

Abstract
A promising design paradigm for small-molecule inhibitors of tubulin polymerization that bind to the colchicine site draws structural inspiration from the natural products colchicine and combretastatin A-4 (CA4). Our previous studies with benzocycloalkenyl and heteroaromatic ring systems yielded promising inhibitors with dihydronaphthalene and benzosuberene analogues featuring phenolic (KGP03 and KGP18) and aniline (KGP05 and KGP156) congeners emerging as lead agents. These molecules demonstrated dual mechanism of action, functioning as both potent vascular disrupting agents (VDAs) and as highly cytotoxic anticancer agents. A further series of analogues was designed to extend functional group diversity and investigate regioisomeric tolerance. Ten new molecules were effective inhibitors of tubulin polymerization (IC50 < 5 µM) with seven of these exhibiting highly potent activity comparable to CA4, KGP18, and KGP03. For one of the most effective agents, dose-dependent vascular shutdown was demonstrated using dynamic bioluminescence imaging in a human prostate tumor xenograft growing in a rat.
Keywords: inhibitors of tubulin polymerization, benzosuberene analogues, vascular disrupting agents, small-molecule synthesis
Introduction
Solid tumors require a functional vasculature to supply oxygen and nutrients to their cells when they exceed 1 mm3 in size.1,2 Unlike normal vasculature, the tumor-associated vascular network tends to expand irregularly, incorporating fragile and chaotic bulges and blind ends.3–5 The primitive character and inherent fragility of tumor-associated vasculature, along with the seminal observations by Denekamp and co-workers that blocking established tumor-associated blood flow leads to tumor regression in mice, positioned tumor-associated vasculature as a promising target for cancer therapy.6–8 Two categories of small-molecule, vascular-targeted therapies have been developed: angiogenesis inhibiting agents (AIAs) that inhibit neovascularization in developing tumors; and, separately, vascular disrupting agents (VDAs) that irreversibly damage established tumor-associated vasculature.9–13 The two major sub-divisions of VDAs include biologics and small-molecule anticancer agents. The majority of small-molecule VDAs function as inhibitors of tubulin polymerization, which destabilize the tubulin-microtubule protein system by binding to the colchicine site on β-tubulin in close proximity to the α,β-tubulin heterodimer interface. The endothelial cells lining microvessels undergo rapid cytoskeletal disruption, manifested by morphological changes (flat to round) in response to inhibition of their tubulin-microtubule protein system cytoskeleton triggered by VDA binding to the colchicine site. This rapid endothelial cell cytoskeletal rearrangement leads to irreversible damage to the tumor-associated vasculature, culminating in tumor necrosis.4,14–17
The natural products colchicine, combretastatin A-4 (CA4), and combretastatin A-1 (CA1), along with the synthetic analogue phenstatin (Fig. 1), are potent colchicine site inhibitors of tubulin polymerization that function as promising VDAs.18–24 These molecules have provided structural inspiration and guidance for the design, synthesis, and biological evaluation of many second-generation (and beyond) molecules in a world-wide effort to identify a small-molecule colchicine site agent with the necessary efficacy coupled with safety to be utilized as a cancer therapeutic in humans. To date, no small-molecule therapeutic agent that interacts with the colchicine site and functions as either an antiproliferative agent or a VDA (or demonstrates a dual mechanism of action) has reached FDA approval. Structural similarities between these natural products include a trimethoxy phenyl ring, a separate hydroxylated p-methoxy aryl moiety, and a bridging functionality connecting the two rings with a comparable centroid-to-centroid distance. Our long-standing program focused on molecular recognition for the colchicine site has led to the discovery of promising synthetic analogues and derivatives,25–28 including stilbenoid,14,29–31 benzo[b]thiophene,32–34 benzofuran,34 dihydronaphthalene,8,35,36 benzosuberene,8,25,35–38 and indole-based39 molecules (Fig. 1). Numerous other studies have investigated a myriad of structural and functional group modifications to the A-ring, the B-ring, and the ethylene bridge of combretastatin A-4; the results of which are conveyed in extensive publications (too numerous to cite herein) and summarized in various review articles, yet none are comprehensive.40–49
Figure 1.
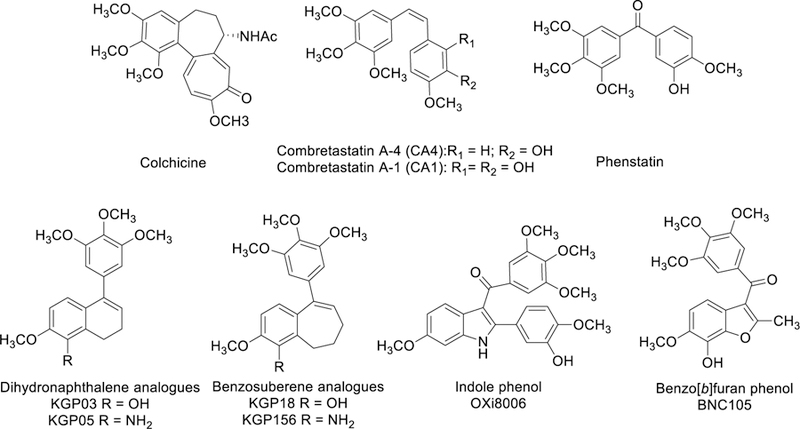
Representative small-molecule inhibitors of tubulin polymerization: colchicine,21 combretastatins (CA4, CA1),15,23 phenstatin, dihydronaphthalene analogues (KGP03, KGP05),35,36 benzosuberene analogues (KGP18, KGP156),8,25,37,38 indole analogue (OXi8006),39,50 and benzo[b]furan analogue (BNC105).51–53
Two benzosuberene-based analogues (referred to as KGP188,25,37 and its amino congener KGP15635,38) emerged from our previous studies as molecules with high relevance as potential preclinical candidates due, in part, to their potent inhibition of tubulin polymerization, pronounced cytotoxicity against human cancer cell lines, and promising activity as VDAs.8,25,38
Studies by us and others have investigated a variety of functional group modifications on both the fused and pendant aryl rings of tubulin-binding benzosuberene and dihydronaphthalene molecular frameworks (Fig. 2).8,25,35,36,54,55 In particular, Maderna and co-workers contributed efficient methodology towards the benzosuberene structural motif and presented a number of important B-ring modified benzosuberene analogues (Fig. 2) along with an evaluation of their biological potency and their drugability characteristics.56,57 Interestingly, a benzosuberene B-ring diene analogue was identified as one of the most potent cytotoxic agents amongst their synthesized series of eleven members,57 and this same molecule (Maderna compound, listed as 68 herein) was obtained during our synthetic campaign as an unexpected product. It is also noteworthy that a new class of benzodiazepines has been reported as inhibitors of tubulin polymerization.58
Figure 2.

Selected KGP18 derivatives as inhibitors of tubulin polymerization with modifications at the C-4 position of the A ring and the C-6, 7, 8 positions of the B ring.25,57
Herein we designed, synthesized and biologically evaluated a series of structurally varied analogues using KGP18 and KGP05 as lead compounds and further adopted our methodology to investigate functional group modifications on the A-ring (C-4 position) and the B-ring (C-6, 7, 8, 9 positions), along with regiochemical translocation of the pendant aryl ring (C-ring) in regard to their influence on inhibition of tubulin polymerization and cytotoxicity against several human cancer cell lines.
Results and Discussion
Molecular Design and Synthesis
Eighteen benzosuberene and dihydronaphthalene analogues (Fig. 3) were prepared by chemical synthesis and evaluated for their cytotoxicity against selected human cancer cell lines and their ability to inhibit tubulin polymerization. Structural modifications included: 1) functional group (R) modifications on the fused aryl ring including the installation of alcohol, aldehyde, nitrile, and ester groups along with ether linkages to facilitate extension of the polar alcohol moiety away from the fused six-seven ring system; 2) R2 and R3 incorporation at the olefinic and allylic positions of the seven-membered ring introduced -Br, -OH, and -NHAc groups; 3) modification (R4 position) of the fused aliphatic ring adjacent to the tertiary alcohol site; 4) olefination and pendant trimethoxy phenyl ring regiochemistry on the fused non-aromatic ring. The synthesis of analogues 3, 6, 8, 11, 13, 15 was initiated from a common intermediate ketone 1 that was readily available utilizing our previous methodolgy.25 Treatment of benzosuberone 1 with trimethoxyphenyllithium (prepared from the corresponding bromide) generated tertiary alcohol 2, which was subsequently converted to diol 3 upon removal of the phenolic TBS protecting group. Separately, tertiary alcohol 2 was converted to its corresponding benzosuberene 4, which underwent treatment with a series of oxidants (m-CPBA, NBS, OsO4) to facilitate epoxidation followed by ring opening and oxidation, bromination, and Upjohn dihydroxylation.59,60 Following the removal of protecting groups, target compounds 6, 13, and 15 were obtained. Similarly, reaction of 4-methylbenzosuberene 12 (prepared by us previously25) with NBS/AIBN afforded vinylbromide 13. Lead compound 7 (referred to as KGP18), also available through this methodology, was directly converted to its corresponding ether analogues 8 and 11 (Scheme 1).25,61
Figure 3.
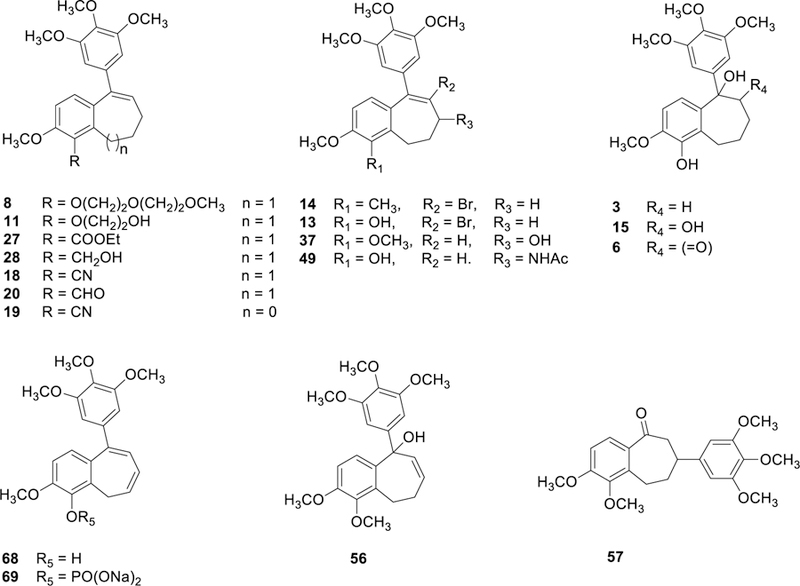
Compilation of synthesized benzosuberene, benzosuberane, benzosuberone, and dihydronaphthalene analogues
Scheme 1.

Synthesis of benzosuberene and benzosuberane intermediates and analogues.
Lead compounds benzosuberene 16 (referred to as KGP156) and its corresponding dihydronaphthalene analogue 17 (referred to as KGP05), which were readily available from our previous synthetic studies, were subjected to a Sandmeyer radical-nucleophilic aromatic substitution protocol to generate nitrile analogues 18 and 19 (Scheme 2).35,38,62,63 The benzosuberene aldehyde analogue 20 was obtained after subsequent reduction of the nitrile analogue (Scheme 2).
Scheme 2.

Synthesis of nitrile- and aldehyde- substituted R position analogues 18–20.
As part of a larger program focused on the use of potently cytotoxic benzosuberene and dihydronaphthalene analogues as payloads in antibody-drug conjugates (ADCs) and as prodrugs targeted for selective release in regions of profound tumor hypoxia, we were intrigued by the possibility of replacing the heteroatom [oxygen (phenol) or nitrogen (aniline)] at the 4-position with a short carbon chain terminating with a primary alcohol (or amino) moiety, thus maintaining its hydrogen-bond donor nature and serving as a viable position for future attachment of various linkers. Accordingly, methylation of phenolic bromo-aldehyde 21 (Scheme 3), followed by Wittig olefination and subsequent hydrogenation (under Ph2S mediation to maintain the aryl bromine group) afforded methyl ester 24.64 Benzosuberone 25 was obtained through an intramolecular Friedel-Crafts annulation facilitated by Eaton’s reagent (7.7% weight percent P2O5 in CH3SO3H), and subsequent treatment of compounds 25 with trimethoxyphenyllithium, followed by reaction work-up under acidic conditions, generated benzosuberene intermediate 26.65,66 We have previously evaluated corresponding fluorine and chlorine benzosuberene analogues.25 Halogen-lithium exchange, followed by reaction with ethyl chloroformate, afforded ethyl ester 27, which was reduced (LiAlH4) to generate benzylic alcohol 28. Notably, while it proved possible to obtain analogue 28 directly from intermediate 26, the isolated yield was quite low (≤ 8%) under these conditions, which was likely due, in part, to the low solubility of paraformaldehyde at low temperature in THF and its low reactivity as a polymer (Scheme 3).
Scheme 3.

Synthesis of ester analogue 27 and primary alcohol analogue 28.
Secondary allylic alcohol 37 and its corresponding N-acetyl congener 49 (Scheme 4) were prepared to investigate structure-activity relationship correlations associated with heteroatom incorporation on the conformationally flexible fused seven-membered ring. Appropriate aldehyde chain elongations were facilitated by Meldrum’s acid (towards 37) and Wittig-ylide methodology (towards 49), and subsequent functional group transformations (including installation of carboxylic acid moieties obtained under saponification conditions) afforded protected alcohol 34 and N-acetamide 45, separately. Lewis acid mediated cyclization to obtain the benzosuberone molecular core was achieved by treatment of the requisite acyl chloride with either tin tetrachloride (to obtain ketone 35) or Eaton’s reagent (to obtain ketone 46). In each case, the pendant ring was installed through reaction with 3,4,5-trimethoxyphenyllithium. The secondary alcohol moiety (target compound 37) was revealed (by deprotection) prior to the organolithium step, while the phenolic moiety (target compound 49) was revealed after the organolithium reaction (Scheme 4).8,67–69 Both compounds 36 and 37 undergo elimination first (allylic alcohol), then demethylation if treated with a Lewis acid such as AlCl3 or BCl3.
Scheme 4.

Synthesis of allylic alcohol analogue 37 and allylic N-acetamide analogue 49.
Translocation of the trimethoxyphenyl group (Scheme 5) was achieved by initial cyclization (Eaton’s reagent) of carboxylic acid 54 with concomitant elimination to afford a,b-unsaturated ketone 55. With the α,β-unsaturated ketone in hand, 1,2- and 1,4- addition reactions were conducted using the appropriate aryl-lithium and Gilman reagents to provide tertiary alcohol analogue 56 (with unsaturated 7-membered ring to maintain rigidity), and separately the Michael adduct, trimethoxy pendant phenyl ring shifted analogue 57 (Scheme 5).70
Scheme 5.

Synthesis of α-unsaturated tertiary alcohol analogue 56 and pendant ring shifted analogue 57.
During the course of an investigation centered on variability in phenolic moiety protecting groups for a subset of benzosuberene analogues, we were surprised by the formation of diene 67, obtained upon 1,2-addition of trimethoxyphenyllithium to ketone 66, followed by reaction work-up. In this case, the secondary alcohol demonstrated a propensity to undergo elimination even under mild acidic or basic conditions such as TBAF deprotection or BCl3 cleavage at lowered temperature. We attempted various combinations of 4-position (phenolic moiety on the fused aryl ring) and allylic alcohol protecting group strategies, which eventually led to the unanticipated formation of diene 68. It is important to note that this diene (68) was previously obtained by Maderna and co-workers.57 Having this compound in hand, and noting its exceptional biological activity (inhibition of tubulin polymerization and cytotoxicity against human cancer cell lines, Table 1),57 motivated us to prepare the corresponding water-soluble phosphate prodrug disodium salt 69 to facilitate in vivo studies in a mouse model of prostate cancer to evaluate the efficacy of this compound as a VDA, as evidenced by bioluminescence imaging (BLI) (Scheme 6).8,25,57,68,69
Table 1.
