

Abstract
Flavin-dependent ‘ene’-reductases (EREDs) are exquisite catalysts for effecting stereoselective reductions. While these reactions typically proceed through a hydride transfer mechanism, we recently found that EREDs can also catalyze reductive dehalogenations and cyclizations via single electron transfer mechanisms. Here we demonstrate that these enzymes can catalyze redox-neutral radical cyclizations to produce enantioenriched oxindoles from α-haloamides. This transformation is a C–C bond forming reaction currently unknown in nature and one for which there are no catalytic asymmetric examples. Mechanistic studies indicate the reaction proceeds via the flavin semiquinone/quinone redox couple, where ground state flavin semiquinone provides the electron for substrate reduction and flavin quinone oxidizes the vinylogous α-amido radical formed after cyclization. This mechanistic manifold was previously unknown for this enzyme family, highlighting the versatility of EREDs in asymmetric synthesis.
Graphical Abstract
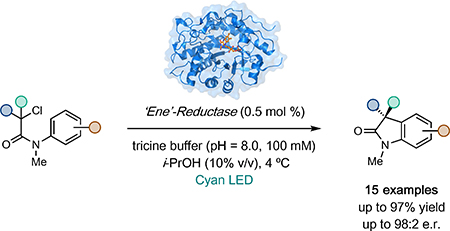
The selective synthesis of stereochemically pure molecules is a goal central to the agrochemical, fragrance, and pharmaceutical industries. Consequently, numerous chiral catalysts have been developed to carry out a diverse range of chemical transformations (1,2). Among these, enzymes are particularly attractive because they can be engineered to provide unparalleled levels of catalyst efficiency and product selectivity (3). Unfortunately, the majority of enzymes are thought to only catalyze a single type of chemical transformation. In contrast, small molecule chiral catalysts are often able to catalyze many mechanistically distinct reactions with good levels of selectivity (4). This generality is due, in part, to their ability to react with a diverse array of substrates. Substrate promiscuity, however, is not a feature exclusive to small molecule catalysts — many enzymes for organic synthesis possess this trait (5). Oxidoreductases, for instance, are renowned for their broad substrate scopes (6,7). We hypothesized that these enzymes could provide prosperous ground on which to discover and develop new reactivity (8,9,10,11). In particular, we are interested in exploiting oxidoreductases to address long-standing selectivity challenges inherent to the reactivity of radicals in chemical synthesis (12,13).
Flavin-dependent ‘ene’-reductases (EREDs) are substrate promiscuous oxidoreductases widely used in chemical synthesis for the stereoselective reduction of activated alkenes (14). Mechanistically, the reduction occurs via hydride transfer from the flavin hydroquinone (FMNhq) to the electrophilic β-position of the alkene (15,16). In other protein scaffolds, such as ferredoxin reductase or P450 reductase, flavin effects the reduction via two single electron transfers (17). We recognized that if this reactivity profile could be achieved with EREDs, they could be used as catalysts in asymmetric radical reactions. In our initial studies, we found that EREDs can catalyze asymmetric radical hydrodehalogenations of α-bromoesters via a single electron transfer (18). Mechanistically, this occurs via electron transfer from FMNhq to the substrate, forming an α-acyl radical, followed by hydrogen atom transfer from the flavin semiquinone (FMNsq). Recently, we demonstrated that these enzymes are also able to catalyze reductive radical cyclizations with high levels of diastero- and enantioselectivity using a related mechanism (19). These examples demonstrate the synthetic versatility of EREDs and highlight their ability to address long-standing challenges in radical-mediated asymmetric synthesis.
