

SUMMARY
Estrogen exerts extensive and diverse effects throughout the body of women. In addition to the classical nuclear estrogen receptors (ERα and ERβ), the G protein-coupled estrogen receptor GPER is an important mediator of estrogen action. Existing ER-targeted therapeutic agents act as GPER agonists. Here, we report the identification of a small molecule, named AB-1, with the previously unidentified activity of high selectivity for binding classical ERs over GPER. AB-1 also possesses a unique functional activity profile as an agonist of transcriptional activity but an antagonist of rapid signaling through ERα. Our results define a class of small molecules that discriminate between the classical ERs and GPER, as well as between modes of signaling within the classical ERs. Such an activity profile if developed into an ER antagonist could represent an opportunity for the development of first-in-class nuclear hormone receptor-targeted therapeutics for breast cancer exhibiting reduced acquired and de novo resistance.
Keywords: Breast Cancer, Estrogen, ERα, ERβ, GPER, GPR30, Receptors, SERM, SERD
eTOC Blurb (In Brief)
ER-selective ligands lacking GPER cross-reactivity remain unknown. Revankar et al. identify an ER-targeted ligand, termed AB-1, with high selectivity for ER over GPER. AB-1 activates ER transcription while antagonizing ER rapid signaling. This activity profile, if converted into an antagonist, could prove beneficial in breast cancer treatment.
Graphical Abstract
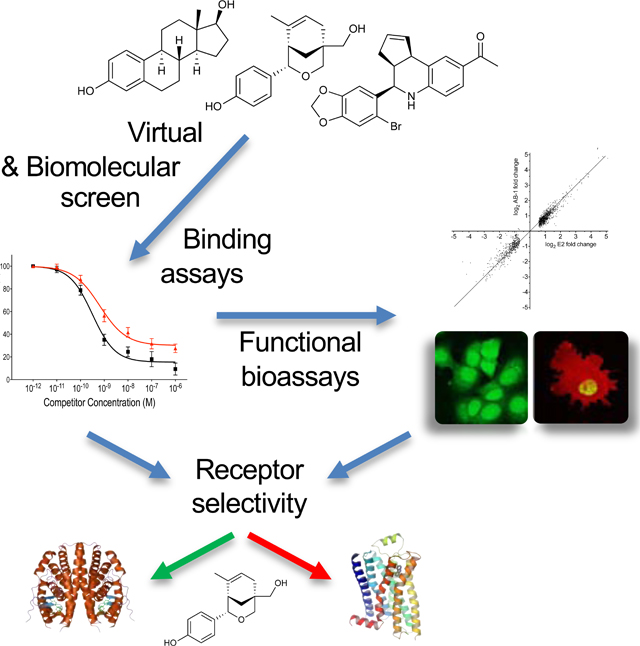
INTRODUCTION
Estrogens (predominantly 17β-estradiol, E2) regulate multiple diverse aspects of physiology throughout the body, particularly during development, puberty and reproduction, but also in metabolic, endocrine, cardiovascular, nervous, musculoskeletal and immune functions (Edwards, 2005). Although many of these effects are traditionally associated with women, E2 also has important roles in male physiology (Lombardi et al., 2001). As a result of these varied actions, targeting E2 pathways has been exploited extensively in the development of therapeutic and preventative approaches (Arnal et al., 2013). For example, E2 and its derivatives have been used for over a half a century as the primary constituent of contraceptive pills (Woutersz, 1991).
E2 and its receptors also play important roles in both health and disease, particularly breast cancer development and treatment. In addition to the classical nuclear estrogen receptors (ERα and ERβ), the 7-transmembrane spanning G protein-coupled estrogen receptor GPER (previously GPR30) has become recognized as an important mediator of E2 action (Barton et al., 2018; Prossnitz and Arterburn, 2015; Prossnitz and Barton, 2014; Prossnitz and Hathaway, 2015; Pupo et al., 2016; Sharma et al., 2018). Although many of the effects of E2 are mediated by ERα and ERβ through transcriptional regulation, rapid signaling pathways (e.g. kinase activation, such as ERK1/2 and Akt, cAMP production and ion fluxes) that occur in the time frame of seconds to minutes are now understood to be activated by both ERα (Levin and Hammes, 2016) and GPER (Barton et al., 2018). Pharmacological approaches have identified families of compounds for breast cancer therapy as well as for managing symptoms of menopause (including osteoporosis) termed selective estrogen receptor modulators and downregulators (SERMs, such as tamoxifen and raloxifene, and SERDs, such as fulvestrant, respectively) (Maximov et al., 2013; McDonnell and Wardell, 2010), based on their (tissue-dependent, in the case of SERMs) transcriptional activities assumed to occur exclusively through ERα; however, tested SERMs and SERDs lack selectivity with respect to GPER, functioning as GPER agonists (Filardo et al., 2000; Petrie et al., 2013; Revankar et al., 2005). In fact, a broad array of xenoestrogens, including synthetic (industrial, agricultural and pharmacological) and natural (phyto- and myco-estrogens), have been shown not only to bind GPER but also to function as GPER agonists (Prossnitz and Arterburn, 2015; Thomas and Dong, 2006; Thomas et al., 2005). This lack of ER/GPER pharmacological discrimination led us and others to seek compounds with the ability to selectively modulate GPER activity, in the absence of ERα/β activity (Bologa et al., 2006; Dennis et al., 2009; Dennis et al., 2011; Lappano et al., 2012). The most widely used GPER-selective ligands are the tetrahydroquinolines G-1 (Bologa et al., 2006) (an agonist), G15 (Dennis et al., 2009) and G36 (Dennis et al., 2011) (both antagonists). G-1 mediates or reproduces many of the salutary effects of E2, particularly those associated with rapid signaling, in rodent models of multiple sclerosis (Blasko et al., 2009; Wang et al., 2009), stroke (Lebesgue et al., 2010; Zhang et al., 2010), cerebral ischemia following cardiac arrest (Kosaka et al., 2012), traumatic brain and spinal cord injury (Hu et al., 2012; Prossnitz, 2012), myocardial infarction (Bopassa et al., 2010), atherosclerosis (Meyer et al., 2014), obesity (Sharma et al., 2018), diabetes (Sharma and Prossnitz, 2017), pancreatic islet survival (Liu et al., 2009) and transplantation (Liu et al., 2013), hypertension (Haas et al., 2009; Lindsey et al., 2009), and diastolic dysfunction (Wang et al., 2012), among others (Prossnitz and Arterburn, 2015; Prossnitz and Hathaway, 2015). In contrast, the GPER antagonists G15 and G36 have been shown to have important applications in carcinogenesis (Mo et al., 2013; Petrie et al., 2013; Scaling et al., 2014) and cardiovascular aging (Meyer et al., 2016), the latter through the regulation of NADPH oxidase-mediated superoxide production (Barton et al., 2019; Prossnitz, 2018).
A similar lack of pharmacological selectivity towards the classical estrogen receptors ERα/β and against GPER has resulted in important experimental and clinical challenges. This is evident as the result of unexpected agonist activities of both SERMs and SERDs via GPER in both experimental systems (Chen et al., 2014; Filardo et al., 2000; Hofmeister et al., 2012; Petrie et al., 2013; Zekas and Prossnitz, 2015) and clinical use of the SERD ICI182,780 (fulvestrant) as an anti-hormone therapy for advanced breast cancer in women where, for example, symptomatic hypotension is a common side effect (Vergote and Abram, 2006), consistent with the GPER-mediated vasodilatory activity of ICI182,780 observed ex vivo (Meyer et al., 2010). There is also evidence suggesting that the acquired resistance observed in women treated with anti-estrogens (SERMs and SERDs) for prolonged periods may result in part from chronic activation of GPER (Ignatov et al., 2010; Ignatov et al., 2011; Mo et al., 2013), potentially through the inactivation of the pro-apoptotic transcription factor FOXO3a (Zekas and Prossnitz, 2015) as recently reviewed (Pepermans and Prossnitz, 2019).
Here we present the discovery of the first truly ER-selective ligand that lacks binding and activity towards GPER, defined as a selective ligand for estrogen receptor proteins (SLERP). We employed a combination of computational and biomolecular screening to identify AB-1, a previously described oxabicyclic compound that binds both ERα and ERβ (Hamann et al., 2005; Hsieh et al., 2006; Sibley et al., 2003). Our extensive characterization revealed unique properties of AB-1 in that it lacks binding and thus rapid signaling via GPER, and although transcriptional activity via ERα is largely similar to that of E2, AB-1 also lacks the ability to initiate multiple rapid signaling events via ERα. Thus, in addition to discriminating between ERα and GPER, AB-1 also discriminates between the classic nuclear transcriptional (genomic) and rapid signaling (non-genomic) activities of ERα, providing the complementary activity profile to compounds that elicit extra-nuclear signaling but not transcriptional activity through ERα (Madak-Erdogan et al., 2016).