Inhibition of tubulin polymerization, percent inhibition of colchicine binding, and cytotoxicity of the benzosuberene and dihydronaphthalene analogues.
| Compound | Inhibition of tubulin polymerization IC50 (μM) ± SD | % Inhibition of colchicine binding ± SD | GI50 (μM) SRB assaya | ||
|---|---|---|---|---|---|
| SK-OV-3 | NCI-H460 | DU-145 | |||
| CA4 | 1.0b | 84 ± 3 (1 μM), 98 ± 0.007 (5 μM) | 0.00455 | 0.00223c | 0.00327c |
| CA4P | >40b | ND | 0.00119 | 0.00194c | 0.00323c |
| KGP18 | 0.85 ± 0.02d | 73 ± 5 (1 μM), 95 ± 0.5 (5 μM) | 0.0000543e | 0.0000418e | 0.0000249e |
| KGP03 | 0.5f | 90 ± 2 (1 μM), 98 ± 0.3 (5 μM) | 0.0029f | 0.0032f | 0.00040f |
| Doxorubicin | ND | ND | 0.0789 | 0.123 | 0.134 |
| Paclitaxel | NR | NR | 0.00134 | 0.00176 | 0.00147 |
| 3 | >20 | ND | 0.394 | 0.173 | 0.0330 |
| 6 | 1.2 ± 0.1 | 72 ± 2 (5 μM) | 0.0314 | 0.0476 | 0.141 |
| 8 | >20 | ND | 6.52 | 1.90 | 5.66 |
| 11 | >20 | ND | 3.22 | 0.855 | 3.76 |
| 13 | 0.39 ± 0.06 | 88 ± 1 (5 μM) | 0.0221 | 0.0353 | 0.0362 |
| 14 | 4.9 ± 0.1 | 35 ± 5 (1 μM) | 0.312 | 0.449 | 0.423 |
| 15 | >20 | 0 (5 μM) | 3.03 | 3.86 | 4.04 |
| 18 | 1.1 ± 0.1 | 62 ± 0.7 (5 μM) | 0.0648 | 0.434 | 0.860 |
| 19 | 0.37 ± 0.08 | 73 ± 0.6 (5 μM) | 0.0384 | 0.0605 | 0.0252 |
| 20 | 3.2 ± 0.08 | 61 ± 3 (5 μM) | 0.0572 | 0.0847 | 0.0402 |
| 27 | 1.0 ± 0.07 | 47 ± 1 (5 μM) | 0.299 | 0.353 | 0.631 |
| 28 | 0.63 ± 0.03 | 76 ± 2 (5 μM) | 0.0403 | 0.0628 | ND |
| 37 | 3.8 ± 0.5 | 28 ± 4 (5 μM) | 0.263 | 0.432 | 0.439 |
| 49 | >20 | 0.4 ± 0.6 (5 μM) | ND | 0.334 | 1.20g |
| 56 | >20 | 0 (5 μM) | 27.2 | 70.5 | 26.0 |
| 57 | >20 | 0 (5 μM) | 5.77 | 2.00 | 4.45 |
| 68 | 0.48 ± 0.08 | 68 ± 1 (0.5 μM), 95 ± 0.8 (5 μM) | 0.00690 | 0.0581 | 0.0976 |
| 69 | 16 ± 0.7 | 45 ± 2 (5 μM) | 0.0153 | 0.0291 | 0.0239 |
Average of n ≥ 3 independent determinations (unless otherwise noted)
For additional data, see ref. 2424
For additional data, see ref. 3737
For additional data, see ref. 88
Average of n = 2 independent determinations (of duplicates)
ND = Not Determined
NR = Not Relevant (paclitaxel enhances microtubule assembly)
Scheme 6.

Synthesis of benzosuberdiene 68 and phosphate salt 69.
Biological Evaluation
Each of the eighteen compounds (Fig. 3) was evaluated for its cytotoxicity against human cancer cell lines [SK-OV-3 (ovarian), NCI-H460 (lung), DU-145 (prostate)] and for the ability to inhibit tubulin polymerization (Table 1). Ten of the evaluated molecules were identified as strong inhibitors of tubulin assembly (IC50 < 5 μM, cell-free assay), while seven of the ten were highly active (IC50 ≤ 1.2 μM). CA4 (IC50 ≈ 1 μM) and KGP18 (compound 7, IC50 ≈ 0.85 μM) were utilized as comparative compounds. Three of the benzosuberene and dihydronaphthalene analogues (compounds 13, 19), along with the Maderna compound (68), were more potent against tubulin in comparison to our dihydronaphthalene lead compound KGP03 (IC50 ≤ 0.5 μM). Excellent IC50 values for inhibition of tubulin polymerization were retained upon alteration of the 4-position phenolic moiety into nitrile, ethyl ester, and CH2OH groups (18, 19, 27, 28), along with modifications to the double bond on the seven-member ring that included replacement with ketone and tertiary alcohol groups, substitution with a bromine group, and increased unsaturation (6, 13, 68). Extension of the alkyl chain (at position 4) through an ether linkage that terminated with a polar alcohol group (compound 11) and separately a methoxy moiety (compound 8) resulted in loss of inhibitory activity. Incorporation of a hydrogen bond donor at the allylic position on the seven-member ring (compounds 37 and 49) and substituted saturation of the double bond (compounds 3 and 15) both reduced inhibition of tubulin polymerization. The lack of tubulin activity observed with compound 56 was unanticipated, since semi-rigidity of the seven-membered ring was maintained through a double bond one carbon removed from the stereogenic center, and its parent benzosuberene analogue (dimethoxy group on the fused aryl ring) demonstrated a moderate degree of inhibition of tubulin polymerization (IC50 = 3.1 μM) reported in our previous study.25 The lack of activity of compound 57 in regard to inhibition of tubulin polymerization suggested that the trimethoxy pendant aryl ring situated at the benzylic position on the fused-ring system was closely correlated to biological efficacy (at least in regard to inhibition of tubulin polymerization). While we have previously described similar pendant ring shifts in related benzosuberene analogues, this β-position substitution with a trimethoxy aryl ring has not been previously investigated.8
Among the eighteen benzosuberene and dihydronaphthalene analogues investigated in this study (Fig. 3), the most cytotoxic agents were compounds 6, 13, 18, 19, 28, 68 (GI50 = 0.0314, 0.0221, 0.0648, 0.0384, 0.0403 μM, and 0.00690 μM, respectively, against the SK-OV-3 ovarian cancer cell line, for example). Judiciously selected structural modifications to the 4, 8, and 9-positions in the parent benzosuberene scaffold accounted for the majority of the highly potent analogues evaluated in this study. While the strong cytotoxicity of this sub-set of molecules is encouraging, it is noteworthy that all molecules proved less cytotoxic than the lead benzosuberene KGP18 and less cytotoxic (with the partial exception of the Maderna compound 68) than the natural product CA4, despite demonstrating similar inhibition of tubulin polymerization (cell free assay). These observations provided an important extension to the known SAR considerations regarding structural modifications to KGP18. As anticipated (and similarly observed for combretastatin A-4 phosphate),15,71 the benzosuberene phosphate prodrug salt 69 was inactive as an inhibitor of tubulin polymerization in this cell-free assay, presumably due to the lack of phosphatase enzymes necessary to cleave the prodrug to its parent phenolic (biologically active) agent. Prodrug 69 was an active cytotoxic agent since non-specific phosphatase activity is present in these cancer cell-based cytotoxicity assays.72,73
Assessment of Vascular Damage
Ultimately, VDAs will be used in vivo, and thus it is crucial to understand both efficacy of vascular disruption and potential off target toxicity in vivo. As a preliminary investigation, the extent of vascular damage was assessed in a human prostate tumor line in rats treated with the water-soluble prodrug salt 69 compared to CA4P as control. Many imaging methods have been developed to assess vascular disruption non-invasively in vivo.11 We favor dynamic bioluminescence imaging (BLI) for initial validation of VDA activity since it provides a fast, non-invasive and easy method and allows comparison of repeat or sequential investigations. BLI does require the use of cells transfected to express luciferase (luc), but these are commonly available and we have used this approach extensively11,25,39,53,75 as have others.76 We used the human prostate cancer PC3 line, in which the tumor suppressor protein DAB2IP had been knocked down and luciferase introduced.77 BLI requires administration of luciferin substrate, which readily crosses membranes and is carried throughout the vasculature. The measurement of light emission dynamics is related to vascular delivery of the luciferin substrate, and thus it provides a measure of vascular patency.78 Disruption of tumor vasculature blocks delivery of the substrate and consequently results in a quantifiable decrease in bioluminescent signal. The extent of vascular shutdown was evaluated using IP doses of 10 and 40 or 80 mg/kg of 69 and compared to a dose of 30 mg/kg of CA4P, which had been shown previously to cause extensive vascular shutdown in rats at this dose.79–81 It should also be noted that the lead benzosuberene KGP18 and the dihydronaphthalene KGP03 (both as their corresponding water-soluble phosphate prodrug salts (KGP265 and KGP04, respectively), along with other structurally modified benzosuberene analogues, demonstrated vascular shutdown (as evidenced by similar BLI imaging studies or color Doppler ultrasound).25,37,75
Prodrug 69 administered at 10 mg/kg resulted in minimal change in light emission. A subsequent dose of 40 mg/kg 69 resulted in substantially diminished light emission (signal reduction 95%; Figs. 4 and 5), but with substantial recovery by 24 h. When CA4P (30 mg/kg) was administered 4 days later it caused a very similar effect in terms of extent and longevity of diminished BLI signal as a surrogate for vascular shutdown. Similar activity was observed when a treatment naïve rat was given 40 mg/kg 69 (Fig. 5) with substantial recovery at 24 h. At 48 h the rats appeared healthy and the signal increased marginally. A dose of 80 mg/kg was also well tolerated, but this higher dose did not show additional vascular disruption.
Figure 4.

BLI assessment of vascular response to VDA. Left) Heat maps overlaid on photographs of male Copenhagen rat with subcutaneous PC3-DAB2IP-luc human prostate tumor xenograft showing light emission about 20 min after administration of D-luciferin (120 mg/kg) at various times with respect to VDA administration IP. Right) Corresponding dynamic light emission curves acquired over about 30 min following luciferin administration. Blue shows baseline; red about 4 h after VDA and green at 24 h. A) 10 mg/kg compound 69 indicating no vascular perturbation, but increased signal at 24 h consistent with rapid tumor growth. B) 6 h later 40 mg/kg compound 69 was administered to the same rat generating about 95% reduced signal at 4 h, consistent with substantial vascular shutdown and showing substantial recovery by 24 h. C) Four days later 30 mg/kg CA4P was administered to this rat eliciting BLI response similar to the BLI response shown in B.
Figure 5.
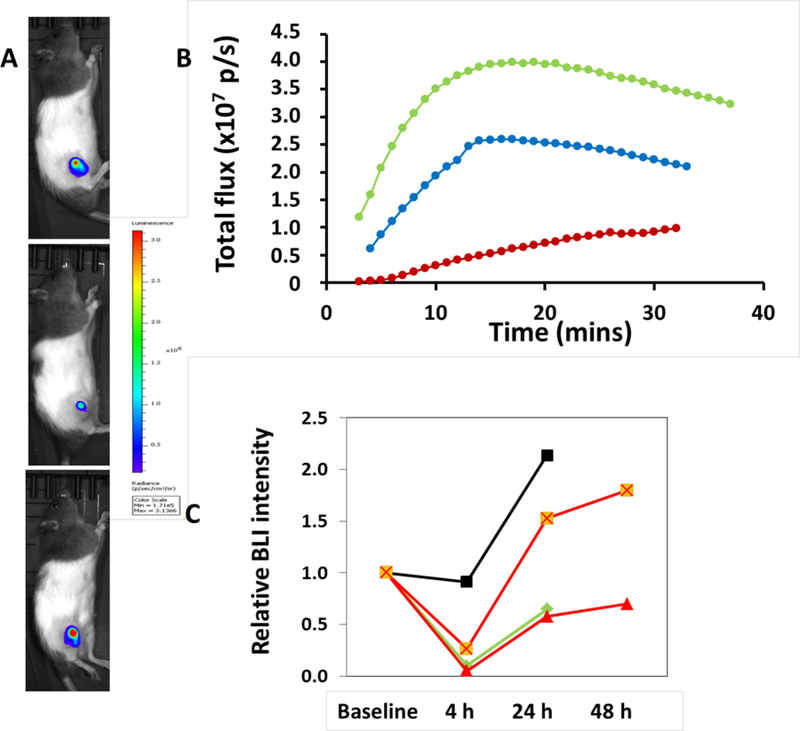
Relative light emission following administration of VDAs. A) Relative signal intensity is shown about 20 min after administration of D-luciferin subcutaneously in the foreback neck region of a rat with a subcutaneous PC3-DAB2IP-luc prostate tumor xenograft in the thigh. Top) baseline (no prior drug), center) 4 h after 40 mg/kg 69, and bottom) 24 h after 69. B) Corresponding light emission dynamic curve at baseline (blue), 4 h after 69 (red) and 24 h after 69 (green). C) Normalized BLI signal at various times for the rat in Fig 4 receiving 69 sequentially at 10 mg/kg (black), 40 mg /kg (red) and 30 mg/kg CA4P (green), together with the treatment naive rat in Fig 5 A, B receiving 40 mg/kg 69 (orange).
Conclusions:
These studies have expanded our SAR knowledge regarding the impact of structural modifications to lead benzosuberene and dihydronaphthalene analogues on inhibition of tubulin polymerization and cytotoxicity against human cancer cell lines. Amongst this group of seventeen new molecules [along with the Maderna compound (68), accessed (in this study) through separate synthesis], emerged several promising analogues (compounds 6, 13, 18, 19, 28, 68) that elicited inhibition (IC50) of tubulin assembly (cell free assay) greater than or comparable to that of the lead natural product CA4 and our lead benzosuberene analogues KGP18 and KGP156. These compounds demonstrated potent cytotoxicity (GI50) against SK-OV-3 (ovarian), NCI-H460 (lung), and DU-145 (prostate) cells typically in the low to mid nM range. Preliminary investigation of water-soluble benzosuberene phosphate prodrug salt 69 at 40 mg/kg in vivo revealed vascular disruption in a PC3-DAB2IP-luc human prostate tumor xenograft based on BLI (Figs. 4 and 5), which was similar to that obtained with CA4P.
Experimental Section
Chemistry
General Materials and Methods
Tetrahydrofuran (THF), carbon tetrachloride, dichloromethane, methanol, dimethylformamide (DMF), and acetonitrile were used in their anhydrous forms. Reactions were performed under nitrogen gas. Thin-layer chromatography (TLC) plates (precoated glass plates with silica gel 60 F254, 0.25 mm thickness) were used to monitor reactions. Purification of intermediates and products was carried out with a Biotage Isolera flash purification system using silica gel (200–400 mesh, 60 Å) or RP-18 pre-packed columns or manually in glass columns. Intermediates and products synthesized were characterized on the basis of their 1H NMR (500 or 600 MHz), 13C NMR (125 or 150 MHz) spectroscopic data using a Varian VNMRS 500 MHz or Bruker DPX 600 MHz instrument. Spectra were recorded in CDCl3, D2O, (CD3)2CO, or CD3OD. All chemicals shifts are expressed in ppm (δ), and peak patterns are reported as broad (br), singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), sextet (sext), septet (sept), double doublet (dd), double double doublet (ddd), and multiplet (m).
