

SUMMARY
HIV-1 Nef enhances virion infectivity by counteracting host restriction factor SERINC5; however, the impact of natural Nef polymorphisms on this function is largely unknown. We characterize SERINC5 downregulation activity of 91 primary HIV-1 subtype B nef alleles, including isolates from 45 elite controllers and 46 chronic progressors. Controller-derived Nef clones display lower ability to downregulate SERINC5 (median 80% activity) compared with progressor-derived clones (median 96% activity) (p = 0.0005). We identify 18 Nef polymorphisms associated with differential function, including two CTL escape mutations that contribute to lower SERINC5 downregulation: K94E, driven by HLA-B*08, and H116N, driven by the protective allele HLA-B*57. HIV-1 strains encoding Nef K94E and/or H116N display lower infectivity and replication capacity in the presence of SERINC5. Our results demonstrate that natural polymorphisms in HIV-1 Nef can impair its ability to internalize SERINC5, indicating that variation in this recently described function may contribute to differences in viral pathogenesis.
Graphical Abstract
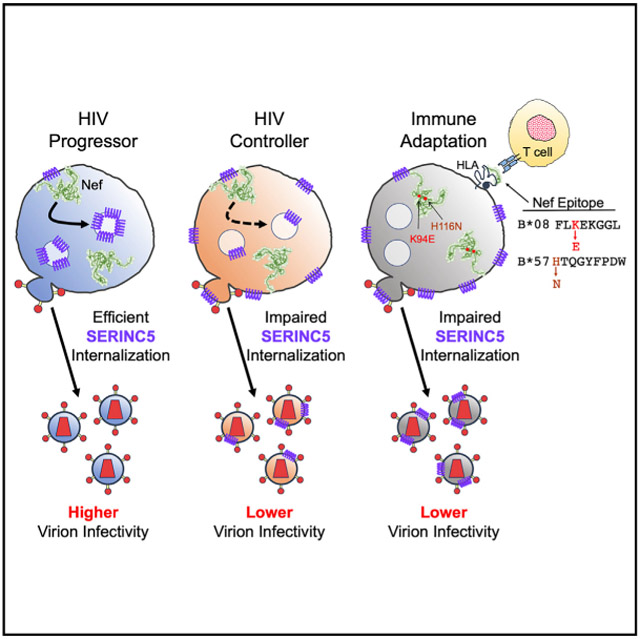
In Brief
HIV-1 Nef counteracts the cellular restriction factor SERINC5, but the importance of this for pathogenesis is unclear. Jin et al. show that Nef clones isolated from HIV controllers display lower ability to antagonize SERINC5, in part because of viral mutations that are selected to evade host T cells.
INTRODUCTION
The HIV-1 Nef protein is crucial for viral pathogenesis (Deacon et al., 1995; Kestler et al., 1991; Kirchhoff et al., 1995), serving to modulate diverse cellular events related to vesicular transport, signal transduction and actin cytoskeletal remodeling that collectively enhance viral infectivity and replication (Abraham and Fackler, 2012; Cheng-Mayer et al., 1989; Chowers et al., 1994; Landi et al., 2011; Miller et al., 1994; Pereira and daSilva, 2016; Terwilliger et al., 1986; Tokarev and Guatelli, 2011). Nef’s well-characterized abilities to internalize CD4 and HLA class I from the infected cell surface also allow HIV-1 to evade antibody-dependent cellular cytotoxicity (ADCC) and cytotoxic T lymphocytes (CTLs) (Aiken et al., 1994; Alsahafi et al., 2015, 2017; Collins et al., 1998; Garcia and Miller, 1991; Schwartz et al., 1996).
Nef-mediated enhancement of viral infectivity was recently shown to be due in part to its ability to counteract members of the serine incorporator (SERINC) family of host restriction factors, of which SERINC5 is most potent (Rosa et al., 2015; Usami et al., 2015). SERINC5 enters into the membrane of progeny virions and inhibits fusion with target cells (Sood et al., 2017). Nef prevents this by internalizing SERINC5 from the cell surface and trafficking it to lysosomes via an endosomal route that is similar to that used to downregulate CD4 (Shi et al., 2018). Several Nef mutations are reported to impair its ability to antagonize SERINC5, including G2A, D123A, and LL165AA, which block myristoylation, dimerization, and interaction with AP-2 trafficking complexes, respectively (Foster et al., 2011); however, while HIV-1 nef exhibits extensive genetic diversity (Brumme et al., 2007; Foster et al., 2001), these mutations are rare in circulating viral strains (all >99% conserved; HIV Sequence Database, https://www.hiv.lanl.gov). Studies to examine the impact of naturally occurring HIV-1 Nef polymorphisms on its ability to counteract SERINC5 have not been conducted. In a prior study, we demonstrated that Nef clones isolated from HIV-1 elite controllers, who spontaneously suppress plasma viremia without therapy (Deeks and Walker, 2007), displayed functional impairments despite the absence of obvious genetic defects (Mwimanzi et al., 2013). Rather, reduced function was linked to natural variation in nef sequences, including mutations selected by the protective HLA allele B*57, indicating that viral adaptation to host immune selection pressure contributed to attenuation in at least some cases.
RESULTS
Primary HIV-1 Nef Alleles Display Variable Abilities to Internalize SERINC5
To assess whether SERINC5 antagonism differs among circulating HIV-1 strains, we characterized the ability of 91 primary subtype B nef alleles (representative isolates collected from 45 elite controllers and 46 chronic progressors during untreated infection) to downregulate SERINC5 using a transfection-based assay (Figure S1; STAR Methods). The function of each Nef clone was normalized to that of a control subtype B Nef isolate (SF2 strain), such that activity better or worse than SF2 Nef is reported as >100% or <100%, respectively. Empty vector and Nef G2A mutant were included as negative controls. We observed that controller-derived clones displayed lower SERINC5 downregulation activity (median 80%, interquartile range [IQR] 38%–95%) compared with progressor-derived clones (median 96%, IQR 75%–100%) (p = 0.0005, Mann-Whitney test; Figure 1A; Table S1). To confirm this, we engineered HIV-1 subtype B reference strain NL4.3 to encode each of 24 nef alleles that were selected to display a range of SERINC5 downregulation phenotypes, as well as control nef sequences, and then produced virions in the absence or presence of SERINC5. We observed that relative viral infectivity, calculated as the ratio of infectivity for each strain produced in the presence versus absence of SERINC5, was variable (Figure 1B). Consistent with prior studies showing that internalization of SERINC5 is a key mediator of Nef’s ability to counteract this restriction factor (Rosa et al., 2015), a strong correlation was found between SERINC5 downregulation function and viral infectivity (Spearman R = 0.62, p = 0.0004; Figure 1C). These data also indicated that ~80% normalized SERINC5 downregulation function was necessary to enhance infectivity in our assays, suggesting that Nef must maintain relatively high SERINC5 internalization activity to be effective and, conversely, that relatively modest impairment of this function may be biologically relevant. While additional studies are needed to assess the activity of each primary Nef allele against a wider range of SERINC5 expression levels, we noted that more than half of controller-derived clones displayed SERINC5 downregulation activity below 80%, compared with one-quarter of progressor-derived clones (p = 0.02, Mann-Whitney test). Together, these results demonstrate that the ability of Nef to internalize SERINC5 varies among circulating isolates and that this function is impaired in clones isolated from elite controllers.
