

Abstract
Recent studies indicate that endothelial Nlrp3 inflammasome is critically involved in the development of cardiovascular complications. However, it remains unknown whether endothelial inflammasome is involved in endothelial barrier dysfunction associated with smoking. This study aims to investigate the role of endothelial Nlrp3 inflammasome in nicotine-induced disruption of inter-endothelial tight junctions and consequent endothelial barrier dysfunction. The confocal microscopic analysis demonstrated that mice treated with nicotine exhibited disrupted inter-endothelial tight junctions as shown by decreased ZO-1 and ZO-2 expression in the coronary arterial endothelium, whereas the decreases in ZO-1/2 were prevented by Nlrp3 gene deficiency. In cultured endothelial cells, nicotine caused Nlrp3 inflammasome complex formation and enhances the inflammasome activity as shown by increased cleavage of pro-caspase-1, and interleukin-1β (IL-1β) production. Further, nicotine disrupted tight junction and increased permeability in an endothelial cell monolayer, and this nicotine-induced effect was prevented by silencing of Nlrp3 gene, inhibition of caspase-1, or blockade of high mobility group box 1 (HMGB1). Nicotine increased endothelial cell lysosomal membrane permeability and triggered the lysosomal release of cathepsin B, whereas these events were prevented by pretreating cells with a lysosome stabilizing agent, dexamethasone. Collectively, our data suggest that nicotine enhances cathepsin B-dependent Nlrp3 inflammasome activation and the consequent production of a novel permeability factor HMGB1, which causes disruption of inter-endothelial tight junctions leading to endothelial hyperpermeability. Instigation of endothelial inflammasomes may serve as an important pathogenic mechanism contributing to the early onset of vasculopathy associated with smoking.
Keywords: Nicotine, Nlrp3 inflammasome, endothelial dysfunction, lysosome permeabilization
1. Introduction
Cigarette smoking is a well-established risk factor for cardiovascular diseases including atherosclerosis, ischemic heart disease, acute coronary thrombosis, and hypertension (Hawkins et al., 2002; Kiyohara et al., 1990). Recent studies highlight nicotine as one of the critical compounds of cigarette that mediates the pathogenesis of various smoking-associated diseases(Mazzone et al., 2010; Sliwinska-Mosson and Milnerowicz, 2017). Nicotine may have direct or indirect effects on the vasculature that could contribute to acute cardiovascular events or accelerated atherogenesis experienced by cigarette smokers. Nicotine can inhibit nitric oxide production and cause endothelial dysfunction and microvascular injury (Koide et al., 2005; Luo et al., 2006). It was also shown that nicotine induces atherogenic genes in human coronary artery endothelial cells (Kudo et al., 2014; Zhang et al., 2001). The indirect effects of nicotine relate to elevated serum cholesterol level that causes endothelial injury or through activation of the sympathetic nervous system (SNS) (Joukar et al., 2012; Suzuki et al., 1973).
The Nlrp3 inflammasomes have been identified as intracellular machinery responsible for activation of the inflammatory response in various tissues or organs (Mariathasan et al., 2004; Strowig et al., 2012; Wen et al., 2012). Moreover, recent studies demonstrated that Nlrp3 inflammasomes are critically involved in cardiovascular diseases (Abais et al., 2013; Chen et al., 2016; Menu et al., 2011; Xu et al., 2012). The Nlrp3 inflammasome consists of a proteolytic complex formed by Nlrp3, the adaptor protein ASC (apoptotic speck-containing protein with a CARD), and pro-caspase-1 (Martinon et al., 2002). Formation of inflammasome complex leads to activation of caspase-1, which proteolytically cleaves IL-1β or IL-18 into their biologically activate form, and releases other damaging factors such damage-associated molecular patterns (DAMPs) (Busso and So, 2010; Dinarello et al., 2010; Martinon et al., 2009; Rathinam et al., 2012; Rodriguez et al., 2009; Zheng et al., 2011). All these caspase-1-dependent factors may turn on inflammatory responses and cause cell dysfunction. Moreover, sustained activation of caspase-1 may trigger pyroptotic cell death or deranged cell metabolism (De Nardo and Latz, 2011; Kepp et al., 2010; Lamkanfi, 2011).
Endothelial dysfunction induced by cardiovascular risk factors like cigarette smoking or nicotine exposure is believed to be an early onset of cardiovascular complications. Inter-endothelial junction complex consists of ZO-1/2 proteins that critically regulate endothelial junction permeability. Losses of ZO-1/2 protein expression are associated with junction disruption and consequent dysfunction of endothelial barrier leading to plasma protein leakage and leucocyte transmigration into interstitium (Hernandez et al., 2007; Vandenbroucke et al., 2008). Recent studies demonstrated that danger factors including free fatty acid and Lactobacillus casei cell wall fragments induce lysosome destabilization that activates endothelial Nlrp3 inflammasomes (Chen et al., 2015a; Wang et al., 2016). It has been suggested that Nlrp3 inflammasome-mediated release of high mobility group box 1 (HMGB1) is a triggering mechanism to mediate the junction disruption, the early onset of endothelial dysfunction, in various disease settings including obesity, diabetes, and coronary arteritis (Chen et al., 2015b; Chen et al., 2016; Wang et al., 2016). Therefore, the present study aims to investigate whether nicotine could disrupt inter-endothelial junctions through the Nlrp3 inflammasome pathway. We determined whether Nlrp3 deficiency could protect against nicotine-induced junction disruption in arterial endothelium of mice. In cultured endothelial cells, we further explored the related mechanism with a focus on the instigation of Nlrp3 inflammasome and release of HMGB1. Our study provides novel insights into the therapeutical potential of targeting inflammasomes for preventing the early onsets of vasculopathy associated with smoking or nicotine exposure.
2. Materials and methods
2.1. Mice
Nlrp3+/+ and Nlrp3−/− male mice (C57BL/6J background) at 8 weeks of age were used (Jackson Laboratory, ME). The genotyping of mice was typed following the vendor’s protocol. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University (Protocol AM10174). Mice were injected with nicotine (i.p., 2 mg/kg/d; Sigma, MO) or phosphate buffer solution (PBS) for two weeks. This dose of nicotine is approximately a dose comparable to the amount of nicotine that a person smoking 50 cigarettes a day (Armitage et al., 1968). Mice were killed after treatment above and heart tissues were collected and frozen in Tissue-Tek OCT.
2.2. Immunofluorescence analysis of proteins in coronary arterial endothelium
Confocal immunofluorescence analysis was preformed to detect junction protein ZO1/2 in the coronary arterial endothelium as previous described (Wei et al., 2013a). Briefly, the hearts collected from above were cut into 10 μm sections using a cryostat and followed by mounting on Superfrost Plus slides. The frozen section slides were then fixed with acetone and incubated with primary antibodies from Invitrogen against ZO-1 (1: 100) or ZO-2 (1:100). To visualize the vascular endothelium, slides were also co-stained with primary antibodies against Von Willebrand Factor (vWF) (1:200, Abcam). After incubation with primary antibodies for 1 h at room temperature, slides were washed with PBS and incubated with corresponding secondary antibodies conjugated with fluorescent cores alexa Fluor-488 or -555 (1:100, Invitrogen, NY) for 1 h at room temperature followed by two washes with PBS and mounted. The immunofluorescence was analyzed using a Fluoview FV1000n Olympus laser scanning confocal microscope. The fluorescence intensity ratio of junction protein ZO-1/2 over vWF was calculated using Image Pro Plus software to represent the colocalization coefficient.
2.3. Culture of MVECs
MVECs (mouse microvascular endothelial cells) were cultured as recently described (Guan et al., 2018 10.1111/bcpt.13146). Briefly, MVECs from ATCC were cultured in DMEM supplemented with fetal calf serum (10%) and 1% penicillin/streptomycin (Gibco, NY). Cells were passaged using Trypsin-EDTA (Sigma, MO) and were maintained in DMEM in a humidified incubator at 37°C with 5% CO2 and 95% air.
