

Abstract
It is accepted that insulin-secreting β-cells release insulin in response to glucose even in the absence of functional ATP-sensitive K+ (KATP)-channels, which play a central role in a ‘consensus model’ of secretion broadly accepted and widely reproduced in textbooks. A major shortcoming of this consensus model is that it ignores any and all anionic mechanisms, known for more than 40 years, to modulate β-cell electrical activity and therefore insulin secretion. It is now clear that, in addition to metabolically regulated KATP-channels, β-cells are equipped with volume-regulated anion (Cl–) channels (VRAC) responsive to glucose concentrations in the range known to promote electrical activity and insulin secretion. In this context, the electrogenic efflux of Cl– through VRAC and other Cl– channels known to be expressed in β-cells results in depolarization because of an outwardly directed Cl– gradient established, maintained and regulated by the balance between Cl– transporters and channels. This review will provide a succinct historical perspective on the development of a complex hypothesis: Cl– transporters and channels modulate insulin secretion in response to nutrients.
Keywords: chloride, chloride channels, chloride transporters, insulin secretion, volume regulation
Introduction
Stimulus-secretion coupling in β-cells is a complex process with multiple facets that cannot be simply incorporated in any single comprehensible model. [1]
In a widely accepted consensus mechanism for insulin secretion, ATP-sensitive K+ (KATP)-channels play a key role [1]. In this model, when the circulating glucose is low, β-cell plasma membrane (PM) KATP-channels remain open allowing a high K+ conductance and maintaining the PM potential (Em) hyperpolarized (approximately –70 mV) [2]. An increase in blood glucose concentration triggers signaling paths summarized as follows: glucose enters β-cells via facilitative transporters, e.g. Slc4a2 (Glut2) and is immediately phosphorylated by glucokinase [3]. Subsequently, glucose is metabolized generating ATP and decreasing ADP through the Krebs’ cycle. This change in the ATP/ADP ratio inactivates/closes KATP-channels, resulting in a gradual β-cell PM depolarization to a threshold where voltage-gated Ca2+ channels’ open and action potentials are triggered. The influx of Ca2+, necessary for β-cell exocytosis, causes the release of granules containing insulin [4]. This model is strengthened by the fact that inactivating mutations affecting either of the two subunits of the human KATP-channel, i.e. ABCC8/Sur1 or KCJN11/Kir6.2, genes [5,6] result in insufficient insulin secretion leading to neonatal diabetes [7]. Conversely, activating mutations in Sur1/Kir6.2 result in unregulated insulin responses independent of the level of glucose present, leading to hyperinsulinemic hypoglycemia [8].
While the simplicity of this model is attractive and presents the essentials of the triggering pathway, it is restricted by failing to include anionic (Cl–) mechanisms known, for more than 40 years, to modulate β-cell electrical activity and insulin secretion [9–11]. Clearly, unless an inward background current exists to drive Em away from the K+ equilibrium potential [12], the simple closure of KATP-channels is not enough to increase PM potential (depolarize) to the activating threshold for voltage-gated Ca2+ channels to open. As originally discussed by Henquin et al. [1] and Best et al. [10], accumulating experimental evidence suggests that glucose triggers insulin secretion by modulating a background inward Cl– current necessary to cause β-cell depolarization when KATP-channels are fully closed. The slow systematic dissection of the complex anionic mechanism(s), modulating β-cell electrical activity, has provided evidence for the regulation of intracellular Cl– concentration ([Cl–]i) and Cl– fluxes by glucose metabolism consistent with a more inclusive hypothesis: Cl– transporters and channels can modulate insulin secretion in response to nutrients.
Overview of β-cell Cl– physiology
Regulation of intracellular Cl– concentration
Some cells actively extrude Cl–, others actively accumulate it, but few cells ignore it. [13]
Insulin-secreting β-cells, like immature neurons, maintain their [Cl–]i higher than expected for passive distribution across the PM [9,14,15] due to the predominance of Cl– loaders relative to extruders. This outwardly directed Cl– gradient in β-cells is likely to be set, maintained and regulated by multiple Cl– transporters and channels [16]. The active movement of Cl– ions, in and out of cells, is carried out by members of the Cl– transporter families, Slc12a, Slc4a, Slc26a, that can maintain [Cl–]i above thermodynamic equilibrium, whereas Cl– channels allow constitutive and regulated fluxes that dissipate the gradient. Theoretically, when a cell without Cl– transporters and an Em = −70 mV is incubated in media with physiological [Cl–]o, i.e. ∼120 mM, Cl– will passively distribute across the PM to reach thermodynamic equilibrium where the net movement of Cl– ions equals zero. Under these conditions, [Cl–]i will settle at ∼10 mM, the concentration predicted by the Nernst equation. In β-cells, however, [Cl–]i is kept above that Nernstian value by the net action of Cl– loaders. Therefore, the opening of any Cl– channel will allow for efflux, rather than influx, of Cl–, as shown in Figure 1. This naturally electrogenic and depolarizing efflux of Cl– is expected to contribute to insulin secretion, even in the absence of functional KATP-channels [17,18].
Figure 1. [Cl–]i β-cell regulation.

β-cells exhibit an [Cl–]i ≈ 34 mM, i.e. ∼3.4-times above the predicted thermodynamic equilibrium. Therefore, the functional prevalence of Cl– loaders over Cl– extruders makes possible the efflux of the anion upon Cl– channel opening. The expression pattern of some of the Cl– transporters and channels already identified and others in β-cells are currently being mapped. Shown are those partially/fully supported by experimental evidence (e.g. Nkccs, Kccs, Ano1/2, Cftr, GABAA, GlyR and VRAC).
Evolution of the anionic mechanism of insulin secretion
Cl– fluxes and insulin secretion
Electrophysiological experiments, performed in the 1970s using mouse β-cells, demonstrated that a reduction in [Cl–]i had a very small effect on their resting Em and insulin secretion in response to glucose [2]. These experiments, and others performed a decade or so later, erroneously assumed that Cl– fluxes in β-cells follow Em, concluding that Cl– passively distributes across the PM [19], while disregarding previous evidence suggesting otherwise. Indeed, Sehlin [9] showed for the first time that Cl– distributes in a non-passive way across mouse β-cells’ PM and suggested that Cl– could be transported into the β-cell against its electrochemical gradient by mechanisms requiring cellular metabolism.
In analogy to GABAergic action on immature neurons, the opening of Cl– channels in β-cells is depolarizing, an early hypothesis supported by data from db/db diabetic β-cells that exhibit an altered regulation of Cl– permeability, Em and insulin secretion [20,21]. Additional support for that hypothesis is provided by experiments using rat islets incubated in very low Cl– concentrations (to reduce [Cl–]i in islet cells) which established a decrease in both secretory phases [22]. As an additional line of support, at least partially, Tamagawa et al. demonstrated an ∼66% reduction in the second phase of glucose-stimulated insulin secretion (GSIS) in rat islets incubated in low Cl– [23]. As it became apparent that [Cl–]i and Cl– fluxes had a role in insulin secretion, it was difficult to reconcile, in a single picture, the complexity of the secretory response using data involving anions, cations and secretagogues [23]. Therefore, a clearer model of insulin secretion mainly focused on K+ channels and few other players rapidly progressed [24–27], while Cl– channels and transporters were altogether disregarded [28].
The role of [Cl–]i in β-cell physiology regained momentum a decade or so after the introduction of the consensus model when Kinard and Satin [29] and Best et al. [30] in 1995 demonstrated that β-cells are equipped with a volume-sensitive anionic (Cl–) conductance, thus providing, for the first time, a functional link between hypotonic β-cell swelling [31,32], transient insulin secretion [31] and β-cell electrical activity [29,30,33,34]. Furthermore, the demonstration of a close relationship between β-cell electrical activity, glucose and [Cl–]i by Best [18] allowed him and collaborators to put forward the ‘VRAC hypothesis’ that a volume-regulated anion channel contributes to PM depolarization, electrical activity and insulin secretion [10]. This now 10-year-old hypothesis has been experimentally tested and elegantly demonstrated at the molecular, functional and in vivo levels very recently [11,35].
Cl– transporters and insulin secretion
Slc12a family of Cl– loaders and extruders
The Slc12a family of genes encode at least seven secondary active cation-Cl– cotransporters [36], all extensively characterized at the molecular, pharmacological and functional levels and considered to be key regulators of cellular volume and [Cl–]i [37].