Figure 1. Lower SERINC5 Downregulation by Nef Isolates from HIV Controllers.

(A) SERINC5 downregulation function was compared between Nef clones from 45 elite controllers (EC; red) and 46 chronic progressors (CP; blue) (p < 0.001, Mann-Whitney U-test) using flow cytometry (see Figure S1; STAR Methods). Mean function of each clone (relative to Nef SF2 strain) is reported, on the basis of triplicate data from three independent experiments. Bars represent median (± interquartile range) of all EC or CP Nef clones.
(B) Relative infectivity of NL4.3 strains encoding 24 nef alleles (14 elite controllers, red; 10 chronic progressors, blue) and 4 controls (G2A, ΔNef, NL4.3 Nef, and SF2 Nef, black) was quantified as the quotient of infectivity for each virus generated in the presence of SERINC5 divided by that of the same virus generated in the absence of SERINC5. All viruses were tested at least twice in independent experiments. Mean results based on triplicate data from one representative experiment are shown.
(C) Correlation between SERINC5 downregulation and viral infectivity is shown for data described in (A) and (B) (Spearman R = 0.62, p = 0.0004).
(D) Eight Nef polymorphisms (see Table 1) were confirmed by mutagenesis. Results for mutants that were anticipated to increase (green) or decrease (red) SERINC5 downregulation function are reported as mean (±SD), on the basis of at least three independent experiments. Significant differences compared with NL4.3 Nef (100%) are indicated by asterisks (unpaired Student’s t test): *p < 0.05, **p < 0.01, and ***p < 0.001.
(E) Nef expression was assessed by western blot, and percentage relative to NL4.3 Nef is indicated.
Nef Polymorphisms Are Associated with SERINC5 Downregulation Function
Analysis of our linked genotype-phenotype dataset (see STAR Methods) identified 18 naturally occurring Nef polymorphisms, located at 14 codons, that were associated with differential SERINC5 downregulation activity (p < 0.05 for all, Mann-Whitney test) (Table 1). The most statistically significant correlation was observed at Nef codon 51, where clones encoding threonine (n = 43) displayed higher activity (median 95.3%) compared with clones that did not (n = 45, typically asparagine) (81%) (p = 0.004). We validated eight polymorphisms by introducing the mutation into a subtype B Nef isolate (NL4.3 strain), each of which resulted in the expected change in function (p < 0.05 for all, Student’s t test) (Figure 1D). Specifically, downregulation function was increased modestly for N51T, I114V, and S163C mutants, each of which displayed ~5% higher activity compared with parental Nef; whereas downregulation was impaired for C55S, K94E, H116N, V148L, and S163R mutants, each of which displayed 5%–50% lower activity compared with parental Nef. Of note, Nef codon 51 is associated with viral infectivity (Carl et al., 2001). Furthermore, Nef codon 163 is located adjacent to the dileucine motif that is critical for SERINC5 antagonism (Rosa et al., 2015; Shi et al., 2018) and has been shown previously to affect Nef binding to clathrin adaptor protein complexes (Coleman et al., 2006). While steady-state expression of these Nef mutants was variable (Figure 1E), SERINC5 downregulation activity did not correlate with our ability to detect Nef by western blot, indicating that changes in Nef function were unlikely to be due to differences in protein stability.
Table 1.
Nef Polymorphisms Associated with SERINC5 Downregulation (N ≥ 5, p < 0.05, q < 0.35)
| Median Nef Activity | Number of Individuals | |||||||
|---|---|---|---|---|---|---|---|---|
| Codona | Amino Acid (AA) | With AA | Without AA | With AA | Without AA | Impactb | p Value | q Value |
| 11 | A | 100.5 | 86.5 | 6 | 75 | +14 | 0.005 | 0.22 |
| 19 | K | 100.7 | 85.6 | 9 | 82 | +15 | 0.011 | 0.25 |
| 28 | D | 85.1 | 93.4 | 47 | 44 | −8 | 0.047 | 0.34 |
| 43* | V | 39.4 | 88.4 | 5 | 85 | −49 | 0.025 | 0.33 |
| 51 | T | 95.3 | 80 | 43 | 45 | +15 | 0.004 | 0.22 |
| 51 | N | 80 | 96 | 42 | 46 | −16 | 0.005 | 0.22 |
| 55 | C | 90.8 | 73.7 | 82 | 9 | +17 | 0.03 | 0.33 |
| 65 | E | 84.9 | 98.3 | 84 | 7 | −13 | 0.01 | 0.25 |
| 94* | E | 50.3 | 88.6 | 5 | 86 | −38 | 0.015 | 0.28 |
| 94 | K | 88.4 | 55.2 | 83 | 8 | +33 | 0.047 | 0.34 |
| 114 | V | 89.8 | 74.9 | 70 | 21 | +15 | 0.037 | 0.34 |
| 116 | H | 95.3 | 80.6 | 59 | 32 | +15 | 0.012 | 0.25 |
| 116* | N | 80.9 | 95.3 | 31 | 60 | −14 | 0.02 | 0.3 |
| 148 | V | 89.8 | 66.8 | 78 | 13 | +23 | 0.041 | 0.34 |
| 163 | C | 97.2 | 83.6 | 27 | 64 | +14 | 0.028 | 0.33 |
| 163 | R | 39.4 | 87.6 | 7 | 84 | −48 | 0.033 | 0.33 |
| 170 | L | 81 | 91.1 | 55 | 36 | −10 | 0.049 | 0.34 |
| 182 | Q | 97.9 | 85.1 | 10 | 81 | +13 | 0.021 | 0.3 |
HXB2-aligned residues; Nef polymorphisms associated with evasion from CD8+ T cells are indicated by asterisks (Brumme et al., 2007).
Median Nef activity with AA – median Nef activity without AA, rounded to nearest whole number.
SERINC5 Downregulation Correlates with CD4 Internalization
The mechanism of Nef-mediated SERINC5 downregulation shares several features with that of CD4 downregulation, including reliance on Nef’s dileucine motif (LL165) (Rosa et al., 2015; Shi et al., 2018) and trafficking of the internalized receptors to lysosomes through endosomal compartments (Shi et al., 2018). To explore potential associations between Nef functions, we compared the SERINC5 downregulation activity of our 91 Nef clones with prior results for CD4 and HLA downregulation as well as protein stability, reported by Mwimanzi et al. (2013) (Figure 2; Table S1). Notably, SERINC5 downregulation correlated more strongly with the ability of Nef clones to internalize CD4 (Spearman R = 0.55, p < 0.0001) compared with HLA (R = 0.31, p = 0.0025). In addition, polymorphisms at S163 that are adjacent to the dileucine motif were similarly associated with differential ability of Nef to downregulate CD4 (Mwimanzi et al., 2013). We observed no correlation between Nef expression levels detected by western blot and downregulation of SERINC5, CD4, or HLA, indicating that associations between these functions were not due simply to differences in protein stability; however, additional studies will be needed to address this. These results suggest that similar polymorphisms in circulating nef alleles contribute to variation in SERINC5 and CD4 downregulation functions, while effects on HLA downregulation activity may be more independent.