2.4. Immunofluorescence microscopic analysis of MVECs
The colocalization of Nlrp3 inflammasome subunits was analyzed to determine the effects of nicotine on the assembly of the Nlrp3 inflammasome complex in MVECs. Cells were stimulated with or without nicotine at indicated concentration for 24 h. After stimulation, cells grown on glass coverslips were terminated by a 15-min fixation in 4 % PFA. Cells were permeabilized in 0.3% Triton X-100 in phosphate-buffer saline (PBS) for 15 min. Cells were washed in PBS followed by 2-h incubation with primary antibodies at 4 °C. The primary antibodies against inflammasome component are from Abcam including Nlrp3 (1:100), ASC (1:100), and caspase-1 (1:200). The primary antibodies against junction protein are from Invitrogen ZO-1 (1:500) and ZO-2 (1:500). The slides were washed with PBS, and incubated with secondary antibodies conjugated with alexa Fluor-488 and alexa Fluor-555 (Invitrogen). The immunofluorescence was analyzed using a Fluoview FV1000n Olympus laser scanning confocal microscope. Pearson’s correlation coefficient was calculated using Image Pro Plus software to represent the colocalization coefficient (Zhang et al., 2014).
2.5. Cell viability assay
The cell viability was assessed by lactate dehydrogenase (LDH) assay as reported (Wang et al., 2019a). LDH activity in the cell culture medium was determined by Piercetm LDH Assay Kit (Thermo, IL, USA) according to the manufacturer’s protocol. The absorbance was spectrophotometrically quantified at 490 nm using a CLARIOstar microplate reader (BMG Labtech, Germany). The maximum LDH activity (LDHmax.) was obtained from samples treated with lysis buffer. The basal level of LDH activity (LDH0) due to spontaneous LDH release was obtained from control samples. The survival was calculated as percentage of (LDHmax-LDHsample) over (LDHmax-LDH0).
2.6. Western blot analysis
MVECs were lysed by incubation with sucrose buffer (pH7.4) containing 20 mM HEPES, 1 mM EDTA, 255 mM sucrose, and cocktail protease inhibitors (Roche, Nutley, NJ) as described(Wang et al., 2019b). Protein samples were denatured in 5×Laemmli SDS-sample buffer with 5-min heat at 95°C. 25 μg of protein samples were separated by SDS-PAGE gel, transferred to PVDF membrane, and blocked with 5% bovine serum albumin (BSA). The membranes were incubated overnight at 4°C with antibodies (1:500) against caspase-1 (Santa Cruz), or against ZO-1/2 (Invitrogen). The membranes were then incubated with secondary antibodies conjugated to horseradish peroxidase (1:5000, Santa Cruz). The target bands were augmented by chemiluminescence reagents (Pierce, Rockford, IL) and visualized on Kodak film. β-actin was detected as a control of protein loading. The band intensity was quantified by NIH provided ImageJ 6.0 software as we described (Zhang et al., 2018).
2.7. Nucleofection
Nucleofection was performed to transfect shRNA plasmids into MVECs using a 4D Nucleofector (Lonza, CA) (Chen et al., 2015b; Wei et al., 2013b). The Nlrp3 shRNA plasmid was purchased from Origene (#TG510752). In brief, cells were dissociated with trypsin and pelleted by 10-min centrifugation at 80×g. Then, for each Nucleofection, cell pellet with 106 cells was resuspended in 100 μl Nucleofection solution P1 (Lonza, CA) and 2 μg plasmid DNA were added into cell mixture. Nucleofection was executed with the program code DS198 and maintained in fresh medium. Flow cytometry analysis showed that 80% of cells Nucleofected with GFP control plasmid under this program are GFP-positive. The silencing efficiency for Nlrp3 shRNA was confirmed as we described previously (Chen et al., 2016).
2.8. IL-1β and HMGB1 quantitation
MVECs were cultured in 6-well plates until 90% confluency and treated with nicotine (0-100 nM) for 24 h. The cell supernatants were collected after cell stimulation and treatment. IL-1β concentration in the cell supernatants was determined by an ELISA kit (R&D System, Minneapolis, MN) (Chen et al., 2015b). Quantification of HMGB1 release in cell supernatants was also determined by using ELISA kit (MyBioSource, USA)(Chen et al., 2015b).
2.9. Analysis of lysosome membrane permeability
Acridine orange staining was used to analyze the lysosomal membrane permeability as we previously described (Chen et al., 2015a). Cells cultured in 8-well chambered coverslips until 90% confluency and were treated with 100 nM nicotine for 24 h in the presence or absence of dexamethasone (100 μM). Then, cells were incubated with 1 μM acridine orange (Sigma, MO) for 20 min at 37°C. Then cells were washed two times with warmed PBS to remove extracellular acridine orange dyes. Cells on the coverslips were immediately photographed using LSM710 fluorescence microscope (Zeiss, Bonn, Germany).
2.10. Cathepsin B release assay
The release of cathepsin B from disrupted lysosomes was determined by CV-(RR)2 assay as described (Nurmi et al., 2013). Cells cultured in a 8-well chambered coverslip were treated with 100 nM nicotine for 24 h in the presence or absence of dexamethasone (100 μM) and stained with a fluorescent cell-permeable selective cathepsin B substrate z-Arg-Arg-cresyl violet (CV-(RR)2) and DAPI with a CV-(RR)2 kit according to the manufacturer’s protocols (Enzo, MI). The CV-(RR)2 fluorescence in live cells were examined using LSM710 fluorescence microscope (Zeiss, Germany).
2.11. Measurement of endothelial barrier function
The barrier function of the endothelial monolayer of MVECs was assessed by determining the transendothelial electrical resistance (TEER) at low frequency (4000 Hz). Briefly, cells were grown on gold electrodes (in 8W10+E arrays) till confluence and maintained in medium with 1% FBS. Cells were challenged with nicotine in the presence or absence of inhibitors as indicated in the Figure legend. TEER was measured using electrical cell-substrate impedance sensing (ECIS) ZΘ system (Applied Biophysics). The changes in TEER for the duration of 12 h were determined.
The barrier function was also confirmed by determining relative permeability of endothelial monolayer to high molecular weight molecule FITC-dextran using a 24-well transwell assay as described (Wang et al., 2016). Briefly, cells were cultured in transwell inserts till confluence and maintained in 1% FBS medium. Cells were challenged with nicotine in the presence or absence of inhibitors as indicated in the Figure legend. After treatment, the inserts were incubated with 200 μl fresh media. Then, in each insert, 100 μl 40 kDa FITC-dextran solution (1mg/ml; Sigma) was added to individual insert. After 2-h incubation at 37 °C, the FITC-dextran molecules (excitation/emission of 485/530 nm) that flow through the endothelial cell monolayer was analyzed by microplate reader (BMG Labtech).
2.12. Statistics
Data are presented as means ± S.D. Significant differences between and within multiple groups were examined using ANOVA for repeated measures, followed by Duncan’s multiple-range test. Student’s t-test was used to evaluate the significance of differences between two groups of observations. P < 0.05 was considered statistically significant.