The presence, in β-cells, of a depolarizing Cl– conductance requires that [Cl–]i be maintained above thermodynamic equilibrium by Cl– transport mechanisms operating in a net uptake mode. In the early 1980s, such Cl– transport mechanisms, sensitive to diuretics such as bumetanide and furosemide, were identified in β-cells [38–45]. These diuretics are extensively used in the clinic and were long suspected to interfere with glucose homeostasis in humans, as summarized by Giugliano et al. [46]. Low concentrations of these diuretics inhibit insulin secretion, Ca2+ and Cl– uptake from β-cells [39,40,43] and impair glucose tolerance in mice [41,42,47]. This early pharmacological evidence supported the existence of Cl– loaders in β-cells. The subsequent demonstration of diuretic-sensitive K+Cl– extruder mechanisms involved in osmotic volume regulation [48,49] and the fact that osmotic β-cell swelling promoted insulin secretion [31] further highlighted the importance of Cl– cotransport systems in mouse β-cells [45]. More recent molecular studies [50–53] have confirmed that β-cells express several splice variants of the prototypical Cl– transporters of the Slc12a family, i.e. loaders Slc12a2 (Nkcc1a-b), Slc12a1 (Nkcc2a) [52], and extruders Slc12a4 (Kcc1), Slc12a5 (Kcc2a-b), Slc12a6 (Kcc3a-d), Slc12a7 (Kcc4) and a Kcc2a variant, Kcc2a-S25, lacking exon 25 [53]. Our recent work has shown that pharmacological inhibition of Kcc2 influences the efficacy of GSIS in vitro [53].
Slc4a and Slc26a families of anion exchangers
β-cell transcriptome profiling and quantitative proteomic analysis identified an assorted repertoire of Cl– transporters [54–56] including members of the Slc4a and Slc26a families. Based on their recognized function in several tissues and cells, some of them can be considered as electroneutral Cl– loaders. Indeed, Slc4a1, Slc4a2, Slc4a3, Slc26a3, Slc26a4, Slc26a6, Slc26a7 or Slc26a9 can function as Cl–/HCO3– exchangers [57,58]. These transporters are functionally sensitive to changes in intracellular pH ([pH]i), thus contributing to its regulation by extruding intracellular bicarbonate in exchange for extracellular Cl–. They also contribute to cell volume homeostasis and Em through modulation of the PM Cl− gradient in many cell types studied [59]. In β-cells, however, virtually nothing is known about the potential physiological roles of any of these electroneutral Cl–/HCO3– exchangers. However, their presence in β-cells might be indirectly inferred by the fact that (i) pHi modulates β-cell electrical activity [60], (ii) glucose increases β-cell pHi [61] and (iii) β-cells do regulate its pHi [61], at least via Slc4a4, which encodes electrogenic Na+/HCO3– cotransporters [62] and through Slc4a8, a Na+-dependent Cl–/HCO3– exchanger [63].
Cl–/H+ exchanger Clc3
The electrogenic 2Cl–/H+ exchanger Clc3 was the first and last of a large family of anion transporters and channels [64] to be associated with insulin secretion [65,66]. Clc3 was considered to be expressed in mouse and human β-cells localized to large insulin-containing dense-core vesicles, where it was proposed to play a physiological role in the maturation and acidification of these vesicles [65–67]. However, the use of knockout-validated Clc3 antibodies previously demonstrated a different Clc3 localization; β-cell synaptic-like macrovesicles [68], thus generating an important controversy [69] that will come to fruition with new experiments. Furthermore, it remains unknown whether β-cells express other members of the Clc family of Cl– channels and exchangers.
Cl– channels and insulin secretion
Volume-regulated anion channel, VRAC
Although inferred from previous results [9], the first description of Cl– channels in β-cells was made by Kinard and Satin [29] and by Best and collaborators [30,33]. Using the whole-cell patch clamp technique, these authors demonstrated swelling (volume)-regulated, ATP-dependent and outwardly rectifying Cl– currents in rodent β-cells. This Cl– current was sensitive to classic anion channel blockers, like 4,4'-di-isothiocyano-2,2'-stilbenedisulfonic acid. Further biophysical characterization demonstrated an ionic selectivity sequence very close to that known for the Cl– channel cystic fibrosis transmembrane conductance regulator (Cftr), but suggested that the volume-activated Cl– current recorded in β-cells was mediated by a different channel or group of channels, all permeable to several anions [34]. Activation of this current in intact β-cells was linked to the generation of a depolarizing Cl–-selective efflux which promoted electrical activity [33,70] and stimulated insulin secretion [33], providing a functional explanation for the early observation that hyposmolarity increases insulin secretion [31]. However, a question remained: Does glucose activate a depolarizing Cl– current? This has been partially answered: glucose and hypotonically induced cell swelling promote a similar pattern of transiently depolarizing Cl– currents [71] and β-cells swell in response to increased glucose concentrations within the physiological range [32,72,73]. Reiterating that KATP-channel closure is not enough to fully depolarize β-cells, unless an inward current existed [12], a new model for insulin secretion was conceived, whereby glucose decreases the magnitude of the outward K+ current due to the inhibition of KATP-channels while increasing the inwardly directed Cl– current, promoting β-cell PM depolarization, electrical activity and secretion [34]. VRAC was the only fairly studied Cl– channel at the end of the 1990s and was the one included in the model. This ‘VRAC’ model was experimentally tested and strengthened after the confirmation that (i) VRAC activity does not depend on that of KATP-channels [74,75], (ii) VRAC carries a depolarizing Cl– current (efflux) which promotes insulin secretion in response to osmotic swelling [17,32,72,76–78], (iii) β-cell electrical activity in response to glucose is reliant on [Cl–]i [18] and (iv) KATP-channel-independent modalities of β-cell electrical activity and secretion are clearly present in β-cells lacking KATP-channels [79]. Indeed, β-cells from knockout mice lacking either subunit of the KATP-channel (Sur1KO or Kir6.2KO) are depolarized and exhibit increased [Ca2+]i, as predicted by the consensus model of insulin secretion. However, these β-cells had well-defined Ca2+ oscillations and modulated [Ca2+]i in response to glucose [79–81], an observation not anticipated by the consensus model providing additional evidence for some of the shortcomings in the model [1]. The new hypothesis, introduced by Best et al., in 2010, thus proposed that glucose metabolism activates VRAC, generating a depolarizing Cl– efflux, which requires [Cl–]i kept above its electrochemical equilibrium [10].
The discovery of the molecular identity of VRAC, i.e. leucine-rich repeat-containing family 8 (Lrrc8a-e) proteins [82,83] paved the road for detailed biophysical and structural characterization of this channel [84–89]. Conditional animal models lacking the essential VRAC subunit Lrrc8a only in β-cells provided a direct test of the VRAC hypothesis [10]. Kang et al. [35] confirmed the original observations by Best et al. [30,33] using shRNA-mediated silencing, CRISPR/Cas9 technology or Cre/Lox-mediated elimination of Lrrc8a in MIN6 or primary β-cells. Loss of Lrrc8a reduced Cl– currents in response to cell swelling and blunted insulin secretion in response to glucose [35]. In more controlled experiments Stuhlmann et al. [11] confirmed that glucose swells β-cells triggering VRAC currents that rely on Lrrc8a to provoke electrical excitation. However, these responses were delayed and insulin secretion was decreased [11], but not abolished, as suggested by the results of Kang et al. [35]. Therefore, VRAC may not be the only Cl– channel β-cells require to reach a PM depolarization threshold for voltage-gated Ca2+ channel-dependent Ca2+ entry and insulin secretion, a concept entirely in line with the notion that VRAC may form part of a ‘Cl– machinery’ composed of many Cl– channels. Once more, Stuhlmann et al. [11] demonstrated heterogeneous expression of the five VRAC subunits in mouse islets with Lrrc8a, Lrrc8c and Lrrc8d relatively abundant, less Lrrc8b and minute amounts of Lrrc8e. These findings may have further physiological relevance; Lrrc8d confers VRAC with permeability to the osmotically active metabolite taurine [90], which may be involved in insulin secretion [91] as well as that of the osmolyte myo-inositol, GABA or lysine [92] which have been also implicated in the secretory response [15,93–95]. Obviously, the physiological importance of VRAC in β-cells has added new tools to study islet biology including the proposed paracrine/autocrine effects [11] of glucose metabolites or neurotransmitters, such as GABA. Finally, the differential stoichiometry of VRAC subunits within the different islet cells may modulate autocrine/paracrine islet responses and the ability of VRAC to act as a Cl– channel.
