Indeed, a model of the tightest binding πφ-LxVx motif peptide (LsPA, from Nup53) bound to CN reveals that the position of the i-1 and i-2 amino acids (both Thr) can provide hydrogen bonding interactions with Gln50 (CNB) and Asn122 (CNB) (Figure 2C). To test the role of Asn122-mediated hydrogen bonds on πφ-LxVx binding, we generated a CNN122A variant. CNN122A and WT-CN have similar melting temperatures: 58.5 vs 59.5 °C, respectively; Figure S3. As predicted, the SPR data show that Nup53 binds CNN122A 4-fold weaker than WT-CN with a KD of 150 μM, confirming the importance of interactions outside the core LxVx motif for πφ-LxVx motif binding (Figure 2D).
Analysis of 119 Novel CN Interactors.
Ninety-one of the 119 newly identified CN-interacting proteins have SBSC-predicted πφ-LxVx and PxIxIT SLiMs. Of the remaining 28 proteins, 3 are predicted to have only a PxIxIT SLiM: Plastin-2 (PLSL), which plays a role in T-cell activation; the cell cycle protein ZWINT, which is part of the MIS12 complex required for kinetochore formation and spindle checkpoint activity; and CP055/SPATA33, whose function is currently unknown. The last 25 identified proteins contain only a πφ-LxVx motif. As the LxVP site is the CSA/FK-506 binding site and the key substrate binding site,36 such single SLiM substrate recruitment might be conceivable. This is in line with the ser/thr protein phosphatase PP2A:B56, where a single SLiM (LxxIxE) is responsible for the recruitment of substrates.12 However, additional regulation (e.g., dynamic charge:charge interactions) might allow for critical modulation of these interactions.37
Next, we examined the functional distribution of the identified CN interactors. The largest subset of interactors (21%; 24 interactors) are involved in transcription and translation (Figure 3A). This is not surprising, as CN has long been known to regulate the activity of multiple transcription factors, especially the NFATs. The second-largest group of interactors is involved in the function, formation, or regulation of the nuclear pore. Interestingly, we identified that ~1/3 of the known nuclear pore complex proteins likely interact with, and are possibly regulated by, CN. Indeed, our data indicate that CN is part of nearly every nuclear pore complex, excluding only the Nup62 complex which forms the nuclear envelope (Figure 3B,C). Finally, we discovered that CN associates with XPO1, an essential regulator of nuclear import/export.38
Figure 3.

Expanded view of CN’s biological functions. (A) Biological function of LxVP and PxIxIT SLiM-containing proteins enriched in CNA pulldowns. (B) Schematic of the nuclear pore and nuclear pore complexes. Proteins enriched in CNA pulldowns are named. (C) GO analysis of cellular compartments of SLiM-containing proteins enriched in CNA pulldowns.
Not surprisingly, a large number of identified CN interactors are involved in signal transduction. Thirteen identified CN interactors participate in intracellular signaling pathways, with eight interacting directly with, and possibly regulating, protein kinases, including GSK3B, the TAK1 adaptor protein TAB3, and Phosphatidylinositol 4-kinase alpha (PI4KA).39–41 Our data showed CN interacting not only with PI4KA but also with its effector proteins FAM126A and TTC7A/B, proteins involved with localizing PI4KA to the plasma membrane. The remaining 48 proteins that bind to CN are involved in more diverse biological processes such as metabolism, cell cycle regulation, and protein degradation (Figure 3A).
Are the Identified CN Interactors Also CN Substrates?