Purity of the final compounds was further analyzed at 25 °C using an Agilent 1200 HPLC system with a diode-array detector (λ = 190−400 nm), a Zorbax XDB-C18 HPLC column (4.6 mm Å~ 150 mm, 5 μm), and a Zorbax reliance cartridge guard-column; Method: solvent A, acetonitrile, solvent B, H2O; gradient, 10% A/ 90% B to 100% A/ 0% B over 0 to 40 min; post-time 10 min; flow rate 1.0mL/min; injection volume 20 μL; monitored at wavelengths of 210, 230, 254, 280, and 320 nm. Purity of target molecules (with reported biological data) was ≥ 95% (as determined by HPLC at one or more scanned wavelengths) with the exception of compound 27 (94.3% at 254 nm). Mass spectrometry was carried out under positive or negative ESI (electrospray ionization) using a Thermo Scientific LTQ Orbitrap Discovery instrument.
1-((Tert-butyldimethylsilyl)oxy)-2-methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-ol (2)25
To an oven dried flask, THF (10 mL) and 3, 4, 5-trimethoxyphenyl bromide (0.89 g, 3.6 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (1.44 mL, 3.60 mmol) was added dropwise to the reaction mixture, which was then stirred at −78 °C for 1 h. TBS-protected ketone (1) (0.77 g, 2.4 mmol) in THF (5 mL) was then added slowly to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was quenched with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 2 (1.05 g, 2.15 mmol, 89%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 7.15 (1H, d, J = 9 Hz), 6.69 (1H, d, J = 9 Hz), 6.50 (2H, s), 3.84 (3H, s), 3.80 (3H, s), 3.75 (6H, s), 3.29 (1H, m), 2.56 (1H, m), 2.26 (1H, m), 2.12 (2H, m), 1.90 (1H, m), 1.75 (2H, m), 0.99 (9H, s), 0.17 (3H, s), 0.15 (3H, s). 13C NMR (125 MHz, CDCl3) δ 153.1, 149.4, 142.0, 141.9, 138.7, 137.3, 132.9, 119.8, 108.0, 104.4, 80.2, 61.0, 56.2, 54.8, 41.4, 27.1, 26.4, 26.2, 25.5, 19.1, −3.8, −4.0.
2-Methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulene-1,5-diol (3)25
TBS-protected tertiary alcohol 2 (0.41 g, 0.84 mmol) was dissolved in THF (6 mL), and TBAF (1.01 mL, 1 M in THF, 1.01 mmol) was added, and the reaction mixture was stirred at room temperature for 4 h. The solution was washed with water and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 3%A / 97%B (1 CV), 3%A / 97%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford phenol (0.11 g, 0.29 mmol, 35%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.04 (1H, d, J = 9 Hz), 6.70 (1H, d, J = 9 Hz), 6.52 (2H, s), 5.79 (1H, s), 3.91 (3H, s), 3.84 (3H, s), 3.76 (6H, s), 3.23 (1H, m), 2.56 (1H, m), 2.35 (1H, m), 2.11 (1H, m), 1.92 (1H, m), 1,75 (2H, m), 1.47 (1H, m). 13C NMR (125 MHz, CDCl3) δ 153.1, 145.6, 142.7, 141.9, 139.4, 137.3, 127.2, 118.2, 107.3, 104.4, 80.2, 61.0, 56.3, 56.0, 41.5, 26.8, 26.3, 24.7. HRMS: Obsvd 397.1623 [M + Na+], Calcd for C21H26O6Na: 397.1622. HPLC: 16.33 min.
Tert-butyl((3-methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)dimethylsilane (4)37
TBS-protected tertiary alcohol 2 (0.64 g, 1.3 mmol) was dissolved in acetic acid (10 mL), and the reaction mixture was stirred at room temperature for 6 h. The unreacted acetic acid was removed under reduced pressure. The resulting reaction mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine, dried over sodium sulfate, evaporated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford a clear oil that solidified as a colorless solid of TBS-protected benzosuberene analogue 4 (0.41 g, 0.87 mmol, 66%). 1H NMR (CDCl3, 500 MHz) δ 6.68 (1H, d, J = 8.5 Hz), 6.61 (1H, d, J = 8.5 Hz), 6.48 (2H, s), 6.32 (1H, t, J = 7 Hz), 3.85 (3H, s), 3.81 (3H, s), 3.79 (6H, s), 2.76 (2H, t, J = 7 Hz), 2.10 (2H, m), 1.95 (2H, m), 1.04 (9H, s), 0.23 (6H, s). 13C NMR (CDCl3, 125 MHz) δ 152.9, 148.8, 143.2, 141.6, 138.8, 137.3, 133.9, 133.4, 127.0, 122.5, 108.5, 105.3, 61.0, 56.2, 54.8, 34.1, 26.3, 25.7, 24.4, 19.2, −3.7.
1-(Tert-butyldimethylsilyl)oxy)-5-hydroxy-2-methoxy-5-(3,4,5-trimethoxyphenyl)-5,7,8,9-tetrahydro-6H-benzo[7]annulen-6-one (5)
To a solution of TBS-protected benzosuberene 4 (0.51 g, 1.1 mmol) dissolved in CH2Cl2 (20 mL) was added m-CPBA (0.36 g, 2.1 mmol) at −5 °C, and the reaction mixture was stirred for 2 h and then at room temperature for 12 h. The solution was washed with saturated Na2S2O3 and saturated NaHCO3 and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 5 (0.256 g, 0.51 mmol, 47%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.26 (1H, d, J = 9 Hz), 6.76 (1H, d, J = 9 Hz), 6.42 (2H, s), 5.01 (1H, s), 3.83 (3H, s), 3.81 (3H, s), 3.75 (6H, s), 3.10 (2H, m), 2.82 (1H, m), 2.70 (1H, m), 1.98 (1H, m), 1.76 (1H, m). 13C NMR (150 MHz, CDCl3) δ 211.1, 153.4, 150.2, 142.1, 138.0, 137.5, 131.4, 131.1, 127.8, 109.1, 105.2, 85.6, 61.0, 56.3, 54.8, 39.7, 26.2, 25.8, 24.2, 19.1, −3.7, −3.9.
1,5-Dihydroxy-2-methoxy-5-(3,4,5-trimethoxyphenyl)-5,7,8,9-tetrahydro-6H-benzo[7]annulen-6-one (6)
TBS-protected benzosuberane 5 (0.17 g, 0.33 mmol) was dissolved in THF (10 mL). TBAF (0.33 mL, 1 M, 0.33 mmol) was added, and the reaction mixture was stirred at room temperature for 1 h at 0 °C. A brine (30 mL) solution was added, and the reaction mixture was extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford phenol 6 (123 mg, 0.320 mmol, 96%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.17 (1H, d, J = 8.4 Hz), 6.77 (1H, d, J = 9 Hz), 6.43 (2H, s), 5.88 (1H, s), 5.00 (1H, s), 3.90 (3H, s), 3.82 (3H, s), 3.74 (6H, s), 3.08 (2H, m), 2.84 (1H, m), 2.68 (1H, m), 1.99 (1H, m), 1.83 (1H, m). 13C NMR (150 MHz, CDCl3) δ 211.0, 153.3, 146.4, 142.8, 138.0, 137.2, 131.6, 125.7, 120.2, 108.4, 105.2, 85.5, 60.9, 56.3, 56.0, 39.4, 25.4, 23.3. HRMS: Obsvd 411.1414 [M + Na+], Calcd for C21H24O7Na: 411.1414. HPLC: 15.75 min.
3-Methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (7)
TBS-protected benzosuberene 4 (0.41 g, 0.87 mmol) was dissolved in THF (10 mL). TBAF (1.13 mL, 1.13 mmol) was added, and the reaction mixture was stirred at room temperature for 1 h. The solution was washed with water and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 3%A / 97%B (1 CV), 3%A / 97%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford phenol 7 (0.25 g, 0.70 mmol, 81%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 6.71 (1H, d, J = 9 Hz), 6.56 (1H, d, J = 9 Hz), 6.50 (2H, s), 6.34 (1H, t, J = 7.5 Hz), 5.74 (1H, s), 3.91 (3H, s), 3.86 (3H, s), 3.80 (6H, s), 2.76 (2H, t, J = 7 Hz), 2.14 (2H, m), 1.97 (2H, m). 13C NMR (CDCl3, 125 MHz) δ 152.9, 145.2, 142.9, 142.4, 138.6, 134.4, 127.9, 127.4, 121.0, 110.1, 107.8, 105.4, 61.1, 56.3, 56.1, 33.7, 25.9, 23.7.
3-Methoxy-4-(2-(2-methoxyethoxy)ethoxy)-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (8)
Phenol 7 (0.11 g, 0.31 mmol) was dissolved in DMF (6 mL), and K2CO3 (0.12 g, 0.86 mmol) was added. The solution was stirred at room temperature for 20 min. 1-Bromo-2-(2-methoxyethoxy) ethane in 90% purity (0.07 mL, 0.5 mmol) was added, the reaction mixture was stirred at room temperature for 15 h, followed by an additional 3 h of stirring at 150 °C. The solution was washed with water and extracted with EtOAc (3 × 40 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 3%A / 97%B (1 CV), 3%A / 97%B → 30%A / 70%B (20 CV), 30%A / 70%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford ether 8 (25 mg, 0.06 mmol, 18%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 6.74 (2H, m), 6.48 (2H, s), 6.32 (1H, t, J = 7.5 Hz), 4.18 (2H, t, J = 5 Hz), 3.87 (2H, t, J = 5.5 Hz), 3.85 (6H, s), 3.79 (6H, s), 3.76 (2H, m), 3.60 (2H, m), 3.40 (3H, s), 2.78 (2H, t, J = 6.5 Hz), 2.13 (2H, m), 1.93 (2H, m). 13C NMR (125 MHz, CDCl3) δ 152.9, 151.5, 145.1, 142.9, 138.5, 137.4, 136.1, 133.8, 127.3, 125.3, 109.3, 105.3, 72.8, 72.2, 70.78, 70.77, 61.0, 59.2, 56.2, 55.7, 34.5, 25.7, 24.2. HRMS: Obsvd 481.2198 [M + Na+], Calcd for C26H34O7Na: 481.2197. HPLC: 21.65 min.
((8-Bromo-3-methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)(tert-butyl)dimethylsilane (9)
To a solution of TBS-protected benzosuberene (102 mg, 0.22 mmol) in CCl4 (30 mL) was added NBS (46 mg, 0.26 mmol) and AIBN (3.6 mg, 0.02 mmol). The solution was heated at reflux for 2 h, followed by the addition of water (20 mL) and extraction with CH2Cl2 (3 × 30 mL). The combined organic phase was dried over sodium sulfate, filtered, and the solvent was removed under reduced pressure. The crude product was obtained as a yellow oil and taken to the next step directly without any further purification.
1-((Tert-butyldimethylsilyl)oxy)-2-methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulene-5,6-diol (10)
To a solution of TBS-protected benzosuberene 4 (1.00 g, 2.12 mmol) in acetone/ water (35 mL/ 15 mL) were added OsO4 (270 mg, 1.06 mmol) and N-methylmorpholine-N-oxide (0.66 mL, 4.8 M, 3.2 mmol) at room temperature, and the reaction mixture was stirred for 12 h. A saturated sodium hydrosulfite (20 mL) solution was added, and the reaction mixture was extracted with EtOAc (5 × 20 mL). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford diol 10 (0.35 mg, 0.69 mmol, 33%) as an orange oil. 1H NMR (600 MHz, CDCl3) δ 7.30 (1H, d, J = 8.4 Hz), 6.74 (1H, d, J = 9 Hz), 6.47 (2H, s), 4.51 (1H, s, b), 3.81 (3H, s), 3.80 (3H, s), 3.72 (6H, s), 3.42 (1H, m), 3.33 (1H, s), 2.15 (1H, m), 1.96 (2H, m), 1.83 (1H, m), 1.62 (1H, m), 1.51 (1H, m), 0.98 (9H, s), 0.15 (6H, d, J = 3.6 Hz). 13C NMR (150 MHz, CDCl3) δ 153.1, 149.8, 142.0, 138.8, 137.7, 133.1, 132.7, 122.3, 108.5, 105.0, 83.1, 76.5, 60.9, 56.2, 54.7, 32.7, 26.2, 25.9, 21.3, 19.0, −3.9, −4.0.
2-((3-Methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)oxy)ethan-1-ol (11)
Phenol 7 (0.14 g, 0.39 mmol) was dissolved in DMF (3 mL), then ethylene carbonate (70 mg, 0.79 mmol) and tetrabutyl ammonium bromide (0.13 g, 0.39 mmol) were added together. The solution was stirred and heated at reflux for 24 h. The reaction mixture was diluted with brine, extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford alcohol 11 (0.11 g, 0.27 mmol, 68%). 1H NMR (500 MHz, CDCl3) δ 6.76 (2H, m), 6.47 (2H, s), 6.33 (1H, t, J = 7.5 Hz), 4.12 (2H, m), 3.89 (2H, m), 3.88 (3H, s), 3.84 (3H, s), 3.79 (6H, s), 2.75 (2H, t, J = 7 Hz), 2.15 (2H, m), 1.95 (2H, m). 13C NMR (125 MHz, CDCl3) δ 152.9, 151.1, 145.0, 142.8, 138.3, 137.4, 136.1, 134.2, 127.3, 125.7, 109.2, 105.3, 76.1, 62.2, 61.0, 56.2, 55.8, 34.6, 25.6, 24.6. HRMS: Obsvd 423.1780 [M + Na+], Calcd for C23H28O6Na: 423.1778. HPLC: 13.77 min.
8-Bromo-3-methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-ol (13)
Brominated benzosuberene 9 (0.12 g, 0.22 mmol, crude) was dissolved in THF (20 mL), and TBAF (0.22 mL, 1 M, 0.22 mmol) was added to the solution at 0 °C. The reaction mixture was stirred for 1 h, washed with brine (20 mL), and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate, filtered, and evaporated under reduced pressure. The resulting material was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford brominated phenol 13 (97 mg, 0.22 mmol, 100% over two steps) as a white crystalline solid. 1H NMR (600 MHz, CDCl3) δ 6.63 (1H, d, J = 8.4 Hz), 6.45 (2H, s), 6.41 (1H, d, J = 8.4 Hz), 5.74 (1H, s), 3.88 (3H, s), 3.87 (3H, s), 3.80 (6H, s), 2.88 (2H, t, J = 7.2 Hz), 2.58 (2H, t, J = 7.2 Hz), 2.26 (2H, m). 13C NMR (150 MHz, CDCl3) δ 152.7, 145.5, 142.5, 140.8, 137.9, 137.3, 135.3, 126.4, 121.5, 121.1, 108.0, 107.5, 61.0, 56.3, 56.1, 38.5, 32.5, 23.2. HRMS: Obsvd 457.0621 [M + Na+], Calcd for C21H23BrO5Na: 457.0621. HPLC: 17.54 min.
8-Bromo-3-methoxy-4-methyl-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (14)
KGP391 12 (68 mg, 0.19 mmol) was dissolved in CCl4 (20 mL), and NBS (37 mg, 0.21 mmol) and AIBN (3.1 mg, 0.02 mmol) were added carefully avoiding shaking or metal spatula since AIBN can be explosive. The reaction mixture was refluxed and stirred for 2 h. The solution was washed with water and extracted by CH2Cl2, the organic phase was further washed by brine and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 6%A / 94%B (1 CV), 6%A / 94%B → 70%A / 30%B (10 CV), 70%A / 30%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford brominated benzosuberene analogue 14 (66 mg, 0.15 mmol, 80% ) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.70 (1H, d, J = 7 Hz), 6.62 (1H, d, J = 7 Hz), 6.47 (2H, s), 3.88 (3H, s), 3.81 (9H, s), 2.81 (2H, m), 2.53 (2H, m), 2.26 (3H, s), 2.24 (2H, m). 13C NMR (150 MHz, CDCl3) δ 156.8, 152.7, 141.4, 140.2, 138.0, 137.2, 134.2, 127.6, 123.5, 120.5, 107.7, 107.4, 61.0, 56.3, 55.6, 38.3, 33.0, 27.5, 11.9. HRMS: Obsvd 457.0808 [M + Na+], Calcd for C22H25BrO4Na: 455.0828. HPLC: 25.38 min.