Cystic fibrosis conductance regulator, CFTR
In addition to Cl– transporters, β-cells also express an impressive array of Cl– channels [54–56] whose characterization has advanced slowly amidst controversy due, in part, to their low expression and perceived functional relevance in the islet. The presence of Cftr in β-cells, as in many other non-epithelial cells, is low and expression is heterogeneous making its study challenging [96,97]. In fact, Cftr is considered not expressed at all in mouse β-cells based on RNAseq analysis [98] or at physiologically irrelevant levels [99–101]. However, it is known that ‘omics’ approaches exclude low expressed genes, particularly in heterogeneous populations of cells [102] like islet β-cells [103,104] or α- and β-cell lines [105]. Although some investigators implicated Cftr in insulin and glucagon secretion [106–108], others did not [99,101]. Similarly, few studies performed in neurons, where Cftr is expressed at low levels, have suggested a role for Cftr in the regulation of [Cl–]i [109,110], including glucose sensing by hypothalamic neurons [111,112]. Importantly and contrary to general awareness, Cftr does not need to be an abundant PM-confined Cl– channel to have a function; Cftr is active in the trans-Golgi network [113], endosomes and lysosomes [114] and in acidic organelles/granules [115]. Moreover, it has been suggested that Cftr regulates exocytosis in β-cells [107], a hypothesis recently tested and extended to a potential role of Cftr in insulin processing [116], a phenomenon of the acidified insulin granule [117]. Indeed, exocytosis events and the number of insulin-containing granules docked at the PM were reduced in β-cells lacking functional Cftr, whereas stimulation of Cftr increased granular acidification [116]. Furthermore, and to keep in mind, Cftr, as many other proteins, may have channel-independent roles, which await discovery. At any rate, the critical analysis of available experimental evidence suggests that Cftr may have a role in the secretory response under certain conditions. It also remains possible that Cftr, as many other islet proteins, may be expressed in a subpopulation of β-cells. At any rate, as ‘absence of evidence is not evidence of absence’ additional experiments are needed to dissect the potential physiological relevance of Cftr in the islet.
Ca2+-activated Cl– channels, Ano1/2
The same issues are applicable to other Cl– channels expressed at low levels in the islet, Ano1/2 are examples. Kozak and Logothetis [118] first described the biophysical properties of a Ca2+-dependent Cl– current in guinea pig β-cells. A current that reversed at –22 mV giving an estimated [Cl–]i of ∼65 mM in these cells, well above that predicted by thermodynamic equilibrium. These biophysical properties are comparable to those of the recently characterized Ca2+-dependent Cl– channels Ano1 and/or Ano2 [119] and now implicated in the secretory response [107,120]. Crutzen et al. established that (i) Ca2+-activated Cl– channels are required to sustain glucose-stimulated membrane potential oscillations and insulin secretion in dispersed β-cells, (ii) Ano1 is expressed at the mRNA and protein levels and (iii) reducing the Cl– driving force by decreasing [Cl–]i through inhibition of Cl– uptake mechanisms impairs Ano1-dependent depolarizing currents [120]. Additionally, Ano1 gene expression is responsive to glucose, whereas siRNA-mediated silencing of Ano1 inhibits insulin secretion in human islets [121,122]. The physiological role(s) and expression of Ano1/Ano2 in the islet have been questioned and used to argue against the specificity TAO1, an inhibitor of Ano1, used to study the involvement of Ca2+-sensitive Cl– channels in β-cells [98]. However, TAO1 [107], like siRNA-mediated Ano1 silencing [121] does inhibit insulin secretion from human islets [107], supporting the hypothesis that Ca2+-activated Cl– channels participate in β-cell secretory responses.
Gamma-aminobutyric acid receptor-A, GABAA
The observation that GABA, which binds and activates pentameric Cl– channels, i.e. the ionotropic GABA receptor-A (GABAA) in other cells, has the potential to modulate the β-cell secretory response is also controversial [123].
We have known, since the early 1970s, that rodent β-cells [124] contain high levels of the neurotransmitter GABA [125–127] distributed in the cytoplasm, and potentially in insulin-containing granules and other cell organelles [128,129]. These results suggested the possibility that β-cells released the neurotransmitter, a concept first demonstrated in rodent β-cells in 1995 [130] and extended to primary human β-cells a decade later [131]. Although it has been already shown that exogenous GABA inhibits first-phase insulin secretion in response to arginine [132] or glucose [133], attributing this effect to the activation of GABAA receptors, which are Cl– channels, would have been complex to interpret based on the hypothesis that opening Cl– channels depolarize/stimulate β-cells [9]. Therefore, if GABAA receptors are not involved in GABA actions in rodent β-cells, then the G-coupled GABA receptor-B (GABAB) might. In support, the GABAB receptor agonist baclofen inhibited islet insulin release [132] but not that of islets lacking GABAB [134]. Furthermore, the GABAA agonist muscimol did not affect insulin release from the isolated and perfused rat pancreas [135] or rodent islets [136], consistent with the proposal that functional GABAA Cl– channels are undetectable in rodent β-cells [98]. However, clonal mouse and rat β-cells do express different subunits of GABAA receptor [137,138]. In fact, GABAA receptor activation in rat INS1 β-cells stimulates or inhibits insulin secretion following changes in glucose levels [138], whereas glucose lowers GABA content in mouse [139] and human islets [140]. Therefore, the role of GABA in rodent islet function remains disputed.
In human β-cells, muscimol activation of GABAA receptors increased insulin secretion in human insulinoma cells [141], whereas the application of GABA to human β-cells resulted in PM depolarization, increased action potential firings and insulin secretion by mechanisms involving GABAA [15]. Taneera et al. [142] subsequently confirmed an active role for GABA in human islets and provided a detailed expression analysis of GABAA subunits. Therefore, it seems that human islets are endowed with a GABA system which can be considered an integral part of the secretory machinery.
Other Cl– channels expressed in the islet
Glycine receptors (GlyRs) are strychnine-sensitive Cl–-selective channels gated by the amino acid and neurotransmitter glycine [143]. GlyRs were originally inferred to be present in pancreatic β-cells by Weaver et al. [144], but their molecular/functional confirmation required two decades [140,145]. Human β-cells do conduct depolarizing Cl– currents and promote action potentials in a GlyR-dependent manner [145]. In addition, human β-cells actively uptake glycine and release this neurotransmitter, whereas insulin appears to enhance GlyR-mediated Cl– currents. Therefore, Yan-Do et al. [145] proposed that β-cells are endowed with an autocrine positive feedback loop that enhances insulin secretion. Although further experiments are needed to corroborate that proposal, it is worth mentioning that activation of GlyR did not promote Ca2+ uptake in a small proportion of human β-cells [145]. These results suggest that a sub-set of these cells may exhibit [Cl–]i below equilibrium. Should that be the case, the opening of a Cl– channel would result in β-cell PM hyperpolarization, decreased Ca2+ uptake and inhibition of insulin secretion in response to glucose. This hypothesis, if tested and demonstrated, would add an extra layer of complexity to the picture of insulin secretion, but would reaffirm the concept that [Cl–]i regulates the secretory response.
Perspectives
The importance of the field: ‘Models are useless if they are not regularly challenged and counterproductive when they become dogmatic" [1]. The current consensus model of insulin secretion is important, but incomplete and must be challenged as new information arises. In Figure 2, we revise the current model to include a version of a 40-year-old hypothesis [9].
Summary of the current thinking: Cl– channels and transporters do contribute to the secretory response complementing KATP-channel function. However, the apparent functional redundancy and cooperativity of the Cl– machinery on the regulation of insulin release, remains far from being understood.
- Future directions: Since global knockout mice did not offer a clear vision on the role that a single Cl– transporter or channel may play in islet biology [146–148], it is now critical to generate conditional mice models harboring β-cell-specific deletion of one, as previously done [11,35], or two, or more components of the machinery involved in the regulation of [Cl–]I and carefully track their phenotypic characterization.
Figure 2. Hypothesis model: Cl– transporters and channels regulate insulin secretion.