To determine if these SLiM-containing CN binding proteins are not only CN interactors but also substrates, we performed in vitro phosphatase assays using recombinant CN (CNA1–391,CNB1–160). We expressed and purified proteins selected for validation (SKP1, GCFC2, RANBP3, NUP53, MCM2, IPO7, and XPO1) and determined their phosphorylation state using LC-MS/MS (Figure 4A). Because we had no prior knowledge of opposing kinases, we relied on endogenous phosphorylation states as the substrate of our analysis. This approach is limited, as not all phosphorylation sites are accessible by mass spectrometry. Furthermore, it is possible that CN opposed sites are low in occupancy due to endogenous CN activity or lack of kinase activity under basal conditions and are not reproducibly detected. For IPO7, we did not detect any phosphorylation sites under our growth conditions. For SKP1 and XPO1, we identified phosphorylation sites but did not detect dephosphorylation by CN. In contrast, for GCFC2, MCM2, NUP53, and RANBP3, we found several CN-dependent phosphorylation sites (Figure 4A). Importantly, for each of these four proteins, we identified phosphorylation sites that were specifically and reproducibly dephosphorylated by CN while other phosphorylation sites were unchanged (Figure 4B). For example, for GCFC2, residue Ser40 is specifically dephosphorylated by CN while Ser180 is not. CN dephosphorylated three sites on MCM2 (Thr25, Ser27, and Ser41) and NUP53 (Ser53, Ser55, and Thr273) and four sites on RANBP3 (Ser126, Thr128, Thr219, and Ser341).
Figure 4.

CN substrate identification. (A) Experimental identification using an in vitro phosphatase assay. (B) Plot of phosphorylation site abundance of indicated protein in CN phosphatase assays. Note, for each protein at least one site was detected that was not dephosphorylated by CN (*p < 0.05). (C) Western blot analysis of RANBP3 phosphorylation on Ser58/Ser126 in untreated and Cyclosporine A (CSA) treated HeLa cells and quantification of protein-corrected RANBP3 phosphorylation (*p-value < 0.005). Plot of Ser126 phosphorylation site abundance of RANBP3 WT, LxVP (AxAP), PxIxIT (AxAxAA), and double (AxAP/AxAxAA) mutants in CSA treated HEK293T cells (D) and in CN in vitro phosphatase assays (E) (*p < 0.05). (F) Immunofluorescence images of HeLa cells treated with or without CSA and probed for SMAD2 and DNA. (G) Quantification of nuclear and total SMAD2 in HeLa cells treated with CSA or not (n = 90 cells in three independent replicates, *p < 0.001).
To determine the importance of one of these dephosphorylation events within a cellular context, we monitored the dephosphorylation of RANBP3 in HeLa cells. Phosphorylation of RERTpSSLTQ in RANBP3 (Ser126 in RANBP3 isoform 1 and 2; Ser58 in RANBP3 isoform 3) by RSK or AKT was previously shown to increase the affinity of RANBP3 for Ran, establishing the Ran gradient.32 Furthermore, RANBP3 Ser58/Ser126 phosphorylation was shown to regulate nuclear export of SMAD2 and SMAD3.42 Upon TGF-β stimulation, PPM1A can dephosphorylate Ser58/Ser126, resulting in increased SMAD2/3 export.42 To determine if CN dephosphorylates RANBP3 Ser58/Ser126 under basal growth conditions, we treated HeLa cells with the CN inhibitor Cyclosporine A (CSA) for 45 min and probed cell lysates with an anti-pSer58/pSer126-RANBP3 antibody. In the presence of CSA, Ser58/Ser126 phosphorylation was significantly increased by approximately 2-fold, showing that CN dephosphorylates RANBP3 under basal conditions (Figure 4C). Furthermore, we immunoprecipitated 3xFLAG-RANBP3 under both conditions from HEK293T cells to compare phosphorylation levels (Figure 4D and Figure S4A). These analyses confirmed that upon CSA treatment, the four sites did increase. To investigate the relative contribution of the LxVP and PxIxIT sites in RANBP3 on the CN ability to dephosphorylate it, we generated single (AxAP and AxAxAA) and double mutants. While in vitro CN was ability to dephosphorylate mutant RANBP3 (Figure 4E and Figure S4B), in cells mutations of the PxIxIT site abrogated dephosphorylation by CN for most sites (Figure 4D and Figure S4A).