3. Results
3.1. Nlrp3 gene deletion prevents nicotine-induced endothelial tight junction disruption in mice
We first examined the effect nicotine on the ZO-1/2 expression in the endothelium of coronary arteries of Nlrp3+/+ and Nlrp3−/− mice. To visualize the ZO-1/2 in coronary arteries, heart frozen sections were stained with ZO-1/2 antibodies and co-stained with endothelium marker vWF. The relative fluorescence intensity ratios of ZO-1/2 over vWF were obtained to represent relative expression of ZO-1/2 in the endothelial cells as we described previously (Chen et al., 2016). It was found that mice treated with nicotine showed decreased the levels of ZO-1 (Fig. 1A) or ZO-2 (Fig. 1B) expression in the vWF+ endothelium layer of coronary arteries from Nlrp3+/+ mice. However, the much lower nicotine-induced expression levels of ZO-1/2 were found in coronary arterial endothelium in Nlrp3−/− mice. The summarized fluorescence intensity ratio also confirmed that Nlrp3 deficiency prevented nicotine-induced down-regulation of ZO-1/2. Therefore, these results suggest that nicotine-induced junction disruption in the endothelium is associated with Nlrp3 inflammasomes.
Fig. 1. Nlrp3 deficiency prevents nicotine-induced tight junction disruption in coronary arterial endothelium of mice.
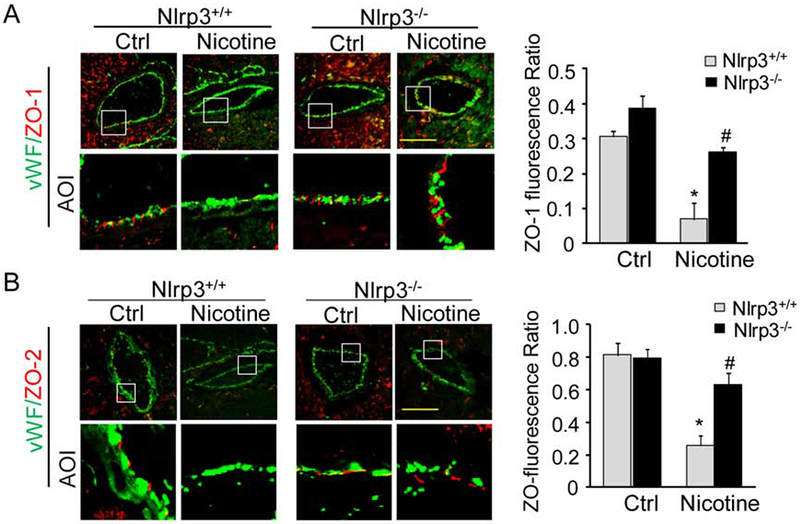
Nlrp3+/+ or Nlrp3−/− mice were treated with nicotine (i.p., 2 mg/kg/d) or vehicle control (PBS) as described in Methods. (A) Fluorescent images show alexa-555-ZO-1 (red) in coronary arteries. The endothelium was visualized by alexa-488-vWF staining (green). The summarized data on the right panel show the fluorescence intensity ratio of ZO-1 over vWF (n=5-7 mice). (B) Fluorescent images show alexa-555-ZO-2 (red) in coronary arteries. The endothelium was visualized by alexa-488-vWF staining (green). The summarized data on the right panel show the fluorescence ratio of ZO-2 over vWF (n=5-8 mice). Each image also includes an image for the area of interest (AOI). Data are presented as means ± S.D. *P < 0.05 vs. Nlrp3+/+ Ctrl; # P < 0.05 vs. Nlrp3+/+ with nicotine. Scale bar = 50 μm.
3.2. Nicotine-induced disruption of inter-endothelial tight junctions
We then examined whether nicotine could directly cause disruption of inter-endothelial junctions. By using fluorescence microscopy, we visualized the protein expression of ZO-1 and ZO-2 in the inter-cellular area of endothelial monolayers of MVECs. To confirm that ZO-1/2 are expressed in cell boundaries, we analyzed the relative fluorescence intensity through the white lines between two cell-cell contacts as previously describe (Chen et al., 2016). As shown in Fig. 2A and 2B, a honeycomb pattern of ZO-1/2 staining was observed in cells under control condition indicating that these proteins were mainly localized in the intercellular junctions of endothelial cell monolayer. Moreover, nicotine stimulation at 100 nM and 1 μM but not 50 nM diminished the immunoreactivity for these proteins and the fluorescence intensity across the cell-cell contacts. We also examined the correlation of the changes in ZO1/2 expression and relative permeability in MVECs. As shown in Fig. 2C, we observed nicotine at 100 nM and 1 μM but not 50 nM could significantly increase the amount of FITC-dextran molecules across the MVEC monolayer. However, LDH cytotoxicity assay showed that nicotine at 1 μM induced cell death (Fig. 2D). Because nicotine at 100 nM could decrease ZO1/2 and increase endothelial permeability without inducing cytotoxicity, we decided to use this optimal dose for other experiments in this study if not particularly mentioned. These results indicate that nicotine indeed disrupts inter-endothelial tight junctions of endothelial cell monolayers, which is associated with increased endothelial permeability.
Fig. 2. Nicotine decreases inter-endothelial tight junction protein expression.
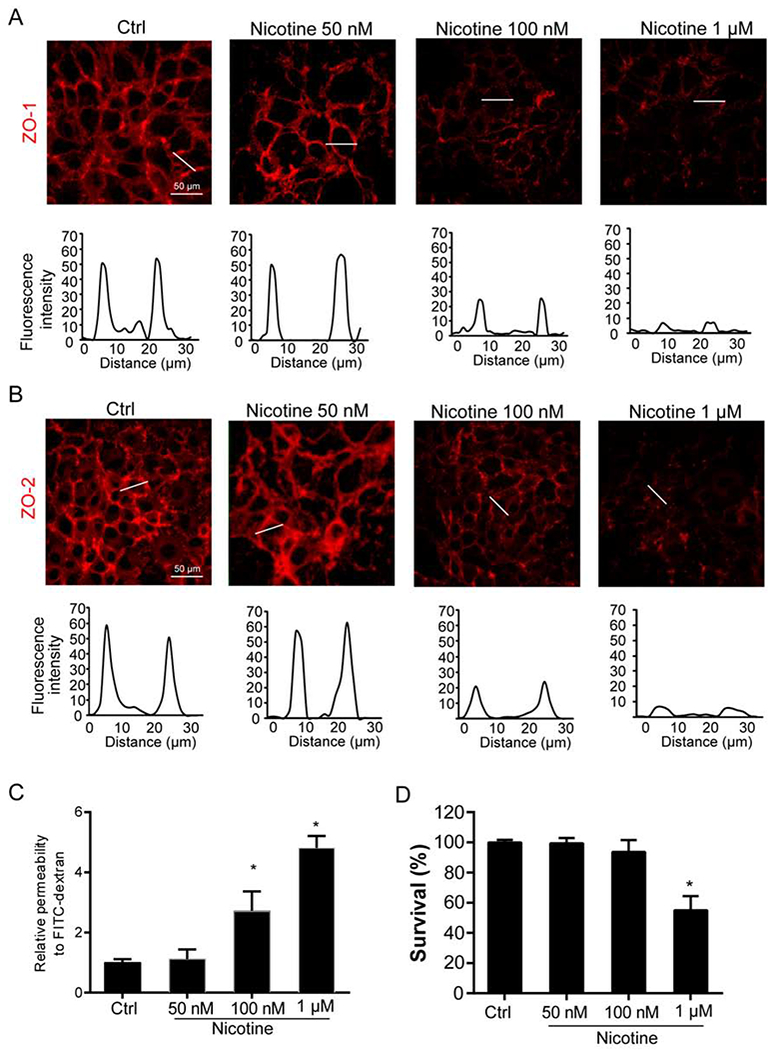
MVECs stimulated with or without nicotine for 24 h. (A) Fluorescence images show the ZO-1 expression from at least three independent experiments. (B) Fluorescence images show the ZO-2 expression from at least three independent experiments. The histograms for fluorescence intensity (RFI) were used to represent the expression of ZO-1/2 at inter-cellular junctions that is indicated by a dotted line across a two cell-cell contacts. (C) EC permeability was determined by measuring the amount of FITC-dextran that crossed the endothelial monolayer cultured on the inserts of transwell (n=5). (D) Cell viability by LDH assay (n=4). Data are presented as means ± S.D. *P < 0.05 vs. untreated control (Ctrl). Scale bar = 50 μm.