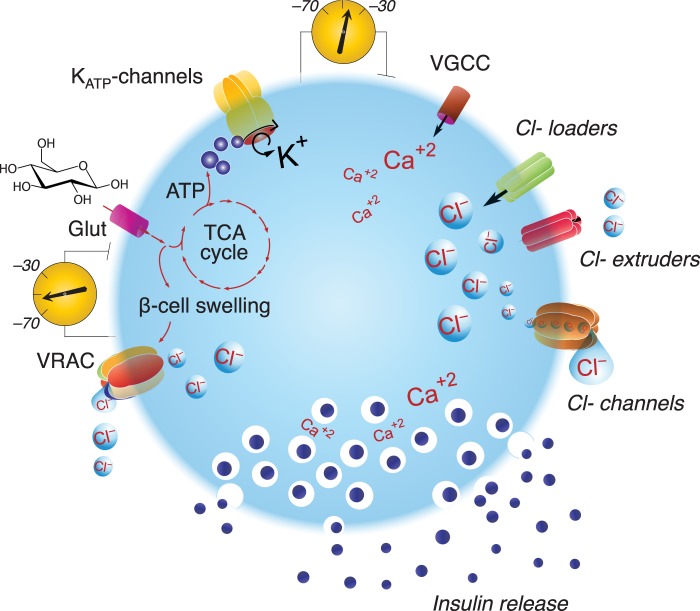 Represented is a β-cell expressing glucose transporter (Glut), KATP-channels, voltage-gated Ca2+ channels (VGCC), Cl– loaders, extruders and channels, including VRAC. Upon facilitative transport of glucose into the β-cell, the sugar is metabolized producing ATP and osmotically active metabolites, including lactate [32], hypothesized to promote the uptake of water to increase cell volume. The former closes KATP-channels, leading to reduced K+ permeability and PM depolarization, and the latter activates a VRAC-mediated inward Cl– current that depolarizes the PM as well. Additional Cl– channels are expected to contribute to Cl– currents, which altogether are sufficient to activate VGCC provoking Ca2+ entry, action potentials, electrical activity and insulin secretion.
Represented is a β-cell expressing glucose transporter (Glut), KATP-channels, voltage-gated Ca2+ channels (VGCC), Cl– loaders, extruders and channels, including VRAC. Upon facilitative transport of glucose into the β-cell, the sugar is metabolized producing ATP and osmotically active metabolites, including lactate [32], hypothesized to promote the uptake of water to increase cell volume. The former closes KATP-channels, leading to reduced K+ permeability and PM depolarization, and the latter activates a VRAC-mediated inward Cl– current that depolarizes the PM as well. Additional Cl– channels are expected to contribute to Cl– currents, which altogether are sufficient to activate VGCC provoking Ca2+ entry, action potentials, electrical activity and insulin secretion.

Acknowledgements
The authors would like to extend sincere thanks to Dr Joseph Bryan for critically reading the manuscript and Drs Len Best and Peter D. Brown for their useful discussions.
Abbreviations
- GlyRs
glycine receptors
- GSIS
glucose-stimulated insulin secretion
- KATP
ATP-sensitive K+
- PM
plasma membrane
- VRAC
volume-regulated anion channel
Author Contribution
L.A.-B. and M.DF. drafted, critically read and edited the article. Both authors approved the final version.
Funding
This work was supported by NIDDK R01 097829 (L.A.-B.), ADA1-17-IBS-258 and R21DK113446-01 (M.D.F.).
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Henquin J.C., Nenquin M., Ravier M.A. and Szollosi A. (2009) Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 11, 168–179 10.1111/j.1463-1326.2009.01109.x [DOI] [PubMed] [Google Scholar]
- 2.Dean P.M. and Matthews E.K. (1970) Electrical activity in pancreatic islet cells: effect of ions. J. Physiol. 210, 265–275 10.1113/jphysiol.1970.sp009208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matschinsky F.M. (2002) Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes 51, S394–S404 10.2337/diabetes.51.2007.S394 [DOI] [PubMed] [Google Scholar]
- 4.Henquin J.C. (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760 10.2337/diabetes.49.11.1751 [DOI] [PubMed] [Google Scholar]
- 5.Aguilar-Bryan L. and Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 PMID: [DOI] [PubMed] [Google Scholar]
- 6.Bryan J., Munoz A., Zhang X., Dufer M., Drews G., Krippeit-Drews P. et al. (2007) ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 453, 703–718 10.1007/s00424-006-0116-z [DOI] [PubMed] [Google Scholar]
- 7.Babenko A.P., Polak M., Cave H., Busiah K., Czernichow P., Scharfmann R. et al. (2006) Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 355, 456–466 10.1056/NEJMoa055068 [DOI] [PubMed] [Google Scholar]
- 8.Thomas P.M., Cote G.J., Wohllk N., Haddad B., Mathew P.M., Rabl W. et al. (1995) Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268, 426–429 10.1126/science.7716548 [DOI] [PubMed] [Google Scholar]
- 9.Sehlin J. (1978) Interrelationship between chloride fluxes in pancreatic islets and insulin release. Am. J. Physiol. 235, E501–E508 [DOI] [PubMed] [Google Scholar]
- 10.Best L., Brown P.D., Sener A. and Malaisse W.J. (2010) Electrical activity in pancreatic islet cells: the VRAC hypothesis. Islets 2, 59–64 10.4161/isl.2.2.11171 [DOI] [PubMed] [Google Scholar]
- 11.Stuhlmann T., Planells-Cases R. and Jentsch T.J. (2018) LRRC8/VRAC anion channels enhance beta-cell glucose sensing and insulin secretion. Nat. Commun. 9, 1974 10.1038/s41467-018-04353-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunne M.J. and Petersen O.H. (1991) Potassium selective ion channels in insulin-secreting cells: physiology, pharmacology and their role in stimulus-secretion coupling. Biochim. Biophys. Acta 1071, 67–82 10.1016/0304-4157(91)90012-L [DOI] [PubMed] [Google Scholar]
- 13.Alvarez-Leefmans F. (2012) Intracellular Chloride Regulation. In Cell Physiology Sourcebook: Essentials of Membrane Biophysics, pp. 221–259, Elsevier/AP, Boston, Amsterdam [Google Scholar]
- 14.Eberhardson M., Patterson S. and Grapengiesser E. (2000) Microfluorometric analysis of Cl- permeability and its relation to oscillatory Ca2+ signaling in glucose-stimulated pancreatic β-cells. Cell. Signal. 12, 781–786 10.1016/S0898-6568(00)00122-4 [DOI] [PubMed] [Google Scholar]
- 15.Braun M., Ramracheya R., Bengtsson M., Clark A., Walker J.N., Johnson P.R. et al. (2010) Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59, 1694–1701 10.2337/db09-0797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Fulvio M., Brown P. D. and Aguilar-Bryan L. (2014) Chloride channels and transporters in β-cell physiology In The Islets of Langerhans (Islam M. S., ed.), pp. 401–451, Springer-Verlag [Google Scholar]
- 17.Best L. (2000) Glucose-sensitive conductances in rat pancreatic beta-cells: contribution to electrical activity. Biochim. Biophys. Acta. 1468, 311–319 10.1016/S0005-2736(00)00272-8 [DOI] [PubMed] [Google Scholar]
- 18.Best L. (2005) Glucose-induced electrical activity in rat pancreatic β-cells: dependence on intracellular chloride concentration. J. Physiol. 568, 137–144 10.1113/jphysiol.2005.093740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddlestone G.T. and Beigelman P.M. (1983) Pancreatic beta-cell electrical activity: the role of anions and the control of pH. Am. J. Physiol. 244, C188–C197 10.1152/ajpcell.1983.244.3.C188 [DOI] [PubMed] [Google Scholar]
- 20.Berglund O., Sehlin J. and Taljedal I.B. (1979) Defective ion transport in diabetic mouse islet cells. Acta Physiol. Scand. 106, 489–490 10.1111/j.1748-1716.1979.tb06431.x [DOI] [PubMed] [Google Scholar]
- 21.Berglund O. (1981) Disturbed fluxes of Rb+(K+) and Cl− in islets of spontaneously diabetic mice (C57BL/KsJ-db/db). Acta Biol. Med. Ger. 40, 23–30 [PubMed] [Google Scholar]
- 22.Somers G., Sener A., Devis G. and Malaisse W.J. (1980) The stimulus-secretion coupling of glucose-induced insulin release. XLV. The anion-osmotic hypothesis for exocytosis. Pflugers Arch. 388, 249–253 10.1007/BF00658490 [DOI] [PubMed] [Google Scholar]
- 23.Tamagawa T. and Henquin J.C. (1983) Chloride modulation of insulin release, 86Rb+ efflux, and 45Ca2+ fluxes in rat islets stimulated by various secretagogues. Diabetes 32, 416–423 10.2337/diab.32.5.416 [DOI] [PubMed] [Google Scholar]
- 24.Cook D.L. and Hales C.N. (1984) Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311, 271–273 10.1038/311271a0 [DOI] [PubMed] [Google Scholar]
- 25.Rorsman P. and Trube G. (1985) Glucose dependent K+-channels in pancreatic beta-cells are regulated by intracellular ATP. Pflugers Arch. 405, 305–309 10.1007/BF00595682 [DOI] [PubMed] [Google Scholar]