2-Methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulene-1,5,6-triol (15)
To a solution of TBS-protected phenol 10 (0.35 g, 0.69 mmol) in THF (20 mL) was added TBAF (0.76 mL, 1 M in THF, 0.76 mmol) at 0 °C. The reaction mixture was stirred for 1 h, subsequently washed with brine (30 mL), and extracted with EtOAc (3 × 30 mL). The resultant organic phase was dried over sodium sulfate, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 100%A / 0%B (10 CV), 100%A / 0%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford diol phenol 15 (138 mg, 0.35 mmol, 51%) as a yellow solid. 1H NMR (600 MHz, CDCl3) δ 7.24 (1H, d, J = 8.4 Hz), 6.78 (1H, d, J = 8.4 Hz), 6.51 (2H, s), 5.82 (1H, s), 4.56 (1H, m), 3.93 (3H, s), 3.83 (3H, s), 3.75 (6H, s), 3.37 (1H, m), 3.21 (1H, s), 2.24 (1H, m), 2.05 (1H, m), 1.96 (1H, m), 1.69 (2H, m). 13C NMR (150 MHz, CDCl3) δ 153.3, 146.1, 142.8, 138.6, 137.9, 133.8, 127.0, 120.6, 107.9, 105.2, 83.3, 76.8, 61.0, 56.3, 56.0, 32.7, 25.1, 21.2. HRMS: Obsvd 413.1571 [M + Na+], Calcd for C21H26O7Na: 413.1571. HPLC: 13.71 min.
3-Methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene-4-carbonitrile (18)38
To KGP156 (0.10 g, 0.28 mmol) in a 2 M HCl/ CH3OH solution (5 mL/ 5 mL) was added NaNO2 (77.7 mg, 1.12 mmol) at 0 °C, and the mixture was stirred for 1 h. CuCN was added (50.4 mg, 0.56 mmol), and the reaction mixture was heated at 60 °C for 2 h. Na2CO3 and NaCN were added (50 mg of each), and the reaction mixture was stirred for 12 h at room temperature. A saturated FeCl3 solution (50 mL) was added to quench the reaction, and the reaction mixture was extracted with EtOAc (3 × 30 mL). The combined organic phase was washed with brine and a saturated NaHCO3 solution, then dried over sodium sulfate and evaporated under reduced pressure. The crude reaction was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford nitrile 18 (41 mg, 0.11 mmol, 40%) as a yellow solid. 1H NMR (600 MHz, CDCl3) δ 7.20 (1H, d, J = 8.4 Hz), 6.80 (1H, d, J = 8.4 Hz), 6.43 (1H, t, J = 7.8 Hz), 6.42 (2H, s), 3.94 (3H, s), 3.86 (3H, s), 3.80 (6H, s), 2.90 (2H, t, J = 7.2 Hz), 2.26 (2H, m), 1.94 (2H, m). 13C NMR (150 MHz, CDCl3) δ 160.7, 153.1, 147.5, 141.6, 137.6, 137.5, 134.9, 133.4, 128.7, 116.1, 108.5, 105.0, 101.7, 61.1, 56.3, 56.2, 34.8, 25.7, 25.4. HRMS: Obsvd 388.1521 [M + Na+], Calcd for C22H23NO4Na: 388.1519. HPLC: 20.75 min.
2-Methoxy-5-(3,4,5-trimethoxyphenyl)-7,8-dihydronaphthalene-1-carbonitrile (19)35
KGP05 (48.6 mg, 0.14 mmol) was dissolved in 2 M HCl/ CH3OH (2 mL/ 2 mL). The solution was cooled to 0 °C, NaNO2 (39.2 mg, 0.56 mmol) was added, and the resultant reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was heated at 60 °C for 2 h before the addition of CuCN (25.5 mg, 0.28 mmol). After the reaction mixture was cooled to room temperature, Na2CO3 and NaCN were added to adjust the pH to 10 and provide more nitrile ions for improving the yield, followed by an additional 12 h of stirring. FeCl3 was added to quench the reaction, followed by extraction with EtOAc (3 × 20 mL). The combined organic phase was washed with brine and a saturated NaHCO3 solution, dried over sodium sulfate, and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B→ 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford nitrile 19 (17 mg, 0.046 mmol, 32%) as a white foam. 1H NMR (600 MHz, CDCl3) δ 7.20 (1H, d, J = 8.4 Hz), 6.69 (1H, d, J = 9 Hz), 6.50 (2H, s), 6.03 (1H, t, J = 4.8 Hz), 3.91 (3H, s), 3.88 (3H, s), 3.84 (6H, s), 3.06 (2H, t, J = 7.8 Hz), 2.43 (2H, m). 13C NMR (150 MHz, CDCl3) δ 160.3, 153.3, 142.9, 138.5, 137.6, 135.8, 130.8, 128.8, 126.1, 115.6, 108.3, 105.8, 101.6, 61.1, 56.3, 56.2, 26.8, 22.7. HRMS: Obsvd 374.1363 [M + Na+], Calcd for C21H21NO4Na: 374.1363. HPLC: 20.92 min.
3-Methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene-4-carbaldehyde (20)
To a solution of nitrile 18 (48 mg, 0.13 mmol) in toluene (15 mL) was added DIBAL-H (0.16 mL, 1 M, 0.16 mmol) at 0 °C, and the resultant solution was stirred for 12 h while warming to room temperature. 1 M HCl (100 mL) was added to the reaction mixture, which was stirred for 30 min at room temperature while the solution color turned to yellow. EtOAc (3 × 50 mL) was used to extract the organic compound. The combined organic phase was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford aldehyde 20 (37.8 mg, 0.10 mmol, 78%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 10.68 (1H, s), 7.18 (1H, d, J = 7.2 Hz), 6.82 (1H, d, J = 8.4 Hz), 6.47 (2H, s), 6.43 (1H, t, J = 7.8 Hz), 3.92 (3H, s), 3.86 (3H, s), 3.81 (6H, s), 2.99 (2H, m), 2.27 (2H, m), 1.91 (2H, m). 13C NMR (150 MHz, CDCl3) δ 193.3, 161.8, 153.2, 145.2, 141.9, 138.2, 137.6, 135.5, 134.7, 128.8, 123.7, 108.7, 105.3, 61.1, 56.3, 55.9, 31.7, 25.7, 22.8. HRMS: Obsvd 391.1519 [M + Na+], Calcd for C22H24O5Na: 391.1516. HPLC: 22.39 min.
2-Bromo-3-methoxybenzaldehyde (22)
To a solution of 2-bromo-3-hydroxybenzaldehyde 21 (2.50 g, 12.4 mmol) in DMF (50 mL) was added CH3I (1.01 mL, 16.2 mmol) and K2CO3 (1.35 g, 13.7 mmol). The reaction mixture was stirred at room temperature for 3 h. The solvent was removed under reduced pressure, and the residue was washed with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic phases were combined and concentrated without further purification to afford 2-bromo-3-methoxybenzaldehyde 22 (2.67 g, 12.4 mmol, 100%) as a brown solid. 1H NMR (600 MHz, CDCl3) δ 10.44 (1H, s), 7.52 (1H, d, J = 7.8 Hz), 7.38 (1H, t, J = 7.8 Hz), 7.13 (1H, d, J = 8.4 Hz), 3.96 (3H, s). 13C NMR (150 MHz, CDCl3) δ 192.4, 156.4, 134.9, 128.5, 121.6, 117.3, 117.1, 56.8.
5-(2-Bromo-3-methoxyphenyl)pent-4-enoic acid (23)
To dissolved 3-(carboxypropyl)triphenyl phosphonium bromide (5.33 g, 12.4 mmol) in THF (250 mL) was added potassium tert-butoxide (3.08 g, 27.3 mmol), and the reaction mixture was stirred at room temperature for 1 h. 2-Bromo-3-methoxybenzaldehyde 22 (2.67 g, 16.2 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. The THF was removed under reduced pressure, and the resulting material was quenched and acidified with 2 M HCl (30 mL) and extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate, filtered, and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 23 (1.74 g, 6.10 mmol, 49%) as a white solid. NMR characterization was performed after the next step.
Methyl 5-(2-bromo-3-methoxyphenyl)pentanoate (24)
To dissolved carboxylic acid 23 (0.69 g, 2.42 mmol) in CH3OH (30 mL) was added 10% palladium on carbon (0.26 g), Ph2S (40 µL, 0.24 mmol), and two balloons with hydrogen gas. After stirring for 24 h, the mixture was filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (CH3OH and EtOAc) was evaporated under reduced pressure. The residue was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford saturated ester 24 (0.48 g, 1.6 mmol, 66%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.54 (1H, t, J = 7.8 Hz), 7.19 (1H, d, J = 8.4 Hz), 7.10 (1H, d, J = 7.8 Hz), 4.24 (3H, s), 4.02 (3H, s), 3.14 (2H, m), 2.72 (2H, m), 2.07 (2H, m), 2.02 (2H, m). 13C NMR (150 MHz, CDCl3) δ 174.2, 156.1, 143.3, 127.8, 122.5, 113.9, 109.5, 56.4, 51.6, 36.1, 34.0, 29.4, 24.8.
1-Bromo-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (25)
To ester 24 (0.48 g, 1.6 mmol) was added Eaton’s reagent (8.5 mL), and the mixture was stirred at room temperature for 12 h. The reaction mixture was then poured over ice and neutralized with sodium carbonate. The aqueous layer was extracted with EtOAc (3 × 40 mL). The combined organic phase was dried over sodium sulfate, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford benzosuberone 25 (0.20 g, 0.74 mmol, 47%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.63 (1H, d, J = 8.4 Hz), 6.82 (1H, d, J = 8.4 Hz), 3.94 (3H, s), 3.17 (2H, m), 2.69 (2H, m), 1.85 (2H, m), 1.75 (2H, m). 13C NMR (150 MHz, CDCl3) δ 205.2, 159.0, 142.1, 133.7, 129.1, 114.1, 109.4, 56.6, 40.5, 31.2, 23.9, 20.7.
4-Bromo-3-methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene (26)
To an oven dried flask, THF (20 mL) and 3, 4, 5-trimethoxyphenyl bromide (1.12 g, 4.53 mmol) were added, and the solution was cooled to −78 °C. n-Buli (1.81 mL, 2.5 M, 4.52 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 45 min. Benzosuberone 25 (0.61 g, 2.3 mmol) was then added to the flask dropwise, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction was quenched with excess HCl (2 M, 50 mL), and stirred for 30 min at room temperature. The reaction mixture was washed with water and extracted with EtOAc (3 × 40 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford brominated benzosuberene 26 (0.46 g, 1.1 mmol, 49%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.97 (1H, d, J = 8.4 Hz), 6.75 (1H, d, J = 8.4 Hz), 6.48 (2H, s), 6.37 (1H, t, J = 7.8 Hz), 3.92 (3H, s), 3.86 (3H, s), 3.81 (6H, s), 2.95 (2H, t, J = 7.2 Hz), 2.17 (2H, m), 1.92 (2H, m). 13C NMR (150 MHz, CDCl3) δ 154.9, 153.1, 143.0, 142.8, 138.0, 137.6, 134.4, 129.1, 127.8, 113.4, 109.1, 105.3, 61.0, 56.4, 56.3, 33.8, 31.9, 25.4.
Ethyl 3-methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulene-4-carboxylate (27)
To a solution of brominated benzosuberene 26 (0.15 g, 0.36 mmol) in THF (20 mL) was added n-Buli (0.34 mL, 1.6 M, 0.54 mmol) dropwise at −78 °C. The reaction mixture was stirred at −78 °C for 30 min, followed by the addition of ethyl chlorofomate (61.5 µL, 0.64 mmol). The reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford benzosuberene ester 27 (51.8 mg, 0.13 mmol, 35%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.03 (1H, d, J = 8.4 Hz), 6.76 (1H, d, J = 9 Hz), 6.47 (2H, s), 6.37 (1H, t, J = 7.8 Hz), 4.43 (2H, q, J = 7.2 Hz), 3.85 (3H, s), 3.84 (3H, s), 3.79 (6H, s), 2.56 (2H, t, J = 6.6 Hz), 2.17 (2H, m), 1.96 (2H, m), 1.41 (3H, t, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 168.8, 155.2, 153.0, 142.3, 140.2, 138.1, 137.5, 133.2, 131.6, 127.7, 123.4, 108.6, 105.2, 61.4, 61.0, 56.3, 55.9, 34.9, 29.6, 25.3, 14.4. HRMS: Obsvd 435.1778 [M + Na+], Calcd for C24H28O6Na: 435.1778. HPLC: 22.42 min.
(3-Methoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-4-yl)methanol (28)
Ester 27 (0.12 g, 0.28 mmol) was dissolved in THF (10 mL), and the solution was cooled to 0 °C. LiAlH4 (77 µL, 4 M in ether, 0.31 mmol) was added to the solution dropwise, and the reaction mixture was stirred while warming to room temperature for 1 h. The reaction mixture was washed with water and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford benzyl alcohol 28 (54.3 mg, 0.15 mmol, 52%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.97 (1H, d, J = 9 Hz), 6.75 (1H, d, J = 9 Hz), 6.50 (2H, s), 6.35 (1H, t, J = 7.8 Hz), 4.87 (2H, s), 3.89 (3H, s), 3.86 (3H, s), 3.81 (6H, s), 2.79 (2H, t, J = 7.2 Hz), 2.16 (2H, p, J = 7.2 Hz), 1.92 (2H, m). 13C NMR (150 MHz, CDCl3) δ 157.1, 153.1, 143.3, 142.4, 138.5, 137.5, 133.8, 130.2, 127.1, 126.0, 107.9, 105.4, 61.1, 57.7, 56.3, 55.7, 35.3, 27.6, 25.4. HRMS: Obsvd 393.1672 [M + Na+], Calcd for C22H26O5Na: 393.1672. HPLC: 19.09 min.
3-(2,3-Dimethoxyphenyl)propanoic acid (30)
To cinnamic acid 29 (5.0 g, 24 mmol) was added methanol (50 mL) and 10% Pd/C (0.8 g). Two hydrogen balloons were installed through the rubber septum, and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was filtered through Celite®, and the Celite® was washed with EtOAc (3 × 50 mL). The organic solvents (CH3OH and EtOAc) were evaporated under reduced pressure to afford carboxylic acid 30 (5.0 g, 24 mmol, quantitative) as a white solid. No further purification was needed. 1H NMR (600 MHz, CDCl3) δ 6.98 (1H, t, J = 7.8 Hz), 6.78 (2H, m), 3.86 (3H, s), 3.84 (3H, s), 2.95 (2H, t, J = 7.8 Hz), 2.66 (2H, t, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3) δ 178.9, 152.8, 147.2, 134.1, 124.1, 121.8, 110.9, 60.7, 55.8, 34.8, 25.4.