RESULTS
Employing computational and virtual screening of a 10,000 compound GPCR-optimized library, we previously identified the GPER-selective agonist G-1 (Bologa et al., 2006), the GPER-selective antagonist G15 (Dennis et al., 2009) and subsequently optimized the even more selective antagonist, G36 (Dennis et al., 2011), as compounds that lack ERα/β-binding (Figure 1). The discovery of these GPER-selective compounds has facilitated a better understanding of the physiological roles of GPER in E2 signaling (Barton and Prossnitz, 2015; Prossnitz and Arterburn, 2015). To further distinguish the roles of ERs and GPER in E2 signaling, we sought to expand our repertoire of selective compounds, this time screening for compounds harboring an inverse profile to that of our GPER-selective compounds (i.e. high selectivity for binding ERα/β over GPER). Employing high-throughput flow cytometry-based biomolecular screening with ERα-GFP- and ERβ-GFP-expressing COS7 cells and the fluorescently labeled E2 derivative E2-Alexa633 as previously described (Bologa et al., 2006), we screened the top 100 virtual hits of our compound library for selective binding activity towards ERα and ERβ. We identified one compound, a phenol-substituted oxabicyclo[3.3.1]nonene, hereafter termed AB-1 (Figure 1), that competed with E2-Alexa633 binding to ERα and ERβ.
Figure 1.

Chemical structures of estrogen (17β-estradiol, E2), GPER-selective ligand G-1 and ER-selective ligand AB-1.
To validate the activity and confirm the chemical identity of our primary hit, we synthesized AB-1 (4-(5-(hydroxymethyl)-8-methyl-3-oxabicyclo[3.3.1]non-7-en-2-yl))-phenol) (Hamann et al., 2005; Sibley et al., 2003), following a modified procedure employing a hafnium(IV) triflate catalyzed Prins cyclization (Nakamura et al., 2009) (see STAR Methods). The compound was fully characterized and was identical to previously reported NMR spectra with characteristic 1H NMR signals observed for the C8-methyl (δ 1.01, 3H) and benzylic hydrogen at C2 (δ 4.50, 1H) (Hamann et al., 2005) (Figures S1–S3).
To confirm our findings and examine selectivity with respect to GPER, we co-expressed ERα-GFP or ERβ-GFP with GPER-mRFP1 in COS7 cells, incubated the cells with E2-Alexa633, and imaged by confocal microscopy. As previously demonstrated, because ERα/β and GPER localization is mutually exclusive, with ERα/β in the nucleus and GPER in the endoplasmic reticulum (i.e. cytosolic), selectivity of E2-Alexa633 binding can be assessed through spatial co-localization with each receptor (Figure 2A) (Bologa et al., 2006). In cells expressing ERα-GFP and GPER-mRFP1, E2-Alexa633 is localized to both ERα and GPER (Figure 2A, top row). Addition of E2 blocked binding of E2-Alexa633 to ERα-GFP (and ERβ-GFP, not shown) as well as GPER (GPER-mRFP1) (Figure 2A, second row), whereas addition of AB-1 blocked the binding of E2-Alexa633 to both ERα-GFP and ERβ-GFP, but not to GPER-mRFP1 (Figure 2A, third and fourth rows). To characterize the binding properties of AB-1 in greater detail, we determined its binding affinity to the individual ERs (Table I). Using a flow cytometry-based competitive binding assay with transiently transfected COS7 cells, we determined that AB-1 blocked E2-Alexa633 binding to ERα and ERβ with IC50 values of 3 nM and 26 nM, respectively (Figure 2B–C). IC50 values for E2 were 0.3 nM and 0.6 nM for ERα and ERβ, respectively. Importantly, AB-1 did not significantly block E2-Alexa633 binding to GPER at concentrations up to 10 μM (Figure 2D). Binding affinities to the purified ligand binding domain (LBD) of ERα and ERβ were also determined employing a TR-FRET-based competitive binding assay, revealing IC50 values for ERα and ERβ LBDs of 38 nM and 24 nM, respectively (with IC50 values for E2 of 0.26 nM and 0.47 nM for ERα and ERβ, respectively) (Figure S4 and Table I). Taken together, these results show that AB-1 selectively binds to ERα and ERβ, but not GPER.
Figure 2.

Ligand Binding properties of AB-1. (A) COS7 cells co-expressing ERα-GFP or ERβ-GFP with GPER-mRFP1 were stained with E2-Alexa633 in the presence or absence of unlabeled E2 (100 nM) or AB-1 (1 μM). AB-1 blocks the binding of E2-Alexa633 to ERα and ERβ, but not to GPER. Confocal images are representative of three independent experiments. White scale bar represents 10 μm. Data are from three independent experiments. (B-D) Binding affinities of E2 and AB-1 for ERα, ERβ and GPER. Competitive ligand binding assays were performed using 2 nM E2-Alexa633 and the indicated concentrations of unlabeled E2 (■) or AB-1 (▲) in COS7 cells transfected with either ERα-GFP (b), ERβ-GFP (c) or GPER-GFP (D). Data are mean ± s.e.m. from three independent experiments. (E) Competitive radio-ligand binding assays of AB-1 (1 μM and 10 μM) for androgen receptor (AR), mineralocorticoid receptor (MR), progesterone receptor B (PR-B), glucocorticoid receptor (GR) and ERα. NHR-specific ligands (see Methods) were used as control competitors. Data are mean values from technical duplicates.
Table I.
Summary of AB-1 Properties
| E2 | AB-1 | |
|---|---|---|
| ERα cell binding (IC50) | 0.30 nM | 3 nM |
| ERβ cell binding (IC50) | 0.65 nM | 26 nM |
| ERα LBD binding (IC50) | 0.26 nM | 38 nM |
| ERβ LBD binding (IC50) | 0.47 nM | 24 nM |
| GPER cell binding | ∼8 nM | >> 10 μM |
| ERE expression (EC50) | 0.08 nM | 15 nM |
| MCF-7 proliferation (EC50) | 0.3 pM | 0.5 nM |
| ERα protein degradation (%) | 54 % | 52 % |
| Calcium signaling ERα (IC50) | N/A* | 33 nM |
| Calcium signaling ERβ (IC50) | N/A | 75 nM |
| Uterine Imbibition (EC50) | ∼3 ng | ∼90 μg |
| Uterine Proliferation (EC50) | ∼5 ng | ∼30 μg |
N/A, not applicable
To further examine the selectivity of AB-1 with respect to other nuclear hormone receptors (NHRs), we evaluated its binding to androgen receptor (AR), mineralocorticoid receptor (MR), progesterone receptor B (PR-B), glucocorticoid receptor (GR) and ERα using competitive radio-ligand binding assays (Figure 2E). AB-1 exhibited no binding to AR, MR, PR-B or GR, but did, as expected, bind to ERα (Figure 2E). Control inhibitors to each of the receptors showed >90% inhibition to the respective NHRs. Taken together with the previous data, these results show that AB-1 is not only selective to the classical ERs over GPER, but also selective to ER over other NHRs.
To assess the functional properties of AB-1, we first examined its effect on ER-mediated transcription in MCF-7 cells stably expressing an ERE-GFP reporter gene (Yamaguchi et al., 2005). Like E2, AB-1 dose-dependently induced ERE activation with an EC50 value of ~15 nM (vs. ~0.08 nM for E2) (Figure 3A). To expand upon its transcriptional activity, we also assessed the effect of AB-1 on global ER-mediated gene transcription compared to that of E2 in MCF-7 cells. Interestingly, AB-1 induced a highly similar transcription profile (both in terms of activation and inhibition) to that of E2 (Figure 3B, r=0.94, p<0.0001), with two of the best characterized E2/ER-stimulated genes [Progesterone Receptor (PGR) and GREB1] showing virtually identical levels of upregulation, implying that AB-1 functions as an ER transcriptional regulator that activates and inhibits expression of ER-target genes largely similar to that of E2. Interestingly, a number of the most E2repressed genes (e.g. PSCA, MYCN and FAM65C) were repressed to a far lesser extent by AB-1 compared to E2, although other genes repressed by about 8–10-fold by E2 were similarly repressed by AB-1. Many additional genes were either induced less or repressed less with AB-1 compared to E2 (by 50% or more), suggesting a contribution of rapid signaling to these transcriptional events, complimenting conclusions reached, employing E2-dendrimers (that lack direct transcriptional regulation by ER in the nucleus), that rapid signaling alone can regulate many E2-responsive genes (Madak-Erdogan et al., 2008).
Figure 3.

Transcriptional activity of AB-1. (A) Ligand-induced expression of GFP in MCF-7 cells. MCF-7 cells stably expressing an ERE-GFP reporter were stimulated with the indicated concentrations of E2 (■) or AB-1 (▲) and GFP expression was measured by flow cytometry. Data indicate mean ± s.e.m. of four independent experiments. (B) Ligand-induced global ER-mediated gene transcription profile. MCF-7/WS8 cells were stimulated with 1 nM E2 or 1 μM AB-1 and gene expression was assessed in duplicate. Gene expression changes of 1231 genes (greater than 1.5-fold) are shown as average log2 fold-change compared to vehicle-treated cells for E2 (x-axis) and AB-1 (y-axis). Expression of GREB1 and PGR are shown with arrows. Correlation factor (R) was 0.94 with a p-value <0.0001. (C) Effect of AB-1 on MCF-7 cell growth. MCF-7 cells were stimulated with the indicated concentrations of E2 (■) or AB-1 (▲) and total cell numbers were analyzed after 5 days. Cell numbers are shown as percentages relative to E2-treated cells (100%). Data points are mean ± s.e.m. of three independent experiments each performed in triplicate. (D) Ligand-induced protein degradation of ERα. MCF-7 cells were cultured with the indicated compounds and ERα levels determined by Western blot. Data is normalized to DMSO-treated samples and is shown as mean ± s.e.m. of at least 4 independent experiments. ***p<0.001 vs. DMSO by one-sample t-test.