3.3. Nicotine increases formation of endothelial Nlrp3 inflammasome complex and its activation
The formation and activation of Nlrp3 inflammasomes were determined by analyzing the assembly of Nlrp3 inflammasome subunits, activation of pro-caspase-1 cleavage into caspase-1, and the production of IL-1β. Nlrp3 inflammasome consists of three major subunits including Nlrp3, ASC, and pro-caspase-1. Upon activation, Nlrp3 inflammasome subunits are assembled and oligomerized into a protein complex, in which pro-caspase-1 is cleaved into active form of caspase-1. Activated caspase-1 then proteolytically cleaves pro-IL-1β or other pro-cytokines into their biologically activate form. In this regard, we analyzed the yellow staining that represents the co-localization of Nlrp3 (green) with ASC (red) and found that nicotine increased the yellow staining (Fig. 3A) and co-localization coefficient (Fig. 3B). A similar increase in co-localization was found between Nlrp3 and caspase-1. It should be noted that nicotine at 100 nM but not 50 nM significantly increased the co-localization for both Nlrp3/ASC and Nlrp3/pro-caspase-1 (Fig. 3B). Consistent with the findings for the formation of Nlrp3 inflammasome complex, we further demonstrated that nicotine increased Nlrp3 inflammasome activity in MVECs as shown by enhanced cleavage of pro-caspase-1 proteins to their bio-active form (p20 subunit) (Fig. 3C), production of important inflammatory cytokines IL-1β (Fig. 3D), and release of danger associated molecule HMGB1 (Fig. 3E).
Fig. 3. Nicotine increases the Nlrp3 inflammasome complex formation, caspase-1 activity and HMGB1 release in MVECs.
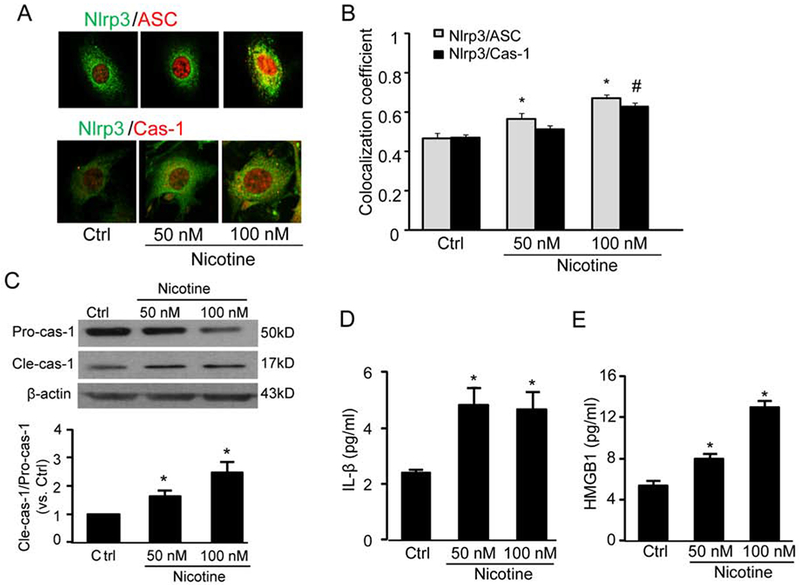
Cells were stimulated with nicotine (0-100 nM) for 24 h. (A) Fluorescence images show the effect of nicotine on the co-localization of Nlrp3 with ASC or Nlrp3 with caspase-1. (B) Colocalization efficiency of Nlrp3 with ASC or Nlrp3 with caspase-1 (n =6). *P < 0.05 vs. control of Nlrp3/ASC; #P < 0.05 vs. control of Nlrp3/caspase-1. (C) Western blot analyses show the effects of nicotine on the expression of pro-caspase-1 (Pro-casp1) or the expression of cleaved caspase-1 (Cle-casp1) (n=4). (D) and (E) IL-1β or HMGB1 release in MVECs (n=6). Data are presented as means ± S.D. *P < 0.05 vs. Ctrl. Scale bar = 50 μm.
3.4. Nlrp3 inflammasome-HMGBl axis is involved in nicotine-induced tight junction disruption
Instigation of Nlrp3 inflammasomes by various danger factors including high glucose and free fatty acids causes disruption of tight junctions, which is attributed to the inflammasome-dependent release of HMGB1, an endothelial permeability factor (Chen et al., 2016; Wang et al., 2016). We previously have shown that gene silencing with Nlrp3 shRNA transfection efficiently downregulates Nlrp3 expression (Chen et al., 2016), and this phenomenon was also confirmed in the present study (Fig. 4A). Moreover, fluorescent microscopy qualitatively showed that Nlrp3 gene silencing reversed the trend of nicotine-induced disassembly of ZO-1 at intercellular junctions (Fig. 4B). Our immunoblot analysis confirmed this trend by showing that nicotine-induced down-regulation of ZO-1 was also reversed by Nlrp3 gene silencing (Fig. 4C). In addition to ZO-1, immunoblot analysis also found Nlrp3 gene silencing attenuated nicotine-induced down-regulation of ZO-2 (Fig. 4C). It should be noted that Nlrp3 gene silencing alone seems to increase ZO-2 expression, however, no statistical significance was observed (Fig. 4C). Similarly, pharmacological inhibition of inflammasome activity by caspase-1 inhibitor WEHD or HMGB1 activity by glycyrrhizin reversed the effects of nicotine on tight junction protein down-regulation as shown by fluorescent imaging studies (Fig. 5A and 5B) and Western blot analysis (Fig. 5C).
Fig. 4. Effects of Nlrp3 gene silencing on the nicotine-induced disruption of tight junction proteins in MVECs.
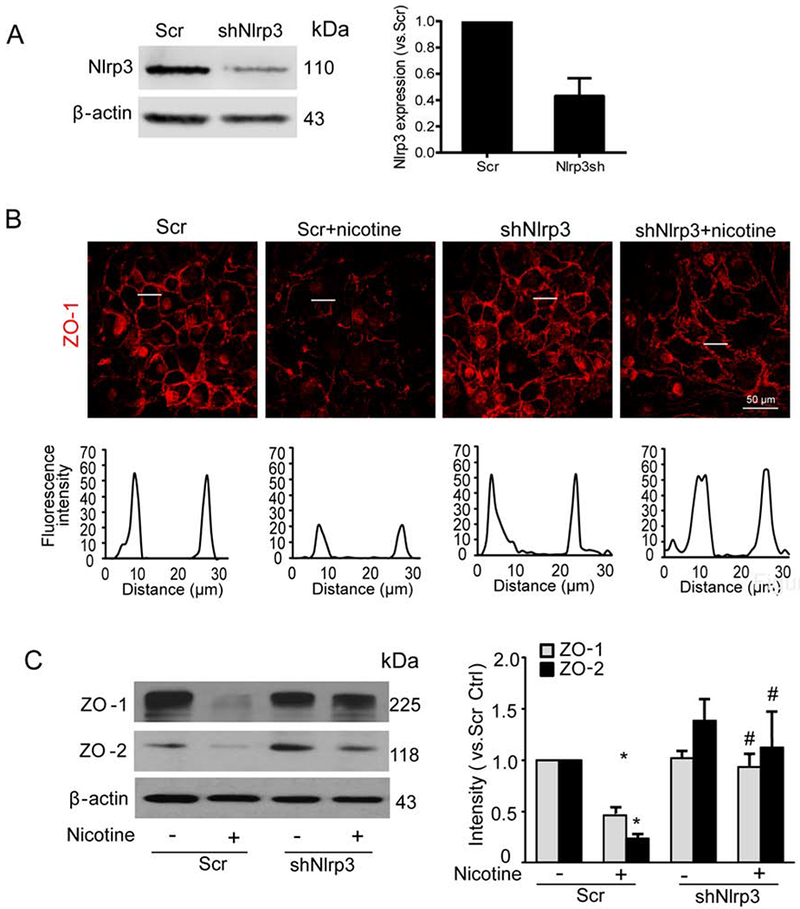
MVECs were silenced with plasmids containing either scramble shRNA (Scr) or Nlrp3 shRNA (shNlrp3). Cells were then treated with or without nicotine (100 nM) for 24 h. (A) Representative Western blot documents and quantified data show the Nlrp3 gene silencing effect (n=2). (B) Fluorescence images show the ZO-1 expression from at least three independent experiments. (C) Representative Western blot documents and summarized data show the effects of nicotine on ZO-1/ZO-2 expression over β-actin (n=4). Data are presented as means ± S.D. *P < 0.05 vs. Scr Ctrl; #<0.05 vs. Scr+Nicotine. Scale bar = 50 μm.