Redox-neutral radical reactions are a type of transformation where the substrate is both oxidized and reduced during the reaction mechanism. Catalysts that move between reducing and oxidizing redox states, such as photoredox catalysts (20), are ideally suited for this family of reactivity. We recognized that flavin could serve as reductant and oxidant within the same enzyme active site. As a model, we targeted the cyclization of α-halo-β-amidoesters to furnish 3,3-disubstituted oxindoles. This motif is prevalent in medicinally valuable molecules and there are no known methods for rendering this radical cyclization asymmetric (21,22). Mechanistically, this cyclization occurs via reduction of the α-halo-β-amidoester to afford an α-acyl radical that can react with the pendant aromatic ring and upon oxidation of the resulting vinylogous α-amido radical, followed by deprotonation, afford product (Fig. 1). Central to the development of this reactivity is the identification of a flavin redox pair where the oxidized form is capable of oxidizing the vinylogous α-amido radical without reducing the α-acyl radical. If successful, this reaction would provide a new biocatalytic strategy for stereoselectively preparing Csp2-Csp3 bonds (23, 24, 25).
Fig. 1. Strategies and Challenges in Using ‘Ene’-Reductases for Redox-Neutral Radical Cyclizations.

(a) The desired redox-neutral radical cyclization to prepare oxindoles from α-haloamides, a transformation currently unknown in nature (b) The central challenge to this reactivity is identifying a flavin redox couple that will favor cyclization over reduction. (c) The viable flavin redox pairs for redox-neutral cyclizations. If FMNhq is responsible for reducing the starting material, FMNsq will need to serve as an oxidant for the desired transformation. FMNsq can also function as a reductant, providing access to an undesired reductive mechanism. However, if FMNsq is used to reduce the starting material, FMN is formed in the active site, which can only function as an oxidant.
Results and Discussion
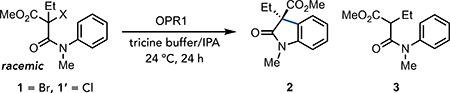
We began by exploring the ability of eight structurally diverse wild-type EREDs to catalyze the cyclization of α-bromo-β-amidoester 1 to oxindole 2 under ambient conditions with NADPH to reduce the ERED to the FMNhq oxidation state (Supplementary Table 1). While most EREDs were inactive, morphinone reductase (MorB), with its large active site (26), proved effective in providing racemic oxindole 2 in good yield with the remaining mass balance being reduced amide 3. Improved enantioselectivity can be achieved with 12-oxophytodienoate reductase (OPR1), although the major product of this reaction is amide 3 (Table 1, entry 1). Under these conditions, we hypothesize the flavin hydroquinone/semiquinone (FMNhq/FMNsq) redox couple to be operative, where FMNhq functions as a reductant and FMNsq serves as the oxidant (Figure 1C). However, FMNsq is also capable of reducing the α-acyl radical, potentially accounting for the formation of amide 3. We recognized that by moving to an orthogonal redox couple, it may be possible to improve the product selectivity without needing to alter the protein sequence.
Table 1.
Reaction Optimization
 | |||||
|---|---|---|---|---|---|
| entry | X | LED Irradiation | yield 2 (%)a | e.r. b | yield 3 (%)a |
| 1c | Br | none | 18 | 89:11 | 37 |
| 2 | Br | Cyan LED | 78 | 80:20 | 19 |
| 3 | Cl | Cyan LED | 94 | 95:5 | 4 |
| 4 | Cl | none | - | - | - |
| 5d | Cl | Cyan LED | - | - | - |
Standard Reaction Conditions. Substrate (10 mM), OPR1 (50 μM), tricine buffer (100 mM, pH = 8.0), 10 (v/v)% IPA (iPrOH), Cyan LEDs, 24 h.
Yield determined via reverse phase HPLC relative to an internal standard.
Enantiomeric ratios determined via HPLC on a chiral stationary phase.
NADP+ (1 mol %), Glucose (1 equiv) and GDH-105 (glucose dehydrogenase) added.
No enzyme added.