To further confirm the agonist nature of AB-1, we tested its ability to induce MCF-7 cell growth, which is not only induced by ER activation, but also dependent on it. AB-1 stimulated cell growth to a similar (in fact, slightly greater) maximal extent compared to E2, with an EC50 of ~0.5 nM (vs. ~0.3 pM for E2) (Figure 3C). Upon binding of both agonists and antagonists (classical SERDs), ERα protein undergoes degradation and ultimately downregulation of its steady state levels (Wijayaratne and McDonnell, 2001). Therefore, to determine whether AB-1 exerts the same effect as E2 on ERα stability and protein levels, we treated MCF-7 cells with E2, AB-1, the SERM 4-hydroxytamoxifen (4-OHT), which stabilizes ERα, or the SERD ICI182,780, which potently downregulates ERα. AB-1 induced a ~50% decrease in ERα levels, similar to that of E2, whereas 4-OHT and the SERD ICI182,780, as expected, moderately increased and potently decreased ERα levels, respectively (Figure 3D) (Wijayaratne and McDonnell, 2001). Together, these data demonstrate that AB-1 acts as an agonist of ERα/β transcriptional activity, stimulating MCF-7 cell growth and inducing ERα degradation.
To determine whether AB-1 mediates rapid signaling as observed for E2, we examined the PI3K/Akt-mediated inactivation of FOXO3a in MCF-7 cells (Zekas and Prossnitz, 2015). FOXO3a is a forkhead box transcriptional activator of pro-apoptotic genes in the absence of survival factors. Growth factors (e.g. EGF) that stimulate the PI3K pathway lead to the Akt-mediated phosphorylation of FOXO3a, which in turn leads to its translocation to the cytoplasm and subsequent proteasomal degradation. To evaluate FOXO3a localization, we employed a FOXO3a-GFP construct that was transiently expressed in MCF-7 cells. Following EGF stimulation, FOXO3a translocated from the nucleus to the cytoplasm (Figure 4A). E2 and the GPER-selective agonist G-1 also stimulated cytosolic translocation, although in a lower percentage of cells (Figure 4B). In contrast, AB-1 had no effect on FOXO3a translocation, nor did it alter the extent of E2- or G-1-mediated translocation (Figure 4B). This result is in fact consistent with our previous observations that the E2-mediated activation of PI3K and Akt, leading to FOXO3a inactivation, is mediated by GPER.
Figure 4.

Ligand-induced intracellular translocation of FOXO3a. (A) Intracellular localization of FOXO3a-GFP. MCF-7 cells transiently expressing FOXO3a-GFP were treated with vehicle (Ctl), E2 (10 nM), G-1 (100 nM), AB-1 (1 μM), EGF (50 ng/mL) or a combination of AB-1 + E2 or AB-1 + G-1 and FOXO3a-GFP localization determined by confocal microscopy. White scale bar represents 10 μm. Data are representative of three independent experiments. (B) Quantification of data in (A) and represent the mean ± s.e.m. of at least 3 independent experiments. *p<0.05 vs. vehicle (Ctl) by one-way ANOVA with Bonferroni post-hoc test.
In order to determine whether AB-1 can also mediate E2-dependent rapid signaling specifically via the classical estrogen receptors, we employed COS7 cells expressing either ERα, ERβ or GPER. We first examined the ability of AB-1 to induce calcium mobilization. Surprisingly, unlike E2, which induced rapid calcium mobilization in COS7 cells expressing ERα, ERβ or GPER (Figure 5A), AB-1 did not induce calcium mobilization in any of these cells (Figure 5B). More importantly, AB-1 dose-dependently inhibited E2-mediated calcium mobilization in COS7 cells expressing either ERα (IC50 = 33 nM) or ERβ (IC50 = 75 nM) (Figure 5C), but did not block E2-mediated calcium mobilization in GPER-expressing COS7 cells (Figure 5B). This result suggests that despite acting as an agonist of transcriptional activation via ERα, AB-1 acts an antagonist or inverse agonist of ER-mediated rapid calcium signaling.
Figure 5.

AB-1 antagonizes classical ER-mediated rapid signaling. (A-C) Ligand-induced effect on intracellular calcium mobilization through individual ERs. COS7 cells transiently expressing ERα-GFP (red curve), ERβ-GFP (blue curve) or GPER-GFP (green curve) were stimulated with either 1 nM E2 (A) or 1 μM AB-1 followed by 1 nM E2 (B). Intracellular calcium mobilization was evaluated using indo1-AM and ligands were added at 20 s or 80 s as indicated. Data is shown as the relative 490nm/400nm ratio change (y-axis) compared to mock-transfected COS7 cells (black curve) and re representative of three independent experiments. (C) Intracellular calcium mobilization dose-response curves for E2-stimulated COS7 cells expressing ERα-GFP (▲) or ERβ-GFP (■), treated with the indicated concentrations of AB-1. Data indicate mean ± s.e.m. of three independent experiments. (D) AB-1 antagonism of PI3K activation through ERα and ERβ. COS7 cells co-expressing PH-mRFP1 and either ERα-GFP (left panel), ERβ-GFP (middle panel) or GPER-GFP (right panel) were stimulated with vehicle (DMSO), 1 nM E2, 1 μM AB-1 or a combination of E2 + AB-1. PI3K activation was assessed by the translocation of the PH-mRFP1 reporter from the cytoplasm to the nucleus as exemplified by E2 treatment of ERα and ERβ-expressing cells. White scale bar represents 10 μm. Confocal images are representative of three independent experiments.
Despite E2-dependent PI3K/Akt activation in MCF-7 cells being mediated by GPER, we have previously shown that E2 can mediate PI3K activation by both classical estrogen receptors (ERα and ERβ) and GPER in transfected COS7 cells (Revankar et al., 2005). Thus, to determine whether the inhibitory effect of AB-1 on rapid calcium signaling extends to other rapid signaling pathways, we next examined whether AB-1 could regulate PI3K activation in COS7 cells transfected with either ERα-GFP, ERβ-GFP or GPER-GFP. Cells were cotransfected with the PH-mRFP1 reporter, which contains the PIP3-binding pleckstrin homology (PH) domain of Akt fused to a red fluorescent protein and thus translocates to sites of PI3K activity and PIP3 accumulation (Revankar et al., 2005). Employing this system, we observed that E2 induced strong nuclear localization of the PH-mRFP1 reporter in COS7 cells expressing ERα, ERβ or GPER (Figure 5D, second row), indicative of PI3K activation, as previously reported (Revankar et al., 2005). However, unlike E2, AB-1 did not induce nuclear translocation of the PH-mRFP1 reporter in COS7 cells expressing ERα, ERβ or GPER (Figure 5D, third row). Furthermore, AB-1 was again able to block E2-mediated signaling via ERα and ERβ, but not through GPER (Figure 5D, bottom row). Together, the calcium and PI3K signaling results not only further confirm the binding selectivity of AB-1 for ERα and ERβ vs. GPER, but more importantly and surprisingly, they reveal that AB-1 acts as an antagonist of rapid signaling via the classical estrogen receptors ERα and ERβ.
In vivo assessment of compound estrogenicity has traditionally been carried out employing the uterotrophic assay, based on highly E2-dependent actions in the uterus. Upon E2 depletion in mice, typically through ovariectomy, the uterus regresses with the epithelium entering a non-proliferative state and the uterine losing electrolytes and water, resulting in substantial weight reduction. Treatment with E2 for 1–3 days leads to an acute stimulation of proliferation within the uterine epithelium and an increase in overall weight due to water uptake, termed imbibition. To investigate the estrogenic effects of AB-1 in vivo, we evaluated the uterotrophic effects of AB-1 compared to E2. Whereas E2 yielded a strong imbibition response at a dose of 10 ng (with an EC50 estimated between 2 and 10 ng), AB-1 displayed imbibition only at a dose of 91 μg (with an almost 2-fold increase in uterine wet weight over that of sham-treated mice), with no effect at doses of 2 and 10 μg, suggesting an EC50 in the 50–90 μg range (Figure 6A). We also examined the effect of AB-1 on the proliferative response of uterine epithelial cells in the same mice used for the uterotrophic assay. AB-1, at the highest dose tested, induced an almost 12-fold increase in epithelial proliferation (measured as Ki-67 positive staining) vs. sham-treated mice, similar to the response observed with 10 ng E2 (Figure 6B). Together, these results demonstrate that AB-1 stimulates multiple murine uterine effects associated with the activities of ERα, though with less potency compared to E2.