Figure 2. Associations between Nef Functions and Protein Stability.

(A and B) The ability of each elite controller (red) or chronic progressor (blue) Nef clone to downregulate SERINC5 was compared with that for CD4 (A) and HLA class I (A*02) (B).
(C–E) Next, the ability of each Nef clone to downregulate CD4 (C), HLA class I (D), or SERINC5 (E) was compared with its steady-state protein expression as detected by western blot.
Correlations were assessed using Spearman rank test. Data for CD4 and HLA downregulation and western blot were reported in Mwimanzi et al., (2013).
Viral Adaptation to T Lymphocytes Contributes to Variability in SERINC5 Downregulation Activity
Notably, three Nef polymorphisms that were associated with a substantial reduction in SERINC5 downregulation function, I43V (–49% activity), K94E (–38%), and H116N (–14%) (Table 1), are viral “escape mutations” selected in CD8+ T cell epitopes restricted by HLA-C*03, B*08, and B*57, respectively (Altfeld et al., 2006; Brumme et al., 2008). This indicates that immune pressure on Nef may attenuate its ability to antagonize SERINC5. Notably, H116N is often selected early following infection in individuals expressing the protective B*57 allele (Brumme et al., 2008), and it is the predominant variant observed for the HW9 epitope (at position 1 of HTQGYFPDW). Thus, we speculate that reduced Nef-mediated SERINC5 downregulation may contribute to lower viremia in these cases. In contrast, K94E is selected more slowly in individuals expressing the non-protective B*08 allele (Brumme et al., 2008) and 94E is not the dominant variant observed for the FL8 epitope (at position 5 of FLKEKGGL) (rather K92R). This suggests that Nef can adapt to B*08-restricted T cell pressure without enduring the detrimental impact of this polymorphism on SERINC5 downregulation. Additional studies will be needed to explore these links between host immune pressure and Nef function in greater detail.
Viral Infectivity and Replication Capacity Are Impaired by K94E and H116N
To assess the impact of K94E and H116N on viral phenotypes, we generated HIV-1 NL4.3-derived strains encoding these mutations in the absence or presence of SERINC5. Consistent with downregulation results, we observed lower SERINC5 surface expression on HEK293T cells producing wild-type NL4.3 compared with cells producing viruses that lacked Nef (ΔNef) or those that encoded Nef mutations that were anticipated to be detrimental to this function (G2A, K94E, H116N, or K94E/H116N) (Figure 3A). Moreover, while viral infectivity was comparable for all NL4.3-derived strains generated in the absence of SERINC5 (Figure 3B, white bars), differences in infectivity were seen among viruses generated in the presence of SERINC5. Specifically, ΔNef virus was 41-fold less infectious compared with wild-type NL4.3, while G2A, K94E, H116N, and double-mutation viruses were 11-, 4-, 3-, and 5.5-fold less infectious than NL4.3, respectively (Figure 3B, black bars) (p < 0.05 for all, unpaired Student’s t test). Infectivity of the ΔNef mutant was modestly impaired in the absence of SERINC5 (1.7-fold lower compared with NL4.3), which may be due to SERINC3 expression by HEK293T cells (Usami et al., 2015). Notably, NL4.3-derived viruses encoding Nef K94E, H116N, or the double mutation also displayed reduced infectivity when they passaged using primary peripheral blood mononuclear cells (PBMCs) (Figure 3C), indicating that these polymorphisms altered viral phenotypes in the presence of endogenous levels of SERINC5. Next, we examined the impact of these Nef mutations on viral replication using Jurkat-derived GFP reporter T cells, which express high endogenous levels of SERINC5 (Rosa et al., 2015). The results of these studies were consistent with those obtained for SERINC5 downregulation and viral infectivity enhancement by each mutant. Representative data from one experiment are shown in Figure 3D. In repeated assays, we observed statistically significant delays in viral replication (measured as the slope of viral cell-to-cell spread over time) for viruses encoding Nef K94E, H116N, or the double mutation compared with wild-type NL4.3 (p < 0.05 for all). To confirm that these differences were due to SERINC5, we generated a SERINC5 knockout (KO) clone of the Jurkat-GFP reporter T cell line (Figure S2) and examined the impact of Nef mutations on viral replication using this line. Representative data from one experiment are shown in Figure 3E. In repeated assays, we observed no significant differences in replication capacity between NL4.3-derived viruses encoding wild-type Nef versus Nef K94E, H116N, or the double mutation in cells lacking SERINC5 (p > 0.05 for all). Unexpectedly, NL4.3 viruses encoding Nef mutants displayed a modest enhancement of replication in our SERINC5 KO cell line, which was significant for the virus lacking Nef (ΔNef) (p < 0.01). We confirmed that this strain lacked Nef (Figure S2) and also obtained similar results using a second independent SERINC5 KO clone (data not shown). While the mechanism for this is unclear, similar results have been observed for some Nef mutant viruses in other studies (Wu et al., 2019), suggesting that other Nef functions modulate viral replication and that these may differ in part depending on cell type (Li et al., 2019; Wu et al., 2019). Finally, to further validate our observations, we performed viral replication assays in PHA-stimulated PBMCs infected with equivalent amounts of each virus (5 ng p24) and measured viral Gag p24 in culture supernatants at days 0, 3, and 6. Representative data from one experiment are shown in Figure 3F. Results from repeated assays were consistent with our prior observations, indicating that viruses encoding Nef K94E, H116N, or the double mutation displayed significantly lower replication capacity in primary cells compared with wild-type NL4.3 (p < 0.01 for all). Together, these studies demonstrate that K94E and H116N impair viral infectivity and replication capacity in the presence of SERINC5.
Figure 3. Impact of K94E and H116N Mutants on Nef Function.

(A) Representative flow cytometry plots display SERINC5 surface expression on cells co-transfected with pSERINC5-iHA and pNL4.3. MFI of SERINC5 in the p24+ gate is indicated.
(B and C) Infectivity of NL4.3-derived viruses produced in the absence (white) or presence (black) of SERINC5-iHA (B) or viruses passaged once in healthy donor PBMCs (C) is shown. Results reflect luminescence (absolute light units [ALUs]) following incubation of TZM-bl reporter cells with a normalized amount of virus. Data are representative of two independent experiments (B) or PBMCs from three independent donors (C). Unpaired Student’s t test was used to compare each mutant to NL4.3 (***p < 0.001).
(D and E) Viral replication was examined using Jurkat LTR-GFP R5 cells (D) or those in which SERINC5 was knocked out using CRISPR/Cas9 (Figure S2; STAR Methods) (E). Viral spread was monitored by flow cytometry. The mean (±SD) fold increase in percentage GFP+ cells (from day 2) is reported for each mutant, on the basis of triplicate infections. Results are representative of three experiments.
(F) Replication was also assessed following infection of PHA-stimulated PBMCs. Supernatant was harvested on days 0, 3, and 6 and viral p24 quantified using ELISA. Results are representative of PBMCs from three donors.
(G) The ability of NL4.3 Nef mutants encoding K94E and/or H116N or EC48 Nef reversion mutants encoding E94K and/or N116H to downregulate SERINC5, CD4, and HLA is shown. Bars represent the mean (±SD) function, normalized to control, in at least three experiments. Unpaired Student’s t test was used to compare each mutant with control (*p < 0.05, **p < 0.01, and ***p < 0.001).