Fig. 5. Inhibition of caspase-1 or HMGB1 activity abolishes nicotine-induced disruption of tight junction proteins in MVECs.
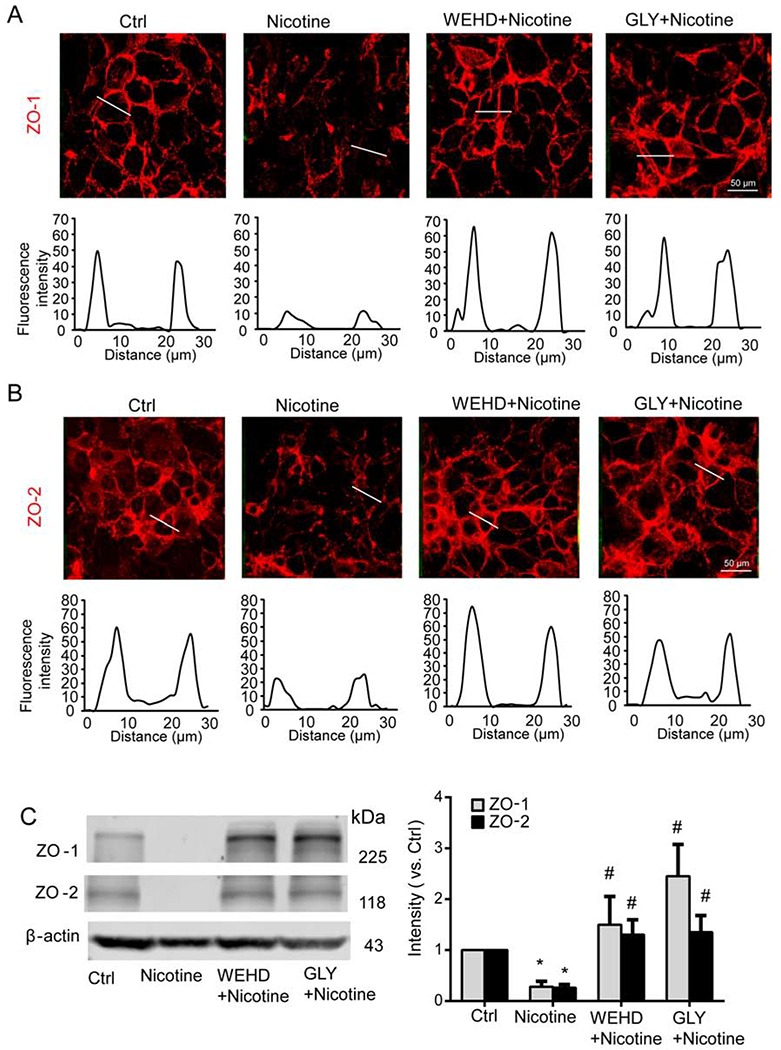
MVECs were treated nicotine (100 nM) for 24 h in the presence of caspase-1 inhibitor WEHD (10 μM) or HMGB1 activity blocker glycyrrhizin (GLY) (130 μM). (A) Representative fluorescence images show the cell membrane fluorescence of ZO-1 from at least three independent experiments. (B) Representative fluorescence images show the cell membrane fluorescence of ZO-2 from at least three independent experiments. The histograms of fluorescence intensity (RFI) were used to represent the expression of ZO-1/2 at inter-cellular junctions that is indicated by a dotted line across a two cell-cell contacts. (C) Representative Western blot documents and summarized data show the effects of nicotine on ZO-1/ZO-2 expression over β-actin (n=4). Data are presented as means ± S.D. *P<0.05 vs. untreated control; #P<0.05 vs. Nicotine alone. Scale bar = 50 μm.
3.5. Activation of Nlrp3 inflammasome by nicotine is associated with lysosome permeabilization
We previously demonstrated that in MVECs, lysosomal destabilization by free fatty acids increases lysosome permeability and results in the release of lysosome protease cathepsin B into the cytosol, where it activates Nlrp3 inflammasomes leading to inter-endothelial junction disruption (Wang et al., 2016). We then determined whether a similar mechanism is responsible for nicotine-induced effects on MVECs. Acridine orange, a lysosomotropic agent, accumulates in acidic organelles such as lysosomes with red fluorescence, whereas in neutral environments such as cytoplasm and nucleus, acridine orange exhibits green fluorescence (Granato et al., 2013). As shown in Fig. 6A, acridine orange staining in the control group was primarily found in normal lysosomes as red fluorescence. In contrast, cells treated with nicotine showed minimal red fluorescence staining but most of the cells had enhanced green fluorescence staining in the nuclei, which suggest that nicotine may cause lysosome permeabilization resulting in lysosome alkalization so that more acridine orange dyes accumulate in the nuclei. We next detected cathepsin B activity by loading the cells with a selective cathepsin B substrate CV-(RR)2. As shown in Fig. 6B, we found that this red fluorescent substrate CV-(RR)2 was mainly found in intracellular organelles suggesting an intact integrity of lysosomes in control group. However, the red fluorescence of CV-(RR)2 disappeared when cells were stimulated by nicotine, suggesting increased lysosome permeabilization and leakage of lysosomal cathepsin B. Dexamethasone was shown to preserve lysosomal membrane integrity and used as a lysosome stabilizer in endothelial cells (Chen et al., 2015a; Hinz and Hirschelmann, 2000). The effects of nicotine on lysosome permeabilization and cathepsin B were inhibited by dexamethasone (Fig. 6A–B).
Fig. 6. Nicotine increases lysosome destabilization and cathepsin B release in MVECs.
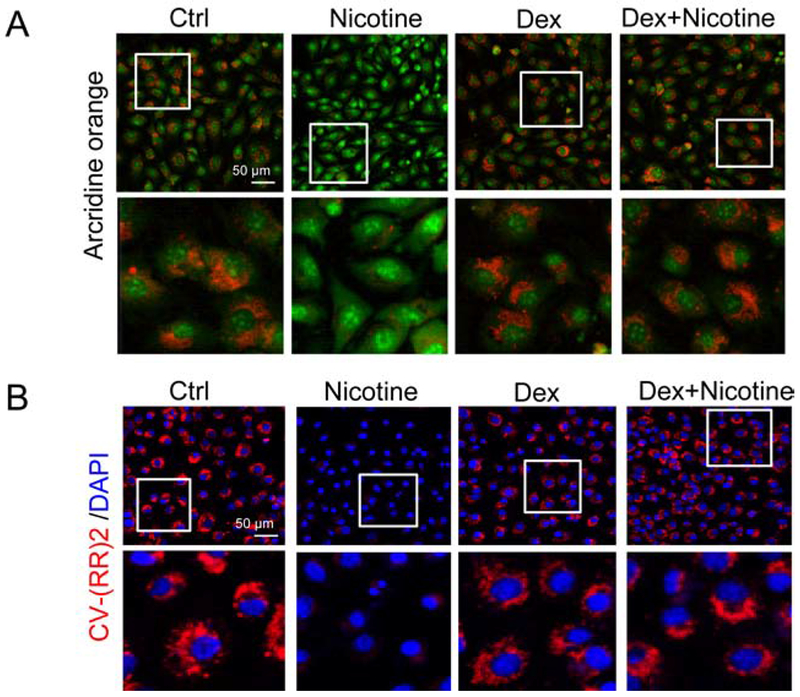
Cells were treated with 100 nM nicotine in the presence or absence of dexamethasone (100 μM). Lysosome membrane permeability and cathepsin B was detected by staining the cells with acridine orange (A) or CV-(RR)2 (B) staining and visualization by fluorescence microscopy. Representative images are shown from at least three independent experiments. Scale bar = 50 μm.