Figure 6.

Estrogenic effects of AB-1 in the mouse uterus. (A) Ligand-induced effect on mouse uterine weight. Ovariectomized mice were treated with vehicle (sham) or the indicated amounts of E2 or AB-1 for 18 h and body weights and uterine wet weights determined. Uterine weights are shown as ratios to total body weights (mean ± s.e.m.). (B) Uterine epithelial cell proliferation. Fixed uterine sections from samples in (A) were assessed for epithelial cell proliferation by staining for Ki-67 expression. Data are the mean ± s.e.m. of 3 mice per group; *p<0.05 vs. sham by one-way ANOVA with Bonferroni post-hoc test.
DISCUSSION
Our understanding of E2 signaling has evolved over the last half century, from the earliest cellular studies of rapid signaling responses (Pietras and Szego, 1975; Szego and Davis, 1967), to the subsequent appreciation of its transcriptional regulation through ERα and later ERβ. With the discovery of GPR30 as an additional estrogen receptor (leading to its designation as GPER) that mediates many of the rapid signaling events in response to E2, the landscape of E2 signaling mediators became more complicated. Pharmacological approaches have traditionally been critical in unravelling the roles of individual receptor subtypes within a family. In the case of the classical ERs and GPER, this approach has been complicated by the high degree of overlap in ligand specificity (Dahlman-Wright et al., 2006). Not only are the ligand binding pockets of ERα and ERβ highly homologous, but to date all tested ER-binding compounds exhibit binding and/or activity towards GPER (Prossnitz and Arterburn, 2015). This is particularly true of the family of SERMs and SERDs, which despite generally inhibiting activity of the classical ERs, act as agonists of GPER. Studies of GPER were facilitated with the identification of the highly selective GPER agonist G-1 (Bologa et al., 2006) and soon thereafter GPER antagonists (G15 and G36) (Dennis et al., 2009; Dennis et al., 2011), all of which exhibit little to no activity towards the classical ERs. Unfortunately, compounds with the inverse selectivity, i.e. binding to ERs but not GPER, have to date not been identified. In this report, we described the identification of the first such compound AB-1, that binds with high affinity to both ERα (and ERβ) but not to GPER, defining AB-1 as a SLERP.
Pharmacological selectivity between the two classical estrogen receptors (ERα and ERβ) has been difficult to achieve, largely due to the extremely high sequence and structural conservation of the ligand binding pockets of these two receptors. Following decades of optimization, the most highly ERα-selective compound PPT (PPT) exhibits only about 400-fold selectivity for ERα over ERβ (Stauffer et al., 2000). Despite this, PPT has been shown to lack selectivity for ERα against GPER, where it acts as an agonist (Petrie et al., 2013). Thus, based on the fact that to date all tested ERα ligands bind to or activate GPER (Prossnitz and Arterburn, 2015), one might speculate that achieving ERα selectivity vs. GPER might be extremely difficult. This is in contrast to the high selectivity (>105 fold) of the GPER-selective agonist G-1 for GPER over ERα (Dennis et al., 2011), which is believed to be due to the fact that G-1 is slightly larger than E2 (Bologa et al., 2006), precluding its occupancy of the ligand binding pocket of ERα or ERβ while allowing its binding to the presumably slightly larger or conformationally more accommodating ligand binding pocket of GPER.
Although “bulky” bicyclic compounds may seem like a poor substitute for the planar E2 molecule, the ability of bicyclic compounds, such as bicyclo[3.3.1]nonanes, to function as ER ligands was reported by Katzenellenbogen in 2003 (Muthyala et al., 2003). Compounds of the oxabicyclo[3.3.1]nonene structural class were first identified as ER ligands through screening campaigns carried out by multiple independent groups in the mid-2000s. In 2003, Sibley et al. at Bayer AG, identified AB-1 (termed compound 2) in a primary screen as an ER ligand (Sibley et al., 2003). In 2005, Hamann et al. at Ligand Pharmaceutical again identified AB-1 (compound 3) in a primary screen (Hamann et al., 2005) and in 2006 Hsieh et al. reported the characterization of AB-1 (termed OBCP-1M) identified from a high-throughput functional screen of the ChemBridge 10,000-compound chemical library (San Diego, CA) (Hsieh et al., 2006). Thus, the inclusion of compounds with the oxabicyclo[3.3.1]nonene scaffold has been a recurring occurrence in the design of chemical libraries, perhaps due to its structural rigidity. Interestingly, our virtual screen for similarity to E2 ranked AB-1 as 53rd, whereas G-1 was ranked as 92nd of the 10,000 compounds in our GPCR-optimized library. Thus, both compounds fell within the top 1% of the library in terms of E2 “similarity”, despite that fact that they display inverse properties with respect to ER and GPER selectivity.
Pharmacological cross-reactivity of NHR ligands is not uncommon, due to a high degree of structural similarities between the various NHR LBDs as well as their ligands (Carson-Jurica et al., 1990; Gao et al., 2005). For example, progesterone, the natural ligand of PR, as well as many synthetic progestins (commonly used in oral contraceptives), binds to MR and other NHRs, leading to unwanted side-effects (Madauss et al., 2007; Oelkers, 1996). Despite these examples of cross-reactivity among NHRs, AB-1 shows no significant binding towards other NHRs, selectivity that could prove to be beneficial for future therapeutic development.
The previous three reports identifying AB-1 as an ER ligand were published prior to the wide acceptance of GPER as an E2 receptor; as a consequence, no evaluation of GPER selectivity, either in terms of binding or function, was performed. Furthermore, none of the reports examined rapid signaling mechanisms such as those observed for E2. Selectivity of ERα vs. ERβ was however examined. Hamann et al, reported, based on transcriptional reporter assays, a 2-fold difference in EC50 of racemic AB-1, favoring ERβ over ERα (Hamann et al., 2005). Whereas the (+) and (−) isomer displayed similar EC50 values for ERβ, similar to the (−) isomer for ERα, the (+) isomer displayed a 20-fold worse EC50 for ERα. Hsieh et al. also observed a selectivity for ERβ employing racemic AB-1, both in terms of binding to purified ER ligand binding domain (~10-fold selectivity) and function (transcriptional reporter assays, ~60-fold) (Hsieh et al., 2006). Interestingly, in permeabilized whole cell ligand binding assays, we observed comparable binding of AB-1 to ERα and ERβ (Table I).
Our results demonstrated a high correlation between the gene expression profiles of E2 and AB-1 in MCF-7 cells. Given the lack of rapid signaling observed for AB-1, this would suggest that rapid signaling has a minimal overall impact on ERα-mediated transcriptional activity of the majority of genes. There were however approximately 4–5% of genes that exhibited lower regulation (less activation or less repression by 50% or more) by AB-1 as compared to E2, indicating a contribution of rapid signaling to “maximal” transcription regulation (defined as that induced by E2). A study employing an E2-dendrimer conjugate (that cannot cross the plasma membrane) that activates rapid (non-genomic) signaling pathways, but not nuclear ER-mediated transcriptional (genomic) pathways revealed that approximately 25% of E2-regulated genes were E2-dendrimer responsive (Madak-Erdogan et al., 2008). Although this result indicates that rapid signaling alone can recapitulate a portion of E2-regulated transcription, it does not imply the converse, that the same genes require rapid signaling. Downregulation of ERK2 (via siRNA) has also been shown to alter the gene expression profile of E2 in MCF-7 cells (Madak-Erdogan et al., 2011), suggesting a role for MAPK signaling in transcriptional activity of ERα. Interestingly, there were also unique genes that were only regulated by E2 in the presence of ERK1/2 knockdown. Overall, these results indicate the extreme complexity of E2-mediated transcriptional regulation. In our gene expression study, MCF-7 cells were deprived of E2 for a total of 4 days prior to stimulation with either E2 or AB-1 for 24 hours. Under these conditions, basal levels of ERK2 activity are expected to be decreased but perhaps not to the same extent as in the presence of ERK2 knockdown, suggesting that basal ERK2 activity may be sufficient to support E2-mediated regulation of transcription. Finally, the overall high concordance between E2- and AB-1-mediated transcriptional regulation suggests that the conformation of ERα induced by AB-1 is very similar to that of E2, resulting in the similar recruitment of co-activators and corepressors.