- 26.Sturgess N.C., Ashford M.L., Cook D.L. and Hales C.N. (1985) The sulphonylurea receptor may be an ATP-sensitive potassium channel. Lancet 2, 474–475 10.1016/S0140-6736(85)90403-9 [DOI] [PubMed] [Google Scholar]
- 27.Dunne M.J. and Petersen O.H. (1986) Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett. 208, 59–62 10.1016/0014-5793(86)81532-0 [DOI] [PubMed] [Google Scholar]
- 28.Ashcroft F.M. and Rorsman P. (1989) Electrophysiology of the pancreatic beta-cell. Prog. Biophys. Mol. Biol. 54, 87–143 10.1016/0079-6107(89)90013-8 [DOI] [PubMed] [Google Scholar]
- 29.Kinard T.A. and Satin L.S. (1995) An ATP-sensitive Cl- channel current that is activated by cell swelling, cAMP, and glyburide in insulin-secreting cells. Diabetes 44, 1461–1466 10.2337/diab.44.12.1461 [DOI] [PubMed] [Google Scholar]
- 30.Best L., Sheader E.A. and Brown P.D. (1996) A volume-activated anion conductance in insulin-secreting cells. Pflugers Arch. 431, 363–370 10.1007/BF02207273 [DOI] [PubMed] [Google Scholar]
- 31.Blackard W.G., Kikuchi M., Rabinovitch A. and Renold A.E. (1975) An effect of hyposmolarity on insulin release in vitro. Am. J. Physiol. 228, 706–713 10.1152/ajplegacy.1975.228.3.706 [DOI] [PubMed] [Google Scholar]
- 32.Best L., Miley H.E., Brown P.D. and Cook L.J. (1999) Methylglyoxal causes swelling and activation of a volume-sensitive anion conductance in rat pancreatic beta-cells. J. Membr. Biol. 167, 65–71 10.1007/s002329900472 [DOI] [PubMed] [Google Scholar]
- 33.Best L., Miley H.E. and Yates A.P. (1996) Activation of an anion conductance and beta-cell depolarization during hypotonically induced insulin release. Exp. Physiol. 81, 927–933 10.1113/expphysiol.1996.sp003993 [DOI] [PubMed] [Google Scholar]
- 34.Best L., Brown P.D. and Tomlinson S. (1997) Anion fluxes, volume regulation and electrical activity in the mammalian pancreatic β-cell. Exp. Physiol. 82, 957–966 10.1113/expphysiol.1997.sp004081 [DOI] [PubMed] [Google Scholar]
- 35.Kang C., Xie L., Gunasekar S.K., Mishra A., Zhang Y., Pai S. et al. (2018) SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat Commun. 9, 367 10.1038/s41467-017-02664-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartmann A.M., Tesch D., Nothwang H.G. and Bininda-Emonds O.R. (2014) Evolution of the cation chloride cotransporter family: ancient origins, gene losses, and subfunctionalization through duplication. Mol. Biol. Evol. 31, 434–447 10.1093/molbev/mst225 [DOI] [PubMed] [Google Scholar]
- 37.Gamba G. (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493 10.1152/physrev.00011.2004 [DOI] [PubMed] [Google Scholar]
- 38.Sehlin J. (1981) Are Cl− mechanisms in mouse pancreatic islets involved in insulin release? Ups. J. Med. Sci. 86, 177–182 10.3109/03009738109179226 [DOI] [PubMed] [Google Scholar]
- 39.Sandstrom P.E. and Sehlin J. (1987) Stereoselective inhibition of chloride transport by loop diuretics in pancreatic beta-cells. Eur. J. Pharmacol. 144, 389–392 10.1016/0014-2999(87)90394-3 [DOI] [PubMed] [Google Scholar]
- 40.Sandstrom P.E. and Sehlin J. (1988) Furosemide reduces insulin release by inhibition of Cl– and Ca2+ fluxes in beta-cells. Am. J. Physiol. 255, E591–E596 [DOI] [PubMed] [Google Scholar]
- 41.Sandstrom P.E. and Sehlin J. (1988) Furosemide-induced glucose intolerance in mice is associated with reduced insulin secretion. Eur. J. Pharmacol. 147, 403–409 10.1016/0014-2999(88)90175-6 [DOI] [PubMed] [Google Scholar]
- 42.Sandstrom P.E. (1988) Evidence for diabetogenic action of bumetanide in mice. Eur. J. Pharmacol. 150, 35–41 10.1016/0014-2999(88)90747-9 [DOI] [PubMed] [Google Scholar]
- 43.Sandstrom P.E. (1990) Bumetanide reduces insulin release by a direct effect on the pancreatic beta-cells. Eur. J. Pharmacol. 187, 377–383 10.1016/0014-2999(90)90365-D [DOI] [PubMed] [Google Scholar]
- 44.Sandstrom P.E. and Sehlin J. (1990) Na+ participates in loop diuretic-sensitive Cl(-)-cation co-transport in the pancreatic beta-cells. Biochim. Biophys. Acta. 1023, 191–196 10.1016/0005-2736(90)90413-I [DOI] [PubMed] [Google Scholar]
- 45.Sandstrom P.E. and Sehlin J. (1993) Evidence for separate Na+, K+, Cl− and K+, Cl− co-transport systems in mouse pancreatic beta-cells. Eur. J. Pharmacol. 238, 403–405 10.1016/0014-2999(93)90875-I [DOI] [PubMed] [Google Scholar]
- 46.Giugliano D., Varricchio M., Cerciello T., Varano R., Saccomanno F. and Giannetti G. (1980) Bumetanide and glucose tolerance in man. Farmaco Prat. 35, 403–408 [PubMed] [Google Scholar]
- 47.Sandstrom P.E. and Sehlin J. (1988) Furosemide causes acute and long-term hyperglycaemia and reduces glucose tolerance in mice. Acta Physiol. Scand. 132, 75–81 10.1111/j.1748-1716.1988.tb08300.x [DOI] [PubMed] [Google Scholar]
- 48.Lindstrom P., Norlund L. and Sehlin J. (1986) Potassium and chloride fluxes are involved in volume regulation in mouse pancreatic islet cells. Acta Physiol. Scand. 128, 541–546 10.1111/j.1748-1716.1986.tb08010.x [DOI] [PubMed] [Google Scholar]
- 49.Engstrom K.G., Sandstrom P.E. and Sehlin J. (1991) Volume regulation in mouse pancreatic beta-cells is mediated by a furosemide-sensitive mechanism. Biochim. Biophys. Acta 1091, 145–150 10.1016/0167-4889(91)90054-2 [DOI] [PubMed] [Google Scholar]
- 50.Majid A., Speake T., Best L. and Brown P.D. (2001) Expression of the Na + K+-2CI− cotransporter in α and β cells isolated from the rat pancreas. Pflugers Arch. 442, 570–576 10.1007/s004240100566 [DOI] [PubMed] [Google Scholar]
- 51.Davies S.L., Roussa E., Le Rouzic P., Thevenod F., Alper S.L., Best L. et al. (2004) Expression of K+-Cl- cotransporters in the alpha-cells of rat endocrine pancreas. Biochim. Biophys. Acta 1667, 7–14 10.1016/j.bbamem.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 52.Alshahrani S., Alvarez-Leefmans F. and Di Fulvio M. (2012) Expression of the Slc12a1 gene in pancreatic β-cells: molecular characterization and in silico analysis. Cell. Physiol. Biochem. 30, 95–112 10.1159/000339050 [DOI] [PubMed] [Google Scholar]
- 53.Kursan S., McMillen T.S., Beesetty P., Dias-Junior E., Almutairi M.M., Sajib A.A. et al. (2017) The neuronal K + Cl- co-transporter 2 (Slc12a5) modulates insulin secretion. Sci. Rep. 7, 1732 10.1038/s41598-017-01814-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Segerstolpe A., Palasantza A., Eliasson P., Andersson E.M., Andreasson A.C., Sun X. et al. (2016) Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 10.1016/j.cmet.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blodgett D.M., Nowosielska A., Afik S., Pechhold S., Cura A.J., Kennedy N.J. et al. (2015) Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64, 3172–3181 10.2337/db15-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waanders L.F., Chwalek K., Monetti M., Kumar C., Lammert E. and Mann M. (2009) Quantitative proteomic analysis of single pancreatic islets. Proc. Natl Acad. Sci. U.S.A. 106, 18902–18907 10.1073/pnas.0908351106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alper S.L. and Sharma A.K. (2013) The SLC26 gene family of anion transporters and channels. Mol. Aspects Med. 34, 494–515 10.1016/j.mam.2012.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker M.D. (2018) Mouse models of SLC4-linked disorders of HCO3(-)-transporter dysfunction. Am. J. Physiol. Cell Physiol. 314, C569–C588 10.1152/ajpcell.00301.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alper S.L. (2009) Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J. Exp. Biol. 212, 1672–1683 10.1242/jeb.029454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pace C.S. and Tarvin J.T. (1983) Ph modulation of glucose-induced electrical activity in B-cells: involvement of Na/H and HCO3/Cl antiporters. J. Membr. Biol. 73, 39–49 10.1007/BF01870339 [DOI] [PubMed] [Google Scholar]