The flavin semiquinone/quinone (FMNsq/FMN) redox couple, where FMNsq would serve as the reductant and FMN as the oxidant, has features that make it ideal for redox-neutral radical cyclizations (Figure 1C). FMNsq (E1/2ox = −0.490 V vs SCE) is a slightly stronger reductant than FMNhq, enabling it to reduce activated α-haloacetanilide (27). Moreover, FMN has a comparable oxidation potential to FMNsq, enabling it to oxidize vinylogous α-amido radicals. Importantly, FMN cannot function as a reductant, consequently, formation of amide 3 should be diminished. Massey had previously demonstrated that FMNsq is formed within the active sites of EREDs by irradiating with visible light in the presence of electron donors, such as EDTA (28, 29, 30). Indeed, we found that by removing NADPH and irradiating a sample containing OPR1 in tricine buffer with cyan light, FMNsq is formed (as determined by EPR). When these conditions were tested with α-bromo-β-amidoester 1, oxindole 2 was formed as the major product, with the remaining mass balance being amide 3 (Table 1, entry 2). Analysis of this reaction by UV-vis spectroscopy revealed formation of FMNhq, providing a mechanism for formation of the dehalogenated amide (Supplementary Figure 3–5). This pathway can be avoided by moving to the less oxidizing α-chloro-β-amidoester 1’, which provides the desired oxindole in 94% yield with 95:5 er favoring the (S)-enantiomer (Table 1, entry 3).
Control experiments confirm both light and enzyme are required to form oxindole (Table 1, entry 4 and 5). A survey of lamps with different emission spectra revealed blue lamps (λmax = 450 nm and 476 nm) to furnish more dehalogenated product than cyan or green ones (λmax = 530 nm) (Supplementary Table 4–5). As the absorption spectrum of OPR1 is red-shifted by comparison to free FMN (Supplementary Figure 6), we hypothesized that photoexcitation of free FMN is facilitating the formation of amide 3. Indeed, when OPR1 is replaced with FMN under the reaction conditions, only dehalogenated amide 3 is observed (Table S6). Transient absorbance spectroscopy indicates that ground state FMNhq, presumably formed via photoreduction of FMN with tricine, is responsible for substrate dehalogenation (Supplementary Figure 9–11).
Next, we probed the flavin oxidation state responsible for reducing 1’ to the α-acyl radical within the active site of OPR1. While we propose this species to be FMNsq, based on our previous studies, we recognized that ground state or photoexcited flavin hydroquinone (FMNhq) could also be responsible for reduction (23). To determine whether FMNhq is capable of effecting cyclization, reactions were run for 2 hours with 2-fold and 40-fold excess (with respect to enzyme) of NADPH. In the absence of light, neither reaction afforded any consumption of chloroamide 1’, confirming that ground state FMNhq is not responsible for the observed reaction (Figure 2A). Next, we considered the photoexcited state of FMNhq. Previously, we found that α-chloroamides can be dehalogenated via photoinduced electron transfer if they form electron donor-acceptor (EDA) complexes with FMNhq in the ERED active site. However, quenching of the excited state (FMNhq*) was not observed in the presence of 1’ (Figure 2B), indicating that photoinduced electron transfer from FMNhq to 1’ is not responsible for radical formation. Attempts to observe an EDA complex were unsuccessful because of flavin oxidization on the time-scale of the experiment.
Fig. 2. Studies to determine the mechanism of oxindole formation.

(A) This experiments demonstrates that ground state FMNhq is not able to initiate the radical cyclization. (B) The fluorescence spectra indicates that the excited state can be accessed, however, this state is not quenched by the substrate, indicating that FMNhq* is not responsible for initiating the reaction. (C) In this experiment, oxidized OPR1 is photoreduced with cyan light and tricine buffer to partially reduce FMN to FMNsq− (as determined by EPR). Then substrate 1’ is introduced to the enzyme in the absence of light. Oxindole is formed under these conditions indicating that gound state FMNsq− is responsible for initiating the reaction. (D) This represents a proposed mechanism where light and tricine buffer are responsible for reducing FMN to FMNsq−, which can reduce the substrate to generate an α-acyl radical and FMN. Cyclization of the radical generates a reducing vinylogous amido radical which can be oxidized by FMN to form product and regenerate FMNsq−.