The ability of E2 to mediate rapid (i.e. non-genomic) signaling has been known for over 50 years, from early studies of E2-mediated cAMP production and calcium (45Ca) mobilization (Pietras and Szego, 1975; Szego and Davis, 1967), to the resurgence of interest in such pathways in the 1990s (Wehling, 1994, 1997). Multiple approaches have been employed over the years to investigate mechanisms of rapid E2-mediated signaling, including the generation of mutant forms of ERα (e.g. membrane- or nuclear-targeted forms of the receptor) (Levin and Hammes, 2016) and pharmacological approaches employing novel ligands, such as E2-dendrimers (Harrington et al., 2006) and small molecule pathway preferential estrogens (PaPEs) that exhibit exceptionally low affinity for ERα, purportedly resulting in the activation of non-genomic signaling but not transcriptional activity (Madak-Erdogan et al., 2016). The advent of GPER-selective ligands further enhanced our understanding of rapid E2-mediated signaling events in multiple cell types and tissues by selectively activating or inhibiting GPER in the absence of ER activity (Prossnitz and Arterburn, 2015). Now, for the first time, we have identified a truly ER-selective compound that displays no binding affinity or activity towards GPER, enabling studies of ER-specific activities in the absence of GPER signaling. Furthermore, the selective profile of AB-1 with respect to ER activity, activating transcription while precluding ER-mediated rapid signaling, provides additional selectivity that will further our understanding of ER function. It should be noted that we only examined two aspects of ER-specific rapid signaling, namely calcium mobilization and PI3K activation, limiting our conclusions to these pathways. Because the mechanisms of ER-mediated signaling are in general poorly understood, it is possible that other aspects of rapid signaling may be preserved. Nevertheless, the ability of AB-1 to regulate ER-mediated gene expression in a highly similar manner to E2, while having no effect on ER-mediated signaling (thus acting as an antagonist of these pathways), represents a previously unidentified pharmacological profile, analogous to the tissue-selective activities of SERMs (Komm and Mirkin, 2014) and the pathway-specific actions of biased agonists of GPCRs (Wacker et al., 2017).
There has been mounting evidence that GPER expression and activation by currently employed anti-estrogens, particularly tamoxifen, play an important role in resistance to these drugs, as suggested by the poor prognosis of breast cancer patients treated only with tamoxifen (Ignatov et al., 2011), increased GPER expression in breast cancer patient biopsies following tamoxifen treatment (Ignatov et al., 2011), enhanced GPER signaling in tamoxifen-resistant MCF-7 cells (Ignatov et al., 2010), inhibition of tamoxifen-resistant breast cancer cell growth by GPER antagonists (Mo et al., 2013) and improved survival of MCF-7 cells in the presence of G-1 (Zekas and Prossnitz, 2015). Based on such results, the development of highly ER-selective antagonists that lack GPER cross-reactivity could be of significant clinical benefit, lowering the occurrence of resistance seen with current antiestrogen therapies (Pepermans and Prossnitz, 2019).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
The lead contact is Dr. Eric R. Prossnitz (eprossnitz@salud.unm.edu). All requests for materials, reagents and resources should be directed to the lead contact. AB-1 generated in this study will be made available on request but may require a payment and/or a completed Materials Transfer Agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mouse Strains
C57BL/6NHsd mice were obtained from Harlan Laboratories (now Envigo).
Cell Lines
COS7 and MCF-7 cells (obtained from the American Type Culture Collection, ATCC) were cultured in Dulbelcco’s Modified Eagle’s Medium (DMEM), supplemented with 10% FBS, 2 mM L-glutamine, 100 units/mL penicillin and 100 ug/mL streptomycin and maintained in a standard tissue culture incubator at 37°C in 5% CO2. MCF-7/WS8 cells, provided by Craig Jordan (MD Anderson), were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, non-essential amino acids, antibiotic/antimycotic (Gibco) and 6 ng/ml of insulin. MCF-7 ERE-GFP cells (Yamaguchi et al., 2005) were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 units/mL penicillin, 100 ug/mL streptomycin and 1 mg/mL G418.
METHODS DETAILS
Cell Transfection
Transient transfection experiments were performed 24 h after seeding cells using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. The expression plasmids have been previously described (Revankar et al., 2005). For E2 deprivation, cells were grown for 24–48 h (with intermediate changes of medium) in phenol red-free medium lacking serum or supplemented with 10% charcoal-stripped FBS, both of which were further supplemented with 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin.
Virtual Screening
A database containing structures of 10,000 molecules (CDLDB) provided by Chemical Diversity Labs Inc (San Diego, CA), to which 17β-estradiol was added, was processed as described previously (Olah et al., 2004). Briefly, using 17β-estradiol as reference point, 2D-based similarity coefficients were computed employing both Daylight and MDL fingerprints using Tanimoto’s symmetric distance-between-patterns (Tanimoto, 1961) and Tversky’s asymmetric contrast model (Tversky, 1977). We also obtained 3D shape similarity coefficients using the Tanimoto (Tanimoto, 1961) and Tversky (Tversky, 1977) formulae using Rapid Overlay of Chemical Structures (Grant et al., 2001). An additional pharmacophore-based 3D similarity metric was derived from ALMOND descriptors (Pastor et al., 2000). The combined similarity score attributed 40% weighting to 2D fingerprints, 40% to the shape-based similarities and 20% to pharmacophore-based similarity. Given this composite score, the top 100 ranked molecules were selected for physical screening employing a fluorescent whole cell ligand-binding assay.
Chemical Synthesis
G-1 was synthesized as previously described (Burai et al., 2010). The compound AB-1 (4-(5-(hydroxymethyl)-8-methyl-3-oxabicyclo[3.3.1]non-7-en-2-yl))-phenol) has been reported previously (Hamann et al., 2005; Sibley et al., 2003), and was synthesized by a modified procedure (Nakamura et al., 2009) and obtained as a diastereomerically pure, racemic mixture of enantiomers. All compounds were synthesized in an efficient fume-hood. All other commercially available solvents and reagents were purchased and used without further purification. Compound identity was verified by comparison of high field 1H NMR (500 MHz) spectra to published values (Hamann et al., 2005), and purity was demonstrated by quantitative analytical HPLC chromatography to be >98%. Preparative chromatography was performed by medium pressure column chromatography using AnaLogix SuperFlash pre-packed columns. 1H NMR spectra were acquired using Varian Oxford 300 MHz, Varian Unity 400 MHz, and 500 MHz spectrometers and 13C NMR were acquired using Varian Oxford 75 MHz, Varian Unity 100 MHz and 125 MHz spectrometers at ambient temperatures (20±2 °C). 1H NMR spectra in CDCl3 and acetone-d6 were referred to TMS. Mass spectra were obtained using an Orbitrap Fusion Mass Spectrometer (Thermo Fisher, San Jose, CA) acquired with funding from NSF MRI #1626468. Spectroscopic data confirming the identification and purity of AB-1 are provided in the supplemental information.
Diethyl 4-methylcyclohex-3-ene-1,1-dicarboxylate
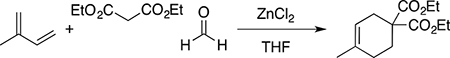
A sealed tube containing a diethylmalonate (0.800 g, 5.0 mmol), paraformaldehyde (0.450 g, 15.0 mmol), 2-methyl-1,3-butadiene (0.408 g, 6.0 mmol) and zinc chloride (0.09 g, 0.66 mmol, 7.5 mol %) in dry tetrahydrofuran (2.5 mL) was stirred at 70 °C for 24 h. The reaction mixture was concentrated under reduced pressure, diluted with dichloromethane (45 mL) and washed successively with saturated aqueous NaHCO3, and H2O (25 mL each), dried over Na2SO4, evaporated in vacuo, and purified by silica gel column chromatography eluting with ethyl acetate/hexanes (1: 99) to obtain the pure product as a colorless oil (0.668 g, 57%). 1H NMR (300 MHz, CDCl3) δ 5.37–5.35 (m, 1H), 4.18 (q, J =7.23 Hz, 4H), 2.53–2.51 (m, 2H), 2.16–2.12 (m, 2H), 2.02–1.94 (m, 2H), 1.63 (bs, 3H), 1.23 (t, J = 7.40 Hz, 6H); FT-IR (Neat), 2960, 1731, 1210, 1151, 503 cm−1).
(4-Methylcyclohex-3-ene-1,1-diyl)dimethanol
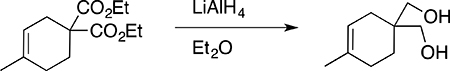
A solution of diethyl 4-methylcyclohex-3-ene-1,1-dicarboxylate (0.68 g, 2.83 mmol) in dry diethylether (5 mL) was added dropwise to a cooled (0°C) suspension of lithium aluminum hydride (0.240 g, 6.32 mmol) in dry diethylether (1 mL) and allowed to warm to ambient temperature with magnetic stirring under a nitrogen atmosphere for 3 h. The reaction mixture was cooled in an ice-bath, and worked up by successive slow addition of water, 10% sodium hydroxide, and three additional portions of water (240 μL each) to yield tractable aluminum salt precipitates that were filtered, and the filtrate was concentrated and dried under vacuum to provide the product (0.327 g, 74 % mp 103–108 °C). 1H NMR (300 MHz, CDCl3) δ 5.30–5.27 (m, 1H), 3.61 (d, J = 5.47 Hz, 4H), 2.13 (t, J = 5.47Hz, 2H), 1.96–1.91 (m, 2H), 1.81–1.77 (m, 2H), 1.66–1.64 (bs, 3H), 1.60 (t, J = 6.64 Hz, 2H); FT-IR (Neat) 3300, 1610, 1518, 1269, 1071 cm−1.