K94E and H116N Selectively Impair Nef-Mediated SERINC5 Downregulation Function
To determine if the effect of K94E or H116N on SERINC5 downregulation was selective, we next tested the ability of Nef mutants encoding these substitutions to internalize CD4 and HLA class I (Figure 3G, left side). Compared with parental NL4.3 Nef, K94E reduced HLA downregulation activity modestly (to 87%) (p < 0.05, unpaired Student’s t test) but it had no effect on CD4 internalization. Conversely, H116N reduced CD4 downregulation activity modestly (to 89%) (p < 0.05), but it had no effect on HLA internalization. All three Nef-mediated downregulation functions were impaired in the mutant encoding both K94E and H116N, indicating that the impact of these polymorphisms can be additive. In particular, SERINC5 downregulation activity was reduced markedly in the Nef double mutant (to 19%) (p < 0.001), while more moderate effects were seen for downregulation of CD4 (69%) and HLA (87%) (p < 0.05 for both). To explore this further, we identified an elite controller-derived Nef clone (EC48) encoding E94 and N116 that displayed poor SERINC5 downregulation activity (32%) but retained moderate CD4 and HLA internalization functions (78% and 92%, respectively). To confirm that E94 and/or N116 were determinants of impaired SERINC5 downregulation function by this primary Nef clone, we reverted these polymorphisms to the consensus residue (Figure 3G, right side). Consistent with results for NL4.3 Nef, reversion of E94K or N116H alone partially rescued SERINC5 downregulation activity (to 69% and 59%, respectively) (p < 0.05 for both); while reversion of both polymorphisms enhanced this activity further (to 89%) (p < 0.001). In contrast to results obtained using NL4.3 Nef, the E94K and double reversion mutant displayed lower HLA downregulation activity compared with the primary Nef clone (56% and 51%, respectively) (p < 0.01), suggesting that unidentified compensatory polymorphisms in the EC48 nef sequence contributed to maintain this function. Together, these results demonstrate that K94E and H116N reduce Nef’s ability to downregulate SERINC5 to a greater extent compared with CD4 or HLA class I and further indicate that their impact on all three Nef functions may be modulated by other sequence variation present in primary nef alleles.
DISCUSSION
We have demonstrated that circulating HIV-1 subtype B nef alleles exhibit substantial variability in SERINC5 downregulation function. Nef clones isolated from elite controllers displayed lower in vitro function compared with those from chronic progressors, suggesting that impaired SERINC5 antagonism may contribute to lower plasma viremia and slower disease progression in this rare group of individuals. Consistent with the observation that even limited amounts of SERINC5 incorporation into viral particles reduces infectivity (Trautz et al., 2016), we found that Nef-mediated infectivity enhancement required ~80% normalized SERINC5 downregulation activity in our assays, a threshold that was not met by 51% of elite controller-derived Nef clones.
While a prior study examined the ability of diverse HIV and SIV Nef isolates to antagonize SERINC5 (Heigele et al., 2016), it assessed relatively few (~15) HIV-1 group M clones. Our analysis of 91 HIV-1 subtype B Nef clones offers greater power to explore functional variation among patient-derived sequences. Here, we identified 18 polymorphisms that were associated with differential SERINC5 downregulation activity. We validated 8 of these polymorphisms, including 2 well-characterized CD8+ T cell escape mutations, K94E and H116N, using site-directed mutagenesis. While mechanisms responsible for impaired SERINC5 internalization by primary nef alleles or mutants have not been defined in this study, it is intriguing that K94E and H116N had only modest effects on Nef’s ability to downregulate CD4 or HLA class I, suggesting that K94 and H116 lie within domains of Nef that are particularly important to antagonize SERINC5 rather than to internalize CD4 or HLA.
Our results indicate that Nef polymorphisms can impair viral infectivity and replication in the presence of SERINC5; however, additional work is needed to assess the importance of Nef-mediated SERINC5 antagonism for HIV-1 pathogenesis. Our ability to address this question is hindered by an incomplete understanding of SERINC5 restriction mechanisms and by Nef’s continuously evolving role as a mediator of viral infectivity (Li et al., 2019; Wu et al., 2019). A limitation of this study is that we did not directly measure SERINC5 incorporation into the virions of strains encoding Nef mutants. Strong correlations between SERINC5 downregulation activity and viral infectivity suggest that internalization of SERINC5 is the most important contribution of Nef in this setting, but we cannot rule out a role for other known (or unknown) Nef functions. In addition, HIV-1 Envelope modulates viral sensitivity to SERINC5 (Beitari et al., 2017), but its relative impact compared with Nef has not been elucidated, and potential functional interactions between primary nef and env alleles have not been studied. Furthermore, Nef’s ability to counteract SERINC5 is dependent on the expression levels of both proteins (Schulte et al., 2018; Trautz et al., 2016), which may not be recapitulated fully by our in vitro assays. While our results using primary cells are consistent with data collected using T cell lines, validation using other cell types is needed. Despite these limitations, our study provides strong evidence that natural variation in Nef-mediated SERINC5 antagonism function contributes to clinical outcomes following HIV-1 infection. Our data highlight the constraints placed on Nef by the interplay between adaptive host immunity and intrinsic host restriction mechanisms, which may result in viral attenuation during the course of natural infection.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Brockman (mark_brockman@sfu.ca).
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human subjects
All HIV-infected participants were recruited from the Boston (USA) area and provided written informed consent. None were receiving antiretroviral therapy at the time of specimen collection. Elite controllers (EC) were defined as having an untreated HIV-1 plasma viral load (pVL) of less than 50 RNA copies/mL for at least 2 years. Chronic progressors (CP) were defined as having an untreated pVL of at least 10,000 RNA copies/mL. The EC cohort (N = 45) displayed a median pVL of 2 RNA copies/mL (interquartile range [IQR] 0.2 –14) and a median CD4 count of 811 cells per mm3 (IQR 612 – 1022). The CP cohort (N = 46) displayed a median pVL of 80,500 RNA copies per mL (IQR 25,121 – 221,250) and a median CD4 count of 293 cells per mm3 (IQR 73 – 440). Information regarding the sex of EC participants (84% male) is indicated in Table S1. Information regarding the sex of CP participants and the age of all participants was not recorded at the time of specimen collection, and therefore this data is unavailable. This study was approved by the Research Ethics Boards at the Massachusetts General Hospital (Boston, MA USA) and Simon Fraser University (Burnaby, BC Canada).
Primary Cells
Human peripheral blood mononuclear cells (PBMC) from healthy HIV-uninfected donors were purchased from StemCell Technologies and maintained in RPMI-1640 media supplemented with 10% FBS, 2mM L-glutamine, 1000 U/mL Penicillin, and 1 mg/mL Streptomycin (all from Sigma-Aldrich), plus 50 U/mL human recombinant IL-2 (NIH AIDS Reagents Program) at 37°C with 5% CO2. All donors were male.