Having ruled out FMNhq as a reductant, we conducted experiments to determine whether FMNsq was responsible for the observed reactivity. First, OPR1 in tricine buffer was irradiated with cyan light and the formation of FMNsq was determined and quantified via EPR spectroscopy (Supplementary Figure 12–14). Next, to determine if ground state FMNsq is responsible for oxindole formation, a Schlenk flask containing OPR1 and tricine buffer was irradiated with cyan light for 1 hr. After FMNsq formation was confirmed and quantified by EPR (Supplementary Figure 13), the reaction was shielded from light and substrate was added. After two hours, oxindole 2 was observed in 3% yield with 95:5 er and negligible formation of the reduced amide 3 (Figure 2C). This experiment indicates that ground FMNsq provides the electron to initiate the cyclization. Moreover, 68 catalyst turnovers are observed (based on FMNsq concentration determined by EPR), suggesting FMNsq is regenerated during the reaction mechanism. Based on these studies, we propose a mechanism in which enzyme-bound FMN is reduced via photoinduced electron transfer to generate FMNsq (24). This species can reduce 1’ to generate an α-acyl radical and FMN. Stereoselective cyclization into the aromatic ring forms a vinylogous α-amidoradical, which is readily oxidized by FMN to form FMNsq and the desired oxindole (Figure 2D)
As light is only required to initiate the reduction of FMN to FMNsq, we questioned why constant irradiation is required to achieve high conversions. Schaller found that OPR1 kinetically stabilizes FMNsq. Consequently, this OPR1⊂FMNsq (⊂indicates binding) can disproportionate to form catalytically inactive FMNhq and FMN (31). Indeed, an EPR spectrum collected at the end of the photoinitiated reaction discussed above (Figure 2C) revealed consumption of FMNsq (Supplementary Figure 13). Moreover, FMNsq concentration steadily decreases over two hours in the dark in the absence of substrate (Supplementary Figure13). Collectively, these results suggest that FMNsq is unstable under the reaction conditions. We considered that the role of irradiation is to oxidize catalytically inactive FMNhq to FMNsq, presumably through oxidation by photoexcited FMN (Supplementary Figure 21). To test this hypothesis, OPR1 is reduced with NADPH to form FMNhq. Upon irradiation with cyan light, formation of an EPR signal consistent with FMNsq is observed, suggesting light facilitates FMNhq oxidation (Supplementary Figure 14) (32). This feature can be observed in a preparative reaction containing excess NADPH (5-fold excess by comparison to enzyme). When run in the dark, no product is formed, presumably because FMNsq is not present (Figure 2A). Upon irradiation with cyan light, oxindole 2 is formed in 6% yield with <1% formation of the dehalogenated product (Supplementary Figure 15). By comparison, when the same reaction is run in the absence of NADPH, the desired product is formed in 26% yield with 1% formation of 3 (Supplementary Figure 14). These experiments demonstrate that photoinduced electron transfer can be used to regenerate flavin oxidation states with limited stability and in doing so, expand the synthetic utility of flavoenzymes beyond what can be achieved using simple chemical oxidants or reductants.