4-(5-(hydroxymethyl)-8-methyl-3-oxabicyclo[3.3.1]non-7-en-2-yl))-phenol [AB-1]
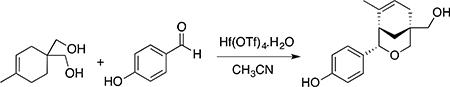
To a solution of the (4-methylcyclohex-3-ene-1,1-diyl)dimethanol (0.161 g, 1.032 mmol) and 4-hydroxybenzaldehyde (0.15 g, 1.23 mmol) in anhydrous acetonitrile (4 mL) was added 5 mol% hafnium(IV) trifluoromethanesulfonate monohydrate (0.040 g, 0.051 mmol). The reaction mixture was stirred at ambient temperature under a nitrogen atmosphere for 18 h. The reaction mixture was quenched with sat. NaHCO3 (10 mL), diluted with water (25 mL) and the product was extracted using CH2Cl2 (3×10 mL), dried over Na2SO4, and evaporated in vacuo. The product was purified by silica gel column chromatography eluted with EtOAc/hexanes (45:55) to isolate the product as white solid (0.23 g, 86%; mp 164–168 °C) (Rf = 0.3). 1H NMR (500 MHz, acetone-d6) δ 8.03 (bs, 1H), 7.10 (d, J = 8.85 Hz, 2H), 6.71 (d, J = 8.85 Hz, 2H), 5.45–5.46 (m, 1H), 4.42 (d, J = 1.83Hz, 1H), 3.83 (dd, J = 10.99, 2.83 Hz, 1H), 3.65 (bs, 1H), 3.53 (d, J = 10.99 Hz, 1H), 3.26 (s, 2H), 2.28–2.26 (m, 1H), 2.22–2.03 (m, 2H), 1.8 (dd, J = 11.6, 2.75 Hz, 1H), 1.65 (m, 1H,), 1.01 (dd, J = 3.97, 2.14 Hz, 3H) (Fig. S1A); 13C NMR (125 MHz, CD3COCD3) δ 157.03, 134.54, 134.16, 127.43, 124.75, 115.36, 80.51, 78.74, 69.90, 44.05, 35.62, 34.85, 34.68, 30.67, 24.4 (Fig. S1B); FT-IR (Neat) 3300, 2975, 1610, 1092, 1051 cm−1. HRMS (m/z) calcd for C16H21O3, 261.1485 [M+ H+]; found, 261.1484 (Fig. S2). The UV absorbance peak areas in the HPLC chromatogram of the AB-1 sample (Fig. S3) were integrated and demonstrated compound purity of 98.6%. Compound identity was verified by comparison of high field 1H NMR (500 MHz) spectra to published values (Hamann et al., 2005), and purity was demonstrated by quantitative analytical HPLC chromatography to be >98%. Full experimental details and spectroscopic data confirming the identification and purity of AB-1 are provided in the supporting information.
Ligand-Binding Assays
Binding assays for ERα ERβ and GPER were performed as previously described (Revankar et al., 2005). Briefly, COS7 cells were transiently transfected with ERα-GFP, ERβ-GFP (Matsuda et al., 2002) or GPER-GFP (Revankar et al., 2005). Following serum starvation for 24 h, cells (~5×104) were incubated with competitor for 20 min prior to addition of an equal volume of 4 nM E2-Alexa633 in saponin-based permeabilization buffer. Following 10 min at 25 °C, cells were washed once with PBS/2%BSA. For flow cytometric analysis, cells were resuspended in 20 μL and 2 μL samples were analyzed on a DAKO Cyan flow cytometer using HyperCyt™ as described (Edwards et al., 2009). For confocal microscopy, cells were stained as above and fixed with 2% PFA in PBS containing 1 mM CaCl2 and 1 mM MgCl2 for 15 min, washed, mounted in Vectashield and analyzed immediately by confocal microscopy using a Zeiss LSM510 confocal fluorescent microscope.
Competitive Radio-Ligand Binding Assays
Competitive radio-ligand binding assays were performed using the NHR Binding Agonist Radioligand Assay (Eurofins) by Eurofins Panlabs Discovery Services. AB-1 selectivity (at 1 μM and 10 μM) was assessed in the presence of [3H]-methyltrienolone (0.5 nM), [3H]-aldosterone (0.4 nM), [3H]-progesterone (0.5 nM), [3H]-dexamethasone (5 nM), [3H]-estradiol (0.5 nM) for AR, MCR, PR-B, GR and ERα, respectively. Control inhibitors for AR, MCR, PR-B, GR and ERα were testosterone (2.1 nM), aldosterone (0.64 nM), promegestone (0.49 nM), dexamethasone (3.8 nM) and diethylstilbestrol (0.77 nM), respectively.
TR-FRET ligand-binding assay
Binding assays for ERα-LBD and ERβ-LBD were performed using the LanthaScreen TR-FRET Competitive Binding Assay by the SelectScreen Biochemical Nuclear Receptor Profiling Service (ThermoFisher Scientific). AB-1 was tested at 300 nM with subsequent 3-fold serial dilutions.
Intracellular Calcium Mobilization
COS7 cells transfected with ERα-GFP, ERβ-GFP or GPER-GFP (5 × 106 cells) were incubated at room temperature in HBSS containing 5 μM indo1-AM and 0.05% pluronic acid for 30 min. Cells were then washed once with HBSS and resuspended in HBSS at a density of 107 cells/mL. Ca++ mobilization was determined ratiometrically using λex 340 nm and λem 400/490 nm at 37°C in a spectrofluorometer (QM-2000–2, Photon Technology International) equipped with a magnetic stirrer and heated sample chamber. The relative 490nm/400nm ratio is plotted as a function of time.
PI3K Activation
The PIP3-binding domain of Akt fused to mRFP1 (PH-mRFP1) was employed to assess cellular PIP3 production and localization as described (Revankar et al., 2005). Briefly, COS7 cells (co-transfected with PH-mRFP1 and either ERα-GFP, ERβ-GFP or GPER-GFP) were plated on coverslips and serum starved for 24 h followed by stimulation with ligands as indicated for 15 min. The cells were fixed with 2% PFA in PBS, washed, mounted in Vectashield and analyzed by confocal microscopy using a Zeiss LSM510 confocal fluorescent microscope.
ER-ERE Transcription
ERα activity via EREs was determined using MCF-7 cells stably transfected with an ERE-GFP reporter construct (Yamaguchi et al., 2005) as previously described (Dennis et al., 2011). Briefly, cells were deprived of E2 for 4 days (with one intermediate medium change) in phenol red-free DMEM/F12 supplemented with 10% charcoal-stripped FBS. Cells (~80,000) were seeded in 24 well plates, and 24 hours later treated with the indicated compounds (dissolved in DMSO, 0.1% final) for 24 hours in triplicate, trypsinized, washed and analyzed for green fluorescence by flow cytometry. Mean fluorescence intensities of gated live cells were determined and normalized to E2 values following subtraction of vehicle control values.
Gene Expression Analysis
MCF-7/WS8 cells, provided by Craig Jordan (MD Anderson), were cultured in RPMI supplemented with 10% FBS, 2 mM L-glutamine, non-essential amino acids, antibiotic/antimycotic and 6 ng/ml of insulin. E2 depletion was carried out by culturing cells in E2-depleted medium with daily medium changes for three days. Cells were seeded sparsely (2×106 cells per 15 cm dish) in E2-depleted medium and treated the following day with 1 nM E2, 1 μM AB-1 or DMSO (vehicle control) for 24 hours. Final DMSO concentrations were 0.01%. Total RNA was isolated using QIAGEN RNeasy minikits following homogenization using QIAshredders and employing the direct lysis protocol for cell monolayers. Total RNA (500ng) was reverse transcribed using a T7 Oligo(dT) primer, followed by second strand synthesis and purification of the double stranded cDNA. In vitro transcription was performed on this product using a mix of biotinylated nucleotides to generate biotin labeled cRNA as described (Ambion/Applied Biosystems Illumina Total Prep RNA Amplification Kit). cRNA samples were hybridized to the BeadChip array, washed, stained with C3-strepavidin following the manufacturer’s protocols (Illumina). The BeadChip was scanned and data analyzed using the Genome Studio Gene Expression Module (Illumina). Samples were normalized using a rank invariant normalization. Missing data were imputed, and Benjamini and Hochberg false discovery rate calculations were applied. The DMSO controls were used as reference samples and the Illumina custom error model was employed.
Cell Proliferation
MCF-7 cells were grown in E2-depleted medium for 4 days (with one intermediate medium change) in phenol red free DMEM/F12 supplemented with 10% charcoal-stripped FBS. Cells were seeded in 96 well plates at low density, and 24 hours later treated with the indicated concentrations of compounds (dissolved in DMSO, 0.1% final) for 3–5 days in triplicate. Cell growth was determined by Alamar Blue staining.
