

Abstract
The opportunistic pathogen Clostridium perfringens possesses the ability to colonize the protective mucin layer in the gastrointestinal tract. To assist this, the C. perfringens genome contains a battery of genes encoding glycoside hydrolases (GHs) that are likely active on mucin glycans, including four genes encoding family 84 GHs: CpGH84A (NagH), CpGH84B (NagI), CpGH84C (NagJ) and CpGH84D (NagK). To probe the potential advantage gained by the expansion of GH84 enzymes in C. perfringens, we undertook the structural and functional characterization of the CpGH84 catalytic modules. Here, we show that these four CpGH84 catalytic modules act as β-N-acetyl-D-glucosaminidases able to hydrolyze N- and O-glycan motifs. CpGH84A and CpGH84D displayed a substrate specificity restricted to terminal β-1,2- and β-1,6-linked N-acetyl-D-glucosamine (GlcNAc). CpGH84B and CpGH84C appear more promiscuous with activity on terminal β-1,2-, β-1,3- and β-1,6-linked GlcNAc; both possess some activity toward β-1,4-linked GlcNAc, but this is dependent upon which monosaccharide it is linked to. Furthermore, all the CpGH84s have different optimum pHs ranging from 5.2 to 7.0. Consistent with their β-N-acetyl-D-glucosaminidase activities, the structures of the four catalytic modules revealed similar folds with a catalytic site including a conserved −1 subsite that binds GlcNAc. However, nonconserved residues in the vicinity of the +1 subsite suggest different accommodation of the sugar preceding the terminal GlcNAc, resulting in subtly different substrate specificities. This structure–function comparison of the four GH84 catalytic modules from C. perfringens reveals their different biochemical properties, which may relate to how they are deployed in the bacterium’s niche in the host.
Keywords: bacterium, carbohydrate, GH84, N-acetyl-glucosaminidase, virulence factor
Introduction
Commonly found on epithelial surfaces, such as those in the respiratory, genitourinary and gastrointestinal tracts, mucins are extensively glycosylated proteins composed of up to 80% carbohydrate (Johansson et al. 2011; McGuckin et al. 2011). These glycoconjugates form a physical barrier to protect the human body from invasion and spread of microbial and viral pathogens (Dhar and McAuley 2019). However, some host adapted bacteria have developed the capacity to degrade protective mucins, such as Clostridium perfringens, which can cause mucosal necrosis in the gastrointenstinal tract (Smedley et al. 2004; Nagahama et al. 2015). Bacteria with this capability possess a variety of carbohydrate-active enzymes (CAZymes), which degrade the glycans comprising the mucin layer or unmask targets for cytosolic toxins (Canard et al. 1994; Flores-Diaz et al. 2005; Sheldon et al. 2006). CAZymes are often considered virulence factors in bacterial pathogens since they are involved in degrading and modifying host glycans, which in turn can influence the progression of infection (Galen et al. 1992; Figura 1997; Shelburne et al. 2008; Hobbs et al. 2018).
A prominent feature associated with C. perfringens virulence is the use of an arsenal of CAZymes, in particular glycoside hydrolases (GHs), to process host glycans. Over 54 open reading frames encoding for putative GHs, falling into 24 known families, have been identified in the C. perfringens genome (Shimizu et al. 2002). One family of GHs that is particularly well represented in this bacterium is the family 84, which comprises exo-β-N-acetyl-D-glucosaminidases (Ficko-Blean and Boraston 2005; Macauley et al. 2005; Sheldon et al. 2006). As exo-β-N-acetyl-D-glucosaminidases, the C. perfringens GH84 enzymes are thought to hydrolyze terminal N-acetyl-glucosamine (GlcNAc) moieties, a sugar found in abundance in both the core and branches of mucin O-linked oligosaccharides as well as in N-glycans (Brockhausen et al. 2009). GH84 enzymes are also found in other commensal and pathogenic bacteria, such as Bacteroides thetaiotaomicron and Streptococcus pyogenes (Dennis et al. 2006; Sheldon et al. 2006). However, unlike these organisms who only possess a single conserved GH84 enzyme each, the C. perfringens (strain ATCC 13124) genome encodes for four GH84 enzymes known as NagH (or μ-toxin), NagI, NagJ and NagK (Shimizu et al. 2002; Myers et al. 2006).
These enzymes have large sizes (110–276 kDa) and extensive modular structures (Figure 1). In keeping with the CAZy classification of GHs (Cantarel et al. 2009), we refer to the catalytic modules of NagH, NagI, NagJ and NagK as CpGH84A, CpGH84B, CpGH84C and CpGH84D, respectively. In addition to the GH84 catalytic modules, all four enzymes contain a combination of ancillary modules, such as family 32 carbohydrate-binding (CBM32s), found-in-various-architectures (FIVAR), fibronectin-like type III (FN3), cohesin (X82) and/or Dockerin (Doc) modules, as well as uncharacterized modules (Figure 1). Like many factors that mediate host–bacterium interactions, these enzymes are predicted to be extracellular. NagI and NagK are predicted to be cell wall attached through LPXTG motifs. However, NagH and NagJ have the capacity to associate tightly via cohesin- and dockerin-domain interaction and are thought to be bound to the bacterial cell surface by the FN3 domain on NagJ.
Fig. 1.
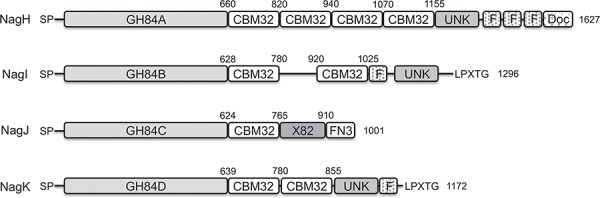
Modular schematic of C. perfringens family 84 GHs. Catalytic modules are shown as GH84A, GH84B GH84C and GH84D in light gray, and carbohydrate binding module 32 as CBM32 in white on the schematic. FIVAR, dockerin, fibronectin-like type III and cohesin modules are shown as F, Doc, FN3 and X82, respectively. SP indicates the presence of a signal peptide. Modules with unknown function (UNK) are also indicated. Module boundaries are shown above each schematic.
NagJ has previously been biochemically and structurally characterized (Rao et al. 2006; Pathak et al. 2008; Ficko-Blean et al. 2009). Its catalytic module, CpGH84C, has been shown to possess β-N-acetyl-D-glucosaminidase activity. The mechanism employed for glycosidic bond hydrolysis is a retaining mechanism using substrate-assistance (Rao et al. 2006). The overall structure of CpGH84C is a classical (α/β)8 fold with a catalytic site similar to that of GH18, GH20 and GH56 families enzymes, which also use a retaining substrate-assisted catalytic mechanism (Markovic-Housley et al. 2000; van Aalten et al. 2001; Pluvinage et al. 2011). NagJ CBM32 binds terminal N-acetyl-D-lactosamine motifs while NagH CBM32-1 binds terminal galactose residues (Ficko-Blean and Boraston 2006, 2009; Grondin et al. 2014). In keeping with typical CBM function (Boraston et al. 2004), these CBM32s are proposed to target the enzymes to specific glycan receptors. The X82 module interacts with Doc modules, such as the NagH Doc module, to form larger enzyme complexes. However, not all X82 modules interact with all Doc modules suggesting other functionalities or selective formation of macromolecular enzyme assemblies (Adams et al. 2008). The FN3 module is proposed to immobilize the enzyme on the bacterial cell surface (Ficko-Blean et al. 2009).