Cell lines
CEM-A*02 cells were derived from CEM (a female human acute lymphoblastic leukemia T cell line) by stably transduction of HLA-A*02:01 using a retroviral vector (murine stem cell virus; Clontech), and maintained in RPMI-1640 media supplemented with 10% FBS, 2 mM L-glutamine, 1000 U/mL Penicillin and 1 mg/mL Streptomycin (all from Sigma-Aldrich) at 37°C with 5% CO2. Jurkat LTR-GFP CCR5+ reporter cells (JLTRG-R5), which are derived from Jurkat (a male human acute T cell leukemia cell line), were maintained using the same conditions. HEK293T cells (a human embryonic kidney cell line) and TZM-bl reporter cells (a female human carcinoma cell line, derived from HeLa) were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 1000 U/mL Penicillin and 1 mg/mL Streptomycin.
METHOD DETAILS
Reagents
The following materials were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 NL4-3 infectious molecular clone (pNL4-3), from Dr. Malcolm Martin (Adachi et al., 1986); Jurkat LTR-GFP CCR5+ cells (JLTRG-R5), from Dr. Olaf Kutsch (Kutsch et al., 2004; Ochsenbauer-Jambor et al., 2006); and TZM-bl cells from Dr. John C. Kappes, and Dr. Xiaoyun Wu (Derdeyn et al., 2000; Platt et al., 2009; Platt et al., 1998; Takeuchi et al., 2008; Wei et al., 2002).
Generation of HIV-1 nef expression and proviral constructs
HIV-1 subtype-B nef alleles were amplified from plasma viral RNA. A single phylogenetically representative nef clone was isolated from each participant in a previous study (Mwimanzi et al., 2013). Nef alleles were transferred into pSELECT-GFPzeo (InvivoGen), which features a composite hEF1-HTLV promoter driving nef and an independent CMV promoter driving expression of GFP. To do this, we modified the multiple cloning site in pSELECT-GFPzeo to incorporate unique Asc I and Sac II sites. Each nef gene was amplified by PCR using degenerate primers incorporating these restriction sites (Fwd: 5′-AGAGCACCGG CGCGCCTCCA CAT ACCTASA AGAATMAGAC ARG-3′, HXB2 nt 8746-8772 underlined, Asc I site in bold; Rev: 5′-GCCTCCGCGG ATCGATCAGG CCACRCCTCC CTGGAAASKC CC-3′, HXB2 nt 9474-9449 underlined, SacII site bolded). The same strategy was used to clone nef from HIV-1 subtype B reference strains SF2 and NL4.3, which served as positive controls. Point mutations were introduced into nef sequences using overlap extension PCR, including G2A substitutions in both reference clones that served as negative controls. All nef clones and mutations were validated by Sanger sequencing.
Nef sequences and mutants were introduced into the HIV-1 subtype B NL4.3 reference strain backbone as described previously (Mwimanzi et al., 2013). Wild-type NL4.3 served as a positive control, while strains encoding G2A or premature stop codons at positions 31 and 33 (referred to as ΔNef) were used as negative controls for infectivity and replication assays.
Nef polymorphisms are reported using HXB2 numbering convention (Korber et al., 1998). Sequences were pairwise-aligned to the reference strain HXB2 (GenBank accession number K03055) and insertions with respect to HXB2 were removed using an in-house alignment algorithm based on the HyPhy platform (Pond et al., 2005).
Generation of SERINC5 knockout Jurkat LTR-GFP reporter cells
Jurkat T cell lines express high endogenous SERINC5 (Rosa et al., 2015). CRISPR/Cas9 methods were used to disrupt the SERINC5 gene in Jurkat LTR-GFP R5 cells. To do this, cells were co-transfected with px330-based plasmids (Cong et al., 2013; Ran et al., 2013) encoding target sequences described by Rosa et al. (2015) along with pMAX-GFP for use as a transfection control. Following transfection, GFP+ cells were isolated as single cells into R20+ media (RPMI-1640 supplemented with 2 mM L-glutamine, 1000 U/ml Penicillin, 1 mg/ml Streptomycin and 20% vol/vol FBS, all from Sigma-Aldrich) and expanded in 96-well flat bottom plates. Stable SERINC5 knockout (KO) clones were validated by western blot using a rabbit polyclonal anti-SERINC5 antiserum (Abcam).
SERINC5 downregulation assays
To assess Nef-mediated internalization of SERINC5 from the cell surface, 1 × 106 CEM-A*02 T cells were co-transfected with 1 μg of pSELECT-GFPzeo encoding nef and 5 μg of pSELECT-SERINC5-internal HA tag (iHA)-ΔGFP (sub-cloned from pBJ5-SERINC5(iHA) (Usami et al., 2015) by electroporation in 150 μL OPTI-mem medium (Thermo Fisher Scientific) using a BioRad GenePulser MXCell™ instrument (square wave protocol: 250 V, 2000 μF, infinite Ω, 25 ms single pulse). Cultures were recovered for 20 hours with 350 μl of R10+ medium (RPMI-1640 supplemented with 2 mM L-glutamine, 1000 U/ml Penicillin, 1 mg/ml Streptomycin and 10% vol/vol FBS, all from Sigma-Aldrich) at 37°C plus 5% CO2. Following this, 2.5 × 105 cells were stained with 0.5 μg of Alexa Fluor® 647 anti-HA.11 (BioLegend) and analyzed by flow cytometry for GFP expression (as a marker for transfected cells) and cell surface HA staining (as an indicator of SERINC5 expression) using a Millipore Guava 8HT instrument. A minimum of 25,000 cells were assessed in each case. The median fluorescence intensity (MFI) values of SERINC5 for each Nef clone were normalized to the positive (pSELECT-nefWT-GFPzeo) and negative (pSELECT-Δnef) controls using the formula: (MFIΔNef – MFICLONE)/(MFIΔNef – MFIWT) × 100, such that Nef function less than or greater than wild-type Nef is represented by values of < 100% or > 100%, respectively. The activity of each primary Nef clone was normalized to Nef (SF2 strain) to be consistent with our previous studies. Since point mutations were introduced into Nef (NL4.3 strain), that strain was used to normalize results from those studies. The SERINC5 downregulation activity of each Nef clone is reported as the mean ± SD based on at least three independent transfection experiments.
CD4 and HLA class I downregulation assays
Selected primary Nef clones and mutants were evaluated for their ability to internalize CD4 and HLA-A*02 (as a representative class I HLA molecule) as described previously (Mwimanzi et al., 2013). Briefly, 2.5 × 105 transfected CEM-A*02 CD4 T cells from the same pool of transfected cells described above were stained with anti-CD4-APC and anti-HLA-A*02-PE (BD Biosciences). The MFI of surface CD4 and HLA-A*02 expression levels in the GFP+ cell subsets were determined by flow cytometry and normalized using the same formula as described for SERINC5. The CD4 or HLA downregulation activity of each Nef clone or mutant is reported as the mean ± SD based on at least three independent transfection experiments.