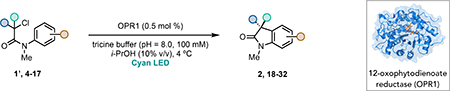
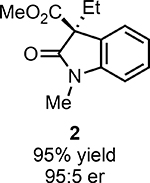
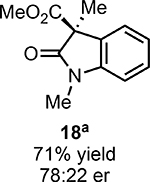
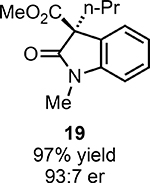
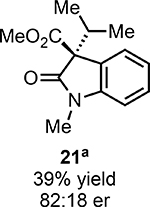
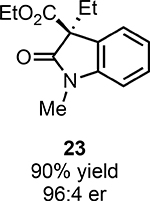
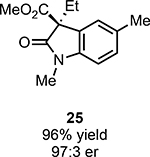
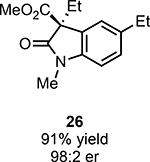
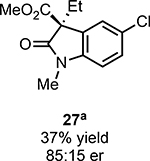
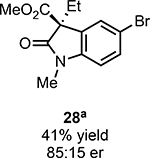
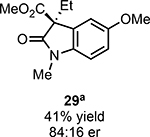
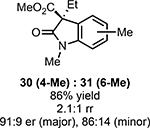
With ideal reaction conditions in hand, we focused our attention toward the scope of this reaction (Table 2). OPR1 accepts a variety of substituents at the α-position of the amide. While the smaller methyl substituent (Table 2, 18) affords product with lower selectivity, longer linear groups, such as propyl, butyl, or allyl (Table 2, 19,20,22), furnish product in good yield with excellent levels of enantioselectivity. Branched substituents, however, deliver product with lower yield and enantioselectivity (Table 2, 21). A variety of ester substituents are accepted, affording the corresponding oxindoles in high yields and enantioselectivities (Table 2, 23,24). With regard to substitution on the aromatic ring, ortho-substituents proved to be unreactive, failing to provide either oxindole or dehalogenated material, suggesting they may be incapable of binding to OPR1. Electron-donating para-substituents, however, are well tolerated with the corresponding oxindoles accessed in good yield with excellent levels of enantioselectivity (Table 2, 26). Arenes with electron-withdrawing substituents are more challenging substrates, requiring elevated reaction temperatures to furnish product (Table 2, 27–29). As these substrates form a larger amount of the hydrodehalogenated product (40% yield of the hydrodehalogenated product in the case of 28), we hypothesize this low product yield is likely due to the kinetic challenge involved in coupling electron poor arenes with electrophilic α-acyl radicals. Electron-withdrawing meta-substituents were found to be poorly reactive substrates, but electron-donating groups are more reactive. For instance, m-methyl amides afford oxindole product as a 1.3:1 ratio of regioisomers (Table 2, 30:31). A preliminary examination of OPR1 variants with mutations made to residues responsible for substrate binding revealed that OPR1-H187N is capable of providing product with modestly improved levels of selectivity (2.1:1), suggesting protein engineering can be used to alter the regioselectivity of the cyclization. Finally, we found that the electron-withdrawing ester group can be removed from the substrate and product formation is still observed, albeit in diminished yields (Table 2, 32). Initial control experiments indicate that ground-state semiquinone is not capable of initiating this reaction (Supplementary Figure 23) and this substrate does not form an EDA complex with FMNhq (Supplementary Figure 24) suggesting this reaction may initiate via a distinct mechanism.
Table 2.
Biocatalytic radical cyclization to form oxindole scope.
 | ||||
|---|---|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
Standard Reaction Conditions. Substrate (10 mM), OPR1 (50 μM), tricine buffer (100 mM, pH = 8.0), 10 (v/v)% IPA, Cyan LEDs, 24 h. Yield determined via reverse phase HPLC relative to an internal standard. Enantiomeric ratios determined via HPLC on a chiral stationary phase.
Reaction run at 24 °C.
‘Ene’-reductases, enzymes traditionally thought to catalyze only reductive reactions, are able to catalyze redox neutral radical cyclizations with high levels of enantioselectivity under photocatalytic conditions. This new function is achieved without any modification to the wild-type sequence. The central mechanism for optimization was to change the flavin redox couple. Moving from FMNhq/FMNsq to FMNsq/FMN redox pair disfavors the mechanistic pathway responsible for dehalogenation. Modulating the flavin oxidation state provides a new handle for biocatalyst optimization and highlights the diversity of reactivity available to substrate-promiscuous wild type enzymes. Moreover, identifying enzymes capable of catalyzing non-natural reactions presents an opportunity to address reactivity and selectivity challenges that small molecule catalysts have thus far failed to address (33).