To better understand the expansion of GH family 84 in C. perfringens, we initiated biochemical and structural studies of the catalytic modules from these enzymes. As CpGH84C is well studied, we focused our additional studies on CpGH84A, CpGH84B and CpGH84D, which are uncharacterized. The experimental results reveal that these catalytic modules display various linkage and aglycon specificities toward N- and O-glycan motifs as well as different pH optima. The X-ray crystal structures of these catalytic modules show a very similar overall fold. Insights into CpGH84 catalytic sites show conserved −1 subsites that accommodate a nonreducing terminal GlcNAc residue. However, nonconserved residues at the apex of long loop located in the vicinity of a +1 subsite seem to contribute to the subtly different specificities observed among C. perfringens GH84s.
Results
Activities of GH84s from Clostridium perfringens
The gene fragments encoding CpGH84A, CpGH84B and CpGH84D were cloned, recombinantly expressed and the protein products purified for biochemical characterization; CpGH84C was cloned and produced previously (Ficko-Blean et al. 2009). A standard colorimetric assay using para-nitrophenyl-β-N-acetyl-D-glucopyranoside (pNP-GlcNAc) was used to confirm the N-acetyl-D-glucosaminidase activity of these enzymes, as well as determine their pH optima (Figure 2). The optimum pH obtained for each of the CpGH84s was different, ranging from 5.2 to 7.0. CpGH84C had optimum activity at the lowest pH of 5.2, while CpGH84D was the most active at pH 7.0. The maximum activity observed for CpGH84A and CpGH84B was at pH values of 5.8 and 5.6, respectively. However, CpGH84A displayed a very broad pH profile and retained >50% of its activity over the entire range of pHs tested (Figure 2).
Fig. 2.
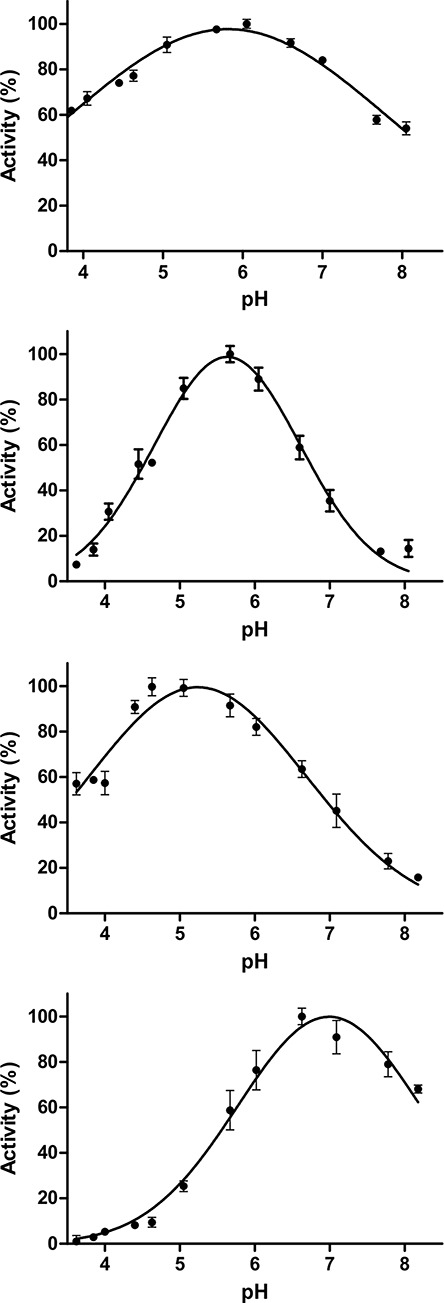
pH optimization curves of (A) CpGH84A, (B) CpGH84B, (C) CpGH84C and (D) CpGH84D. Error bars calculated from the experiment in triplicate are also indicated.
N-acetyl-glucosaminidase efficiencies of GH84s from C. perfringens
To assess the catalytic efficiencies of the different CpGH84s at their optimum pH, initial steady-state kinetics experiments were performed using pNP-GlcNAc. The Michaelis–Menten kinetic parameters (Km and kcat) appeared quite different when comparing the four CpGH84s together (Table I). CpGH84C exhibited the lowest Km for pNP-GlcNAc at 0.13 mM, while the Km value for CpGH84A is approximately five times higher at 0.65 mM. CpGH84D and CpGH84B exhibited relatively poor Km values that are 17- and 37-fold higher, respectively, than the one observed for CpGH84C. In terms of catalytic turnover, CpGH84B exhibited the highest turnover and CpGH84A the lowest, with a 7-fold lower kcat (Table I). Nevertheless, these data translate into very similar and relatively low catalytic efficiencies (kcat/Km) for CpGH84A, CpGH84B and CpGH84D compared to CpGH84C, which has a catalytic efficiency for pNP-GlcNAc more than 25-fold higher than for the other enzymes (Table I).
Table I.
CpGH84s kinetic parameters using para-nitrophenyl-β-D-N-acetyl-glucosamine (pNP-GlcNAc) as a substratea
| GH84A | GH84B | GH84C | GH84D | |
|---|---|---|---|---|
| Km (mM) | 0.65 ± 0.05 | 4.85 ± 0.32 | 0.13 ± 0.01 | 2.23 ± 0.18 |
| V max (μM min−1) | 1.37 ± 0.03 | 9.85 ± 0.40 | 6.98 ± 0.09 | 3.63 ± 0.13 |
| k cat (s−1) | 0.72 ± 0.00 | 5.50 ± 0.22 | 3.83 ± 0.06 | 2.00 ± 0.06 |
| k cat/Km (mM−1 s−1) | 1.11 ± 0.09 | 1.13 ± 0.09 | 29.46 ± 2.31 | 0.90 ± 0.08 |
indicates standard errors calculated from experiments done in triplicate.
Linkage specificities of CpGH84s
The potential redundancy in the activity of the C. perfringens GH84s led us to hypothesize that these enzymes may be active on different substrates. Hence, substrates comprising GlcNAc β-linked motifs commonly found in N- and O-glycans were used to assess the substrate specificities of the CpGH84 enzymes. Thin-layer chromatography (TLC) analyses of their activities toward each substrate reveal that the four CpGH84s present slightly different, though largely overlapping, substrate specificities (Table II, Figure S1). CpGH84A and CpGH84D both possess the same specificity, which is restricted to GlcNAcβ-1,2 linked to mannose (Man) and β-1,6 linked to galactose (Gal) or N-acetyl-D-galactosamine (GalNAc). But, CpGH84B displays a broader substrate specificity with the capability to fully cleave GlcNAc-β-1,2-Man, GlcNAc-β-1,3-GalNAc, GlcNAc-β-1,6-Gal and GlcNAc-β-1,6-GalNAc as well as partially hydrolyze GlcNAc-β-1,4-GlcNAc. However, CpGH84B is not active when GlcNAc is β-1,4 linked to GalNAc or N-acetyl-D-muramic acid (MurNAc), indicating that these carbohydrates are not accommodated in the catalytic site (Table II). CpGH84C appears more promiscuous being able to fully cleave all the substrates tested except GlcNAc-β-1,4-MurNAc (Table II). This indicates that MurNAc is, like for CpGH84B, not accommodated in CpGH84C catalytic site. Overall, however, the general aglycon specificity of the CpGH84s still appears rather “loose” allowing Man, Gal, GalNAc and GlcNAc as aglycon (Table II, Figure S1). Furthermore, the four CpGH84s appear to fully hydrolyze the β-linkage between GlcNAc and a serine residue as the aglycon (Table II, Figure S1), which is in keeping with the relationship of these enzymes to the human GH84 enzyme O-GlcNAcase (Macauley et al. 2005).