Next, we tested if differential phosphorylation of RANBP3 affects the ratio of cytoplasmic versus nuclear SMAD2. For this, we compared SMAD2 localization in HeLa cells treated with or without CSA for 45 min by immunofluorescence (Figure 4F). Cytoplasmic and nuclear SMAD2 staining intensity was quantified in three independent replicates (n = 90 cells for each treated and control condition) and revealed a significant increase of nuclear SMAD2 staining (~15%) upon 45 min CSA treatment, while total SMAD2 levels were not changed (Figure 4G). These data suggest a possible role of CN in regulating phosphorylation-dependent nuclear-cytoplasmic transport.
DISCUSSION
CN plays a central role in a diverse set of biological processes;7 however, through which substrates these functions are achieved is largely unknown. Indeed, recent data suggest that many additional CN functions are currently not widely recognized.18 Here, we develop a combinatorial approach that integrates structural, biophysical, bioinformatics, and mass spectrometry data to identify novel CN interactors and substrates. While peptide-array based approaches for SLiM sequence identification have been useful,22 these studies are of limited use for short SLiMs such as the πφ-LxVx motif, especially when the SLiMs are largely anchored by a single residue (here the “L” of the LxVx). We show that this is because the binding on-rate (kon) of the πφ-LxVx motif, more than the identity of the residues that comprise the motif, is the central driver of binding strength. Specifically, the more readily the motif and its adjacent residues facilitate the sampling of the bound-state conformation, the more tightly the πφ-LxVx motif will bind CN, as its binding on-rate will be faster. Similarly, if the adjacent residues hinder the sampling of the elongated/bound-state conformation, the weaker the πφ-LxVx motif will bind CN, as its binding on-rate will be slower. We find that charged residues adjacent to the core motif or proline substitution at the i+2 and i+3 residues restrict the conformational space of the ensemble, resulting in weaker interactions (higher KD).
The combination of biophysical and structural data allowed for the novel description of the πφ-LxVx motif, that is, πϕ[LIVF]x[VPLIHA]x, that was used in this analysis. By combining affinity-mass spectrometry and SBSC, we identified 119 proteins that we predict interact directly with CN via a πφ-LxVx motif, a PxIxIT motif, or both. Canonical CN interactors such as the CN B-subunit and calmodulin, CN inhibitor such as Cabin1 and RCAN1/2/3, and CN substrates such as the NFAT transcription factors, were readily identified. This is important as it confirms that our novel methodology (MS coupled with SBSC for SLiM identification) identifies well-known CN regulators and substrates. Of the 119 SLiM motif-containing proteins identified to interact directly with CN, 3 contained only a PxIxIT sequence, while 25 contained only an πφ-LxVx sequence. This is reminiscent of the scaffolding protein AKAP79, which was originally shown to bind CN via a PxIxIT motif alone.23 However, it was later discovered that AKAP79 does have an πφ-LxVx motif.43 It will be important to determine if PxIxIT- or LxVx-only CN interactors exist, or if they contain atypical PxIxIT or πφ-LxVx motifs that are not captured with our current approach. Further, alternative binding interactions for CN recruitment toward its substrates may exist.
While the presence of both the PxIxIT and πφ-LxVx motifs are known to enhance the binding affinity of CN regulators and substrates,15,35 a detailed understanding of how CN function is controlled in both a spatial and temporal manner by these SLiMs has not been established. Recently, we have directly shown that these interactions are critical to regulate NHE1.44 Mutation of the πφ-LxVx motif in NHE1 resulted in an increase in dephosphorylation of specific phosphorylation sites compared with others within NHE1; thus, the πφ-LxVx motif is a critical substrate specifier for NHE1. Interestingly, mutation of the PxIxIT site decreased the rate of dephosphorylation, and disruption of both SLiMs resulted in a loss of enzymatic specificity, indicated by an increase in the dephosphorylation rate of the other sites. This suggests that the PxIxIT motif is important for anchoring/targeting CN to discrete cellular compartments, thereby driving the spatial component. The πφ-LxVx, at least in the case of NHE1, controls substrate specificity and thus plays a role in the temporal aspect of phosphatase signaling. We expect that both of these mechanisms play a role in other CN substrate interactions but will be tuned by the strength of the πφ-LxVx interaction and its location relative to both the PxIxIT SLiM and the phospho-serine/threonine that is the direct substrate of CN.