- 61.Shepherd R.M. and Henquin J.C. (1995) The role of metabolism, cytoplasmic Ca2+, and pH-regulating exchangers in glucose-induced rise of cytoplasmic pH in normal mouse pancreatic islets. J. Biol. Chem. 270, 7915–7921 10.1074/jbc.270.14.7915 [DOI] [PubMed] [Google Scholar]
- 62.Soyfoo M.S., Bulur N., Virreira M., Louchami K., Lybaert P., Crutzen R. et al. (2009) Expression of the electrogenic Na+-HCO3—cotransporters NBCe1-A and NBCe1-B in rat pancreatic islet cells. Endocrine 35, 449–458 10.1007/s12020-009-9175-1 [DOI] [PubMed] [Google Scholar]
- 63.Wang C.Z., Yano H., Nagashima K. and Seino S. (2000) The Na+-driven Cl–/HCO3- exchanger. Cloning, tissue distribution, and functional characterization. J. Biol. Chem. 275, 35486–35490 10.1074/jbc.C000456200 [DOI] [PubMed] [Google Scholar]
- 64.Jentsch T.J. and Pusch M. (2018) CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev. 98, 1493–1590 10.1152/physrev.00047.2017 [DOI] [PubMed] [Google Scholar]
- 65.Deriy L.V., Gomez E.A., Jacobson D.A., Wang X., Hopson J.A., Liu X.Y. et al. (2009) The granular chloride channel ClC-3 is permissive for insulin secretion. Cell Metab. 10, 316–323 10.1016/j.cmet.2009.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li D.Q., Jing X., Salehi A., Collins S.C., Hoppa M.B., Rosengren A.H. et al. (2009) Suppression of sulfonylurea- and glucose-induced insulin secretion in vitro and in vivo in mice lacking the chloride transport protein ClC-3. Cell Metab. 10, 309–315 10.1016/j.cmet.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 67.Barg S., Huang P., Eliasson L., Nelson D.J., Obermuller S., Rorsman P. et al. (2001) Priming of insulin granules for exocytosis by granular Cl(–) uptake and acidification. J. Cell Sci. 114, 2145–2154 [DOI] [PubMed] [Google Scholar]
- 68.Maritzen T., Keating D.J., Neagoe I., Zdebik A.A. and Jentsch T.J. (2008) Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J. Neurosci. 28, 10587–10598 10.1523/JNEUROSCI.3750-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jentsch T.J., Maritzen T., Keating D.J., Zdebik A.A. and Thevenod F. (2010) ClC-3–a granular anion transporter involved in insulin secretion? Cell Metab. 12, 307–308; author reply 309–310 10.1016/j.cmet.2010.08.014 [DOI] [PubMed] [Google Scholar]
- 70.Britsch S., Krippeit-Drews P., Gregor M., Lang F. and Drews G. (1994) Effects of osmotic changes in extracellular solution on electrical activity of mouse pancreatic B-cells. Biochem. Biophys. Res. Commun. 204, 641–645 10.1006/bbrc.1994.2507 [DOI] [PubMed] [Google Scholar]
- 71.Best L. (1997) Glucose and alpha-ketoisocaproate induce transient inward currents in rat pancreatic beta cells. Diabetologia 40, 1–6 10.1007/s001250050635 [DOI] [PubMed] [Google Scholar]
- 72.Miley H.E., Sheader E.A., Brown P.D. and Best L. (1997) Glucose-induced swelling in rat pancreatic beta-cells. J. Physiol. 504, 191–198 10.1111/j.1469-7793.1997.00191.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miley H.E., Holden D., Grint R., Best L. and Brown P.D. (1998) Regulatory volume increase in rat pancreatic beta-cells. Pflugers Arch. 435, 227–230 10.1007/s004240050505 [DOI] [PubMed] [Google Scholar]
- 74.Best L. and Benington S. (1998) Effects of sulphonylureas on the volume-sensitive anion channel in rat pancreatic beta-cells. Br. J. Pharmacol. 125, 874–878 10.1038/sj.bjp.0702148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Best L. (2002) Evidence that glucose-induced electrical activity in rat pancreatic β-cells does not require KATP channel inhibition. J. Membr. Biol. 185, 193–200 10.1007/s00232-001-0114-1 [DOI] [PubMed] [Google Scholar]
- 76.Drews G., Zempel G., Krippeit-Drews P., Britsch S., Busch G.L., Kaba N.K. et al. (1998) Ion channels involved in insulin release are activated by osmotic swelling of pancreatic B-cells. Biochim. Biophys. Acta. 1370, 8–16 10.1016/S0005-2736(97)00240-X [DOI] [PubMed] [Google Scholar]
- 77.Best L. (1999) Cell-attached recordings of the volume-sensitive anion channel in rat pancreatic beta-cells. Biochim. Biophys. Acta. 1419, 248–256 10.1016/S0005-2736(99)00071-1 [DOI] [PubMed] [Google Scholar]
- 78.Best L., Yates A.P., Decher N., Steinmeyer K. and Nilius B. (2004) Inhibition of glucose-induced electrical activity in rat pancreatic beta-cells by DCPIB, a selective inhibitor of volume-sensitive anion currents. Eur. J. Pharmacol. 489, 13–19 10.1016/j.ejphar.2004.02.030 [DOI] [PubMed] [Google Scholar]
- 79.Seghers V., Nakazaki M., DeMayo F., Aguilar-Bryan L. and Bryan J. (2000) Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J. Biol. Chem. 275, 9270–9277 10.1074/jbc.275.13.9270 [DOI] [PubMed] [Google Scholar]
- 80.Szollosi A., Nenquin M., Aguilar-Bryan L., Bryan J. and Henquin J.C. (2007) Glucose stimulates Ca2+ influx and insulin secretion in 2-week-old beta-cells lacking ATP-sensitive K+ channels. J. Biol. Chem. 282, 1747–1756 10.1074/jbc.M609875200 [DOI] [PubMed] [Google Scholar]
- 81.Ravier M.A., Nenquin M., Miki T., Seino S. and Henquin J.C. (2009) Glucose controls cytosolic Ca2+ and insulin secretion in mouse islets lacking adenosine triphosphate-sensitive K+ channels owing to a knockout of the pore-forming subunit Kir6.2. Endocrinology 150, 33–45 10.1210/en.2008-0617 [DOI] [PubMed] [Google Scholar]
- 82.Voss F.K., Ullrich F., Munch J., Lazarow K., Lutter D., Mah N. et al. (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638 10.1126/science.1252826 [DOI] [PubMed] [Google Scholar]
- 83.Qiu Z., Dubin A.E., Mathur J., Tu B., Reddy K., Miraglia L.J. et al. (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458 10.1016/j.cell.2014.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ullrich F., Reincke S.M., Voss F.K., Stauber T. and Jentsch T.J. (2016) Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. J. Biol. Chem. 291, 17040–17048 10.1074/jbc.M116.739342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaitan-Penas H., Gradogna A., Laparra-Cuervo L., Solsona C., Fernandez-Duenas V., Barrallo-Gimeno A. et al. (2016) Investigation of LRRC8-mediated volume-regulated anion currents in xenopus oocytes. Biophys. J. 111, 1429–1443 10.1016/j.bpj.2016.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Konig B. and Stauber T. (2019) Biophysics and structure-function relationships of LRRC8-formed volume-regulated anion channels. Biophys. J. 116, 1185–1193 10.1016/j.bpj.2019.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kasuya G., Nakane T., Yokoyama T., Jia Y., Inoue M., Watanabe K. et al. (2018) Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat. Struct. Mol. Biol. 25, 797–804 10.1038/s41594-018-0109-6 [DOI] [PubMed] [Google Scholar]
- 88.Kefauver J.M., Saotome K., Dubin A.E., Pallesen J., Cottrell C.A., Cahalan S.M. et al. (2018) Structure of the human volume regulated anion channel. eLife 7, e38461 10.7554/eLife.38461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deneka D., Sawicka M., Lam A.K.M., Paulino C. and Dutzler R. (2018) Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558, 254–259 10.1038/s41586-018-0134-y [DOI] [PubMed] [Google Scholar]