Methyl 5-(2,3-dimethoxyphenyl)-3-oxopentanoate (31)
To dissolved carboxylic acid 30 (5.05 g, 24.0 mmol) in dichloromethane (96 mL) were added oxalyl chloride (4.12 mL, 47.2 mmol) and a catalytic amount of DMF (0.15 mL). The reaction mixture was stirred at room temperature for 1 h, at which time an additional catalytic amount of DMF (0.15 mL) was added, and the reaction solution stirred for 1 h at room temperature. The solvent and unreacted oxalyl chloride were removed under reduced pressure to afford acyl chloride as a yellow crystalline solid, which was re-dissolved in dichloromethane (50 mL) and cooled to 0° C. Meldrum’s acid (3.47 g, 24.1 mmol) and pyridine (4.33 mL, 53.8 mmol) were added, and the reaction mixture was stirred for 30 min at 0° C, then 1 h at room temperature. The mixture was diluted with dichloromethane (50 mL), and washed with 2 M HCl (20 mL), followed by brine (30 mL). The organic layer was dried over sodium sulfate and concentrated in vacuo. The residue was dissolved in CH3OH (50 mL) and heated at reflux for 3 h. The solvent was removed under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford the ester 31 (3.57 g, 13.4 mmol, 56%) as a pale-yellow oil. 1H NMR (600 MHz, CDCl3) δ 6.96 (1H, t, J = 7.8 Hz), 6.77 (1H, d, J = 9 Hz), 6.74 (1H, d, J = 7.8 Hz), 3.84 (3H, s), 3.81 (3H, s), 3.71 (3H, s), 3.44 (2H, s), 2.89 (2H, m), 2.83 (2H, m). 13C NMR (150 MHz, CDCl3) δ 202.1, 167.6, 152.7, 147.0, 134.2, 124.0, 121.8, 110.6, 60.5, 55.6, 52.3, 49.0, 43.6, 24.2.
Methyl 5-(2,3-dimethoxyphenyl)-3-hydroxypentanoate (32)
To a well-stirred solution of ketone 31 (0.50 g, 1.9 mmol) in CH3OH (8 mL) at 0 °C, sodium borohydride (24 mg, 0.63 mmol) was added in one aliquot. The reaction mixture was initially stirred at 0 °C for 1 h, and then stirred at room temperature for another 1 h. The solvent was removed under reduced pressure. The residue was washed with water (10 mL) and extracted with diethyl ether (3 × 10 mL). The combined organic phase was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A/ 93%B (3 CV), 7%A/ 93%B → 60%A/ 40%B (10 CV), 60%A/ 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] to afford alcohol 32 (0.40 g, 1.5 mmol, 79%). 1H NMR (600 MHz, CDCl3) δ 6.98 (1H, t, 7.8 Hz), 6.78 (2H, m), 3.97 (1H, m), 3.85 (3H, s), 3.82 (3H, s), 3.69 (3H, s), 2.76 (2H, t, J = 7.8 Hz), 2.48 (2H, m), 1.78 (2H, m). 13C NMR (150 MHz, CDCl3) δ 173.3, 152.8, 147.2, 135.4, 124.2, 122.1, 110.5, 67.2, 60.8, 55.8, 51.8, 41.4, 37.6, 25.9.
Methyl 3-((tert-butyldiphenylsilyl)oxy)-5-(2,3-dimethoxyphenyl)pentanoate (33)
To a solution of alcohol 32 (0.38 g, 0.14 mmol) and imidazole (0.16 g, 2.3 mmol) in DMF (2.6 mL) at room temperature was added TBDPSCl (0.55 mL, 2.1 mmol) in one aliquot. The reaction mixture was stirred for 12 h, diluted with brine (10 mL), and extracted with Et2O (3 × 10 mL). The organic extracts were combined and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] to afford ester 33 (0.35 g, 0.69 mmol, 49%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.72 (3H, m), 7.67 (1H, m), 7.38 (6H, m), 6.91 (1H, t, J = 7.8 Hz), 6.73 (1H, d, J = 9.6 Hz), 6.55 (1H, d, J = 9 Hz), 4.29 (1H, m), 3.83 (3H, s), 3.71 (3H, s), 3.54 (3H, s), 2.58 (4H, m), 1.76 (2H, m), 1.06 (9H, s). 13C NMR (150 MHz, CDCl3) δ 172.0, 152.8, 147.1, 136.1, 136.0, 135.9, 135.3, 134.9, 134.2, 134.1, 129.8, 129.7, 127.9, 127.7, 127.6, 123.9, 121.8, 110.2, 70.5, 60.7, 55.8, 51.5, 41.9, 38.2, 27.1, 26.7, 25.4.
3-((Tert-butyldiphenylsilyl)oxy)-5-(2,3-dimethoxyphenyl)pentanoic acid (34)
To a solution of ester 33 (0.67 g, 1.3 mmol) in CH3OH/THF (2.2 mL/ 1.1 mL) at 0 °C was added 2.5 M NaOH (1.76 mL). The reaction mixture was stirred for 1 h at 0 °C, and then 13 h at room temperature, acidified by 2 M HCl (10 mL), and extracted with Et2O (3 × 10 mL). The organic extracts were combined and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 34 (0.26 g, 0.53 mmol, 40%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.67 (4H, m), 7.41 (6H, m), 6.89 (1H, t, J = 8.4 Hz), 6.72 (1H, d, J = 8.4 Hz), 6.52 (1H, m), 4.20 (1H, m), 3.83 (3H, s), 3.69 (3H, s), 2.50 (4H, m), 1.80 (2H, m), 1.06 (9H, s). 13C NMR (150 MHz, CDCl3) δ 152.6, 146.94, 146.93, 135.9, 135.8, 129.9, 129.8, 129.77, 127.7, 127.6, 123.81, 123.80, 121.6, 110.2, 70.2, 60.5, 55.6, 40.8, 37.6, 26.9, 25.3, 19.3.
7-((Tert-butyldiphenylsilyl)oxy)-1,2-dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (35)
To a solution of carboxylic acid 34 (5.37g, 10.9 mmol) in dichloromethane (40 mL) was added oxalyl chloride (4.5 mL, 52 mmol) and 3 drops of DMF as catalyst at room temperature. The resultant reaction mixture was stirred for 2 h at 0 °C. The solvent and unreacted oxalyl chloride were removed under reduced pressure. The residue acyl chloride was dissolved in dichloromethane (50 mL). The solution was cooled to −10 °C, at which point SnCl4 (3.63 mL, 1 M in CH2Cl2, 3.63 mmol) was added, followed by stirring at −10 °C for 30 min. The reaction was quenched with cold water and extracted with EtOAc (3 × 50 mL). The organic extracts were combined and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] to afford cyclized ketone 35 (2.80 g, 5.90 mmol, 54%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.63 (4H, m), 7.56 (1H, d, J = 8.4 Hz), 7.39 (6H, m), 6.81 (1H, d, J = 9 Hz), 6.29 (1H, m), 3.90 (3H, s), 3.78 (3H, s), 3.17 (1H, m), 3.03 (2H, m), 2.88 (1H, m), 1.98 (1H, m), 1.84 (1H, m), 1.03 (9H, s). 13C NMR (150 MHz, CDCl3) δ 199.5, 155.8, 146.2, 137.9, 136.0, 135.9, 134.1, 133.9, 133.1, 129.9, 129.8, 127.81, 127.77, 125.7, 109.6, 68.3, 60.9, 55.9, 50.4, 36.2, 27.0, 21.3, 19.3.
7-Hydroxy-1,2-dimethoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (36)
To a solution of ketone 35 (0.73 g, 1.5 mmol) in THF (10 mL) was added TBAF (3.1 mL 1 M in THF, 3.1 mmol), and the reaction mixture was stirred for 30 min at 0 °C and 16 h at room temperature. The reaction was quenched with brine (10 mL) and extracted with EtOAc (3 × 20 mL). The organic extracts were combined and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 20%A / 80%B (10 CV), 20%A / 80%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] to afford alcohol 36 (0.16 g, 0.66 mmol, 49%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.60 (1H, d, J = 8.4 Hz), 6.84 (1H, d, J = 8.4 Hz), 4.33 (1H, m), 3.91 (3H, s), 3.80 (3H, s), 3.08 (3H, m), 2.99 (1H, m), 1.89 (2H, m). 13C NMR (150 MHz, CDCl3) δ 199.3, 156.0, 146.3, 137.7, 132.6, 125.8, 109.8, 67.3, 60.9, 56.0, 50.3, 35.8, 21.4.
3,4-Dimethoxy-9-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[7]annulen-7-ol (37)
To a solution of 3, 4, 5-trimethoxyphenyl bromide (0.49 g, 2.0 mmol) in THF (20 mL) at −78 °C was added n-BuLi (1.85 mL, 1.6 M in hexanes, 2.98 mmol), and the reaction mixture was stirred for 1 h. Benzosuberone 36 (0.16 g, 0.66 mmol) in THF (5 mL) was added slowly. The reaction mixture was stirred at 0 °C for 20 h. 2 M HCl (20 mL) was added, and the mixture was extracted with EtOAc (4 × 20 mL). The combined organic phase was further washed by brine and dried over sodium sulfate, filtered, and concentrated under reduced pressure and purified by flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford alcohol 37 (0.10 g, 0.26 mmol, 39% ) as a brown solid. 1H NMR (600 MHz, CDCl3) δ 6.75 (2H, m), 6.51 (2H, s), 6.28 (1H, d, J 4.8 Hz), 4.18 (1H, m), 3.877 (3H, s), 3.875 (3H, s), 3.86 (3H, s), 3.80 (6H, s), 3.16 (1H, m), 2.53 (1H, m), 2.43 (1H, m), 2.15 (1H, m). 13C NMR (150 MHz, CDCl3) δ 153.1, 152.0, 146.1, 139.4, 137.8, 137.4, 135.7, 132.9, 131.5, 125.5, 109.6, 105.5, 70.0, 61.4, 61.1, 56.3, 55.8, 43.2, 22.4. HRMS: Obsvd 409.1621 [M + Na+], Calcd for C22H26O6Na: 409.1622. HPLC: 16.79 min.
2-((Tert-butyldimethylsilyl)oxy)-3-methoxybenzaldehyde (39)
To a well-stirred solution of 2-hydroxy-3-methoxybenzaldehyde 38 (0.50 g, 3.3 mmol) in dichloromethane (30 mL) was added TBSCl (0.74 g, 4.9 mmol), DMAP (0.12 g, 0.99 mmol), and Et3N (0.69 mL, 4.9 mmol). The reaction mixture was stirred for 12 h at room temperature, at which point brine (50 mL) was added, and the reaction mixture was extracted with dichloromethane (3 × 40 mL). The organic extracts were combined and dried over sodium sulfate, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] to afford protected aldehyde 39 (0.50 g, 1.86 mmol, 57%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ 10.51 (1H, s), 7.36 (1H, d, J = 7.8 Hz), 7.03 (1H, d, J = 7.8 Hz), 6.94 (1H, t, J = 8.4 Hz), 3.81 (3H, s), 0.99 (9H, s), 0.20 (6H, s). 13C NMR (150 MHz, CDCl3) δ 190.4, 150.8, 149.2, 127.9, 121.2, 119.1, 117.0, 55.2, 26.0, 19.0, 4.1.
Ethyl 5-(2-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)-3-oxopent-4-enoate (40)
To dissolved ethyl 3-oxo-4-(triphenylphophoranylidene) butanoate (3.22 g, 8.26 mmol) in THF (20 mL) was added protected aldehyde 39 (2.2 g, 8.3 mmol), and the reaction mixture was heated at reflux and stirred for 17 h. The solvent was removed under reduced pressure, and the residue was taken up as a slurry and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford ester 40 (2.50 g, 6.59 mmol, 80%) as an off white solid. NMR characterization was conducted after the next step.
Ethyl 5-(2-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)-3-oxopentanoate (41)
To dissolved ester 40 (2.50 g, 6.59 mmol) in methanol (60 mL) was added 10% palladium on carbon (0.54 g), and hydrogen gas was introduced with a balloon. The reaction mixture was stirred at room temperature for 12 h and filtered through Celite®, and the Celite® was washed with EtOAc (3 × 40 mL). The combined organic phase (CH3OH and EtOAc) was evaporated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford saturated ester 41 (1.15 g, 3.02 mmol, 46%) as a light-yellow oil. 1H NMR (600 MHz, CDCl3) δ 6.81(1H, m), 6.71 (1H, d, J = 7.8 Hz), 4.16 (2H, q, J = 7.2 Hz), 3.76 (3H, s), 3.39 (2H, s), 2.90 (2H, m), 2.83 (2H, m), 1.25 (3H, t, J = 7.2 Hz), 0.98 (9H, s), 0.18 (6H, s). 13C NMR (150 MHz, CDCl3) δ 202.3, 167.2, 150.0, 142.8, 131.8, 121.9, 121.0, 109.7, 61.4, 54.8, 49.4, 43.2, 26.2, 24.8, 18.9, 14.2, −3.7.
Ethyl (Z)-3-amino-5-(2-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)pent-2-enoate (42)
To keto-ester 41 (1.10 g, 2.89 mmol) dissolved in methanol (15 mL) was added dry ammonium acetate (1.11 g, 14.5 mmol). The reaction mixture was stirred at 35 °C for 16 h. The methanol was removed under vacuum, and the residue was suspended in EtOAc (30 mL) and filtered. The filtrate was washed with EtOAc (4 × 20 mL). The combined organic layer was dried over sodium sulfate and concentrated under reduced pressure to afford amine 42 (1.02 g, 2.69 mmol, 93%) as pale yellow crystals. No further purification was performed. 1H NMR (600 MHz, CDCl3) δ 6.82 (1H, m), 6.72 (2H, m), 4.58 (1H, s), 4.11 (2H, m), 3.78 (3H, s), 2.87 (2H, m), 2.40 (2H, m), 1.26 (3H, t, J = 7.2 Hz), 1.00 (9H, s), 0.19 (6H, s). 13C NMR (150 MHz, CDCl3) δ 170.7, 163.6, 150.0, 142.8, 131.8, 122.0, 121.1, 109.8, 83.5, 58.7, 54.8, 36.7, 29.3, 26.3, 19.0, 14.7, −3.6.
Ethyl (Z)-3-acetamido-5-(2-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)pent-2-enoate (43)
To amine 42 (4.02 g, 10.6 mmol) dissolved in THF (50 mL) was added pyridine (1.71 mL, 21.2 mmol) and acetic anhydride (6.00 mL, 63.6 mmol). The reaction mixture was stirred for 48 h under reflux. The THF was removed under vacuum, and the residue was dissolved in EtOAc (50 mL) and washed with water (50 mL), 2 M HCl (20 mL), saturated NaHCO3 (50 mL) and brine (50 mL). The combined organic layer was dried over sodium sulfate and concentrated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford N-acetamide 43 (2.17 g, 5.15 mmol, 49%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 6.80 (2H, m), 6.70 (1H, m), 4.90 (1H, s), 4.14 (2H, m), 3.76 (3H, s), 3.00 (2H, m), 2.86 (2H, m), 2.15 (3H, s), 1.26 (3H, m), 0.98 (9H, s), 0.17 (6H, s). 13C NMR (150 MHz, CDCl3) δ 169.3, 168.3, 158.2, 149.8, 142.7, 132.0, 122.2, 120.9, 109.5, 96.2, 59.8, 54.7, 34.5, 28.7, 26.2, 25.3, 18.9, 14.3, −3.8.