Critically, we confirmed our expanded πφ-LxVx motif predictions both in vitro and in cells, demonstrating the robustness of the MS-SBSC methodology. We then extended the work to show that a subset of these interactors are bona fide CN-specific substrates by identifying CN-dependent phosphorylation sites. Together, these data confirm that the CN SLiM predictions are of high quality and can be employed for future studies to understand the underlying biological processes controlled by CN. For example, our data suggest CN has major roles in regulating transcription by modulating the activity of a variety of transcription factors and signaling pathways. For the latter, it is interesting to discover that CN seems to interact directly with kinases and thus likely influences and may control their activity. Furthermore, CN clearly has significant roles in the regulation of the nuclear pore. Here we show CN is likely involved in nuclear pore export through its interaction with and dephosphorylation of RANBP3.42 CN inhibition had only a limited effect on RANBP3 phosphorylation and SMAD2 localization under basal conditions, suggesting that either CN only contributes but does not drive the regulation of nuclear export or that CN’s regulates this process under specific cellular or signaling conditions.
Indeed, Gene Ontology enrichment analysis on CN interactors identified PxIxIT and/or πφ-LxVx motif(s) and found a strong enrichment in proteins that constitute the nuclear pore and envelope. These proteins include components of the cytoplasmic filaments and ring, central pore, and nuclear basket, supporting the notion that CN more broadly functions in protein import and export from the nucleus.
Ultimately, our list of MS confirmed CN interactors lays the critical foundation for studying the CN interactome and proteome and, in turn, CNs involvement in a wide variety of biological functions. As we have shown with the regulation of NHE1 by CN, while SLiM interactions are vital, regulation of CN activity and especially substrate specificity is much more complex. Our list of 119 MS confirmed CN interactors and the SBSC predicted SLiMs represents the perfect platform to further our understanding and analysis of each of these interactions for its underlying biological function.
METHODS
Immunoprecipitation.
CNA, Nup53, RanBP3, XPO1, and MCM2 were cloned into the p3xFLAG-CMV10 vector. For proteomics analysis, HEK293T cells were transfected with empty p3xFLAG-CMV10 or p3xFLAG-CMV10-CNA and selected with G418. For reciprocal immunoprecipitations, HEK293T cells were transiently transfected, and control cells were nontransfected cells. For immunoprecipitations, cells were lysed in chilled lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 0.5% Triton X-100, 5 mM sodium beta-glycerophosphate, 1 mM CaCl2, 1 mM DTT, and 1 Complete EDTA-free protease inhibitor tablet (Roche) per 10 mL buffer). Lysates were sonicated (3 bursts for 15 s at 50% input with intermittent cooling), cleared by centrifugation, and incubated with 20 μL of anti-FLAG M2 resin (SIGMA) for 3 h, rotating at 4 °C. Beads were collected by centrifugation, lysate removed, and 1 mL for fresh lysis buffer was added (wash) and collected again by centrifugation. The wash step was performed for a total of three times. Proteins were eluted with 150 μg/mL 3xFLAG peptide (SIGMA). Elutes were separated by SDS-PAGE and visualized by Coomassie staining for MS analysis or analyzed by Western blotting. Western blot intensities were quantified in ImageJ Fiji.45 For Coomassie-stained gels, each lane was cut into four sections, destained, dehydrated, and digested with Trypsin (Promega) (1:100 w/w) overnight. The next day, peptides were extracted with 50% ACN (Burdick & Jackson)/5% formic acid (Burdick & Jackson), and dried. Peptides were analyzed by LC-MS/MS as previously described.46 Statistical analysis of protein quantification was carried out in Perseus47 by two-tailed Student’s t-test. Missing values in controls were imputed in Perseus47 from a normal distribution to enable visualization by Volcano plot. Protein–protein interactions of specifically identified proteins were extracted from the STRING database and analyzed in Cytoscape.48 Gene ontology analysis was carried out using the BinGO plug-in in Cytoscape.
Bioinformatics.