3.6. Nicotine increases endothelial barrier dysfunction
The effects of nicotine on endothelial barrier function were examined using two methods: ECIS and FITC-dextran transwell assay. First, ECIS was used to measure the changes in TEER in response to nicotine. As shown in Fig. 7A and 7B, nicotine significantly reduced the TEER, which was inhibited by Nlrp3 shRNA transfection, cathepsin B inhibitor Ca074Me, caspase-1 inhibitor WEHD, or HMGB1 inhibitor glycyrrhizin. Second, we used the FITC-dextran transwell assay to confirm the increased permeability of endothelial cell monolayers to high-molecular-weight molecules. As shown in Fig. 7C and 7D, we observed that nicotine significantly increased the amount of FITC-dextran molecules across the endothelial monolayer, which was also inhibited by Nlrp3 shRNA transfection, or treatment of inhibitors (Ca074Me, WEHD, glycyrrhizin).
Fig. 7. Nicotine-induced endothelial hyperpermeability depends on cathepsin B-Nlrp3 inflammasome-HMGB1 axis.
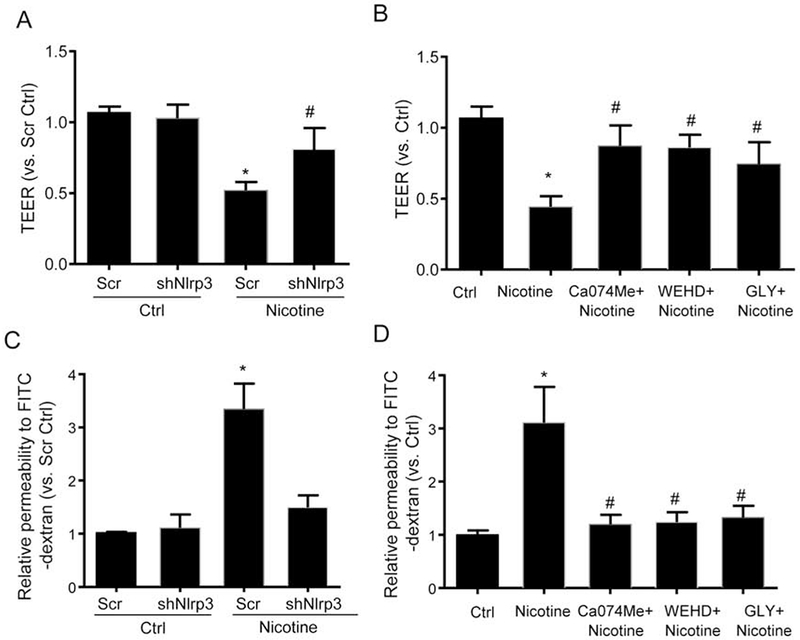
MVECs were transfected with scramble shRNA plasmid (Scr) or Nlrp3 shRNA plasmid (shNlrp3). Cells were left alone or treated with nicotine (100 nM) for 12 h. MVECs were also treated with nicotine (100 nM) in the presence of PBS (Ctrl) or Ca074Me (5 μM), WEHD (10 μM) or glycyrrhizin (GLY, 130 μM) for 12 h. (A) and (B) EC permeability was measured by transendothelial electrical resistance (TEER) (n=6). (C) and (D) EC permeability was determined by measuring the amount of FITC-dextran that crossed the endothelial monolayer cultured on the inserts of transwell (n=6). Data are presented as means ± S.D. from six batches of cells and three independent experiments. *P<0.05 vs. untreated control; #P<0.05 vs. Nicotine alone
4. Discussion
The goal of the present study was to investigate the effect of nicotine exposure on the inter-endothelial junction integrity and identify the mechanisms underlying this effect. This would broaden the understanding of the triggering mechanism for nicotine-induced endothelial dysfunction in the early stage of vascular diseases associated with smoking.
Cigarette smoking promotes endothelial cell damage and causes vascular endothelial dysfunction. The deleterious effect of cigarette smoke including endothelial dysfunction is believed to be partly mediated by actions of nicotine (Benowitz, 2003; Fang et al., 2004; Koide et al., 2005; Mayhan and Patel, 1997). Endothelium is the lining of endothelial cells in the inner lumen of blood vessels. Under physiological condition, endothelium functions as a barrier that prevents the fluid and proteins moving from the intravascular compartment to the interstitium. In addition, the endothelium also regulates diverse functions including vasomotor response, host-defense, thrombosis, vascular repair, and fluid balance (Koide et al., 2005; Lee and Cooke, 2012; Mayhan and Patel, 1997; Mehta and Malik, 2006). Given the critical role of endothelial cells in modulating vascular homeostasis, endothelial dysfunction due to damages to the endothelium is normally the earliest event in the development of vascular diseases under various cardiovascular risk factors. Inter-endothelial junction consists of many junction proteins including tight junction protein ZO-1/2 (Dejana et al., 1995). Decreased expression of junction proteins may cause the loss of inter-endothelial junction integrity and thereby is considered as is a marker event for developing endothelium barrier dysfunction and endothelial hyperpermeability. Endothelial hyperpermeability is associated with many pathological consequences including leakage of plasma molecules and proteins to the interstitial space and leukocyte transmigration in the vasculature (Hernandez et al., 2007; Vandenbroucke et al., 2008). In the present study, we observed that nicotine significantly reduced ZO-1/2 expressions in the coronary endothelium (Figure 1). It should be noted that the mice were treated with a dose comparable to the amount of nicotine that a person smoking 50 cigarettes a day (Armitage et al., 1968). Our first demonstration of injurious effects of nicotine on tight junctions in coronary arteries is consistent with previous findings showing that nicotine alters cerebral microvascular tight junction protein distribution and increases in vivo blood-brain barrier permeability in rats (Hawkins et al., 2004). Consistently, our in vitro data demonstrated that nicotine stimulation could directly decrease the expression of ZO-1/2 in the inter-endothelial regions of cultured endothelial monolayers of MVECs (Figure 2). The concentration of nicotine used (50 nM-1 μM) in our in vitro studies is within the range of plasma concentration of nicotine in smokers (10−8 M and 10−5 M) (Benowitz, 1996). Thus, our in vivo and in vitro data together with previous findings suggest that nicotine, in similar concentrations to those experienced by smokers, induces structural and functional changes in endothelial cells, which could be an initial step in the development of vasculopathy. Using Nlrp3-null mice, we further demonstrated that Nlrp3 gene deficiency could significantly attenuate nicotine-induced decreases in ZO-1/2. Thus, our findings for the first time reveal that Nlrp3 inflammasome is implicated in the nicotine-induced loss of inter-endothelial junction integrity.