Table II.
CpGH84s substrate specificities
| Substrate | GH84A | GH84B | GH84C | GH84D |
|---|---|---|---|---|
| GlcNAc-β-1,2-Man | + | + | + | + |
| GlcNAc-β-1,3-GalNAc-α-pNP | − | + | + | − |
| GlcNAc-β-1,4-GlcNAc | − | +/− | + | − |
| GlcNAc-β-1,4-MurNAc | − | − | − | − |
| GlcNAc-β-1,4-GalNAc-α-pNP | − | − | + | − |
| GlcNAc-β-1,6-Gal | + | + | + | + |
| GlcNAc-β-1,6-GalNAc-α-pNP | + | + | + | + |
| GlcNAc-β-1,6-(Gal-β-1,3)GalNAc-α-pNP | + | + | + | + |
| GlcNAc-β-O-L-Ser | + | + | + | + |
+/− indicates possible activity.
X-ray crystal structures of CpGH84s
Toward deciphering the molecular recognition of substrates by the CpGH84 enzymes, we determined the 3D structures of these four β-N-acetyl-D-glucosaminidases. The structure of the CpGH84C catalytic module has already been solved and studied (Rao et al. 2006; Ficko-Blean et al. 2009). Therefore, this structure (PDB code 2V5C) was used search model for molecular replacement to solve the structures of CpGH84A, CpGH84B and CpGH84D catalytic modules to resolutions of 2.20, 2.18 and 2.65 Å, respectively, and the structure of CpGH84C was further used for structural comparison/analysis. The overall structures are very similar with root-mean-square deviations (RMSD) lower than 2.7 Å over at least 332 residues. The structures of CpGH84A and CpGH84D were a similar pair with a RMSD value of 0.94 Å (over 494 residues), while the structures of CpGH84B and CpGH84C had a RMSD of 0.73 Å (over 386 residues). This is consistent with the relatively high amino acid sequence identity observed between CpGH84A and CpGH84D, and between CpGH84B and CpGH84C (Table SI). Overall, the CpGH84 structures comprise three distinct modules (Figure 3): a N-terminal accessory module (approximately 145 residues) composed of seven-stranded β-sandwich flanked by three helices, two helices on one side of the β-sheet and one on the other; a C-terminal module (approximately 155 residues) consisting of an elongated five α-helix bundle; and a central module (approximately 295 residues) corresponding to the catalytic module and adopting a (α/β)8 barrel fold that lacks the seventh helix, which is replaced by a loop.
Fig. 3.
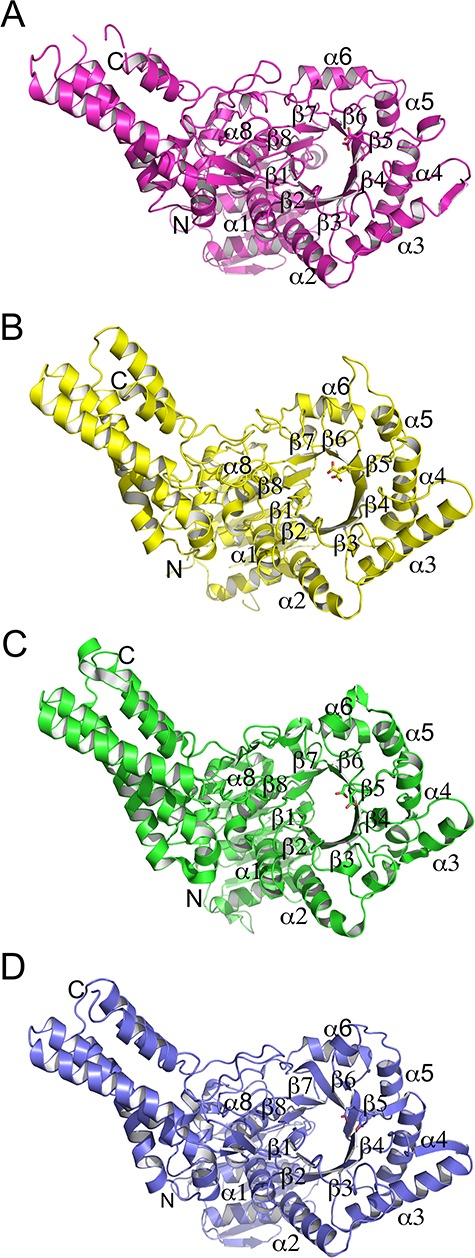
Overall fold of (A) CpGH84A, (B) CpGH84B, (C) CpGH84C and (D) CpGH84D. The structures are represented as cartoon with the catalytic module α-helices and β-strands labeled, as well as the N- and C-termini.
CpGH84 catalytic sites
The previous detailed analysis of the CpGH84C structure and its catalytic site has revealed that two aspartic acids as catalytic residues. D298 acts as catalytic acid/base, while D297 aids in positioning the acetamido oxygen of the GlcNAc residue in the −1 subsite for nucleophilic attack on the anomeric carbon (Rao et al. 2006). Therefore, the structural alignment of CpGH84C with the novel CpGH84 structures, in conjunction with sequence alignment (Figure S2), allowed the identification of their catalytic residues. The following residues were identified: D305 and D306 for CpGH84A, D281 and D282 for CpGH84B, and D310 and D311 for CpGH84D. All these residues appear to be positioned on a loop of ~10-residue long located between the β4 strand and the α4 helix (Figure 4). A comparison of the apo structure of CpGH84C (PDB code 2V5C) with its structure in complex with the inhibitor O-(2-Acetamido-2-deoxy-D-glucopyranosylidenamino) N-phenylcarbamate (PugNAc) bound in the catalytic site (PDB code 2CBJ) reveals that both catalytic residues are oriented away from the −1 subsite in the apo structure, while with the ligand bound in the active site, both catalytic residues are within interaction distance. Therefore, the binding of PugNAc seemed to provoke the engagement of D297 and D298 through their movements of ~1.4 and ~3.4 Å, respectively (Figure 4A). Similarly, the structural alignment of CpGH84C complexed with PugNAc with CpGH84A shows that D305 and D306 (from CpGH84A) would have to move in ~3.2 and 4.4 Å, respectively, toward the catalytic pocket to be within interacting distances of a ligand or substrate (Figure 4B). In CpGH84D, D310 and D311 would have to move ~2.8 and ~5.2 Å, respectively, to engage a bound ligand or substrate (Figure 4C). However, the structure of CpGH84B appears to have been obtained with catalytic site in a more closed conformation, whereby D281 and D282 would require relatively small movements to interact with a ligand or substrate (Figure 4D). This suggests that the β4–α4 loop, where the catalytic residues are present, is quite mobile.