ScanProsite49 was used to identify the CN-specific πϕLxVP SLiM in proteins from the MS derived data set(s). Definitions of the search sequences were based on the experimental three-dimensional CN:NFATc1LxVP complex structure18 and experimentally confirmed LxVP motifs. Filter 1: Disorder prediction via IUPRED50 (IUPRED ≥ 0.4). Filter 2: PxIxIT SLiM (ScanProsite).49 Two PxIxIT definitions were used: [P]-x-x-x-[IV]-[TDHSNQ] NFAT/PVIVIT-like;51 or [P]-x-x-x-x-[IV]-[TDHSNQ] AKAP79-like using an extended CN binding pocket;22 [IV]-[TDHSNQ] are based on experimentally confirmed PxIxIT sequences sites14,15 and an analysis of PxIxIT sites.19 Filter 3: Presence of phosphorylated Ser/Thr residues (using PhosphoSitePlus).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Page, Peti, and Kettenbach laboratories for helpful discussion and comments. The work was supported by grant R01NS091336 from the National Institute of Neurological Disorders and Stroke to W.P., grant R01GM098482 from the National Institute of General Medicine to R.P., and grants R35GM119455 and P20GM113132 from the National Institute of General Medicine to A.N.K. Shared resources at Norris Cotton Cancer Center are supported by grant P30CA023108 from the National Cancer Institute. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00606.
Supplementary table (XLSX)
Peptides used for SPR measurements and their associated affinities, CN interaction network based on the STRING database for PxIxIT and LxVx motif containing proteins, SPR traces for peptides in Table S1, data for measurement of unfolding stability measurements (TM) for CNWT and CNN122A, plot of phosphorylation site abundance of RANBP3 WT, LxVP (AxAP), PxIxIT (AxAxAA), and double (AxAP/AxAxAA) mutants in CSA treated HEK293T cells, and supplementary methods (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Davey NE, and Morgan DO (2016) Building a regulatory network with short linear sequence motifs - lessons from the degrons of the anaphase-promoting complex. Mol. Cell 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ivarsson Y, and Jemth P (2019) Affinity and specificity of motif-based protein-protein interactions. Curr. Opin. Struct. Biol 54, 26–33. [DOI] [PubMed] [Google Scholar]
- (3).Brautigan DL, and Shenolikar S (2018) Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem 87, 921–964. [DOI] [PubMed] [Google Scholar]
- (4).Peti W, Nairn AC, and Page R (2013) Structural Basis for Protein Phosphatase 1 Regulation and Specificity. FEBS J 280, 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Peti W, and Page R (2013) Molecular basis of MAP kinase regulation. Protein Sci 22, 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rusnak F, and Mertz P (2000) Calcineurin: Form and Function. Physiol. Rev 80, 1483–1521. [DOI] [PubMed] [Google Scholar]
- (7).Aramburu J, Rao A, and Klee CB (2001) Calcineurin: From structure to function In Current Topics in Cellular Regulation (Stadtman ER, and Chock PB, Eds.), pp 237–295, Academic Press. [DOI] [PubMed] [Google Scholar]
- (8).Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, Gastinel LN, Habuka N, Chen X, Maldonado F, Barker JE, Bacquet R, and Villafranca JE (1995) Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 378, 641–644. [DOI] [PubMed] [Google Scholar]
- (9).Miller ML, Jensen LJ, Diella F, Jørgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BE, Yaffe MB, Brunak S, and Linding R (2008) Linear Motif Atlas for Phosphorylation-Dependent Signaling. Sci. Signaling 1, No. ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kumar GS, Choy MS, Koveal DM, Lorinsky MK, Lyons SP, Kettenbach AN, Page R, and Peti W (2018) Identification of the substrate recruitment mechanism of the muscle glycogen protein phosphatase 1 holoenzyme. Sci. Adv 4, No. eaau6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Choy MS, Hieke M, Kumar GS, Lewis GR, Gonzalez-DeWhitt KR, Kessler RP, Stein BJ, Hessenberger M, Nairn AC, Peti W, and Page R (2014) Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc. Natl. Acad. Sci. U. S. A 111, 4097–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang X, Bajaj R, Bollen M, Peti W, and Page R (2016) Expanding the PP2A Interactome by Defining a B56-specific SLiM. Structure 24, 2174–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lipinszki Z, Lefevre S, Savoian MS, Singleton MR, Glover DM, and Przewloka MR (2015) Centromeric binding and activity of Protein Phosphatase 4. Nat. Commun 6, 5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li H, Zhang L, Rao A, Harrison SC, and Hogan PG (2007) Structure of Calcineurin in Complex with PVIVIT Peptide: Portrait of a Low-affinity Signalling Interaction. J. Mol. Biol 369, 1296–1306. [DOI] [PubMed] [Google Scholar]
- (15).Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, and Peti W (2013) The Molecular Mechanism of Substrate Engagement and Immunosuppressant Inhibition of Calcineurin. PLoS Biol. l 11, No. e1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Martínez-Martínez S, Rodríguez A, López-Maderuelo MD, Ortega-Pérez I, Vázquez J, and Redondo JM (2006) Blockade of NFAT Activation by the Second Calcineurin Binding Site. J. Biol. Chem 281, 6227–6235. [DOI] [PubMed] [Google Scholar]
- (17).Davey NE, Seo M-H, Yadav VK, Jeon J, Nim S, Krystkowiak I, Blikstad C, Dong D, Markova N, Kim PM, and Ivarsson Y (2017) Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J 284, 485–498. [DOI] [PubMed] [Google Scholar]
- (18).Sheftic SR, Page R, and Peti W (2016) Investigating the human Calcineurin Interaction Network using the πϕLxVP SLiM. Sci. Rep 6, 38920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nguyen HQ, Roy J, Harink B, Damle NP, Latorraca NR, Baxter BC, Brower K, Longwell SA, Kortemme T, Thorn KS, Cyert MS, and Fordyce PM (2019) Quantitative mapping of protein-peptide affinity landscapes using spectrally encoded beads. eLife 8, e40499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Roy J, and Cyert MS (2009) Cracking the Phosphatase Code: Docking Interactions Determine Substrate Specificity. Sci. Signaling 2, No. re9. [DOI] [PubMed] [Google Scholar]
- (21).Roy J, Li H, Hogan PG, and Cyert MS (2007) A Conserved Docking Site Modulates Substrate Affinity for Calcineurin, Signaling Output, and In Vivo Function. Mol. Cell 25, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li H, Pink MD, Murphy JG, Stein A, Dell’Acqua ML, and Hogan PG (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat. Struct. Mol. Biol 19, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nygren PJ, and Scott JD (2016) Regulation of the phosphatase PP2B by protein-protein interactions. Biochem. Soc. Trans 44, 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ryeom S, Greenwald RJ, Sharpe AH, and McKeon F (2003) The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat. Immunol 4, 874–881. [DOI] [PubMed] [Google Scholar]
- (25).Jain J, McCafffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, and Rao A (1993) The T-cell transcription factor NFAT p is a substrate for calcineurin and interacts with Fos and Jun. Nature 365, 352–355. [DOI] [PubMed] [Google Scholar]
- (26).Li H, Rao A, and Hogan PG (2011) Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol 21, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shenkman M, and Lederkremer GZ (2019) Compartmentalization and Selective Tagging for Disposal of Misfolded Glyco-proteins. Trends Biochem. Sci 44, 827–836. [DOI] [PubMed] [Google Scholar]
- (28).Sun L, Youn H-D, Loh C, Stolow M, He W, and Liu JO (1998) Cabin 1, A Negative Regulator for Calcineurin Signaling in T Lymphocytes. Immunity 8, 703–711. [DOI] [PubMed] [Google Scholar]
- (29).Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li S-J, Wang J, Ma L, Lu C, Wang J, Wu J-W, and Wang Z-X (2016) Cooperative autoinhibition and multi-level activation mechanisms of calcineurin. Cell Res 26, 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Benanti JA (2012) Coordination of Cell Growth and Division by the Ubiquitin-Proteasome System. Semin. Cell Dev. Biol 23, 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yoon S-O, Shin S, Liu Y, Ballif BA, Woo MS, Gygi SP, and Blenis J (2008) Ran-binding protein 3 phosphorylation links the Ras and PI3-kinase pathways to nucleocytoplasmic transport. Mol. Cell 29, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Strambio-De-Castillia C, Niepel M, and Rout MP (2010) The nuclear pore complex: bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol 11, 490–501. [DOI] [PubMed] [Google Scholar]