Ethyl 3-acetamido-5-(2-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)pentanoate (44)
Unsaturated N-acetamide 43 (2.17 g, 5.15 mmol) was dissolved in CH3OH (30 mL). Palladium (10%) on carbon (0.53 g) and a hydrogen gas balloon were introduced, and the solution was stirred at room temperature for 60 h and filtered through Celite®. The Celite® was washed with EtOAc (3 × 50 mL). The combined organic phase (CH3OH and EtOAc) was evaporated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford saturated N-acetamide 44 (0.96 g, 2.3 mmol, 44%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.83 (1H, t, J = 7.8 Hz), 6.71 (2H, m), 6.03 (1H, d, J = 9 Hz), 4.29 (1H, m), 4.11 (2H, q, J = 7.2 Hz), 3.77 (3H, s), 2.74 (1H, m), 2.62 (1H, m), 2.59 (1H, m), 2.51 (1H, m), 1.96 (3H, s), 1.82 (2H, m), 1.24 (3H, t, J = 7.2 Hz), 1.00 (9H, s), 0.17 (6H, d, J = 10.8 Hz). 13C NMR (150 MHz, CDCl3) δ 172.1, 169.6, 150.0, 142.7, 132.7, 121.9, 121.0, 109.4, 60.7, 54.8, 46.3, 38.8, 34.3, 27.6, 26.3, 23.7, 19.0, 14.3, −3.6, −3.7.
3-Acetamido-5-(2-hydroxy-3-methoxyphenyl)pentanoic acid (45)
To dissolved unsaturated ester 44 (0.96 g, 2.3 mmol) in methanol (5 mL) was added 1 M KOH (7.48 mL). The reaction was stirred from 0 °C to room temperature over 3 h. The methanol was removed under vacuum, and 2 M HCl (5 mL) was added to the residue, which was then extracted with EtOAc (3 × 20 mL). The combined organic phase was evaporated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 100%A / 0%B (35 CV), 100%A / 0%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 45 (0.38 g, 1.8 mmol, 58%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.74 (3H, m), 6.34 (1H, d, J = 9 Hz), 4.25 (1H, m), 3.85 (3H, s), 2.64 (4H, m), 1.97 (3H, s), 1.91 (2H, m). 13C NMR (150 MHz, CDCl3) δ 175.6, 171.0, 146.6, 143.5, 127.3, 122.5, 119.8, 108.9, 56.2, 46.7, 38.9, 34.1, 26.8, 23.5.
N-(1-hydroxy-2-methoxy-5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-yl)acetamide (46)
Carboxylic acid 45 (0.70 g, 2.5 mmol) was dissolved in Eaton’s reagent (14 mL), and the reaction mixture was stirred at room temperature for 12 h. Ice was added to the reaction mixture, which generated a significant amount of heat. A saturated sodium carbonate solution was added until neutral pH was achieved. The mixture was extracted with dichloromethane (4 × 30 mL). The organic phase was further washed by brine and dried over sodium sulfate, filtered, and concentrated under reduced pressure and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: CH3OH; solvent B: CH2Cl2; gradient: 1%A / 99%B (1 CV), 1%A / 99%B → 10%A / 90%B (10 CV), 10%A / 90%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford cyclized ketone 46 (0.37 g, 1.4 mmol, 57% ) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.34 (1H, d, J = 8.4 Hz), 6.79 (1H, d, J = 9 Hz), 5.91 (1H, s), 4.48 (1H, m), 3.94 (3H, s), 3.23 (1H, m), 3.13 (1H, m), 2.83 (2H, m), 2.73 (1H, m), 2.44 (1H, m), 1.96 (3H, s). 13C NMR (150 MHz, CDCl3) δ 201.1, 169.6, 149.3, 142.9, 133.1, 129.0, 121.1, 108.2, 56.3, 47.0, 45.7, 32.9, 23.6, 22.6.
N-(1-((tert-butyldimethylsilyl)oxy)-2-methoxy-5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-yl)acetamide (47)
To a solution of cyclized ketone 46 (0.37 g, 1.4 mmol) in dichloromethane (20 mL) at room temperature was added TBSCl (0.32 g, 2.1 mmol), DMAP (52 mg, 0.42 mmol) and trimethylamine (0.30 mL, 2.1 mmol), and the resultant reaction mixture was stirred for 12 h. The reaction mixture was subsequently washed with brine (30 mL), and extracted with dichloromethane (3 × 40 mL). The combined organic layer was dried over sodium sulfate and concentrated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: CH3OH; solvent B: CH2Cl2; gradient: 0%A / 100%B (1 CV), 0%A / 100%B → 5%A / 95%B (10 CV), 5%A / 95%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford protected ketone 47 (0.36 g, 0.95 mmol, 68%) as an off-white solid. 1H NMR (600 MHz, CDCl3) δ 7.41 (1H, d, J = 8.4 Hz), 6.78 (1H, d, J = 9 Hz), 5.61 (1H, br), 4.46 (1H, m), 3.84 (3H, s), 3.26 (1H, m), 3.16 (1H, m), 2.80 (2H, m), 2.71 (1H, m), 2.45 (1H, m), 1.96 (3H, s), 1.00 (9H, s), 0.17 (6H, d, J = 14.4 Hz). 13C NMR (150 MHz, CDCl3) δ 201.1, 169.6, 153.4, 142.4, 134.7, 129.0, 122.6, 109.1, 55.1, 46.9, 45.8, 40.0, 33.2, 26.2, 23.6, 19.1, −3.7, −3.8.
N-(1-((tert-butyldimethylsilyl)oxy)-2-methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-yl)acetamide (48)
To an oven-dried flask, THF (20 mL) and 3, 4, 5-trimethoxyphenyl bromide (0.16 g, 0.64 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (1.6 M, 0.59 mL, 0.94 mmol) was added to the reaction mixture slowly, which was then stirred at −78 °C for 45 min. Benzosuberone 47 (80 mg, 0.21 mmol) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. 2 M HCl (20 mL) was added, and the reaction mixture was stirred for 10 min, then extracted with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 20 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 100%A / 0%B (10 CV), 100%A / 0%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford cross-coupling product benzosuberene 48 (63.6 mg, 0.120 mmol, 57%) as a crystalline white solid. 1H NMR (600 MHz, CDCl3) δ 6.68 (1H, d, J = 9 Hz), 6.58 (1H, d, J = 8.4 Hz), 6.47 (2H, s), 5.98 (1H, d, J = 6 Hz), 5.54 (1H, d, J = 8.4 Hz, br), 4.39 (1H, m), 3.85 (3H, s), 3.80 (3H, s), 3.79 (9H, s), 3.20–2.46 (4H, m), 1.95 (3H, s), 1.03 (9H, s), 0.25 (3H, s), 0.22 (3H). 13C NMR (150 MHz, CDCl3) δ 169.0, 152.8, 149.1, 141.7, 141.5, 137.9, 137.6, 132.7, 132.3, 128.4, 122.9, 108.9, 105.4, 60.9, 56.2, 54.7, 47.8, 41.0, 26.2, 23.6, 22.9, 19.0, −3.6, −3.9.
N-(1-hydroxy-2-methoxy-5-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-yl)acetamide (49)
Protected benzosuberene N-acetamide 48 (63.6 mg, 0.12 mmol) was dissolved in THF (2 mL) and cooled to 0 °C. TBAF (0.24 mL, 0.24 mmol) was added, and the reaction mixture was stirred at 0 °C for 2 h. The solution was washed with water and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over sodium sulfate, filtered, and concentrated under reduced pressure and purified b2y flash chromatography using a pre-packed 25 g silica column [solvent A: CH3OH; solvent B: CH2Cl2; gradient: 5%A / 95%B (1 CV), 10%A / 90%B → 10%A / 90%B (10 CV), 10%A / 90%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford benzosuberene N-acetamide 49 (22 mg, 0.05 mmol, 44%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.71 (1H, d, J = 9 Hz), 6.54 (1H, d, J = 8.4 Hz), 6.48 (2H, s), 5.98 (1H, d, J = 6 Hz), 5.76 (1H, s), 5.56 (1H, d, J = 9 Hz, br), 4.38 (1H, m), 3.91 (3H, s), 3.86 (3H, s), 3.80 (6H, s), 3.17–2.43 (4H, m), 1.96 (3H, s). 13C NMR (150 MHz, CDCl3) δ 169.2, 153.0, 145.7, 142.5, 141.4, 137.83, 137.75, 133.3, 128.8, 126.9, 121.5, 108.3, 105.6, 61.1, 56.4, 56.2, 48.1, 40.8, 23.8, 22.3. HRMS: Obsvd 436.1730 [M + Na+], Calcd for C23H29NO6Na: 436.1731. HPLC: 9.92 min.
Ethyl 5-(2,3-dimethoxyphenyl)-3-oxopentanoate (52)
To a solution of 2, 3-dimethoxybenzaldehyde 50 (1.06 g, 6.38 mmol) in THF (50 mL) were added ethyl 3-oxo-4-(triphenylphosphoranylidene) butyrate (3.00 g, 7.65 mmol) and potassium t-butoxide (1.73 g, 15.3 mmol) at room temperature, and the reaction mixture was stirred for 12 h. The THF was removed under reduced pressure, and the resulting material was quenched with 2 M HCl (20 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were evaporated under reduced pressure, and the crude product (51) was dissolved in CH3OH (40 mL). To this solution was added 10% palladium on carbon (0.24 g) and balloons with hydrogen gas. The reaction mixture was stirred at room temperature for 12 h and filtered through Celite®, and the Celite® was washed with EtOAc (3 × 30 mL). The combined organic phase (CH3OH and EtOAc) was evaporated under reduced pressure. The resulting organic material was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford saturated ester 52 (1.45 g, 5.17 mmol, 81%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.97 (1H, t, J = 7.8 Hz), 6.78 (1H, d, J = 9 Hz), 6.75 (1H, d, J = 7.8 Hz), 4.17, (2H, q, J = 7.2 Hz), 3.85 (3H, s), 3.82 (3H, s), 3.43 (2H, s), 2.90 (2H, m), 2.84 (2H, m), 1.26 (3H, t, J = 6.6 Hz). 13C NMR (150 MHz, CDCl3) δ 202.4, 167.3, 152.9, 147.2, 134.5, 124.1, 122.0, 110.8, 61.5, 60.7, 55.8, 49.5, 43.8, 24.5, 14.2.
Ethyl 5-(2,3-dimethoxyphenyl)-3-hydroxypentanoate (53)
To a solution of ketone 52 (1.45 g, 5.17 mmol) in CH3OH (20 mL) at 0 °C was added NaBH4 (110 mg, 2.91 mmol), and the reaction mixture was stirred for 1 h at 0 °C, then stirred for 3 h at ambient temperature. The CH3OH was removed under reduced pressure, and the resulting material was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was concentrated under reduced pressure to afford crude alcohol product 53 (1.20 g, 4.25 mmol, 82%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.98 (1H, t, J = 7.8 Hz), 6.78 (2H, m), 4.15 (2H, m), 3.97 (1H, m), 3.85 (3H, s), 3.83 (3H, s), 2.76 (2H, m), 2.47 (2H, m), 1.80 (1H, m), 1.72 (1H, m), 1.25 (3H, t, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 173.0, 152.8, 147.2, 135.5, 124.2, 122.1, 110.4, 67.3, 60.8, 55.8, 41.5, 37.6, 25.9, 14.3.
5-(2,3-Dimethoxyphenyl)-3-hydroxypentanoic acid (54)
To a solution of ester 53 in CH3OH/ THF (8 mL/ 4mL) was added a 2.5 M NaOH aqueous solution (6 mL) at 0 °C, and the reaction mixture was stirred for 1 h at 0 °C, then warmed to room temperature over 12 h. The CH3OH and THF were removed under reduced pressure, and the resulting material was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic extract was dried over Na2SO4, filtered and concentrated under pressure to afford carboxylic acid 54 (0.88 g, 3.3 mmol, 77%) as an orange oil (no further purification was needed). 1H NMR (600 MHz, CDCl3) δ 7.00 (1H, t, J = 8.4 Hz), 6.78 (2H, dd, J = 6.6 Hz, 6.6 Hz), 3.92 (1H, m), 3.86 (3H, s), 3.84 (3H, s), 2.76 (2H, m), 2.51 (2H, m), 1.82 (1H, m), 1.73 (1H, m). 13C NMR (150 MHz, CDCl3) δ 176.8, 152.7, 146.9, 134.9, 124.5, 122.2, 110.6, 66.9, 61.0, 55.8, 41.2, 37.6, 25.6.
1,2-Dimethoxy-8,9-dihydro-5H-benzo[7]annulen-5-one (55)
To carboxylic acid 54 (0.88 g, 3.3 mmol) was added Eaton’s reagent (15.7 mL), and the mixture was stirred at room temperature for 14 h. The mixture was then poured over ice and neutralized with sodium carbonate. The aqueous layer was extracted with EtOAc (3 × 40 mL). The combined organic phase was dried over sodium sulfate, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] to afford unsaturated cyclized ketone 55 (0.360 g, 1.65 mmol, 50%) as a yellow crystalline solid. 1H NMR (600 MHz, CDCl3) δ 7.55 (1H, d, J = 9 Hz), 6.83 (1H, d, J = 9 Hz), 6.71 (1H, td, J = 4.8 Hz, 12 Hz), 6.23 (1H, td, J = 1.8 Hz, 12 Hz), 3.91 (3H, s), 3.78 (3H, s), 3.16 (2H, m), 2.55 (2H, m). 13C NMR (150 MHz, CDCl3) δ 193.9, 156.2, 147.0, 145.2, 134.4, 134.3, 132.7, 126.7, 110.0, 61.2, 55.9, 29.6, 25.1.
1,2-Dimethoxy-5-(3,4,5-trimethoxyphenyl)-8,9-dihydro-5H-benzo[7]annulen-5-ol (56)
To an oven-dried flask, THF (30 mL) and 3,4,5-trimethoxyphenyl bromide (0.87 g, 3.5 mmol) were added,, and the solution was cooled to −78 °C. n-BuLi (1.41 mL, 2.5 M, 3.52 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 30 min. Unsaturated cyclized ketone 55 (0.35 g, 1.6 mmol) in THF (5 mL) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 12 h. The reaction mixture was washed with 2 M HCl (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 60%A / 40%B (10 CV), 60%A / 40%B (5 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford tertiary alcohol 56 (0.457 g, 1.18 mmol, 74%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.54 (1H, d, J = 9 Hz), 6.82 (1H, d, J = 8.4 Hz), 6.59 (2H, s), 6.07 (1H, d, J = 12 Hz), 5.77 (1H, td, J = 1.2 Hz, 12 Hz), 3.90 (3H, s), 3.82 (3H, s), 3.78 (6H, s), 3.75 (3H, s), 3.05 (1H, m), 2.48 (1H, m), 2.43 (1H, m), 2.15 (1H, m). 13C NMR (150 MHz, CD3OD) δ 153.9, 153.2, 147.5, 145.1, 141.7, 138.3, 137.2, 134.1, 130.8, 121.6, 109.9, 106.0, 78.6, 61.4, 61.1, 56.5, 56.1, 30.0, 23.4. HRMS: Obsvd 409.1622 [M + Na+], Calcd for C22H26O6Na: 409.1622. HPLC: 17.36 min.
1,2-Dimethoxy-7-(3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (57)
To an oven-dried flask, THF (30 mL) and 3,4,5-trimethoxyphenyl bromide (0.47 g, 1.9 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (0.76 mL, 2.5 M, 1.9 mmol) was slowly added to the reaction mixture, which was stirred at −78 °C for 45 min then moved to a −10 °C bath. CuI (0.181 g, 0.95 mmol) was added in one aliquot to the flask, and the reaction mixture was stirred at −10 °C for 1 h. Unsaturated ketone 55 (0.104 g, 0.47 mmol) in THF (10 mL) was then added dropwise to the flask, and the reaction mixture was stirred while warming from −78 °C to room temperature over 7 h. A saturated NH4Cl solution and ammonium hydroxide (20 mL/ 20mL), were added followed by stirring for 30 min at room temperature and subsequent extraction with EtOAc (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 110 mL/min; monitored at 254 and 280 nm] to afford Michael addition ketone 57 (81.7 mg, 0.21 mmol, 44%) as a light white solid. 1H NMR (600 MHz, CDCl3) δ 7.56 (1H, d, J = 8.4 Hz), 6.88 (1H, d, J = 9 Hz), 6.41 (2H, s), 3.93 (3H, s), 3.83 (6H, s), 3.824 (3H, s), 3.819 (3H, s), 3.34 (1H, td, J = 4.8 Hz, 15 Hz), 3.09 (1H, m), 3.04 (1H, m), 2.94 (2H, m), 2.16 (1H, m), 1.97 (1H, m). 13C NMR (150 MHz, CDCl3) δ 203.0, 156.4, 153.4, 146.1, 141.4, 136.6, 135.5, 132.5, 125.7, 110.1, 104.0, 61.3, 61.0, 56.2, 56.0, 47.9, 39.9, 34.6, 23.4. HRMS: Obsvd 409.1624 [M + Na+], Calcd for C22H26O6Na: 409.1622. HPLC: 18.77 min.