Fig. 4.
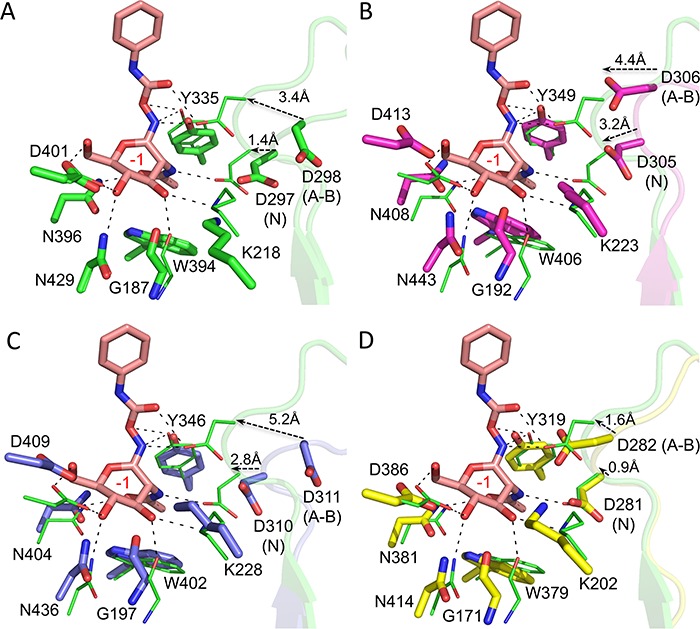
Structures of CpGH84s catalytic site. The residues forming the active site and the α4–β4 loop bearing the two catalytic residues (annotated with N for nucleophile and A–B for acid–base) are presented as sticks and semitransparent cartoon, respectively, in green for (A) CpGH84C, in purple for (B) CpGH84A, in blue for (C) CpGH84D and in yellow for (D) CpGH84B. All apo structures listed above were aligned with the CpGH84C (in green lines) in complex with PugNAc (in pink sticks, PDB code 2CBJ). The distances between the catalytic residue from the apo and complexed structures are indicated as well as the −1 subsite in red.
The catalytic residues are part of a pocket that creates the −1 subsite, where GlcNAc is expected to bind. As shown by the structural overlay of PugNAc from the CpGH84C complex (PDB code 2CBJ), the catalytic pockets of CpGH84A, CpGH84B and CpGH84D accommodate the acetamido-pyranosyl moiety via the same set of interactions in the −1 subsite as was described previously for CpGH84C (Rao et al. 2006) (Figure 4). These interactions consist of hydrogen bonds between the hydroxyl groups and N-acetyl moiety from PugNAc pyranosyl ring with the following residues: G192, K223, D413 and N443, and W406 and N408, respectively (numbering according to CpGH84A, Figure 4).
Further examination of the CpGH84 structures in complex with unhydrolyzed substrates is necessary to better understand the linkage and aglycon specificities. Unfortunately, attempts to obtain structures of catalytically inactive mutants in complex with carbohydrates were unsuccessful.
Discussion
C. perfringens interacts with its host, in part, through the use of a battery of GHs that are capable of processing host glycans. As opposed to other characterized bacteria such as B. thetaiotaomicron and S. pyogenes, which possess one family GH84 enzyme (Dennis et al. 2006; Sheldon et al. 2006), the C. perfringens genome encodes for four GH84s, known as the NagH (or μ-toxin), NagI, NagJ and NagK (Shimizu et al. 2002; Myers et al. 2006). The characterization of their catalytic modules, CpGH84A, CpGH84B, CpGH84C and CpGH84D, respectively, indicates these enzymes exhibit clear exo-β-N-acetyl-D-glucosaminidase activity on the synthetic pNP-GlcNAc substrate. Kinetic experiments show that the Km obtained for CpGH84C (Km = 0.13 mM) with pNP-GlcNAc is very similar to the one previously reported (Km = 0.12 mM) (Rao et al. 2006) and is roughly of the same order of magnitude as the one determined for GH84 from B. thetaiotaomicron (BtGH84; Km = 0.28 mM) (Dennis et al. 2006). Concerning the catalytic efficiencies, CpGH84C (29.46 mM−1 s−1) appears substantially more efficient than CpGH84A (1.11 mM−1 s−1), CpGH84B (1.13 mM−1 s−1) and CpGH84D (0.90 mM−1 s−1). Interestingly, the catalytic efficiencies of the latter three enzymes on pNP-GlcNAc are very similar to the efficiency observed for the human O-glcNAcase (1.00 mM−1 s−1), which has been shown to be very comparable to other characterized hexosaminidases (Wells et al. 2002). Therefore, the C. perfringens GH84 enzymes appear as relatively potent exo-β-N-acetyl-D-glucosaminidases with CpGH84A and CpGH84D being slightly more specialized enzymes compared to CpGH84B and CpGH84C, with CpGH84C having activity on every β-linked GlcNAc substrate tested except that found in peptidoglycan. A caveat to this, however, is that our kinetic analyses utilize isolated catalytic modules and a small soluble synthetic substrate, which assesses the intrinsic capacity of the catalytic modules to hydrolyze a glycosidic bond. In the biological setting, however, the intact enzymes possess CBMs (and other ancillary modules), while the substrate is most likely a glycan presented in a complex matrix (i.e. the mucin layer). These features, particularly the role of CBMs in enzyme targeting and immobilization (Boraston et al. 2004), would most likely have an influence on efficiency of substrate hydrolysis in vivo. Nevertheless, as a group, these enzymes would furnish C. perfringens with considerable capacity to process a wide variety of glycans terminating in β-linked GlcNAc residues.
While some GH84 enzymes are annotated as hyaluronidases, including NagH and NagJ, there is little compelling evidence their target substrate is indeed hyaluronan, a polymer of alternating β-linked GlcNAc and D-glucuronic acid. To date, all of the characterized GH84 enzymes are exo-acting enzymes and, because of their substrate assisted catalytic mechanism, must therefore act on terminal β-linked GlcNAc residues. While a GH84 enzyme with loose aglycon specificity may be able to remove a β-1,4-linked nonreducing end GlcNAc residue from a hyaluronan chain it would not be able to remove the following β-1,3-linked glucuronic acid residue. It is formally possible that a GH84 enzyme, such as those from C. perfringens, may work in concert with other enzymes to exo-hydrolytically degrade hyaluronan fragments, but this remains to be determined. However, the noted specificity of CBMs in both NagH and NagJ for galactose residues argues against hyaluronidase activity as galactose is not present in hyaluronan (Ficko-Blean and Boraston 2006, 2009; Grondin et al. 2014).