Viral infectivity assays
Virus stocks were generated by co-transfecting 8 × 105 HEK293T cells seeded in one well of a 6-well plate with 2.5 μg of pNL4.3 and either 30 ng of pSELECT-SERINC5(iHA)-ΔGFP or empty pSELECT-ΔGFP vector using DNAfectin 2100 (Applied Biological Materials). Culture supernatants were harvested 48 hours post-transfection, aliquoted and stored at –80°C prior to use. To assess SERINC5 surface expression on virus-producing cells, the transfected cells were collected using Trypsin/EDTA (Sigma-Aldrich) and dissociated cells were prepared for analysis by flow cytometry. Briefly, cells were stained with 0.5 μg of Alexa Fluor® 647 anti-HA.11 (BioLegend), washed to remove unbound antibody, treated with Fix/Perm solution (BD Biosciences), and then stained with PE anti-Gag/p24 (KC57; Beckman Coulter). A minimum of 10,000 cells was assessed in each case. Virus particles in the supernatant were quantified by p24 ELISA (XpressBio) and viral infectivity was determined by exposing 1 × 104 TZM-bl reporter cells to a standardized amount of each virus (1 or 5 ng p24) on a 96-well flat bottom plate. Luminescence activity (absolute light units, ALU) of TZM-bl cells was measured 48 hours later using the Steady-Glo® Luciferase Assay (Promega) and a Tecan Infinite M200 PRO plate reader. The relative infectivity of each viral strain (i.e., the difference between viruses generated in the presence versus absence of SERINC5) was calculated using the following formula: (ALU of virus produced by SERINC5-expressing HEK293T cells) / (ALU of virus produced by SERINC5-negative HEK293T). The infectivity of each viral strain produced by HEK293T cells was assessed in at least three independent experiments.
To determine the infectivity of viruses produced by primary cells, VSV-g pseudotyped viruses were generated using HEK293T cells in the absence of SERINC5, as described above and in Kinloch et al. (2018), and then used to infect PBMC isolated from HIV-uninfected donors. Briefly, 5 × 105 activated PBMC (generated by pre-stimulation with 5 μg/ml PHA for 72 hours) were incubated with 5 ng p24 of each virus in 96-well U bottom plates by spinoculation (800 × g for 1 hour at room temperature). PBMC were subsequently incubated for 8 hours at 37°C and unbound virus was removed by washing with PBS. Cells were then resuspended in R10+ media supplemented with 100 U/mL human IL-2 (NIH AIDS Reagent Program). Culture supernatants were collected at 48 and 72 hours post-infection and newly produced viruses were quantified using p24 ELISA. Viral infectivity was measured by exposing TZM-bl reporter cells to a standardized amount of each virus, as described above. The infectivity of each viral strain produced by PBMC was assessed in at least two independent experiments.
Viral replication capacity assays
Replication capacity was determined using viruses produced in the absence of SERINC5 by infecting 1 × 106 Jurkat LTR-GFP CCR5+ and Jurkat SERINC5 KO LTR-GFP CCR5+ cells at a multiplicity of infection (MOI) of 0.003. The proportion of GFP+ cells in culture was measured on days 2 to 8 and results are shown as the fold-increase in %GFP+ cells based on day 2 values. Replication capacity of each viral strain was assessed in at least three (Jurkat LTR-GFP R5) or two (SERINC5 KO cells) independent experiments. To examine potential differences in replication more robustly, we calculated the natural log slope of viral spread during the exponential phase of growth for each virus, as described in Brockman et al. (2006). The resulting slope values from independent experiments were then compared to wild-type NL4.3 using the unpaired Student’s t test to identify statistically significant differences.
To examine replication capacity in primary cells, VSV-g pseudotyped viruses produced using HEK293T cells in the absence of SERINC5, as described above and in Kinloch et al. (2018), were used to infect PBMC isolated from HIV-uninfected donors. Briefly, 5 × 105 activated PBMC (generated by pre-stimulation with 5 μg/ml PHA for 72 hours) were incubated with 5 ng p24 of each virus in 96-well U bottom plates by spinoculation (800 × g for 1 hour at room temperature). PBMC were subsequently cultured for 8 hours at 37°C in R10+ media and unbound virus was removed by washing cells with PBS. Cells were then resuspended in R10+ media supplemented with 100 U/mL human IL-2 (NIH AIDS Reagent Program). Culture supernatants were collected on days 0, 3, 6, and 9, and newly produced viruses were quantified using p24 ELISA. The replication capacity of each viral strain in PBMC was assessed in three independent donors. To examine potential differences in replication more robustly, we calculated the natural log slope of viral spread during the exponential phase of growth for each virus, as described in Brockman et al. (2006). The resulting slope values from independent experiments were then compared to wild-type NL4.3 using the unpaired Student’s t test to identify statistically significant differences.
Western blot
To assess steady-state protein expression of primary Nef alleles and mutants, 5 × 106 CEM-A*02 CD4 T cells were transfected with 10 μg of pSELECT-nef-GFPzeo alone via the electroporation settings as described above. After 24 hours, cells were pelleted, lysed and prepared as described previously (Mwimanzi et al., 2013). Nef was labeled using a polyclonal rabbit serum (NIH AIDS Reagent Program) (1:2,000) followed by staining with donkey anti-rabbit HRP-conjugated secondary antibody (GE Healthcare) (1:30,000). To validate CRISPR/Cas9-mediated knockout of SERINC5 in the Jurkat LTR-GFP cells, 2 × 106 parental or KO cells were pelleted and lysed. SERINC5 protein expression was detected using rabbit polyclonal anti-SERINC5 antibody (Abcam ab204400) at a 1:300 dilution, followed by staining with donkey anti-rabbit HRP-conjugated secondary antibody (GE Healthcare) (1:30,000). Proteins were detected using Clarity Western ECL substrate (Bio-Rad) and visualized on an ImageQuant LAS 4000 imager (GE healthcare).
QUANTIFICATION AND STATISTICAL ANALYSIS
Nef polymorphisms associated with differential in vitro SERINC5 downregulation function were identified using a custom Perl script. For every Nef polymorphism present at > 5% in our dataset (i.e., 5 or more unique HXB-aligned sequences), clones were repeatedly grouped according to the presence versus absence of the observed variant and the nonparametric Mann-Whitney U-test was then used to compare median SERINC5 downregulation function between groups. Multiple comparisons were addressed using q-values, the p value analog of the false discovery rate (FDR). The FDR is the expected proportion of false positives among results deemed significant at a given p value threshold (e.g., at a q ≤ 0.2, we expect 20% of identified associations to be false positives).
All other statistical analyses were performed using Prism v.7 (Graphpad). Results of two-tailed tests were considered significant if the p value was less than 0.05. The nonparametric Mann-Whitney U test was used to compare differences in median Nef function between EC and CP cohorts. The parametric Students t test was used to compare differences in the mean Nef function of viral mutants. For parametric tests, normality of the data was assessed using the Kolmogorov-Smirnov test. Multiple comparisons were addressed using q-values, the p value analog of the false discovery rate (FDR). The FDR is the expected proportion of false positives among results deemed significant at a given p value threshold (e.g., at a q ≤ 0.2, we expect 20% of identified associations to be false positives). Following normalization of data positive and negative controls, we used the unpaired t test to determine if the observed function of each Nef mutant was significantly different from that of the wild-type Nef.