Methods
General procedure for biocatalytic radical cyclization to form oxindoles
Under anaerobic atmosphere, a 4 mL shell vial was charged with 100 mM tricine pH 8 and OPR-1 (0.01 μmol, 0.005 equiv.) to reach a total volume of 1.8 mL. To this mixture was added chloroamide substrate 1 (0.02 mmol, 1.0 equiv.) in 200 μL i-PrOH to reach a final total volume of 2 mL. The reaction mixture was capped with a septum and placed on a stir plate in a 4ºC walk in refrigerator (unless otherwise stated). The reaction mixture was irradiated with a 497 nm cyan LED for 48 hours. After 48 hours elapsed, 1 mL acetonitrile containing 0.01 mmol internal standard (either α,α,α−trifluorotoluene or 1,3,5-tribromobenzene) was added, followed by an additional 3 mL acetonitrile to precipitate enzyme. Yield was calculated vs. internal standard on LC-MS. In a scintillation vial, 2 mL quenched reaction mixture was diluted with 10 mL EtOAc, and evaporated to dryness in vacuo. The resulting residue was redissolved in 1:1 Hexanes : i-PrOH and injected on HPLC to determine enantioselectivity.
Supplementary Material
Acknowledgments
Funding: Financial support was provided by the NIHGMS (R01 GM127703), Searle Scholar Award (SSP-2017-1741), the Princeton Catalysis Initiative, and Princeton University. B.K, acknowledges National Science Foundation Graduate Research Fellowship (DGE-1656466). D.G.O acknowledges support from the Postgraduate Scholarships Doctoral Program of NSERC. We acknowledge support from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant No. DE-SC0019370
Footnotes
Competing interest: The authors declare no competing interest.
Data availability: Data are available in the supplementary materials or from the corresponding author upon request.
References
- 1.de Meijere A, Diederich F Metal-catalyzed Cross-coupling Reactions (Wiley-VCH, 2004). [Google Scholar]
- 2.Scheffler U, Mahrwald R Recent advances in organocatalytic methods for asymmetric C–C bond formation. Chem. Eur. J 19, 14346–14396 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Yoon TP, Jacobsen EN Privileged chiral catalysts. Science 299, 1691–1693 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Pandya C, Farelli JD, Dunaway-Mariano D, Allen KN Enzyme Promiscuity: Engine of Evolutionary Innovation. J. Biol. Chem 289, 30229–30236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martínez AT et al. Oxidoreductases on their way to industrial biotransformations. Biotech. Adv 35, 815–831 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Dong J, Fernández-Fueyo E, Hollmann F, Paul CE, Pesic M, Schmidt S, Wang Y, Younes S, Zhang W Angew. Chem. Int. Ed 57, 9238–9261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kazlauskas RJ, Bornscheuer U Enzyme catalytic promiscuity: expanding the catalytic action of enzymes to new reactions. In: Carreira EM, Yamamoto H (eds). Comprehensive Chirality. 7, 465–480 (2012). [Google Scholar]
- 9.Bornscheuer UT, Kazlauskas R Angew. Chem. Int. Ed 43 6032–6040 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Emmanuel ME, Greenberg NR, Oblinsky DG, Hyster TK Accessing non-natural reactivity by irradiating nicotinamide-dependent ketoreductases with light. Nature 540, 414–417 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Biegasiewicz KF, Cooper SJ, Emmanuel MA, Miller DC Catalytic promiscuity enabled by photoredox catalysis in nicotinamide-dependent oxidoreductases. Nat. Chem 10, 770–775 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Sibi MP, Porter NA Enantioselective Free Radical Reactions. Acc. Chem. Res 32, 163–171 (1999). [Google Scholar]