The structural analysis of the catalytic module of C. perfringens GH84s reveals an overall fold characteristic of the GH84 family comprising three distinct modules. Indeed, the overall structures of the four enzymes are also very similar to other known structures of family GH84 as attested by low RMSD values (values lower than 2.6 Å over at least 209 residues) (Dennis et al. 2006; Schimpl et al. 2010). Furthermore, the absence of the seventh helix observed in C. perfringens GH84 enzymes has also been reported for GH84 enzymes originating from other organisms (Dennis et al. 2006; Ficko-Blean and Boraston 2009; Slamova et al. 2014). CpGH84 catalytic pockets feature two conserved aspartic acid residues as catalytic residues, which are also conserved throughout the GH84 family (Wells et al. 2002; Dennis et al. 2006; Cantarel et al. 2009; Schimpl et al. 2010). The structural study of CpGH84C revealed that the apo structure appears to be in an “open” conformation while the complexed structure defines a “closed” conformation (Figure 4A). In the latter, the mobile β4–α4 loop brings the catalytic residues into the vicinity of the PugNAc pyranosyl ring N1, corresponding to where GlcNAc O1 would be located in an actual substrate, and the acetamido group by moving D297 and D298 of CpGH84C within interacting distances (Figure 4A). Similarly, the catalytic residues of CpGH84A, CpGH84B and CpGH84D are also located on a similar β4–α4 loop that in the absence of a substrate or ligand appears to be in an open conformation, which would presumably undergo a similar transition to a closed and catalytically competent conformation upon substrate or ligand binding. A similar β4–α4 loop carrying the catalytic residues has also been observed in the structure of other GH84s, such as BtGH84 and Oceanicola granuluosus GH84 (OgGH84) (Dennis et al. 2006; Schimpl et al. 2010). Though the transition between an open and closed conformation with a substrate bound has only been hinted at in the literature for the human GH84 (Pathak et al. 2008), this structural accommodation appears to be a characteristic of the GH84 family.
CpGH84s catalytic sites comprise a well-defined and conserved −1 subsite that binds a nonreducing terminal GlcNAc residue (Figures 4 and S2). This carbohydrate moiety is recognized through interactions involving the same conserved residues as the ones found in other GH84 enzymes (Wells et al. 2002; Dennis et al. 2006; Schimpl et al. 2010). To date, no structure of a GH84 in complex with a nonhydrolyzed glycan substrate has been reported in the literature and thus potential interactions between the substrate and + 1 subsite remain unclear. However, CpGH84C in complex with PugNAc showed that W490 makes a stacking interaction with the phenyl carbamate ring of the inhibitor, which is located roughly where the aglycon should sit in a substrate (Rao et al. 2006). This would therefore represent the region where one would expect a +1 subsite. This tryptophan is located at the apex of a long loop (17 residues) situated between the catalytic domain and the C-terminal α-helices bundle. A similar loop of 18 amino acids is also found in CpGH84A and CpGH84D; however, the residues at their apex are not conserved (Figures 5 and S2). Indeed, CpGH84A presents an aspartate and an arginine residue (D510 and R512), while CpGH84D displays a tryptophan with an alternate conformation and a histidine (W496 and H497). Interestingly, CpGH84B possesses a shorter loop carrying an aspartate residue (D471) at its apex, which appears too distant to interact with the substrate at the +1 subsite. The substrate binding at the +1 subsite would, therefore, vary between the CpGH84s, providing each with their own subtly different substrate specificities. An analysis of amino acid conservation in the active site of each catalytic module indicates retention of the −1 subsite but greater variability of the +1 region among a wider representation of GH84 family members, possibly reflecting the general adaptability of the +1 subsite to accommodate different aglycons (Figure S3).
Fig. 5.
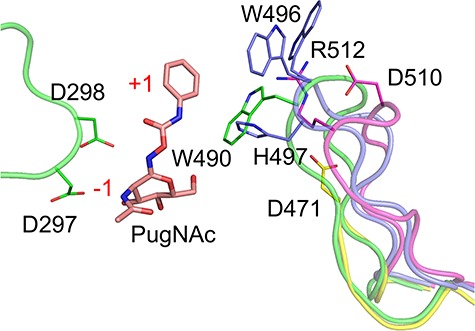
Cartoon representation of CpGH84 potential +1 subsites. CpGH84C (in green) in complex with PugNAc (in pink, PDB code 2CBJ) has been aligned with CpGH84A (in purple), CpGH84B (in yellow) and CpGH84D (in blue). The residues for CpGH84s potentially involved in the description of a + 1 subsite are shown as sticks, as well as CpGH84C catalytic residues. The subsites are labeled in red.
Our original hypothesis for the expansion of GH84 enzymes in C. perfringens was that the different enzymes would have distinct specificities, thus as a group conferring a wider capacity to process GlcNAc containing glycans. However, our observation that the specificities are only subtly different for each enzyme, but largely overlapping, with a single quite promiscuous enzyme (CpGH84C) being the most active enzyme does not support this simple hypothesis. In this light, we proposed the following, alternative possibilities. The first regards the pH vs. activity profiles of the enzymes. The gastrointestinal tract of animals, including humans, is a common site of habitation and/or infection by C. perfringens and the pH can vary widely in this environment. Outside of the gastric environment, the pH can vary between around 3 to above 7, depending on the location in small and large intestines (Fallingborg et al. 1993; Nugent et al. 2001; Koziolek et al. 2015). It is possible that each enzyme, or pair of enzymes, is adapted for activity in particular environmental pH. For example, at pH 4 only CpGH84A and CpGH84C would have significant activity, while above pH 7, CpGH84A and CpGH84D would be most active. Alternatively, the four isozymes may be under different transcriptional control and therefore respond to distinct environmental cues that require deployment of a relatively nonspecific exo-β-N-acetyl-D-glucosaminidase. Indeed, one cue may even be the pH of the environment. Finally, the expansion of GH84 encoding genes may be related to the multimodularity of the encoded enzymes. For example, NagI and NagK are predicted to be localized to the bacterial cell-surface via their LPXTG motifs. In contrast, though NagJ is proposed to interact with the cell-surface via its FN3 module and NagH is proposed to interact with other cell-surface attached enzymes via Doc-X82 (cohesin) interactions, the noncovalent nature of these interactions lends to the likelihood that soluble forms of NagJ and NagK are present free in the extracellular environment (Adams et al. 2008; Ficko-Blean et al. 2009). This partitioning of activities between the cell-wall and extracellular environment, combined with potential CBM-mediated targeting of the enzymes to regions rich in particular glycans, may also aid the bacterium in optimizing its glycan degrading capacity and, therefore, contribute to the expansion of GH84 encoding genes in C. perfringens.
Overall, the GH84 enzymes from C. perfringens likely work in concert with the arsenal of other glycan processing enzymes encoded in the genome, with some enzymes uncapping GlcNAc residues (e.g. sialidases or β-galactosidases (Ashida et al. 2001; Li and McClane 2018)) or others working on the terminal residues revealed by GH84 activity (e.g. core 1 endo-α-N-acetylgalactosaminidase (Ashida et al. 2008)). However, unlike many other human gut microbes, particularly the Bacteroidetes, C. perfringens does not organize its CAZymes into polysaccharide utilization loci making functional associations between the CAZymes more difficult to predict (Grondin et al. 2017). Furthermore, a large proportion of the predicted C. perfringens CAZymes are uncharacterized, so the precise roles of the GH84 enzymes in the enzymatic cascade of glycan degradation await further study.