DATA AND CODE AVAILABILITY
The Nef downregulation data that support this study are provided in Table S1. Nef sequences are available at GenBank (accession numbers JX171199-JX171243 for elite controllers; JX440926-JX440971 for chronic progressors). The code for scripts that were used to align nef sequences and to analyze the linked Nef sequence-function dataset is available by contacting the Lead Author.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Beta-Actin, Mouse clone AC-15 | Sigma-Aldrich | Cat # A1978; RRID: AB_476692 |
| CD4, Mouse Clone RPA-T4 (APC) | BD Biosciences | Cat # 555349; RRID: AB_398593 |
| HA.11 Epitope Tag antibody (Alexa Fluor® 647) | BioLegend | Cat # 682404; RRID:AB_2566616 |
| HIV-1 Core Antigen, Mouse clone KC57 (RD1) | Beckman Coulter | Cat # 6604667 |
| Human HLA-A2 Antibody, Mouse clone BB7.2 (R-PE) | BD Biosciences | Cat # 558570; RRID: AB_647220 |
| Mouse IgG, Goat polyclonal, minimal x-reactivity (HRP) | BioLegend | Cat # 405306; RRID: AB_315009 |
| Nef, Rabbit polyclonal | NIH AIDS Reagents Program, gift of R Swanstrom (Shugars et al., 1993) | Cat # 2949 |
| Rabbit IgG, Donkey polyclonal, minimal x-reactivity (HRP) | BioLegend | Cat # 406401; RRID: AB_2099368 |
| SERINC5, Rabbit polyclonal | Abcam | Cat # ab204400 |
| Bacterial and Virus Strains | ||
| E. cloni 10G SOLOs Chemically Competent Cells | Lucigen | Cat # 0106-2 |
| One Shot Stbl3 Chemically Competent E. coli | Invitrogen | Cat # C737303 |
| Biological Samples | ||
| Human Peripheral Blood Mononuclear Cells, Frozen | Stem Cell Technologies | Cat # 70025 |
| HIV-1 subtype B Nef clones | Mwimanzi et al., 2013 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lectin from Phaseolus vulgaris Erythrohemagglutinin (PHA-E) | Sigma-Aldrich | Cat # L8629 |
| Human Recombinant IL-2 | StemCell Technologies, Inc. | Cat # 78036 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat # 554714 |
| DNAfectin 2100 Transfection Reagent | Applied Biological Materials | Cat # G2100 |
| TrypLE Express Enzyme (1X), phenol red | GIBCO | Cat # 12605010 |
| Critical Commercial Assays | ||
| Gene Pulser MXcell Electroporation System | Bio-Rad | Cat # 165–2670 |
| 96-Well Electroporation Plates | Bio-Rad | Cat # 1652681 |
| HIV-1 p24 ELISA Kit | XpressBio | Cat # XB-1000 |
| Steady-Glo® Luciferase Assay System | Promega | Cat # E2550 |
| Deposited Data | ||
| HIV-1 subtype B Nef sequences | Mwimanzi et al., 2013 | GenBank accession JX171199-JX171243 (controllers) and JX440926-JX440971 (progressors) |
| Experimental Models: Cell Lines | ||
| CEM-A*02, human T cell | Anmole et al., 2015 | N/A |
| HEK293T, human kidney cell | American Type Culture Collection | Cat # CRL-3216 RRID: CVCL_0063 |
| Jurkat LTR-GFP CCR5+ (JLTRG-R5), human T cell | NIH AIDS Reagents Program, gift of O Kutsch (Kutsch et al., 2004) | Cat # 11586 |
| Jurkat LTR-GFP CCR5+ (JLTRG-R5) SERINC5 knockout, human T cell | This paper | N/A |
| TZM-bl, human carcinoma cell, derived from HeLa | NIH AIDS Reagents Program, gift of JC Kappes and X Wu (Derdeyn et al., 2000; Platt et al., 1998; Wei et al., 2002) | Cat # 8129-442; RRID: CVCL_B478 |
| Oligonucleotides | ||
| Nef_Forward Primer (AGAGCACCGG CGCGCCTCCA CATACCTASA AGAATMAGAC ARG) | This paper | N/A |
| Nef_Reverse Primer (GCCTCCGCGG ATCGATCAGG CCACRCCTCC CTGGAAASKC CC) | This paper | N/A |
| Recombinant DNA | ||
| pHEF-VSVG | NIH AIDS Reagent Program, gift of L-J Chang (Chang et al., 1999) | Cat # 4693 |
| pMaxFP-Green-N | Amaxa | Cat # VDF-1012 |
| pNL4-3, infectious HIV-1 molecular clone | NIH AIDS Reagents Program, gift of M Martin (Adachi et al., 1986) | Cat # 114 |
| pSELECT-GFPzeo-mcs | InvivoGen | Cat # psetgz-mcs |
| pSELECT-ΔGFP-zeo-mcs | This paper | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene, provided by F Zhang (Cong et al., 2013; Ran et al., 2013) | Cat # 42230; RRID: Addgene_42230 |
| Software and Algorithms | ||
| FlowJo 9.9.5 | FlowJo, LLC | FlowJo |
| Prism 7 | Graphpad | Prism |
Highlights.
HIV-1 Nef clones from elite controllers display impaired SERINC5 antagonism
Nef polymorphisms associated with variable SERINC5 antagonism are identified
HLA B*57 and B*08-associated Nef mutations reduce SERINC5 antagonism
ACKNOWLEDGMENTS
We thank F. Pereyra and The International HIV Controllers Study (http://hivcontrollers.org/hivcontrollers), supported by the Mark and Lisa Schwarz Foundation, the Bill and Melinda Gates Foundation, and the AIDS Healthcare Foundation. Funding was provided by the Canadian Institutes for Health Research (CIHR) (MOP-93536, HOP-115700, and PJT-148621 to Z.L.B. and M.A.B., foundation grant 352417 to A.F.) and the NIH (UM1-AI126617 to Z.L.B. and M.A.B., R01-AI127263 to H.G.). S.W.J. and X.T.K. held Frederick Banting and Charles Best Graduate Scholarships from CIHR. N.A. received a King Abdullah scholarship from the Saudi Government. Z.L.B. is the recipient of a Scholar Award from the Michael Smith Foundation for Health Research. M.A.B. and A.F. are supported by the Canada Research Chairs Program. The funders played no role in determining the content of the manuscript or the authors’ decision to publish.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.007.