- (34).Riera A, Barbon M, Noguchi Y, Reuter LM, Schneider S, and Speck C (2017) From structure to mechanism—understanding initiation of DNA replication. Genes Dev 31, 1073–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Park S, Uesugi M, and Verdine GL (2000) A second calcineurin binding site on the NFAT regulatory domain. Proc. Natl. Acad. Sci. U. S. A 97, 7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Blumenthal DK, Takio K, Hansen RS, and Krebs EG (1986) Dephosphorylation of cAMP-dependent protein kinase regulatory subunit (type II) by calmodulin-dependent protein phosphatase. Determinants of substrate specificity. J. Biol. Chem 261, 8140–8145. [PubMed] [Google Scholar]
- (37).Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, Kragelund BB, Best RB, and Schuler B (2018) Extreme disorder in an ultra-high-affinity protein complex. Nature 555, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Forbes DJ, Travesa A, Nord MS, and Bernis C (2015) Reprint of “Nuclear transport factors: global regulation of mitosis. Curr. Opin. Cell Biol 34, 122–134. [DOI] [PubMed] [Google Scholar]
- (39).Liu Q, Busby JC, and Molkentin JD (2009) Novel interaction between the TAK1-TAB1-TAB2 and the RCAN1-calcineurin regulatory pathways defines a signaling nodal control point. Nat. Cell Biol 11, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Murphy LLS, and Hughes CCW (2002) Endothelial Cells Stimulate T Cell NFAT Nuclear Translocation in the Presence of Cyclosporin A: Involvement of the wnt/Glycogen Synthase Kinase-3β Pathway. J. Immunol 169, 3717–3725. [DOI] [PubMed] [Google Scholar]
- (41).Baskin JM, Wu X, Christiano R, Oh MS, Schauder CM, Gazzerro E, Messa M, Baldassari S, Assereto S, Biancheri R, Zara F, Minetti C, Raimondi A, Simons M, Walther TC, Reinisch KM, and De Camilli P (2016) The leukodystrophy protein FAM126A/Hyccin regulates PI4P synthesis at the plasma membrane. Nat. Cell Biol 18, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dai F, Shen T, Li Z, Lin X, and Feng X-H (2011) PPM1A dephosphorylates RanBP3 to enable efficient nuclear export of Smad2 and Smad3. EMBO Rep 12, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nygren PJ, Mehta S, Schweppe DK, Langeberg LK, Whiting JL, Weisbrod CR, Bruce JE, Zhang J, Veesler D, and Scott JD (2017) Intrinsic disorder within AKAP79 fine-tunes anchored phosphatase activity toward substrates and drug sensitivity. eLife 6, e30872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hendus-Altenburger R, Wang X, Sjøgaard-Frich LM, Pedraz-Cuesta E, Sheftic SR, Bendsøe AH, Page R, Kragelund BB, Pedersen SF, and Peti W (2019) Molecular basis for the binding and selective dephosphorylation of Na+/H+ exchanger 1 by calcineurin. Nat. Commun 10, 3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lyons SP, Jenkins NP, Nasa I, Choy MS, Adamo ME, Page R, Peti W, Moorhead GB, and Kettenbach AN (2018) A Quantitative Chemical Proteomic Strategy for Profiling Phosphoprotein Phosphatases from Yeast to Humans. Mol. Cell. Proteomics 17, 2448–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- (48).Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003) Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, and Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34, W362–W365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dosztányi Z, Csizmok V, Tompa P, and Simon I (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434. [DOI] [PubMed] [Google Scholar]
- (51).Takeuchi K, Roehrl MHA, Sun Z-YJ, and Wagner G (2007) Structure of the Calcineurin-NFAT Complex: Defining a T Cell Activation Switch Using Solution NMR and Crystal Coordinates. Structure 15, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.