2-Isopropoxy-3-methoxybenzaldehyde (58)
To a solution of 2-hydroxy-3-methoxybenzaldehyde 38 (5.00 g, 32.9 mmol) in DMF (100 mL) were added K2CO3 (14.97 g, 98.58 mmol) and 2-iodopropane (6.54 mL, 65.7 mmol), and the reaction mixture was stirred at 50 °C for 20 h. The DMF was removed under reduced pressure, and the resulting material was washed with water (100 mL) to remove the excess salt and extracted with EtOAc (3 × 100 mL). The combined organic phase was dried over sodium sulfate and concentrated to afford protected aldehyde 58 (6.12 g, 31.6 mmol, 96%) as a colorless oil without further purification. 1H NMR (600 MHz, CDCl3) δ 10.44 (1H, s), 7.41 (1H, d, J = 7.8 Hz), 7.10 (2H, m), 4.62 (1H, m), 3.86 (3H, s), 1.31 (6H, d, J = 6 Hz). 13C NMR (150 MHz, CDCl3) δ 191.0, 153.4, 150.7, 131.0, 123.7, 119.0, 118.0, 76.3, 56.1, 22.4.
Ethyl 5-(2-isopropoxy-3-methoxyphenyl)-3-oxopent-4-enoate (59)
To dissolved ethyl 3-oxo-4-(triphenylphosphoranylidene) butanoate (0.85 g, 2.2 mmol) in THF (50 mL) were added potassium tert-butoxide (0.50 g, 4.4 mmol) and aldehyde 58 (0.35 g, 1.8 mmol), and the resultant reaction mixture was stirred at room temperature for 12 h. The THF was removed under reduced pressure, and the resulting material was neutralized with 2 M HCl (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were evaporated under reduced pressure, and the crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford unsaturated ester 59 (0.40 g, 1.3 mmol, 72%) as a yellow oil. NMR characterization was conducted after the next step.
Ethyl 5-(2-isopropoxy-3-methoxyphenyl)-3-oxopentanoate (60)
To dissolved unsaturated ester 59 (0.39 g, 1.3 mmol) in CH3OH (20 mL) was added 10% palladium on carbon (0.2 g) and balloons with hydrogen gas. The reaction mixture was stirred at room temperature for 12 h, followed by filtration through Celite®, and the Celite® was washed with EtOAc (3 × 20 mL). The combined organic phase (CH3OH and EtOAc) was evaporated under reduced pressure to afford saturated ester 60 (0.23 g, 0.73 mmol, 57%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.93 (1H, t, J = 7.8 Hz), 6.75 (2H, m), 4.51 (1H, sept, J = 6 Hz), 4.17 (2H, q, J = 7.2 Hz), 3.81 (3H, s), 3.41 (2H, s), 2.92 (2H, t, J = 7.8 Hz), 2.83 (2H, t, J = 8.4 Hz), 1.25 (6H, d, J = 6 Hz), 1.25 (3H, t, 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 202.5, 167.2, 152.9, 144.9, 135.0, 123.5, 121.9, 110.7, 74.6, 61.4, 55.7, 49.4, 43.6, 25.0, 22.7, 14.2.
Ethyl 3-hydroxy-5-(2-isopropoxy-3-methoxyphenyl)pentanoate (61)
To dissolved ketone 60 (2.32 g, 7.52 mmol) in CH3OH (30 mL) was added NaBH4 (96 mg, 2.5 mmol) at 0 °C. The reaction was stirred for 1 h and then returned to room temperature for an additional 1 h. The CH3OH was removed under reduced pressure, and the residue was washed with water and extracted with EtOAc (3 × 30 mL). The combined organic phase was evaporated under reduced pressure, and the crude reaction product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford alcohol 61 (1.80 g, 5.80 mmol, 77%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.96 (1H, t, J = 8.4 Hz), 6.77 (1H, d, J = 6.6 Hz), 6.75 (1H, d, J = 7.8 Hz), 4.52 (1H, sept, J = 6.6 Hz), 4.13 (2H, q, J = 7.2 Hz), 3.91 (1H, m), 3.82 (3H, s), 2.82 (1H, m), 2.74 (1H, m), 2.45 (1H, m), 2.44 (1H, m), 1.78 (1H, m), 1.71 (1H, m), 1.28 (3H, t, J = 6.6 Hz), 1.26 (3H, t, J =6.6 Hz), 1.25 (3H, t, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 172.8, 152.9, 144.8, 136.0, 123.7, 122.1, 110.3, 74.9, 67.1, 60.7, 55.7, 41.6, 37.5, 41.6, 37.5, 26.3, 22.9, 22.6, 14.3.
Ethyl 3-((tert-butyldiphenylsilyl)oxy)-5-(2-isopropoxy-3-methoxyphenyl)pentanoate (62)
To dissolved alcohol 61 (0.77 g, 2.5 mmol) in DMF (10 mL) were added tert-butyl(chloro)diphenylsilane (TBDPSCl) (0.96 mL, 3.7 mmol) and imidazole (0.280 g, 3.98 mmol) at room temperature, and the solution was stirred for 12 h at room temperature. The DMF was removed under reduced pressure, and the resulting material was washed with brine (50 mL) and extracted with diethyl ether (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford protected alcohol 62 (0.960 g, 1.75 mmol, 71%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.70 (4H, m), 7.39 (6H, m), 6.87 (1H, t, J = 7.8 Hz), 6.70 (1H, d, J = 8.4 Hz), 6.52 (1H, d, J = 7.8 Hz), 4.41 (1H, m), 4.29 (1H, m), 4.00 (2H, m), 3.80 (3H, s), 2.55 (4H, m), 1.76 (2H, m), 1.19 (6H, m), 1.18 (3H, t, J = 7.2 Hz), 1.08 (9H, s). 13C NMR (150 MHz, CDCl3) δ 171.6, 152.9, 144.9, 136.5, 136.1, 135.3, 135.0, 134.3, 129.8, 129.7, 127.9, 127.6, 123.3, 121.6, 110.1, 74.4, 70.6, 60.4, 55.8, 42.1, 37.8, 26.7, 25.6, 22.7, 19.5, 19.2, 14.2.
3-((Tert-butyldiphenylsilyl)oxy)-5-(2-isopropoxy-3-methoxyphenyl)pentanoic acid (63)
To dissolved protected alcohol 62 (7.80 g, 14.2 mmol) in CH3OH/ THF (60 mL/ 30 mL) was added 2.5 M NaOH (20 mL) at 0 °C, and the solution was stirred for 1 h and then at room temperature for 12 h. The organic solvents (CH3OH and THF) were removed under reduced pressure, and water (30 mL) was added to the resulting suspension, followed by extraction with diethyl ether (3 × 50 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 40%B (15 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford carboxylic acid 63 (3.49 g, 6.70 mmol, 46%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.67 (4H, m), 7.39 (6H, m), 6.86 (1H, t, J = 7.8 Hz), 6.70 (1H, d, J = 8.4 Hz), 6.50 (1H, d, J = 7.2 Hz), 4.41 (1H, m), 4.20 (1H, m), 3.79 (3H, s), 2.56 (4H, m), 1.82 (2H, m), 1.19 (6H, t, J = 6 Hz), 1.06 (9H, s). 13C NMR (150 MHz, CDCl3) δ 175.5, 152.9, 144.8, 136.0, 133.7, 133.6, 129.9, 127.8, 123.4, 121.6, 110.2, 74.5, 70.5, 55.8, 41.2, 37.5, 27.1, 25.7, 22.7, 19.4.
7-((Tert-butyldiphenylsilyl)oxy)-1-isopropoxy-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (64)
To dissolved carboxylic acid 63 (3.49 g, 6.70 mmol) in dichloromethane (30 mL) were added oxalyl chloride (2.77 mL, 31.8 mmol) and DMF (0.1 mL) as catalyst at room temperature, and the solution was stirred for 2 h at room temperature. The solvent and unreacted oxalyl chloride were removed under reduced pressure. The yellow acyl chloride was then dissolved in dichloromethane (40 mL) and cooled to −10 °C. To this solution, 1 M SnCl4 in a dichloromethane solution (7.4 mL, 7.4 mmol) was added, and the reaction mixture was stirred for 30 min at −10 °C. The reaction was quenched by the addition of water, followed by extraction with dichloromethane (3 × 40 mL). The combined organic phase was dried over sodium sulfate and concentrated. The crude product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford ketone 64 (2.19 g, 4.50 mmol, 67%) as a pale-yellow gel. 1H NMR (600 MHz, CDCl3) δ 7.66 (4H, m), 7.54 (1H, d, J = 9.6 Hz), 7.38 (6H, m), 6.79 (1H, d, J = 9 Hz), 4.36 (1H, m), 4.28 (1H, m), 3.86 (3H, s), 3.14 (1H, m), 3.03 (2H, m), 2.88 (1H, m), 1.94 (2H, m), 1.27 (6H, m),1.03 (9H, s). 13C NMR (150 MHz, CDCl3) δ 199.7, 156.0, 144.1, 138.5, 136.1, 136.0, 135.9, 134.2, 133.9, 133.1, 129.9, 129.8, 127.9, 127.8, 127.7, 125.3, 109.5, 75.2, 68.4, 55.9, 50.4, 36.1, 27.0, 22.7, 22.6, 22.0, 19.3.
7-Hydroxy-1-isopropoxy-2-methoxy-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (65)
Ketone 64 (2.19 g, 4.50 mmol) was dissolved in THF (20 mL), TBAF (9.00 mL, 9.00 mmol) was added at 0 °C, and the reaction mixture was stirred for 30 min, followed by additional stirring at room temperature for 6 h. Brine (30 mL) was added, and the resultant solution was extracted with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford alcohol 65 (0.41 g, 1.7 mmol, 37%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.57 (1H, d, J = 8.4 Hz), 6.8 (1H, d, J = 8.4 Hz), 4.41 (1H, m), 4.31 (1H, m), 3.88 (3H, s), 3.11 (2H, m), 3.06 (1H, m), 2.99 (1H, m), 2.15 (1H, m), 1.87 (1H, m), 1.29 (6H, m). 13C NMR (150 MHz, CDCl3) δ199.6, 156.3, 144.2, 138.4, 132.6, 125.4, 109.7, 75.3, 67.3, 55.9, 50.3, 35.8, 22.7, 22.6, 22.1.
1-Isopropoxy-2-methoxy-7-((trimethylsilyl)oxy)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (66)
To a solution of alcohol 65 (0.35 g, 1.33 mmol) in DMF (20 mL) were added imidazole (0.27 g, 6.4 mmol) and TMSCl (4.26 mmol) at room temperature, and the reaction mixture was stirred for 12 h. The solvent was removed under reduced pressure, and brine (20 mL) was added, followed by extraction with EtOAc (3 × 30 mL). The organic extract was dried over sodium sulfate, filtered, and concentrated under reduced pressure, and the residure was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford TMS protected ketone 66 (0.19 g, 0.56 mmol, 43%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.54 (1H, d, J = 10.2 Hz), 6.79 (1H, d, J = 10.8 Hz), 4.39 (1H, m), 4.19 (1H, m), 3.86 (3H, s), 3.22 (1H, m), 2.99 (2H, m), 2.90 (1H, m), 1.91 (2H, m), 1.27 (6H, d, J = 9 Hz), 0.11 (9H, s). 13C NMR (125 MHz, CDCl3) δ 199.7, 156.1, 144.0, 138.6, 132.6, 125.3, 109.5, 75.1, 67.3, 55.8, 51.0, 36.4, 22.6, 22.5, 21.7.
4-Isopropoxy-3-methoxy-9-(3,4,5-trimethoxyphenyl)-5H-benzo[7]annulene (67)
To an oven-dried flask, THF (20 mL) and 3,4,5-trimethoxyphenyl bromide (0.21 g, 0.85 mmol) were added, and the solution was cooled to −78 °C. n-BuLi (0.34 mL, 2.5 M, 0.85 mmol) was slowly added to the reaction mixture, which was then stirred at −78 °C for 45 min. Ketone 66 (0.19 g, 0.56 mmol) was then added dropwise to the flask, and the reaction mixture was stirred (−78 °C to 0 °C) over 4 h, and at 0 °C for an additional 1 h. 2 M HCl (20 mL) was added at °C followed by stirring for 10 min and extraction with EtOAc (3 × 30 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford diene 67 (60 mg, 0.15 mmol, 27%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 6.81 (1H, d, J = 8.4 Hz), 6.71 (1H, d, J = 9 Hz), 6.61 (2H, s), 6.60 (1H, d, J = 5.4 Hz), 6.17 (1H, m), 5.87 (1H, m), 4.48 (1H, p, J = 6.6 Hz), 3.91 (3H, s), 3.86 (6H, s), 3.85 (3H, s), 3.26 (2H, m, b), 1.38 (6H, d, J = 6 Hz). 13C NMR (150 MHz, CDCl3) δ 153.8, 153.0, 145.6, 142.0, 140.4, 137.5, 134.5, 131.7, 128.5, 126.9, 125.9, 124.8, 109.1, 106.7, 75.0, 61.0, 56.3, 55.8, 26.7, 22.8.
3-Methoxy-9-(3,4,5-trimethoxyphenyl)-5H-benzo[7]annulen-4-ol (68)
Isopropyl protected phenol 67 (60 mg, 0.15 mmol) was dissolved in CH2Cl2 (10 mL), to which BCl3 (0.17 mL, 1 M, 0.17 mmol) was added, and the reaction mixture was stirred at 0 °C for 1 h. The solution was washed with water and 2 M HCl and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over sodium sulfate and evaporated under reduced pressure. The crude reaction was purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] to afford phenol 68 (40 mg, 0.11 mmol, 75%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 6.67 (1H, d, J = 8.4 Hz), 6.62 (1H, d, J = 9 Hz), 6.60 (2H, s), 6.59 (1H, d, J = 5.4 Hz), 6.15 (1H, m), 5.94 (1H, m), 3.89 (6H, s), 3.84 (6H, s), 3.23 (2H, m, b). 13C NMR (150 MHz, CDCl3) δ153.0, 147.1, 145.5, 140.6, 140.3, 137.5, 132.2, 128.3, 127.0, 126.2, 126.0, 120.1, 107.6, 106.7, 61.1, 56.3, 56.2, 25.7. HRMS: Obsvd 377.1361 [M + Na+], Calcd for C21H22O5Na: 377.1359. HPLC: 19.52 min.