Materials and methods
Materials
N,N′-Diacetylchitobiose (GlcNAc-β-1,4-GlcNAc) and 2-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-D-mannose (GlcNAc-β-1,2-Man) were purchased from Toronto Research Chemical (Toronto, Canada). 2-acetamido-4-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-2-deoxy-D-muramic acid (GlcNAc-β-1,4-MurNAc), 6-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-D-galactopyranose (GlcNAc-β-1,6-Gal), 2-acetamido-2-deoxy-D-glucopyranosyl serine (GlcNAc-β-O-L-Ser), 4-nitrophenyl 2-acetamido-4-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-2-deoxy-α-D-galactopyranoside (GlcNAc-β-1,4-GalNAc-α-pNP), 4-nitrophenyl 2-acetamido-3-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-2-deoxy-α-D-galactopyranoside (GlcNAc-β-1,3-GalNAc-α-pNP), 4-nitrophenyl 2-acetamido-6-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-2-deoxy-α-D-galactopyranoside (GlcNAc-β-1,6-GalNAc-α-pNP) and 4-nitrophenyl 2-acetamido-6-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-3-O-(β-D-galactopyranosyl)-2-deoxy-α-D-galactopyranoside (GlcNAc-β-1,6-(Gal-β-1,3)-GalNAc-α-pNP) were purchased from Carbosynth (Compton, UK). All other reagents and chemicals were purchased from Sigma unless otherwise specified.
Cloning
Gene fragments encoding the catalytic module of NagH, NagI and NagK, referred to as CpGH84A, CpGH84B and CpGH84D, respectively, (amino acid boundaries: 31–660, 31–628 and 36–639, respectively) were cloned from Clostridium perfringens ATCC 13124 using specific primers (Table SII) as previously described for the cloning of CpGH84C catalytic module (boundaries: 31–624) (Ficko-Blean and Boraston 2005). Briefly, the fragments were amplified via PCR using Platinum Pfx polymerase (Invitrogen). The amplified DNA fragments were then cloned into pET28a(+) (Novagen) via engineered NheI and XhoI restriction sites using standard molecular biology procedures. The resulting gene fusions encoded an N-terminal His6 tag fused to the protein of interest by an intervening thrombin protease cleavage site. Bidirectional DNA sequencing was used to verify the fidelity of each construct.
Protein expression and purification
Recombinant expression vectors for CpGH84A, CpGH84B, CpGH84C and CpGH84D were transformed into Escherichia coli BL21 Star (DE3) cells (Invitrogen) and the resulting strains cultured in LB or 2xYT media supplemented with kanamycin (50 μg mL−1). Transformed bacteria were grown at 37°C with shaking to an OD600 nm of ~0.8. Protein expression was then induced by the addition of isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.5 mM; incubation was continued overnight at 16°C with shaking. Bacterial cells were harvested by centrifugation and disrupted by chemical lysis (Pluvinage et al. 2013). The recombinant proteins were purified from the cleared cell-lysate by Ni2+-immobilized metal affinity chromatography followed by size exclusion chromatography using a Sephacryl S-200 column (GE Healthcare) in 20 mM Tris pH 8.0. Purified proteins were then concentrated using a stirred-cell ultrafiltration device with a 10 K molecular weight cut-off membrane (Millipore).
Protein concentrations were determined by measuring the absorbance at 280 nm and using calculated extinction coefficients of 113,805 M−1 cm−1 for CpGH84A, 98,780 M−1 cm−1 for CpGH84B, 89,395 M−1 cm−1 for CpGH84C and 111,730 M−1 cm−1 for CpGH84D (Gasteiger et al. 2003).
Enzyme activities and kinetics
The pH dependence of CpGH84A, CpGH84B, CpGH84C and CpGH84D activities was determined using 30 nM of enzyme, pNP-GlcNAc at 0.5 mM and bovine serum albumin at 1% in McIlvaine’s buffer (0.2 M disodium phosphate, 0.1 M citric acid) over a range of pH from 2.2 to 8.0 (McIlvaine 1921). The reactions were stopped after 3 min by addition of sodium hydroxide at 0.1 M.
Reaction mixtures for the determination of kinetic constants were set up in triplicate at the optimal pH in 100 μL volumes containing 30 nM of enzyme, 1% BSA and 0–6 mM pNP-GlcNAc in citric acid-sodium citrate buffer (50 mM) for CpGH84A and CpGH84B, sodium acetate buffer (50 mM) for CpGH84C and citric acid-sodium phosphate buffer (50 mM) for CpGH84D. The formation of para-nitrophenolate (pNP−) was followed at 400 nm over an 8-min time period using a SpectraMax5 plate reader (Molecular Devices). The rate of release was determined by linear regression using the linear part of the curve and a pNP− extinction coefficient of 18,300 M−1 cm−1. Michaelis–Menten parameters were determined by nonlinear curve fitting by using Graphpad Prism5.
Linkage specificity analysis
CpGH84s specificities were assessed by thin-layer chromatography. Each substrate (2 mM, see Table II) was incubated at 37°C in presence of CpGH84A, CpGH84B, CpGH84C or CpGH84D (1 μM) for 2 h in McIlvaine buffer pH 5.8, 5.6, 5.2 or 7.0, respectively. The reactions were then stopped by incubation at 90°C for 30 min. Reaction products were spotted onto pre-coated POLYGRAM SIL G/UV254 TLC sheets (Thermo Fisher Scientific, Waltham, MA), separated in a solvent of 2:1:1 butanol:acetic acid: H2O and developed using DPA stain (1 g diphenylamine, 1 mL aniline, 5 mL 85% orthophosphoric acid, 50 mL acetone) at 120°C for 30 min.
Crystallization conditions
Crystals were grown using sitting-drop vapor diffusion for screening and hanging drop vapor diffusion for optimization, all at 18°C. Crystals of CpGH84A (30 mg mL−1) were obtained in 14% (w/v) polyethylene glycol (PEG) 3350, 0.15 M calcium chloride and 20 mM octyl-β-D-glucopyranoside. CpGH84B (18 mg mL−1) was crystallized in 0.1 M Tris HCl pH 8.5 and 2.0 M ammonium sulphate ((NH4)2SO4). CpGH84D crystals (25 mg mL−1) grew in 25% (w/v) PEG 4000, 0.2 M (NH4)2SO4 and 0.1 M sodium citrate: HCl pH 5.6. Prior to data collection, single crystals were flash cooled with liquid nitrogen in crystallization solution supplemented with 20% (v/v) ethylene glycol.
General crystallography procedures
Diffraction data were collected either on the beamline 12-2 of the Stanford Linear Accelerator Center (SLAC, Stanford Synchrotron Radiation Lightsource (SSRL), CA), or beamline 08ID-1 of the Canadian Light Source (CLS, Saskatoon, Saskatchewan) as indicated in Table III. All diffraction data were processed using MOSFLM or XDS and SCALA (CollaborativeComputationalProject 1994; Powell 1999; Kabsch 2010). All data collection and processing statistics are shown in Table III. All structures were solved by molecular replacement (PHASER (McCoy et al. 2007)) using a CHAINSAW (Stein 2008) model of CpGH84C (PDB code 2V5C) to obtain an initial model that was built using BUCANEER (Cowtan 2006) followed by COOT (Emsley and Cowtan 2004). Refinement of atomic coordinates was then performed with REFMAC (Murshudov et al. 1997). The addition of water molecules was performed in COOT with FINDWATERS and manually checked after refinement. In all data sets, refinement procedures were monitored by flagging 5% of all observations as “free” (Brunger 1992). Model validation was performed with MOLPROBITY (Davis et al. 2007). Finally, the models obtained were represented using PyMOL (Delano 2002). All model statistics are shown in Table III. Coordinates and structure factors have been deposited in the PDB under the codes 6PV4 for the CpGH84A, 6PV5 for CpGH84B and 6PWI for CpGH84D.