- 90.Planells-Cases R., Lutter D., Guyader C., Gerhards N.M., Ullrich F., Elger D.A. et al. (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 34, 2993–3008 10.15252/embj.201592409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ribeiro R.A., Bonfleur M.L., Batista T.M., Borck P.C. and Carneiro E.M. (2018) Regulation of glucose and lipid metabolism by the pancreatic and extra-pancreatic actions of taurine. Amino Acids 50, 1511–1524 10.1007/s00726-018-2650-3 [DOI] [PubMed] [Google Scholar]
- 92.Lutter D., Ullrich F., Lueck J.C., Kempa S. and Jentsch T.J. (2017) Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci. 130, 1122–1133 [DOI] [PubMed] [Google Scholar]
- 93.Pace C.S. and Clements R.S. (1981) Myo-inositol and the maintenance of beta-cell function in cultured rat pancreatic islets. Diabetes 30, 621–625 10.2337/diab.30.8.621 [DOI] [PubMed] [Google Scholar]
- 94.Li S.Y.T., Cheng S.T.W., Zhang D. and Leung P.S. (2017) Identification and functional implications of sodium/Myo-inositol cotransporter 1 in pancreatic beta-cells and type 2 diabetes. Diabetes 66, 1258–1271 10.2337/db16-0880 [DOI] [PubMed] [Google Scholar]
- 95.Sener A., Blachier F., Rasschaert J., Mourtada A., Malaisse-Lagae F. and Malaisse W.J. (1989) Stimulus-secretion coupling of arginine-induced insulin release: comparison with lysine-induced insulin secretion. Endocrinology 124, 2558–2567 10.1210/endo-124-5-2558 [DOI] [PubMed] [Google Scholar]
- 96.Farinha C.M., Penque D., Roxo-Rosa M., Lukacs G., Dormer R., McPherson M. et al. (2004) Biochemical methods to assess CFTR expression and membrane localization. J Cyst Fibros. 3, 73–77 10.1016/j.jcf.2004.05.017 [DOI] [PubMed] [Google Scholar]
- 97.Mendes F., Farinha C.M., Roxo-Rosa M., Fanen P., Edelman A., Dormer R. et al. (2004) Antibodies for CFTR studies. J Cyst Fibros. 3, 69–72 10.1016/j.jcf.2004.05.016 [DOI] [PubMed] [Google Scholar]
- 98.Rorsman P. and Ashcroft F.M. (2018) Pancreatic beta-cell electrical activity and insulin secretion: of mice and men. Physiol. Rev. 98, 117–214 10.1152/physrev.00008.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Olivier A.K., Yi Y., Sun X., Sui H., Liang B., Hu S. et al. (2012) Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J. Clin. Invest. 122, 3755–3768 10.1172/JCI60610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun X., Yi Y., Xie W., Liang B., Winter M.C., He N. et al. (2017) CFTR influences beta cell function and insulin secretion through non-cell autonomous exocrine-derived factors. Endocrinology 158, 3325–3338 10.1210/en.2017-00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hart N.J., Aramandla R., Poffenberger G., Fayolle C., Thames A.H., Bautista A. et al. (2018) Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight. 3 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hwang B., Lee J.H. and Bang D. (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 50, 96 10.1038/s12276-018-0071-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benninger R.K.P. and Hodson D.J. (2018) New understanding of beta-cell heterogeneity and in situ islet function. Diabetes 67, 537–547 10.2337/dbi17-0040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnston N.R., Mitchell R.K., Haythorne E., Pessoa M.P., Semplici F., Ferrer J. et al. (2016) Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 24, 389–401 10.1016/j.cmet.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boom A., Lybaert P., Pollet J.F., Jacobs P., Jijakli H., Golstein P.E. et al. (2007) Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocrine 32, 197–205 10.1007/s12020-007-9026-x [DOI] [PubMed] [Google Scholar]
- 106.Guo J.H., Chen H., Ruan Y.C., Zhang X.L., Zhang X.H., Fok K.L. et al. (2014) Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nat Commun. 5, 4420 10.1038/ncomms5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edlund A., Esguerra J.L., Wendt A., Flodstrom-Tullberg M. and Eliasson L. (2014) CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med. 12, 87 10.1186/1741-7015-12-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Edlund A., Pedersen M.G., Lindqvist A., Wierup N., Flodstrom-Tullberg M. and Eliasson L. (2017) CFTR is involved in the regulation of glucagon secretion in human and rodent alpha cells. Sci. Rep. 7, 90 10.1038/s41598-017-00098-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morales F.R., Silveira V., Damian A., Higgie R. and Pose I. (2011) The possible additional role of the cystic fibrosis transmembrane regulator to motoneuron inhibition produced by glycine effects. Neuroscience 177, 138–147 10.1016/j.neuroscience.2010.12.034 [DOI] [PubMed] [Google Scholar]
- 110.Ostroumov A., Simonetti M. and Nistri A. (2011) Cystic fibrosis transmembrane conductance regulator modulates synaptic chloride homeostasis in motoneurons of the rat spinal cord during neonatal development. Dev. Neurobiol. 71, 253–268 10.1002/dneu.20855 [DOI] [PubMed] [Google Scholar]
- 111.Chalmers J.A., Jang J.J. and Belsham D.D. (2014) Glucose sensing mechanisms in hypothalamic cell models: glucose inhibition of AgRP synthesis and secretion. Mol. Cell. Endocrinol. 382, 262–270 10.1016/j.mce.2013.10.013 [DOI] [PubMed] [Google Scholar]
- 112.Murphy B.A., Fakira K.A., Song Z., Beuve A. and Routh V.H. (2009) AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 297, C750–C758 10.1152/ajpcell.00127.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barasch J., Kiss B., Prince A., Saiman L., Gruenert D. and al-Awqati Q. (1991) Defective acidification of intracellular organelles in cystic fibrosis. Nature 352, 70–73 10.1038/352070a0 [DOI] [PubMed] [Google Scholar]
- 114.Biwersi J. and Verkman A.S. (1994) Functional CFTR in endosomal compartment of CFTR-expressing fibroblasts and T84 cells. Am. J. Physiol. 266, C149–C156 10.1152/ajpcell.1994.266.1.C149 [DOI] [PubMed] [Google Scholar]
- 115.Krishnan V., Maddox J.W., Rodriguez T. and Gleason E. (2017) A role for the cystic fibrosis transmembrane conductance regulator in the nitric oxide-dependent release of Cl(–) from acidic organelles in amacrine cells. J. Neurophysiol. 118, 2842–2852 10.1152/jn.00511.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Edlund A., Barghouth M., Huhn M., Abels M., Esguerra J., Mollet I. et al. (2019) Defective exocytosis and processing of insulin in a cystic fibrosis mouse model. J. Endocrinol 241, 45–57 10.1530/JOE-18-0570 [DOI] [PubMed] [Google Scholar]
- 117.Davidson H.W., Rhodes C.J. and Hutton J.C. (1988) Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic beta cell via two distinct site-specific endopeptidases. Nature 333, 93–96 10.1038/333093a0 [DOI] [PubMed] [Google Scholar]
- 118.Kozak J.A. and Logothetis D.E. (1997) A calcium-dependent chloride current in insulin-secreting beta TC-3 cells. Pflugers Arch. 433, 679–690 10.1007/s004240050332 [DOI] [PubMed] [Google Scholar]
- 119.Pedemonte N. and Galietta L.J. (2014) Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94, 419–459 10.1152/physrev.00039.2011 [DOI] [PubMed] [Google Scholar]