To observe the direct effects of nicotine on endothelial inflammasomes, the present study determined whether nicotine could stimulate Nlrp3 inflammasomes leading to tight junction disassembly in cultured endothelial cells. We found that nicotine increased the colocalization of Nlrp3 inflammasome components suggesting that nicotine induces aggregation and assembly of Nlrp3 inflammasome complex. The increased formation of Nlrp3 inflammasome complex is accompanied by increases in caspase-1 activity and IL-1β release in cultured endothelial cells. Together, these findings revealed that nicotine stimulation could directly activate the Nlrp3 inflammasomes in cultured endothelial cells. Endothelial Nlrp3 inflammasomes can be directly stimulated by different cardiovascular risk factors or under pathological conditions such as adipokine visfatin, hyperglycemia, or disturbed shear stress (Chen et al., 2016; Gombault et al., 2012; Xia et al., 2014; Zhou et al., 2014). In line with these previous findings, the present study demonstrates that nicotine is another independent danger factor that activates endothelial Nlrp3 inflammasomes. More importantly, in accordance with our in vivo data, we also demonstrated that Nlrp3 gene silencing or inflammasome inhibition attenuated nicotine-induced tight junction disruption and hyperpermeability in endothelial cells in vitro. It should be noted that the findings from the present study could not exclude a secondary effect of Nlrp3 inflammasomes from other cellular sources (e.g. macrophages) to endothelial dysfunction. Nonetheless, our in vivo and in vitro data suggest that instigation of Nlrp3 inflammasome by nicotine may be a triggering mechanism for endothelial dysfunction during smoking or nicotine exposure.
HMGB1 can be released by immune cells into the extracellular space upon infection or sterile inflammatory stimulation, or passively released from dying cells (Lu et al., 2012; Srikrishna and Freeze, 2009). In immune cells, activation of Nlrp3 inflammasome could cause HMGB1 secretion that is sufficient to mediate inflammation. (Andersson and Tracey, 2011; Lu et al., 2012). Recent studies also showed that the Nlrp3 inflammasome promotes the HMGB1 release in endothelial cells under various cardiovascular risk factors (Chen et al., 2015b; Chen et al., 2016; Wang et al., 2016). HMGB1 is showed as a vascular permeability factor that could lead to inter-endothelial junction disruption under various pathological conditions (Chen et al., 2015a; Chen et al., 2015b; Lv et al., 2017; Sappington et al., 2002; Wolfson et al., 2011). Recent studies demonstrated that HMGB1 enhances endothelial permeability, at least in part, through the action of the receptor for advanced glycation end products (RAGE)-mediated signaling (Chen et al., 2015a; Chen et al., 2015b; Huang et al., 2012; Wolfson et al., 2011). The present study demonstrated that nicotine increased HMGB1 release and inhibition of HMGB1 activity by glycyrrhizin reversed nicotine-induced junction disruption and hyperpermeability indicating a key role of Nlrp3-dependent HMGB1 release in this process.
Another finding of the present study is that nicotine-induced Nlrp3 inflammasome activation is associated with lysosome permeabilization-cathepsin B signaling pathway. Lysosomal damage and the leakage of cathepsin B from lysosomes have been implicated as upstream events of the Nlrp3 inflammasome activation stimulated by many danger factors including free fatty acids, cholesterol crystals, and Lactobacillus casei wall components (Chen et al., 2015a; Wang et al., 2016). To elucidate whether lysosome permeabilization is involved in the effects of nicotine on endothelial cells, cells were stained with acridine orange. Acridine orange accumulates in acidic vacuoles, such as lysosomes, and emits red fluorescence signal, and this signal is lost when lysosomal integrity is compromised. Our data (Figure 7A) showed that incubation of MVECs with nicotine resulted in reduced acridine orange staining, indicating loss of lysosomal integrity. However, when the cells were treated with nicotine in the presence of lysosome membrane stabilizer dexamethasone, there was no clear attenuation of the acidic fluorescent signal, suggesting that dexamethasone reduces the nicotine-induced lysosomal destabilization. Cathepsin B is a lysosomal cysteine protease that is maximally active at the acidic pH, however, when it is released into a cytoplasm with neutral pH, cathepsin B is inactivated. We then studied whether nicotine affected the release of cathepsin B by staining MVECs with a cell-permeable cathepsin B substrate CV-(RR)2, which emits a red fluorescent signal when cleaved by cathepsin B. Cathepsin B activity (red) was observed in the unstimulated endothelial cells. Upon addition of nicotine to the cells, the staining was lost reflecting leakage of cathepsin B into the cytoplasm with ensuing inactivation. In line with the above-described studies with acridine orange, dexamethasone inhibited the nicotine-induced leakage of cathepsin B into the cytoplasm. These results suggest that nicotine induces lysosomal damage and the ensuing release of cathepsin B from intact lysosomes. Our findings are consistent with previous studies showing that cigarette smoke is a potent inducer of cathepsin B activity (Chang et al., 1986). Increased lysosome permeabilization was shown to be associated with the release of lipid mediators, free radicals or other cathepsins, which may also contribute to nicotine-induced effects (Campden and Zhang, 2019; Wang et al., 2018; Wu et al., 2018). Our data further demonstrated that inhibition of cathepsin B prevented nicotine-induced hyperpermeability. Thus, these results implicate that nicotine-induced endothelial barrier dysfunction is, at least in part, due to lysosome permeabilization and cathepsin B-dependent Nlrp3 inflammasomes.
In summary, this study demonstrated that nicotine causes the tight junction disruption through Nlrp3 inflammasomes aggregation and activation and inflammasome-dependent release HMGB1. This stimulatory effect of nicotine on Nlrp3 inflammasomes is associated with lysosome permeabilization-cathepsin B pathway. Activation of the Nlrp3 inflammasome-HMGBl signaling axis may serve as an important triggering mechanism in the development of early onsets of smoking-associated vascular complications.