Table III.
X-ray data collection and structure statistics
| CpGH84A | CpGH84B | CpGH84D | |
|---|---|---|---|
| Data collection | |||
| Beamline | CLS—08ID-1 | SSRL BL 12-2 | SSRL BL 12-2 |
| Wavelength (Å) | 0.97965 | 1.00000 | 0.97910 |
| Space Group | P1 | H32 | P65 |
| Cell dimensions | |||
| a, b, c (Å) | 92.1, 92.1, 103.1 | 171.4, 171.4, 128.8 | 195.7, 195.7, 106.5 |
| α, β, γ (°) | 95.7, 110.5, 119.7 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 78.00–2.20 (2.32–2.20)a | 39.21-2.18 (2.30–2.18) | 38.89–2.65 (2.71–2.65) |
| R merge | 0.167 (0.386) | 0.079 (0.491) | 0.095 (0.477) |
| R pim | 0.113 (0.262) | 0.027 (0.168) | 0.046 (0.243) |
| CC(1/2) | 0.929 (0.454) | 0.998 (0.943) | 0.995 (0.778) |
| <I/σI> | 7.7 (5.6) | 18.5 (5.8) | 14.3 (3.5) |
| Completeness (%) | 98.1 (97.3) | 100.0 (99.8) | 98.8 (99.3) |
| Redundancy | 2.7 (2.7) | 9.7 (9.4) | 5.7 (5.6) |
| No. of reflections | 353,529 | 367,422 | 382,487 |
| No. unique | 129,154 | 37,842 | 66,655 |
| Refinement | |||
| Resolution (Å) | 2.20 | 2.18 | 2.65 |
| R work/Rfree | 0.20/0.25 | 0.19/0.24 | 0.17/0.21 |
| No. of atoms | |||
| Protein | 4668 (A), 4820 (B), 4826 (C), 4757 (D) | 4385 | 4794 (A), 4746 (B) |
| Ligand | 19 (CA), 92 (EDO) | 20 (EDO) | 52 (EDO) |
| Water | 1727 | 234 | 512 |
| B-factors | |||
| Protein | 14.0 (A), 14.4 (B), 14.6 (C), 14.4 (D) | 45.5 | 31.9 (A), 43.1 (B) |
| Ligand | 29.7 (CA), 23.3 (EDO) | 50.6 (EDO) | 49.0 (EDO) |
| Water | 19.7 | 45.6 | 31.4 |
| RMSD | |||
| Bond lengths (Å) | 0.005 | 0.005 | 0.003 |
| Bond angles (°) | 1.198 | 1.296 | 1.037 |
| Ramachandran (%) | |||
| Preferred | 95.7 | 95.2 | 96.4 |
| Allowed | 4.2 | 4.8 | 3.6 |
| Disallowed | 0.1 | 0.0 | 0.0 |
Values for highest resolution shells are shown in parenthesis.
Supplementary Material
Acknowledgements
We thank the beamline staff at the Stanford Synchrotron Research Laboratory. SSRL is a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. DOE Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences. Research described in this paper was performed using beamline 08B1-1 at the Canadian Light Source, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada and the Canadian Institutes of Health Research.
Abbreviations
CAZymes, carbohydrate active enzymes
CBM, carbohydrate-binding module
Doc, Dockerin module
FIVAR, find-in-various-architectures module
FN3, fibronectin-like type III module
GalNAc, N-acetyl-galactosamine
GHs, glycoside hydrolases
GlcNAc, N-acetyl-glucosamine
Man, mannose
(NH4)2SO4, ammonium sulphate
PEG, polyethylene glycol
pNP-GlcNAc, para-nitrophenyl-β-D-GlcNAc
RMSD, root mean square deviations
X82, cohesin-like family X82 module
Funding
Canadian Institutes of Health Research Project (PJT 159786).
Conflict of interest statement
None declared.
References
- Adams JJ, Gregg K, Bayer EA, Boraston AB, Smith SP. 2008. Structural basis of Clostridium perfringens toxin complex formation. Proc Natl Acad Sci USA. 105:12194–12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida H, Anderson K, Nakayama J, Maskos K, Chou CW, Cole RB, Li SC, Li YT. 2001. A novel endo-beta-galactosidase from Clostridium perfringens that liberates the disaccharide GlcNAcalpha 1-->gal from glycans specifically expressed in the gastric gland mucous cell-type mucin. J Biol Chem. 276:28226–28232. [DOI] [PubMed] [Google Scholar]
- Ashida H, Maki R, Ozawa H, Tani Y, Kiyohara M, Fujita M, Imamura A, Ishida H, Kiso M, Yamamoto K. 2008. Characterization of two different endo-alpha-N-acetylgalactosaminidases from probiotic and pathogenic enterobacteria, Bifidobacterium longum and Clostridium perfringens. Glycobiology. 18:727–734. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. 2004. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem J. 382:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhausen I, Schachter H, Stanley P. 2009. O-GalNAc glycans In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2nd ed. The publisher is Cold Spring Harbor Laboratory Press. p. 115–127. [PubMed]
- Brunger AT. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature. 355:472–475. [DOI] [PubMed] [Google Scholar]
- Canard B, Garnier T, Saint-Joanis B, Cole ST. 1994. Molecular genetic analysis of the nagH gene encoding a hyaluronidase of Clostridium perfringens. Mol Gen Genet. 243:215–224. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CollaborativeComputationalProject 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 50:760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan K. 2006. The buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 62:1002–1011. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS et al. 2007. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL. 2002. The PyMOL Molecular Graphics System.