- 120.Crutzen R., Virreira M., Markadieu N., Shlyonsky V., Sener A., Malaisse W.J. et al. (2016) Anoctamin 1 (Ano1) is required for glucose-induced membrane potential oscillations and insulin secretion by murine beta-cells. Pflugers Arch. 468, 573–591 10.1007/s00424-015-1758-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xu Z., Lefevre G.M., Gavrilova O., Foster St. Claire M.B., Riddick G. and Felsenfeld G. (2014) Mapping of long-range INS promoter interactions reveals a role for calcium-activated chloride channel ANO1 in insulin secretion. Proc. Natl Acad. Sci. U.S.A. 111, 16760–16765 10.1073/pnas.1419240111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jian X. and Felsenfeld G. (2018) Insulin promoter in human pancreatic beta cells contacts diabetes susceptibility loci and regulates genes affecting insulin metabolism. Proc. Natl Acad. Sci. U.S.A. 115, E4633–E4641 10.1073/pnas.1803146115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Franklin I.K. and Wollheim C.B. (2004) GABA in the endocrine pancreas: its putative role as an islet cell paracrine-signalling molecule. J. Gen. Physiol. 123, 185–190 10.1085/jgp.200409016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garry D.J., Sorenson R.L., Elde R.P., Maley B.E. and Madsen A. (1986) Immunohistochemical colocalization of GABA and insulin in beta-cells of rat islet. Diabetes 35, 1090–1095 10.2337/diab.35.10.1090 [DOI] [PubMed] [Google Scholar]
- 125.Briel G., Gylfe E., Hellman B. and Neuhoff V. (1972) Microdetermination of free amino acids in pancreatic islets isolated from obese-hyperglycemic mice. Acta Physiol. Scand. 84, 247–253 10.1111/j.1748-1716.1972.tb05175.x [DOI] [PubMed] [Google Scholar]
- 126.Okada Y., Taniguchi H. and Schimada C. (1976) High concentration of GABA and high glutamate decarboxylase activity in rat pancreatic islets and human insulinoma. Science 194, 620–622 10.1126/science.185693 [DOI] [PubMed] [Google Scholar]
- 127.Taniguchi H., Okada Y., Shimada C. and Baba S. (1977) GABA in pancreatic islets. Arch. Histol. Jpn. 40, 87–97 10.1679/aohc1950.40.Supplement_87 [DOI] [PubMed] [Google Scholar]
- 128.Garry D.J., Sorenson R.L. and Coulter H.D. (1987) Ultrastructural localization of gamma amino butyric acid immunoreactivity in B cells of the rat pancreas. Diabetologia 30, 115–119 [DOI] [PubMed] [Google Scholar]
- 129.Thomas-Reetz A.C. and De Camilli P. (1994) A role for synaptic vesicles in non-neuronal cells: clues from pancreatic beta cells and from chromaffin cells. FASEB J. 8, 209–216 10.1096/fasebj.8.2.7907072 [DOI] [PubMed] [Google Scholar]
- 130.Gaskins H.R., Baldeon M.E., Selassie L. and Beverly J.L. (1995) Glucose modulates gamma-aminobutyric acid release from the pancreatic beta TC6 cell line. J. Biol. Chem. 270, 30286–30289 10.1074/jbc.270.51.30286 [DOI] [PubMed] [Google Scholar]
- 131.Braun M., Wendt A., Birnir B., Broman J., Eliasson L., Galvanovskis J. et al. (2004) Regulated exocytosis of GABA-containing synaptic-like microvesicles in pancreatic beta-cells. J. Gen. Physiol. 123, 191–204 10.1085/jgp.200308966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gu X.H., Kurose T., Kato S., Masuda K., Tsuda K., Ishida H. et al. (1993) Suppressive effect of GABA on insulin secretion from the pancreatic beta-cells in the rat. Life Sci. 52, 687–694 10.1016/0024-3205(93)90229-V [DOI] [PubMed] [Google Scholar]
- 133.Shi Y., Kanaani J., Menard-Rose V., Ma Y.H., Chang P.Y., Hanahan D. et al. (2000) Increased expression of GAD65 and GABA in pancreatic beta-cells impairs first-phase insulin secretion. Am. J. Physiol. Endocrinol. Metab. 279, E684–E694 10.1152/ajpendo.2000.279.3.E684 [DOI] [PubMed] [Google Scholar]
- 134.Bonaventura M.M., Catalano P.N., Chamson-Reig A., Arany E., Hill D., Bettler B. et al. (2008) GABAB receptors and glucose homeostasis: evaluation in GABAB receptor knockout mice. Am. J. Physiol. Endocrinol. Metab. 294, E157–E167 10.1152/ajpendo.00615.2006 [DOI] [PubMed] [Google Scholar]
- 135.Robbins M.S., Grouse L.H., Sorenson R.L. and Elde R.P. (1981) Effect of muscimol on glucose-stimulated somatostatin and insulin release from the isolated, perfused rat pancreas. Diabetes 30, 168–171 10.2337/diab.30.2.168 [DOI] [PubMed] [Google Scholar]
- 136.Gilon P., Bertrand G., Loubatieres-Mariani M.M., Remacle C. and Henquin J.C. (1991) The influence of gamma-aminobutyric acid on hormone release by the mouse and rat endocrine pancreas. Endocrinology 129, 2521–2529 10.1210/endo-129-5-2521 [DOI] [PubMed] [Google Scholar]
- 137.Xu E., Kumar M., Zhang Y., Ju W., Obata T., Zhang N. et al. (2006) Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 3, 47–58 10.1016/j.cmet.2005.11.015 [DOI] [PubMed] [Google Scholar]
- 138.Dong H., Kumar M., Zhang Y., Gyulkhandanyan A., Xiang Y.Y., Ye B. et al. (2006) Gamma-aminobutyric acid up- and downregulates insulin secretion from beta cells in concert with changes in glucose concentration. Diabetologia 49, 697–705 10.1007/s00125-005-0123-1 [DOI] [PubMed] [Google Scholar]
- 139.Li C., Nissim I., Chen P., Buettger C., Najafi H., Daikhin Y. et al. (2008) Elimination of KATP channels in mouse islets results in elevated [U-13C]glucose metabolism, glutaminolysis, and pyruvate cycling but a decreased gamma-aminobutyric acid shunt. J. Biol. Chem. 283, 17238–17249 10.1074/jbc.M709235200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li C., Liu C., Nissim I., Chen J., Chen P., Doliba N. et al. (2013) Regulation of glucagon secretion in normal and diabetic human islets by gamma-hydroxybutyrate and glycine. J. Biol. Chem. 288, 3938–3951 10.1074/jbc.M112.385682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Glassmeier G., Hopfner M., Buhr H., Lemmer K., Riecken E.O., Stein H. et al. (1998) Expression of functional GABAA receptors in isolated human insulinoma cells. Ann. N. Y. Acad. Sci. 859, 241–248 10.1111/j.1749-6632.1998.tb11138.x [DOI] [PubMed] [Google Scholar]
- 142.Taneera J., Jin Z., Jin Y., Muhammed S.J., Zhang E., Lang S. et al. (2012) γ-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 55, 1985–1994 10.1007/s00125-012-2548-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lynch J.W. (2004) Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 84, 1051–1095 10.1152/physrev.00042.2003 [DOI] [PubMed] [Google Scholar]
- 144.Weaver C.D., Partridge J.G., Yao T.L., Moates J.M., Magnuson M.A. and Verdoorn T.A. (1998) Activation of glycine and glutamate receptors increases intracellular calcium in cells derived from the endocrine pancreas. Mol. Pharmacol. 54, 639–646 PMID: [PubMed] [Google Scholar]
- 145.Yan-Do R., Duong E., Manning Fox J.E., Dai X., Suzuki K., Khan S. et al. (2016) A glycine-insulin autocrine feedback loop enhances insulin secretion from human beta-cells and is impaired in type 2 diabetes. Diabetes 65, 2311–2321 10.2337/db15-1272 [DOI] [PubMed] [Google Scholar]
- 146.Alshahrani S., Almutairi M.M., Kursan S., Dias-Junior E., Almiahuob M.M., Aguilar-Bryan L. et al. (2015) Increased Slc12a1 expression in β-cells and improved glucose disposal in Slc12a2 heterozygous mice. J. Endocrinol. 227, 153–165 10.1530/JOE-15-0327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alshahrani S. and Di Fulvio M. (2012) Enhanced insulin secretion and improved glucose tolerance in mice with homozygous inactivation of the Na + K + 2Cl- co-transporter 1. J. Endocrinol. 215, 59–70 10.1530/JOE-12-0244 [DOI] [PubMed] [Google Scholar]
- 148.Kelly L., Almutairi M.M., Kursan S., Pacheco R., Dias-Junior E., Castrop H. et al. (2019) Impaired glucose tolerance, glucagon, and insulin responses in mice lacking the loop diuretic-sensitive Nkcc2a transporter. Am. J. Physiol. Cell Physiol. 317, C843–C856 10.1152/ajpcell.00144.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]