REFERENCES
- Abraham L, and Fackler OT (2012). HIV-1 Nef: a multifaceted modulatorof T cell receptor signaling. Cell Commun. Signal 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, and Martin MA (1986). Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol 59, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C, Konner J, Landau NR, Lenburg ME, and Trono D (1994). Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76, 853–864. [DOI] [PubMed] [Google Scholar]
- Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, and Finzi A (2015). Nef proteins from HIV-1 elite controllers are inefficient at preventing antibody-dependent cellular cytotoxicity. J. Virol 90, 2993–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsahafi N, Richard J, Prévost J, Coutu M, Brassard N, Parsons MS, Kaufmann DE, Brockman M, and Finzi A (2017). Impaired downregulation of NKG2D ligands by Nef proteins from elite controllers sensitizes HIV-1-infected cells to antibody-dependent cellular cytotoxicity. J. Virol 91, e00109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, et al. (2006). HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3, e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anmole G, Kuang XT, Toyoda M, Martin E, Shahid A, Le AQ, Markle T, Baraki B, Jones RB, Ostrowski MA, Ueno T, Brumme ZL, and Brockman MA (2015). A robust and scalable TCR-based reporter cell assay to measure HIV-1 Nef-mediated T cell immune evasion. J. Immunol. Meth 426, 104–13. [DOI] [PubMed] [Google Scholar]
- Beitari S, Ding S, Pan Q, Finzi A, and Liang C (2017). Effect of HIV-1 Env on SERINC5 antagonism. J. Virol 91, e02214–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Tanzi GO, Walker BD, and Allen TM (2006). Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131, 134–142. [DOI] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, Kadie C, Bhattacharya T, Chui C, Szinger J, et al. (2007). Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 3, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Carlson J, Streeck H, John M, Eichbaum Q, Block BL, Baker B, Kadie C, Markowitz M, et al. (2008). Marked epitope-and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol 82, 9216–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl S, Greenough TC, Krumbiegel M, Greenberg M, Skowronski J, Sullivan JL, and Kirchhoff F (2001). Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J. Virol 75, 3657–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LJ, Urlacher V, Iwakuma T, Cui Y, and Zucali J (1999). Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Mol Ther 6, 715–728. [DOI] [PubMed] [Google Scholar]
- Cheng-Mayer C, Iannello P, Shaw K, Luciw PA, and Levy JA (1989). Differential effects of nef on HIV replication: implications for viral pathogenesis in the host. Science 246, 1629–1632. [DOI] [PubMed] [Google Scholar]
- Chowers MY, Spina CA, Kwoh TJ, Fitch NJ, Richman DD, and Guatelli JC (1994). Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol 68, 2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman SH, Madrid R, Van Damme N, Mitchell RS, Bouchet J, Servant C, Pillai S, Benichou S, and Guatelli JC (2006). Modulation of cellular protein trafficking by human immunodeficiency virus type 1 Nef: role of the acidic residue in the ExxxLL motif. J. Virol 80, 1837–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KL, Chen BK, Kalams SA, Walker BD, and Baltimore D (1998). HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, et al. (1995). Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991. [DOI] [PubMed] [Google Scholar]
- Deeks SG, and Walker BD (2007). Human immunodeficiencyvirus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27, 406–416. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, and Hunter E (2000). Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol 74, 8358–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JL, Molina RP, Luo T, Arora VK, Huang Y, Ho DD, and Garcia JV (2001). Genetic and functional diversity of human immunodeficiency virus type 1 subtype B Nef primary isolates. J. Virol 75, 1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JL, Denial SJ, Temple BR, and Garcia JV (2011). Mechanisms of HIV-1 Nef function and intracellular signaling. J. Neuroimmune Pharmacol 6, 230–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JV, and Miller AD (1991). Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350, 508–511. [DOI] [PubMed] [Google Scholar]
- Heigele A, Kmiec D, Regensburger K, Langer S, Peiffer L, Stürzel CM, Sauter D, Peeters M, Pizzato M, Learn GH, et al. (2016). The potency of Nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestler HW 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, and Desrosiers RC (1991). Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65, 651–662. [DOI] [PubMed] [Google Scholar]
- Kinloch NN, Lee GQ, Carlson JM, Jin SW, Brumme CJ, Byakwaga H, Muzoora C, Bwana MB, Cobarrubias KD, Hunt PW, et al. (2018). Genotypic and mechanistic characterization of subtype-specific HIV adaptation to host cellular immunity. J. Virol 93, e01502–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, and Desrosiers RC (1995). Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med 332, 228–232. [DOI] [PubMed] [Google Scholar]
- Korber B, Foley BT, Kuiken C, Pillai SK, and Sodroski JG (1998). Numbering positions in HIV relative to HXB2CG In Human Retroviruses and AIDS, Korber B, Kuiken CL, Foley B, Hahn B, McCutchan F, Mellors JW, and Sodroski J, eds., pp. III-102-111, (Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM: ). [Google Scholar]
- Kutsch O, Levy DN, Bates PJ, Decker J, Kosloff BR, Shaw GM, Priebe W, and Benveniste EN (2004). Bis-anthracycline antibiotics inhibit human immunodeficiency virus type 1 transcription. Antimicrob. Agents Chemother 48, 1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi A, Iannucci V, Nuffel AV, Meuwissen P, and Verhasselt B (2011). One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr. HIV Res 9, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Waheed AA, Yu J, Zeng C, Chen HY, Zheng YM, Feizpour A, Reinhard BM, Gummuluru S, Lin S, et al. (2019). TIM-mediated inhibition of HIV-1 release is antagonized by Nef but potentiated by SERINC proteins. Proc. Natl. Acad. Sci. U S A 116, 5705–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Warmerdam MT, Gaston I, Greene WC, and Feinberg MB (1994). The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med 179, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwimanzi P, Markle TJ, Martin E, Ogata Y, Kuang XT, Tokunaga M, Mahiti M, Pereyra F, Miura T, Walker BD, et al. (2013). Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, and Kutsch O (2006). T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques 40, 91–100. [DOI] [PubMed] [Google Scholar]
- Pereira EA, and daSilva LL (2016). HIV-1 Nef: taking control of protein trafficking. Traffic 17, 976–996. [DOI] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, and Kabat D (1998). Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol 72, 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Bilska M, Kozak SL, Kabat D, and Montefiori DC (2009). Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J. Virol 83, 8289–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SL, Frost SD, and Muse SV (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, et al. (2015). HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte B, Selyutina A, Opp S, Herschhorn A, Sodroski JG, Pizzato M, and Diaz-Griffero F (2018). Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 515, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O, Maréchal V, Le Gall S, Lemonnier F, and Heard JM (1996). Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med 2, 338–342. [DOI] [PubMed] [Google Scholar]
- Shi J, Xiong R, Zhou T, Su P, Zhang X, Qiu X, Li H, Li S, Yu C, Wang B, et al. (2018). HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the endosome/lysosome system. J. Virol 92, e00196–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, and Swanstrom R (1993). Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol 67, 4639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood C, Marin M, Chande A, Pizzato M, and Melikyan GB (2017). SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem 292, 6014–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, McClure MO, and Pizzato M (2008). Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J. Virol 82, 12585–12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger E, Sodroski JG, Rosen CA, and Haseltine WA (1986). Effects of mutations within the 3′ orf open reading frame region of human T-cell lymphotropic virus type III (HTLV-III/LAV) on replication and cytopathogenicity. J. Virol 60, 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarev A, and Guatelli J (2011). Misdirection of membrane trafficking by HIV-1 Vpu and Nef: Keys to viral virulence and persistence. Cell. Logist 1, 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautz B, Pierini V, Wombacher R, Stolp B, Chase AJ, Pizzato M, and Fackler OT (2016). The antagonism of HIV-1 Nef to SERINC5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J. Virol 90, 10915–10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami Y, Wu Y, and Göttlinger HG (2015). SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, and Kappes JC (2002). Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother 46, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Olety B, Weiss ER, Popova E, Yamanaka H, and Göttlinger H (2019). Potent enhancement of HIV-1 replication by Nef in the absence of SERINC3 and SERINC5. MBio 10, e01071–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Nef downregulation data that support this study are provided in Table S1. Nef sequences are available at GenBank (accession numbers JX171199-JX171243 for elite controllers; JX440926-JX440971 for chronic progressors). The code for scripts that were used to align nef sequences and to analyze the linked Nef sequence-function dataset is available by contacting the Lead Author.