- Dennis RJ, Taylor EJ, Macauley MS, Stubbs KA, Turkenburg JP, Hart SJ, Black GN, Vocadlo DJ, Davies GJ. 2006. Structure and mechanism of a bacterial beta-glucosaminidase having O-GlcNAcase activity. Nat Struct Mol Biol. 13:365–371. [DOI] [PubMed] [Google Scholar]
- Dhar P, McAuley J. 2019. The role of the cell surface mucin MUC1 as a barrier to infection and regulator of inflammation. Front Cell Infect Microbiol. 9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- Fallingborg J, Christensen LA, Jacobsen BA, Rasmussen SN. 1993. Very low intraluminal colonic pH in patients with active ulcerative colitis. Digest Dis Sci. 38:1989–1993. [DOI] [PubMed] [Google Scholar]
- Ficko-Blean E, Boraston AB. 2005. Cloning, recombinant production, crystallization and preliminary X-ray diffraction studies of a family 84 glycoside hydrolase from Clostridium perfringens. Acta Crystallogr Sect F Struct Biol Cryst Commun. 61:834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficko-Blean E, Boraston AB. 2006. The interaction of a carbohydrate-binding module from a Clostridium perfringens N-acetyl-beta-hexosaminidase with its carbohydrate receptor. J Biol Chem. 281:37748–37757. [DOI] [PubMed] [Google Scholar]
- Ficko-Blean E, Boraston AB. 2009. N-acetylglucosamine recognition by a family 32 carbohydrate-binding module from Clostridium perfringens NagH. J Mol Biol. 390:208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficko-Blean E, Gregg KJ, Adams JJ, Hehemann JH, Czjzek M, Smith SP, Boraston AB. 2009. Portrait of an enzyme, a complete structural analysis of a multimodular {beta}-N-acetylglucosaminidase from Clostridium perfringens. J Biol Chem. 284:9876–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figura N. 1997. Helicobacter pylori factors involved in the development of gastroduodenal mucosal damage and ulceration. J Clin Gastroenterol. 25(Suppl 1):S149–S163. [DOI] [PubMed] [Google Scholar]
- Flores-Diaz M, Alape-Giron A, Clark G, Catimel B, Hirabayashi Y, Nice E, Gutierrez JM, Titball R, Thelestam M. 2005. A cellular deficiency of gangliosides causes hypersensitivity to Clostridium perfringens phospholipase C. J Biol Chem. 280:26680–26689. [DOI] [PubMed] [Google Scholar]
- Galen JE, Ketley JM, Fasano A, Richardson SH, Wasserman SS, Kaper JB. 1992. Role of Vibrio cholerae neuraminidase in the function of cholera toxin. Infect Immun. 60:406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondin JM, Chitayat S, Ficko-Blean E, Houliston S, Arrowsmith CH, Boraston AB, Smith SP. 2014. An unusual mode of galactose recognition by a family 32 carbohydrate-binding module. J Mol Biol. 426:869–880. [DOI] [PubMed] [Google Scholar]
- Grondin JM, Tamura K, Dejean G, Abbott DW, Brumer H. 2017. Polysaccharide utilization loci: Fueling microbial communities. J Bacteriol. 199:e00860–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs JK, Pluvinage B, Boraston AB. 2018. Glycan-metabolizing enzymes in microbe-host interactions: The Streptococcus pneumoniae paradigm. FEBS letters. 592:3865–3897. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Ambort D, Pelaseyed T, Schutte A, Gustafsson JK, Ermund A, Subramani DB, Holmen-Larsson JM, Thomsson KA, Bergstrom JH et al. 2011. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 68:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr. 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziolek M, Grimm M, Becker D, Iordanov V, Zou H, Shimizu J, Wanke C, Garbacz G, Weitschies W. 2015. Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the Intellicap((R)) system. J Pharm Sci. 104:2855–2863. [DOI] [PubMed] [Google Scholar]
- Li J, McClane BA. 2018. NanI sialidase can support the growth and survival of Clostridium perfringens strain F4969 in the presence of sialyated host macromolecules (mucin) or Caco-2 cells. Infect Immun. 86:e00547–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. 2005. O-GlcNAcase uses substrate-assisted catalysis: Kinetic analysis and development of highly selective mechanism-inspired inhibitors. J Biol Chem. 280:25313–25322. [DOI] [PubMed] [Google Scholar]
- Markovic-Housley Z, Miglierini G, Soldatova L, Rizkallah PJ, Muller U, Schirmer T. 2000. Crystal structure of hyaluronidase, a major allergen of bee venom. Structure. 8:1025–1035. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr. 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuckin MA, Linden SK, Sutton P, Florin TH. 2011. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 9:265–278. [DOI] [PubMed] [Google Scholar]
- McIlvaine T. 1921. A buffer solution for colorimetric comparison. J Biol Chem. 49:183–186. [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 53:240–255. [DOI] [PubMed] [Google Scholar]
- Myers GS, Rasko DA, Cheung JK, Ravel J, Seshadri R, DeBoy RT, Ren Q, Varga J, Awad MM, Brinkac LM et al. 2006. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama M, Ochi S, Oda M, Miyamoto K, Takehara M, Kobayashi K. 2015. Recent insights into Clostridium perfringens beta-toxin. Toxins (Basel). 7:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent SG, Kumar D, Rampton DS, Evans DF. 2001. Intestinal luminal pH in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut. 48:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak S, Dorfmueller HC, Borodkin VS, van Aalten DM. 2008. Chemical dissection of the link between streptozotocin, O-GlcNAc, and pancreatic cell death. Chem Biol. 15:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluvinage B, Hehemann JH, Boraston AB. 2013. Substrate recognition and hydrolysis by a family 50 exo-beta-agarase, Aga50D, from the marine bacterium Saccharophagus degradans. J Biol Chem. 288:28078–28088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluvinage B, Higgins MA, Abbott DW, Robb C, Dalia AB, Deng L, Weiser JN, Parsons TB, Fairbanks AJ, Vocadlo DJ et al. 2011. Inhibition of the pneumococcal virulence factor StrH and molecular insights into N-glycan recognition and hydrolysis. Structure. 19:1603–1614. [DOI] [PubMed] [Google Scholar]
- Powell HR. 1999. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr D Biol Crystallogr. 55:1690–1695. [DOI] [PubMed] [Google Scholar]
- Rao FV, Dorfmueller HC, Villa F, Allwood M, Eggleston IM, van Aalten DM. 2006. Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 25:1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimpl M, Schuttelkopf AW, Borodkin VS, van Aalten DM. 2010. Human OGA binds substrates in a conserved peptide recognition groove. Biochem J. 432:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, Davenport MT, Keith DB, Musser JM. 2008. The role of complex carbohydrate catabolism in the pathogenesis of invasive streptococci. Trends Microbiol. 16:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon WL, Macauley MS, Taylor EJ, Robinson CE, Charnock SJ, Davies GJ, Vocadlo DJ, Black GW. 2006. Functional analysis of a group A streptococcal glycoside hydrolase Spy1600 from family 84 reveals it is a beta-N-acetylglucosaminidase and not a hyaluronidase. Biochem J. 399:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci USA. 99:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamova K, Kulik N, Fiala M, Krejzova-Hofmeisterova J, Ettrich R, Kren V. 2014. Expression, characterization and homology modeling of a novel eukaryotic GH84 beta-N-acetylglucosaminidase from Penicillium chrysogenum. Protein Expr Purif. 95:204–210. [DOI] [PubMed] [Google Scholar]
- Smedley JG 3rd, Fisher DJ, Sayeed S, Chakrabarti G, McClane BA. 2004. The enteric toxins of Clostridium perfringens. Rev Physiol Biochem Pharmacol. 152:183–204. [DOI] [PubMed] [Google Scholar]
- Stein N. 2008. CHAINSAW: A program for mutating pdb files used as templates in molecular replacement. J. Appl. Cryst. 41:641–643. [Google Scholar]
- Aalten DM, Komander D, Synstad B, Gaseidnes S, Peter MG, Eijsink VG. 2001. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc Natl Acad Sci USA. 98:8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L, Gao Y, Mahoney JA, Vosseller K, Chen C, Rosen A, Hart GW. 2002. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J Biol Chem. 277:1755–1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.