ERα Degradation
MCF-7 cells were seeded (500,000 cells/well) in 6-well plates in complete culture medium. The following day, cells were transferred to medium containing charcoal-stripped serum for 48 h (with one intermediate change of medium) and subsequently treated with the indicated compounds (0.01% DMSO final) for 24 h. Cells were washed once with ice-cold PBS, lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 1% Na-deoxycholate and 0.1% SDS) containing 50 mM NaF, 1 mM Na3VO4 and protease cocktail (1x) and passed through a 20G needle (10–16 times). Lysates were cleared by centrifugation (13,000 rpm for 15 min at 4˚C) and protein concentrations determined using the Pierce™ BCA Protein Assay Kit. Samples (20 ug) were resolved by SDS-PAGE (4–12% Bis-Tris gel), transferred to nitrocellulose membranes and subjected to Western blot analysis. Membranes were probed overnight with a rabbit anti-ERα antibody (Cell Signaling, 1:1000) in 4% BSA-TBST at 4˚C followed by a secondary HRP-linked goat anti-rabbit antibody (1:5000) for 1 h at RT. Bands were visualized by chemiluminescence. To detect actin, membranes were stripped (30 min at RT) and probed with a mouse anti-actin antibody (Millipore, 1:5000) for 1 h at RT followed by a secondary HRP-linked goat anti-mouse antibody (1:2500) for 1 h at RT. Bands were quantified using ImageJ software (NIH).
FOXO3a Translocation
FOXO3a localization assays were performed as described (Zekas and Prossnitz, 2015). Briefly, MCF-7 cells were seeded on 12 mm coverslips in a 24-well plate one day before transfection. Cells were transfected with 0.3 μg FOXO3a-GFP plasmid (Jacobs et al., 2003) using the Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s protocol. Twenty-four hours post-transfection, cells were serum starved for 24 h prior to treatment. Cells were fixed in 2 % PFA, washed with PBS and mounted in Vectashield on coverslips. Coverslips were imaged on a Zeiss LSM800 microscope and localization determined from 10 fields per condition.
Mouse Uterine Estrogenicity
C57Bl6 female mice (Harlan) were ovariectomized at 10 weeks of age. E2 and AB-1 were dissolved in absolute ethanol at 1 mg/mL and diluted in ethanol. For treatment, 10 μL of diluted E2 or AB-1 was added to 90 μL aqueous vehicle (0.9% NaCl with 0.1% albumin and 0.1% Tween-20). Ethanol alone (10 μL) was added to 90 μL aqueous vehicle as control (sham). Twelve days post-ovariectomy, mice were injected subcutaneously at 5:00 pm with 100 μL sham, E2 or AB-1. Eighteen hours after injection, mice were killed, weighed and uteri removed and weighed (normalizing to body weight) after the mesometrium and any attached adipose tissue was trimmed away. Uteri were then fixed in 4% paraformaldehyde, and embedded in paraffin. Five-micron sections were placed on slides, and proliferation in uterine epithelia was quantitated by immunofluorescence using anti-Ki-67 antibody (LabVision) followed by goat anti-mouse IgG conjugated to Alexa488 (Invitrogen). Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). At least 4 animals per treatment were analyzed, and the Ki-67 immunodetection was repeated three times per mouse. Percent Ki-67 positive cells = (number of Ki-67 positive cells / total number of DAPI-stained luminal epithelial cells) x 100 for three different fields per sample.
QUANTIFICATION AND STATISTICAL ANALYSES
Data were quantified as described above and analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test, by two-tailed, unpaired Student’s t-test or by one-sample t-test as appropriate. Non-linear regression curves were determined using a variable slope fit. Values are expressed as mean ± s.e.m.; n equals the number of assay replicates or animals used. Differences were considered to be significant when P < 0.05. All analyses were carried out using Prism versions 5–7 for Macintosh, GraphPad Software.
DATA AVAILABILITY
Raw and processed data from the gene expression analysis (Figure 3B) are available upon request to the Lead Contact (eprossnitz@salud.unm.edu).
Supplementary Material
SIGNIFICANCE.
Cross-activation of G protein-coupled estrogen receptor (GPER) by estrogen receptor (ER)-targeted therapeutic antagonists, such as tamoxifen and fulvestrant, has been implicated in the development of endocrine resistance in breast cancer. To date, truly ER-selective ligands lacking such GPER cross-reactivity have not been identified. Here, Revankar et al. report the identification and characterization of a small ligand, termed AB-1, that binds with high selectivity to ERα/β over GPER. Although AB-1 acts as an agonist of transcription through ERα, the unique selectivity profile of AB-1 provides new opportunities for the future development of next-generation ER-targeted antagonists that truly lack GPER cross-reactivity, thereby decreasing or delaying the development of endocrine resistance in breast cancer patients.
Highlights.
Identification of first ER-selective ligand (AB-1) that lacks GPER cross-reactivity
AB-1 binds with high affinity to ERα and ERβ, but not to GPER
GPER-mediated signaling pathways are not activated by AB-1
AB-1 initiates ER-mediated transcription but not rapid signaling by ER
ACKNOWLEDGMENTS
We thank Chelin Hu and Daniel Cimino for expert technical assistance, V. Craig Jordan for insightful discussions and Alexander Ivachtchenko for initial synthesis of AB-1. The authors were supported by NIH R01 grants CA127731, CA163890 and CA194496 (E.R.P), the Cowboys for Cancer Research Foundation, the Center of Biomedical Research Excellence in Autophagy, Inflammation and Metabolism (P20 GM121176) and the University of New Mexico Comprehensive Cancer Center (P30 CA118100), which provided Development Pilot and Bridging Funds and supports the Analytical & Translational Genomics, Flow Cytometry & Single Cell Analytics, Fluorescence Microscopy & Cell Imaging, and Animal Models Shared Resources.
Footnotes
DECLARATION OF INTERESTS
E.R.P., T.I.O., C.G.B., J.B.A., L.A.S. and C.R. are inventors on U.S. patents 7,875,721 and 8,487,100 for GPER-selective ligands and imaging agents. E.R.P., C.G.B., and J.B.A. are inventors of a pending patent for the matter in this publication and related compounds.
ADDITIONAL INFORMATION
Supplemental and chemical compound information are available with the online version of the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arnal JF, Gourdy P, and Lenfant F (2013). In vivo dissection of the estrogen receptor alpha: uncoupling of its physiological effects and medical perspectives. Ann Endocrinol (Paris) 74, 82–89. [DOI] [PubMed] [Google Scholar]
- Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M, and Prossnitz ER (2018). Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J Steroid Biochem Mol Biol 176, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton M, Meyer MR, and Prossnitz ER (2019). Nox1 Downregulators: A New Class of Therapeutics. Steroids, 108494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton M, and Prossnitz ER (2015). Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol Metab 26, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, Dennis MK, Prossnitz ER, Karpus WJ, and Horuk R (2009). Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol 214, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, et al. (2006). Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol 2, 207–212. [DOI] [PubMed] [Google Scholar]
- Bopassa JC, Eghbali M, Toro L, and Stefani E (2010). A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 298, H16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burai R, Ramesh C, Shorty M, Curpan R, Bologa C, Sklar LA, Oprea T, Prossnitz ER, and Arterburn JB (2010). Highly efficient synthesis and characterization of the GPR30-selective agonist G-1 and related tetrahydroquinoline analogs. Org Biomol Chem 8, 2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson-Jurica MA, Schrader WT, and O’Malley BW (1990). Steroid receptor family: structure and functions. Endocr Rev 11, 201–220. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li Z, He Y, Shang D, Pan J, Wang H, Chen H, Zhu Z, Wan L, and Wang X (2014). Estrogen and pure antiestrogen fulvestrant (ICI 182 780) augment cell-matrigel adhesion of MCF-7 breast cancer cells through a novel G protein coupled estrogen receptor (GPR30)-to-calpain signaling axis. Toxicol Appl Pharmacol 275, 176–181. [DOI] [PubMed] [Google Scholar]
- Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, and Gustafsson JA (2006). International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev 58, 773–781. [DOI] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, et al. (2009). In vivo effects of a GPR30 antagonist. Nat Chem Biol 5, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, et al. (2011). Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol 127, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BS, Young SM, Ivnitsky-Steele I, Ye RD, Prossnitz ER, and Sklar LA (2009). High-content screening: flow cytometry analysis. Methods Mol Biol 486, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DP (2005). Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol 67, 335–376. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, and Frackelton AR Jr. (2000). Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14, 1649–1660. [DOI] [PubMed] [Google Scholar]
- Gao W, Bohl CE, and Dalton JT (2005). Chemistry and structural biology of androgen receptor. Chem Rev 105, 3352–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant JA, Pickup BT, and Nicholls A (2001). A smooth permittivity function for Poisson-Boltmann solvation methods. J. Comput. Chem, 608–640. [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, et al. (2009). Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res 104, 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann LG, Meyer JH, Ruppar DA, Marschke KB, Lopez FJ, Allegretto EA, and Karanewsky DS (2005). Structure-activity relationships and sub-type selectivity in an oxabicyclic estrogen receptor alpha/beta agonist scaffold. Bioorg Med Chem Lett 15, 1463–1466. [DOI] [PubMed] [Google Scholar]
- Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, and Katzenellenbogen BS (2006). Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20, 491–502. [DOI] [PubMed] [Google Scholar]
- Hofmeister MV, Damkier HH, Christensen BM, Olde B, Fredrik Leeb-Lundberg LM, Fenton RA, Praetorius HA, and Praetorius J (2012). 17beta-Estradiol induces nongenomic effects in renal intercalated cells through G protein-coupled estrogen receptor 1. Am J Physiol Renal Physiol 302, F358–368. [DOI] [PubMed] [Google Scholar]
- Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, Mrksich M, and Greene GL (2006). Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J Biol Chem 281, 17909–17919. [DOI] [PubMed] [Google Scholar]
- Hu R, Sun H, Zhang Q, Chen J, Wu N, Meng H, Cui G, Hu S, Li F, Lin J, et al. (2012). G-protein coupled estrogen receptor 1 mediated estrogenic neuroprotection against spinal cord injury. Crit Care Med 40, 3230–3237. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Ignatov T, Roessner A, Costa SD, and Kalinski T (2010). Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat 123, 87–96. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, Roessner A, Costa SD, and Kalinski T (2011). G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat 128, 457–466. [DOI] [PubMed] [Google Scholar]
- Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, and Smidt MP (2003). FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem 278, 35959–35967. [DOI] [PubMed] [Google Scholar]
- Komm BS, and Mirkin S (2014). An overview of current and emerging SERMs. J Steroid Biochem Mol Biol 143, 207–222. [DOI] [PubMed] [Google Scholar]
- Kosaka Y, Quillinan N, Bond C, Traystman R, Hurn P, and Herson P (2012). GPER1/GPR30 activation improves neuronal survival following global cerebral ischemia induced by cardiac arrest in mice. Transl Stroke Res 3, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappano R, Rosano C, Santolla MF, Pupo M, De Francesco EM, De Marco P, Ponassi M, Spallarossa A, Ranise A, and Maggiolini M (2012). Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr Cancer Drug Targets 12, 531–542. [DOI] [PubMed] [Google Scholar]
- Lebesgue D, Traub M, De Butte-Smith M, Chen C, Zukin RS, Kelly MJ, and Etgen AM (2010). Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PLoS One 5, e8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ER, and Hammes SR (2016). Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol 17, 783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, and Chappell MC (2009). Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 150, 3753–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Kilic G, Meyers MS, Navarro G, Wang Y, Oberholzer J, and Mauvais-Jarvis F (2013). Oestrogens improve human pancreatic islet transplantation in a mouse model of insulin deficient diabetes. Diabetologia 56, 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, Korach KS, and Mauvais-Jarvis F (2009). Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 58, 2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi G, Zarrilli S, Colao A, Paesano L, Di Somma C, Rossi F, and De Rosa M (2001). Estrogens and health in males. Mol Cell Endocrinol 178, 51–55. [DOI] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, and Katzenellenbogen BS (2008). Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol 22, 2116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Kim SH, Gong P, Zhao YC, Zhang H, Chambliss KL, Carlson KE, Mayne CG, Shaul PW, Korach KS, et al. (2016). Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal 9, ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Lupien M, Stossi F, Brown M, and Katzenellenbogen BS (2011). Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol 31, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madauss KP, Stewart EL, and Williams SP (2007). The evolution of progesterone receptor ligands. Med Res Rev 27, 374–400. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Ochiai I, Nishi M, and Kawata M (2002). Colocalization and ligand-dependent discrete distribution of the estrogen receptor (ER)alpha and ERbeta. Mol Endocrinol 16, 2215–2230. [DOI] [PubMed] [Google Scholar]
- Maximov PY, Lee TM, and Jordan VC (2013). The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol 8, 135–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, and Wardell SE (2010). The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol 10, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Baretella O, Prossnitz ER, and Barton M (2010). Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology 86, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Fredette NC, Daniel C, Sharma G, Amann K, Arterburn JB, Barton M, and Prossnitz ER (2016). Obligatory role for GPER in cardiovascular aging and disease. Sci Signal 9, ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, Amann K, Arterburn JB, Barton M, and Prossnitz ER (2014). G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep 4, 7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Liu M, Yang F, Luo H, Li Z, Tu G, and Yang G (2013). GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res 15, R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthyala RS, Carlson KE, and Katzenellenbogen JA (2003). Exploration of the bicyclo[3.3.1]nonane system as a template for the development of new ligands for the estrogen receptor. Bioorg Med Chem Lett 13, 4485–4488. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Niiyama K, and Yamakawa T (2009). Versatile method for the synthesis of 4-substituted 6-methyl-3-oxabicyclo[3.3.1]non-6-ene-1-methanol derivatives: Prins-type cyclization reaction catalyzed by hafnium triflate. Tetrahedron Letters 50, 6462–6465. [Google Scholar]
- Oelkers WK (1996). Effects of estrogens and progestogens on the renin-aldosterone system and blood pressure. Steroids 61, 166–171. [DOI] [PubMed] [Google Scholar]
- Olah M, Bologa C, and Oprea TI (2004). An automated PLS search for biologically relevant QSAR descriptors. J Comput Aided Mol Des 18, 437–449. [DOI] [PubMed] [Google Scholar]
- Pastor M, Cruciani G, McLay I, Pickett S, and Clementi S (2000). GRid-INdependent descriptors (GRIND): a novel class of alignment-independent three-dimensional molecular descriptors. J Med Chem 43, 3233–3243. [DOI] [PubMed] [Google Scholar]
- Pepermans RA, and Prossnitz ER (2019). ERα-targeted endocrine therapy, resistance and the role of GPER. Steroids 152, 108493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, Hathaway HJ, and Prossnitz ER (2013). G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int 2013, 472720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras RJ, and Szego CM (1975). Endometrial cell calcium and oestrogen action. Nature 253, 357–359. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER (2012). G protein-coupled estrogen receptor: a new therapeutic target in stroke and traumatic brain/spinal cord injury? Crit Care Med 40, 3323–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER (2018). GPER modulators: Opportunity Nox on the heels of a class Akt. J Steroid Biochem Mol Biol 176, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, and Arterburn JB (2015). International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev 67, 505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, and Barton M (2014). Estrogen biology: New insights into GPER function and clinical opportunities. Mol Cell Endocrinol 389, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, and Hathaway HJ (2015). What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol 153, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupo M, Maggiolini M, and Musti AM (2016). GPER Mediates Non-Genomic Effects of Estrogen. Methods Mol Biol 1366, 471–488. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, and Prossnitz ER (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630. [DOI] [PubMed] [Google Scholar]
- Scaling AL, Prossnitz ER, and Hathaway HJ (2014). GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer 5, 146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Mauvais-Jarvis F, and Prossnitz ER (2018). Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J Steroid Biochem Mol Biol 176, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, and Prossnitz ER (2017). G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. Adv Exp Med Biol 1043, 427–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley R, Hatoum-Mokdad H, Schoenleber R, Musza L, Stirtan W, Marrero D, Carley W, Xiao H, and Dumas J (2003). A novel estrogen receptor ligand template. Bioorg Med Chem Lett 13, 1919–1922. [DOI] [PubMed] [Google Scholar]
- Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, and Katzenellenbogen JA (2000). Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J Med Chem 43, 4934–4947. [DOI] [PubMed] [Google Scholar]
- Szego CM, and Davis JS (1967). Adenosine 3’,5’-monophosphate in rat uterus: acute elevation by estrogen. Proc Natl Acad Sci U S A 58, 1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto TT (1961). Non-linear model for a computer assisted medical diagnostic procedure. Trans. NY Acad. Sci 23, 576–580. [Google Scholar]
- Thomas P, and Dong J (2006). Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol 102, 175–179. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, and Dong J (2005). Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146, 624–632. [DOI] [PubMed] [Google Scholar]
- Tversky A (1977). Features of Similarity. Psychological Review 84, 327–352. [Google Scholar]
- Vergote I, and Abram P (2006). Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Ann Oncol 17, 200–204. [DOI] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, and Roth BL (2017). How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, and Offner H (2009). Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through upregulation of programmed death 1. J Immunol 182, 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, and Groban L (2012). Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res 94, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M (1994). Nongenomic actions of steroid hormones. Trends Endocrinol Metab 5, 347–353. [DOI] [PubMed] [Google Scholar]
- Wehling M (1997). Specific, nongenomic actions of steroid hormones. Annu Rev Physiol 59, 365–393. [DOI] [PubMed] [Google Scholar]
- Wijayaratne AL, and McDonnell DP (2001). The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 276, 35684–35692. [DOI] [PubMed] [Google Scholar]
- Woutersz TB (1991). Benefits of oral contraception: thirty years’ experience. Int J Fertil 36 Suppl 3, 26–31. [PubMed] [Google Scholar]
- Yamaguchi Y, Takei H, Suemasu K, Kobayashi Y, Kurosumi M, Harada N, and Hayashi S (2005). Tumor-stromal interaction through the estrogen-signaling pathway in human breast cancer. Cancer Res 65, 4653–4662. [DOI] [PubMed] [Google Scholar]
- Zekas E, and Prossnitz ER (2015). Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER. BMC Cancer 15, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H, et al. (2010). Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol 184, 4087–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data from the gene expression analysis (Figure 3B) are available upon request to the Lead Contact (eprossnitz@salud.unm.edu).