Acknowledgments
This study was supported by the faculty development funds of the College of Pharmacy at the University of Houston, National Institutes of Health (HL122937, HL122769), and National Natural Science Foundation of China (NO.81603110).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCE
- Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL, 2013. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxidants & redox signaling 18, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ, 2011. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29, 139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage AK, Hall GH, Morrison CF, 1968. Pharmacological basis for the tobacco smoking habit. Nature 217, 331–334. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, 1996. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol 36, 597–613. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, 2003. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Prog Cardiovasc Dis 46, 91–111. [DOI] [PubMed] [Google Scholar]
- Busso N, So A, 2010. Mechanisms of inflammation in gout. Arthritis Res Ther 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campden RI, Zhang Y, 2019. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys. [DOI] [PubMed] [Google Scholar]
- Chang JC, Lesser M, Yoo OH, Orlowski M, 1986. Increased cathepsin B-like activity in alveolar macrophages and bronchoalveolar lavage fluid from smokers. Am Rev Respir Dis 134, 538–541. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL., 2015a. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta 1853, 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pitzer AL, Li X, Li PL., Wang L, Zhang Y, 2015b. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med 19, 2715–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang L, Pitzer AL, Li X, Li PL., Zhang Y, 2016. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. Journal of molecular medicine 94, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D, Latz E, 2011. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 32, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Corada M, Lampugnani MG, 1995. Endothelial cell-to-cell junctions. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 9, 910–918. [PubMed] [Google Scholar]
- Dinarello CA, Donath MY, Mandrup-Poulsen T, 2010. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17, 314–321. [DOI] [PubMed] [Google Scholar]
- Fang Q, Sun H, Mayhan WG, 2004. L-arginine prevents impaired endothelium-dependent cerebral arteriolar dilatation during acute infusion of nicotine. Nicotine Tob Res 6, 1009–1014. [DOI] [PubMed] [Google Scholar]
- Gombault A, Baron L, Couillin I, 2012. ATP release and purinergic signaling in NLRP3 inflammasome activation. Frontiers in immunology 3, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato M, Lacconi V, Peddis M, Lotti LV, Renzo LD, Gonnella R, Santarelli R, Trivedi P, Frati L, D’Orazi G, Faggioni A, Cirone M, 2013. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis 4, e730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Li X, Umetani M, Boini KM, Li PL., Zhang Y, 2018. 10.1111/bcpt.13146. Tricyclic antidepressant amitriptyline inhibits autophagic flux and prevents tube formation in vascular endothelial cells. Basic Clin Pharmacol Toxicol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, Davis TP, 2004. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res 1027, 48–58. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Brown RC, Davis TP, 2002. Smoking and ischemic stroke: a role for nicotine? Trends Pharmacol Sci 23, 78–82. [DOI] [PubMed] [Google Scholar]
- Hernandez S, Chavez Munguia B, Gonzalez-Mariscal L, 2007. ZO-2 silencing in epithelial cells perturbs the gate and fence function of tight junctions and leads to an atypical monolayer architecture. Exp Cell Res 313, 1533–1547. [DOI] [PubMed] [Google Scholar]
- Hinz B, Hirschelmann R, 2000. Dexamethasone megadoses stabilize rat liver lysosomal membranes by non-genomic and genomic effects. Pharm Res 17, 1489–1493. [DOI] [PubMed] [Google Scholar]
- Huang W, Liu Y, Li L, Zhang R, Liu W, Wu J, Mao E, Tang Y, 2012. HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation 35, 350–362. [DOI] [PubMed] [Google Scholar]
- Joukar S, Shahouzehi B, Najafipour H, Gholamhoseinian A, Joukar F, 2012. Ameliorative effect of black tea on nicotine induced cardiovascular pathogenesis in rat. EXCLI J 11, 309–317. [PMC free article] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Zitvogel L, Kroemer G, 2010. Pyroptosis - a cell death modality of its kind? Eur J Immunol 40, 627–630. [DOI] [PubMed] [Google Scholar]
- Kiyohara Y, Ueda K, Fujishima M, 1990. Smoking and cardiovascular disease in the general population in Japan. J Hypertens Suppl 8, S9–15. [PubMed] [Google Scholar]
- Koide M, Nishizawa S, Yamamoto S, Yamaguchi M, Namba H, Terakawa S, 2005. Nicotine exposure, mimicked smoking, directly and indirectly enhanced protein kinase C activity in isolated canine basilar artery, resulting in enhancement of arterial contraction. J Cereb Blood Flow Metab 25, 292–301. [DOI] [PubMed] [Google Scholar]
- Kudo M, Matsui O, Izumi N, Iijima H, Kadoya M, Imai Y, 2014. Surveillance and diagnostic algorithm for hepatocellular carcinoma proposed by the Liver Cancer Study Group of Japan: 2014 update. Oncology 87 Suppl 1, 7–21. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, 2011. Emerging inflammasome effector mechanisms. Nature reviews. Immunology 11, 213–220. [DOI] [PubMed] [Google Scholar]
- Lee J, Cooke JP, 2012. Nicotine and pathological angiogenesis. Life Sci 91, 1058–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ, 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HL, Zang WJ, Lu J, Yu XJ, Lin YX, Cao YX, 2006. The protective effect of captopril on nicotine-induced endothelial dysfunction in rat. Basic Clin Pharmacol Toxicol 99, 237–245. [DOI] [PubMed] [Google Scholar]
- Lv ZH, Phuong TA, Jin SJ, Li XX, Xu M, 2017. Protection by simvastatin on hyperglycemia-induced endothelial dysfunction through inhibiting NLRP3 inflammasomes. Oncotarget 8, 91291–91305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM, 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J, 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J, 2009. The inflammasomes: guardians of the body. Annu Rev Immunol 27, 229–265. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Patel KP, 1997. Effect of nicotine on endothelium-dependent arteriolar dilatation in vivo. Am J Physiol 272, H2337–2342. [DOI] [PubMed] [Google Scholar]
- Mazzone P, Tierney W, Hossain M, Puvenna V, Janigro D, Cucullo L, 2010. Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: expanding the awareness of smoking toxicity in an underappreciated area. Int J Environ Res Public Health 7, 4111–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Malik AB, 2006. Signaling mechanisms regulating endothelial permeability. Physiological reviews 86, 279–367. [DOI] [PubMed] [Google Scholar]
- Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J, 2011. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis 2, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi K, Virkanen J, Rajamaki K, Niemi K, Kovanen PT, Eklund KK, 2013. Ethanol inhibits activation of NLRP3 and AIM2 inflammasomes in human macrophages--a novel anti-inflammatory action of alcohol. PLoS One 8, e78537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Fitzgerald KA, 2012. Regulation of inflammasome signaling. Nat Immunol 13, 333–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G, Mago N, Rosa F, 2009. [Role of inflammation in atherogenesis]. Invest Clin 50, 109–129. [PubMed] [Google Scholar]
- Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP, 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123, 790–802. [DOI] [PubMed] [Google Scholar]
- Sliwinska-Mosson M, Milnerowicz H, 2017. The impact of smoking on the development of diabetes and its complications. Diab Vasc Dis Res 14, 265–276. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Freeze HH, 2009. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strowig T, Henao-Mejia J, Elinav E, Flavell R, 2012. Inflammasomes in health and disease. Nature 481, 278–286. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Funasaka T, Matsumoto H, 1973. The contractive mechanism of nicotine in isolated aorta of rabbits, with special reference to catecholamine release. Kobe J Med Sci 19, 111–125. [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB, 2008. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 1123, 134–145. [DOI] [PubMed] [Google Scholar]
- Wang F, Gomez-Sintes R, Boya P, 2018. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931. [DOI] [PubMed] [Google Scholar]
- Wang L, Chen Y, Li X, Zhang Y, Gulbins E, 2016. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 7, 73229–73241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Chen J, Li X, Umetani M, Chen Y, Li PL, Zhang Y, 2019a. Contribution of Transcription Factor EB to AdipoRon-induced Inhibition of Arterial Smooth Muscle Cell Proliferation and Migration. Am J Physiol Cell Physiol. 10.1152/ajpcell.00294.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Li X, Chen J, McConnell BK, Chen L, Li PL, Chen Y, Zhang Y, 2019b. Activation of TFEB ameliorates dedifferentiation of arterial smooth muscle cells and neointima formation in mice with high-fat diet. Cell Death Dis 10, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YM, Li X, Xiong J, Abais JM, Xia M, Boini KM, Zhang Y, Li PL, 2013a. Attenuation by statins of membrane raft-redox signaling in coronary arterial endothelium. J Pharmacol Exp Ther 345, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YM, Li X, Xu M, Abais JM, Chen Y, Riebling CR, Boini KM, Li PL, Zhang Y, 2013b. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem 31, 925–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Ting JP, O’Neill LA, 2012. A role for the NLRP3 inflammasome in metabolic diseases--did Warburg miss inflammation? Nature immunology 13, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RK, Chiang ET, Garcia JG, 2011. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res 81, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, Lin Y, Bai X, Liu X, Chen X, Yang H, Xu C, Yang B, 2018. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis 9, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL, 2014. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. The American journal of pathology 184, 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Zhang Y, Xia M, Li XX, Ritter JK, Zhang F, Li PL, 2012. NAD(P)H oxidase-dependent intracellular and extracellular O2*-production in coronary arterial myocytes from CD38 knockout mice. Free Radic Biol Med 52, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Guan Y, Chen J, Li X, McConnell BK, Zhou W, Boini KM, Zhang Y, 2018. Contribution of p62/SQSTM1 to PDGF-BB-induced myofibroblast-like phenotypic transition in vascular smooth muscle cells lacking Smpd1 gene. Cell Death Dis 9, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Day I, Ye S, 2001. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis 154, 277–283. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL, 2014. Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res 102, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Gardner SE, Clarke MC, 2011. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol 31, 2781–2786. [DOI] [PubMed] [Google Scholar]
- Zhou J, Li YS, Chien S, 2014. Shear stress-initiated signaling and its regulation of endothelial function. Arteriosclerosis, thrombosis, and vascular biology 34, 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]