- 13.Meggers E Asymmetric catalysis activated by visible light. Chem. Commun 51, 3290–3301 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Toogood HS, Scrutton NS Discovery, characterization, engineering, and application of ene-reductases for industrial biocatalysis. ACS Catal. 8, 3532–3549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winkler CK, Faber K, Hall M Biocatalytic reduction of activated C=C-bonds and beyond: emerging trends. Curr. Opin. Chem. Biol 43, 97–105 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Heckenbichler K, et al. Asymmetric reductive carbocyclization using engineered ene reductases. Angew. Chem. Int. Ed 57, 7240–7244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura R Versatility and specificity in flavoenzymes: control mechanisms of flavin reactivity. Chem. Rec 1, 183–194 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Sandoval BA, Meichan AJ, Hyster TK Enantioselective hydrogen atom transfer: discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc 139, 11313–11316 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Biegasiewicz KF, Cooper SJ, Gao X, Oblinsky DG, Kim JH, Garfinkle SE, Joyce LA, Sandoval BA, Scholes GD, Hyster TK Photoexcitation of a Flavoenzyme Enables a Stereocontrolled Radical Cyclization. Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw MH, Twilton J, MacMillan DWC, Photoredox Catalysis in Organic Chemistry. J. Org. Chem 81, 6898–6926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju X, Liang Y, Jia P, Li W, Yu W Synthesis of oxindoles via visible light photoredox catalysis. Org. Biomol. Chem 10, 498–501 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Zhou F, Liu Y-L, Zhou J Catalytic Asymmetric Synthesis of Oxindoles Bearing a Tetrasubstituted Stereocenter at the C-3 Position, Adv. Synth. Catal 352, 1381–1407 (2010). [Google Scholar]
- 23.Tang M-C, Zou Y, Watanabe K, Walsh CT, Tang Y Oxidative cyclization in natural product biosynthesis. Chem. Rev 117, 5226–5333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh CT, Wencewicz TA Flavoenzymes: versatile catalysts in biosynthetic pathways. Nat. Prod. Rep 30, 175–200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh CT, Tang Y Recent advances in enzymatic complexity generations: cyclization reactions. Biochemistry 57, 3087–3104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barna T, Messiha HL, Petosa C, Bruce NC, Scrutton NS, Moody PCE Crystal structure of bacterial morphinone reductase and properties of the C191A Mutant Enzyme. J. Biol. Chem 277, 30976–30983. [DOI] [PubMed] [Google Scholar]
- 27.Steward RC, Massey V Potentiometric Studies of Native and Flavin-substituted Old Yellow Enzyme. J. Biol. Chem 260, 13639–13647 (1985). [PubMed] [Google Scholar]
- 28.Massey V, Stankovich M, Hemmerich P Light-mediated reduction of flavoproteins with flavins as catalysts. Biochemistry 17, 1–8 (1978). [DOI] [PubMed] [Google Scholar]
- 29.Taglieber A, Schulz F, Hollmann F, Rusek M, Reetz MT Light-driven biocatalytic oxidation and reduction reactions: scope and limitations. ChemBioChem 9, 565–572 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Peers MK, et al. Light-driven biocatalytic reduction of α,β-unsaturated compounds by ene reductases employing transition metal complexes as photosensitizers. Catal. Sci. Technol 6, 169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strassner J, Fürholz A, Macheroux P, Amrhein N, Schaller A A homolog of old yellow enzyme in tomato. Spectral properties and substrate specificity of the recombinant protein. J. Biol. Chem 274, 35067–35073 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Murthy YVSN & Massey V Synthesis and Properties of 8-CN-flavin Nucleotide Analogs and Studies with Flavoproteins. J. Biol. Chem 273, 8975–8982 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Knight AM, Kan SBJ, Lewis RD, Bradenberg OF Chen K, Arnold FH Diverse engineered heme proteins enable stereodivergent cyclopropanation of unactivated alkenes. ACS Cent. Sci 4, 372–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.