Sodium 3-methoxy-9-(3,4,5-trimethoxyphenyl)-5H-benzo[7]annulen-4-yl phosphate (69)
To phenol 68 (95 mg, 0.27 mmol) dissolved in CH2Cl2 (25 mL) was added POCl3 (0.11 mL, 1.1 mmol) and pyridine (0.078 mL, 0.97 mmol), and the reaction mixture was stirred from 0 °C to room temperature for 8 h. The CH2Cl2 was removed under reduced pressure, the residue was dissolved in H2O/ THF (10 mL/ 5 mL) at room temperature, and the solution was stirred for 1 h. A NaOH solution (0.1 M) was added to the reaction mixture to adjust to pH = 10 at 0 °C, and the solution was stirred at 0 °C for 30 min. Water was removed under reduced pressure. The crude product was purified by flash chromatography using a pre-packed 12 g C-18 column [solvent A: acetonitrile; solvent B: water; gradient: 0%A / 100%B (1 CV), 0%A / 100%B → 10%A / 90%B (10 CV), 10%A / 90%B (2 CV); flow rate: 12 mL/min; monitored at 254 and 280 nm] to afford phosphate salt 69 (65.8 mg, 0.14 mmol, 51%) as a yellow solid. 1H NMR (500 MHz, D2O) δ 6.65 (2H, s), 6.60 (2H, m), 6.53 (1H, d, J = 6 Hz), 6.10 (1H, m), 5.98 (1H, m), 3.78 (3H, s), 3.72 (3H, s), 3.70 (6H, s), 3.29 (2H, b).13C NMR (125 MHz, D2O) δ 153.0, 152.1, 144.5, 140.8, 138.4, 136.0, 134.5, 131.1, 129.8, 126.2, 126.1, 124.2, 109.1, 106.7, 60.9, 55.9, 55.7, 26.9. 31P NMR (200 MHz, D2O) δ 0.81. HRMS: Obsvd 479.0841 [M + H+], Calcd for C21H22O8Na2P+: 479.0842. HPLC: 14.22 min
Confirmation that target molecules are not PAINS molecules
Each of the target molecules and several common motifs were screened using the zinc15 docking algorithm (http://zinc15.docking.org/patterns/home), which provided confirmation that these benzosuberene-based molecules are not Pan Assay Interference compoundS (PAINS). A molecule previously identified as a PAIN molecule (Capuzzi et al., J. Chem. Inf. Model. 2017, 57, 417–427) was screened to provide positive confirmation that the algorithm was functioning accurately.
Biology
Cell lines and sulforhodamine B (SRB) assay
The sulforhodamine B (SRB) assay was used to assess growth inhibition of human cancer cells, as previously described.29,82,83 DU-145, SK-OV-3, and NCI-H460 cancer cell lines (obtained from ATCC) were maintained in T75 flasks (Corning) using high glucose DMEM supplemented with 10% fetal bovine serum (Gibco)/ 1% gentamicin sulfate for a maximum of 15 passages. For these experiments, cells were trypsinized, counted, and plated at 7000–8000 cells/well into 96-well plates (Corning) and incubated for 24 h at 37 °C in a humidified incubator in a 5% CO2 atmosphere. Compounds to be tested were dissolved in DMSO to generate a 10 mg/mL stock solution, and serial dilutions added in media to the plates. Doxorubicin (Sigma-Aldrich) and paclitaxel (Tokyo Chemical) were used as positive controls. After a 48 h treatment, the cells were fixed with trichloroacetic acid (10% final concentration), washed, dried, stained with SRB dye, washed to remove excess dye, and dried. SRB dye was solubilized, and absorbances were measured at wavelength 540 nm and normalized to values at wavelength 630 nm using an automated Biotek plate reader. A growth inhibition of 50% (GI50 or the drug concentration causing 50% reduction in the net protein increase) was calculated from the absorbance data.
84. Colchicine binding assay.84
Inhibition of [3H]colchicine binding to tubulin was measured in 0.1 mL reaction mixtures, each containing 1.0 µM tubulin, 5.0 µM [3H]colchicine (Perkin-Elmer), 5% (v/v) dimethyl sulfoxide, compounds at 1.0 or 5.0 µM, as indicated, and components that stabilize the colchicine binding activity of tubulin (1.0 M monosodium glutamate [adjusted to pH 6.6 with HCl in a 2.0 M stock solution], 0.5 mg/mL bovine serum albumin, 0.1 M glucose-1-phosphate, 1.0 mM MgCl2, and 1.0 mM GTP). Incubation was for 10 min at 37 °C, when in control reaction mixtures colchicine binding is 40–60% complete. Reactions were stopped with 2.0 mL of ice-cold water, with the reaction mixtures being placed on ice. Each diluted sample was poured onto a stack of two DEAE-cellulose filters (GE Biomedical), followed by 3 successive 2 mL aliquots of ice-cold water. A reduced vacuum was used to remove excess water from the filters, which were washed three times with 2 mL water and placed into vials containing 5 mL of Biosafe II scintillation cocktail. Samples were counted 18 h later in a Beckman scintillation counter. Samples with inhibitors were compared to samples with no inhibitor, and percent inhibition was determined, correcting all values for the amount of radiolabel bound to the filters in the absence of tubulin.
85. Inhibition of tubulin polymerization.85
Tubulin polymerization was evaluated in 0.25 mL reaction mixtures (final volume) containing 1 mg/mL (10 µM) purified bovine brain tubulin, 0.8 M monosodium glutamate (pH 6.6), 4% (v/v) dimethyl sulfoxide, 0.4 mM GTP, and different compound concentrations. All components except GTP were preincubated for 15 min at 30 °C in 0.24 mL. The assay mixtures were cooled to 0 °C, and 10 µL of 0.01 M GTP was added to each sample. Reaction mixtures were transferred to cuvettes held at 0 °C in Beckman DU-7400 and DU-7500 spectrophotometers equipped with electronic temperature controllers. The temperature was increased to 30 °C, over about 30 s, and polymerization was followed turbidimetrically at 350 nm for 20 min. The IC50 was defined as the compound concentration inhibiting extent of polymerization by 50% after 20 min.
In vivo vascular disruption
The human prostate cancer cell line (PC3) was modified by knock down of a tumor suppressor protein PC3-DAB2IP and further modified by transfection with a firefly luciferase reporter, as described in detail previously.77 Male Copenhagen rats (originally from Charles River and bred at UT Southwestern) were inoculated subcutaneously with 5 X 105 PC3-DAB2IP-luc cells mixed with 30% Matrigel® in the right thigh. These rats were approximately 6 weeks old and weighed between 100–120 g on date of implantation. Tumors were allowed to grow to a diameter of at least 1 cm before treatment commenced. Three rats were treated with VDA prodrug 69 (25 mg/mL in saline) administered IP. Rat 1 received 10 mg/kg followed by 40 mg/kg 24 h later and CA4P (30 mg/kg at 20 mg/mL in saline) was administered IP as a control 4 days later. Rat 2 received a single dose of 40 mg/kg of VDA prodrug 69, and rat 3 received 80 mg/kg of 69. Bioluminescence imaging (BLI) was performed before treatment (baseline), and at 4 and 24 h after treatment using an IVIS Spectrum® (Perkin-Elmer). Briefly, anesthetized rats (breathing oxygen with 3% isoflurane, Henry Schein Inc.) were injected subcutaneously in the foreback neck region with 120 mg/kg D-luciferin sodium salt (Gold Biotechnology, St. Louis, MO) and imaged over a time course of about 30 min. Fresh luciferin was administered at each time point. The resulting light intensity time curves were analyzed using Living Image® Software and light emission measured for a region of interest encompassing the tumor. All animal procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health as well as the Institutional Animal Care and Use Committee approved protocols (APN 2017–102168) at the University of Texas Southwestern Medical Center.
Supplementary Material
Acknowledgements
The authors are grateful to the Cancer Prevention and Research Institute of Texas (CPRIT, Grant RP140399 to K.G.P., M.L.T., R.P.M.; Grant RP170696 to K.G.P., M.L.T.; Grant RP140285 to R.P.M.; Grant RP120670-C3 to D.S.), the National Cancer Institute of the National Institutes of Health (Grant 5R01CA140674 to K.G.P., M.L.T., R.P.M.), Mateon Therapeutics, Inc. (grant to K.G.P., M.L.T.), and the University Research Committee (URC) and the Vice Provost for Research at Baylor University (M.L.T.) for financial support of these projects and further acknowledge resources associated with the UT Southwestern Small Animal Imaging Resource (SW-SAIRP), supported in part by the Harold C. Simmons Cancer Center through an NCI Cancer Center Support Grant, 1P30CA142543, and the Department of Radiology. The IVIS Spectrum was acquired with the assistance of NIH Shared Instrumentation Grant S10RR024757. Disclaimer: The content of this article is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Abbreviations Used:
- VDA
(vascular disrupting agent)
- AIAs
(angiogenesis inhibiting agents)
- TBDPSCl
(tert-butyldiphenylsilyl chloride)
- GI50
(half maximal growth inhibition)
- BLI
(bioluminescence imaging)
- ND
(not determined)
- NR
(not reported)
- DEAE
(diethylaminoethyl)
Footnotes
Supporting Information:
Supporting Information: 1H NMR, 13C NMR, 31P NMR, HRMS, HPLC for final target compounds and intermediates (1H NMR, 13C NMR only) associated with this article, a table of biological data including standard deviations, and a separate table of biological data including molecular formula strings (as SMILES) can be found, in the online version, at http://10.1021/acs.jmedchem.9b00551.
References:
- (1).Salmon BA; Siemann DW Characterizing the Tumor Response to Treatment with Combretastatin A4 Phosphate. Int. J. Radiat. Oncol. Biol. Phys 2007, 68 (1), 211–217. 10.1016/j.ijrobp.2006.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hanahan D; Weinberg RA Hallmarks of Cancer: The Next Generation. Cell 2011, 144 (5), 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- (3).Siemann DW The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev 2011, 37 (1), 63–74. 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tozer GM; Kanthou C; Baguley BC Disrupting Tumour Blood Vessels. Nat. Rev. Cancer 2005, 5 (6), 423–435. 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- (5).Kanthou C; Tozer GM Tumour Targeting by Microtubule-Depolymerizing Vascular Disrupting Agents. Expert Opin. Ther. Targets 2007, 11 (11), 1443–1457. 10.1517/14728222.11.11.1443. [DOI] [PubMed] [Google Scholar]
- (6).Denekamp J Endothelial Cell Proliferation as a Novel Approach to Targeting Tumour Therapy. Br. J. Cancer 1982, 45 (1), 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Denekamp J Review Article: Angiogenesis, Neovascular Proliferation and Vascular Pathophysiology as Targets for Cancer Therapy. Br. J. Radiol 1993, 66 (783), 181–196. 10.1259/0007-1285-66-783-181. [DOI] [PubMed] [Google Scholar]
- (8).Sriram M; Hall JJ; Grohmann NC; Strecker TE; Wootton T; Franken A; Trawick ML; Pinney KG Design, Synthesis and Biological Evaluation of Dihydronaphthalene and Benzosuberene Analogs of the Combretastatins as Inhibitors of Tubulin Polymerization in Cancer Chemotherapy. Bioorg. Med. Chem 2008, 16 (17), 8161–8171. 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- (9).Horsman MR; Bohn AB; Busk M Vascular Targeting Therapy: Potential Benefit Depends on Tumor and Host Related Effects. Exp. Oncol 2010, 32 (3), 143–148. [PubMed] [Google Scholar]
- (10).Siemann DW; Bibby MC; Dark GG; Dicker AP; Eskens FALM; Horsman MR; Marmé D; LoRusso PM Differentiation and Definition of Vascular-Targeted Therapies. Clin. Cancer Res 2005, 11 (2 I), 416–420. [PubMed] [Google Scholar]
- (11).Mason RP; Zhao D; Liu L; Trawick ML; Pinney KG A Perspective on Vascular Disrupting Agents That Interact with Tubulin: Preclinical Tumor Imaging and Biological Assessment. Integr. Biol 2011, 3 (4), 375–387. 10.1039/C0IB00135J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dougherty GJ; Chaplin DJ Development of Vascular Disrupting Agents. In Vascular Disruptive Agents for the Treatment of Cancer; Springer, New York, NY, 2010; pp 1–27. 10.1007/978-1-4419-6609-4_1. [DOI] [Google Scholar]
- (13).Siemann DW Tumor Vasculature: A Target for Anticancer Therapies. In Vascular-Targeted Therapies in Oncology; Siemann DW, Ed.; Chichester, 2006; pp 1–8. [Google Scholar]
- (14).Monk KA; Siles R; Hadimani MB; Mugabe BE; Ackley JF; Studerus SW; Edvardsen K; Trawick ML; Garner CM; Rhodes MR; Pettit GR; Pinney KG Design, Synthesis, and Biological Evaluation of Combretastatin Nitrogen-Containing Derivatives as Inhibitors of Tubulin Assembly and Vascular Disrupting Agents. Bioorg. Med. Chem 2006, 14 (9), 3231–3244. 10.1016/j.bmc.2005.12.033. [DOI] [PubMed] [Google Scholar]
- (15).Pettit GR; Singh SB; Boyd MR; Hamel E; Pettit RK; Schmidt JM; Hogan F Antineoplastic Agents. 291. Isolation and Synthesis of Combretastatins A-4, A-5, and A-6. J. Med. Chem 1995, 38 (10), 1666–1672. 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- (16).McGown AT; Fox BW Differential Cytotoxicity of Combretastatins A1 and A4 in Two Daunorubicin-Resistant P388 Cell Lines. Cancer Chemother. Pharmacol 1990, 26 (1), 79–81. [DOI] [PubMed] [Google Scholar]
- (17).Pettit GR; Moser BR; Boyd MR; Schmidt JM; Pettit RK; Chapuis J-C Antineoplastic Agents 460. Synthesis of Combretastatin A-2 Prodrugs. Anticancer. Drug Des 2001, 16 (4–5), 185–193. [PubMed] [Google Scholar]
- (18).Pettit GR; Singh SB; Hamel E; Lin CM; Alberts DS; Garcia-Kendal D Isolation and Structure of the Strong Cell Growth and Tubulin Inhibitor Combretastatin A-4. Experientia 1989, 45 (2), 209–211. 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- (19).Dark GG; Hill SA; Prise VE; Tozer GM; Pettit GR; Chaplin DJ Combretastatin A-4, an Agent That Displays Potent and Selective Toxicity Toward Tumor Vasculature. Cancer Res 1997, 57 (10), 1829–1834. [PubMed] [Google Scholar]
- (20).Pettit GR; Toki B; Herald DL; Verdier-Pinard P; Boyd MR; Hamel E; Pettit RK Antineoplastic Agents. 379. Synthesis of Phenstatin Phosphate. J. Med. Chem 1998, 41 (10), 1688–1695. 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- (21).Boyland E; Boyland ME Studies in Tissue Metabolism: The Action of Colchicine and B. Typhosus Extract. Biochem. J 1937, 31 (3), 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Woods JA; Hadfield JA; Pettit GR; Fox BW; McGown AT The Interaction with Tubulin of a Series of Stilbenes Based on Combretastatin A-4. Br. J. Cancer 1995, 71 (4), 705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pettit GR; Singh SB; Niven ML; Hamel E; Schmidt JM Isolation, Structure, and Synthesis of Combretastatins A-1 and B-1, Potent New Inhibitors of Microtubule Assembly, Derived from Combretum Caffrum. J. Nat. Prod 1987, 50 (1), 119–131. 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- (24).Pettit GR; Grealish MP; Herald DL; Boyd MR; Hamel E; Pettit RK Antineoplastic Agents. 443. Synthesis of the Cancer Cell Growth Inhibitor Hydroxyphenstatin and Its Sodium Diphosphate Prodrug. J. Med. Chem 2000, 43 (14), 2731–2737. 10.1021/jm000045a. [DOI] [PubMed] [Google Scholar]
- (25).Herdman CA; Devkota L; Lin C-M; Niu H; Strecker TE; Lopez R; Liu L; George CS; Tanpure RP; Hamel E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG Structural Interrogation of Benzosuberene-Based Inhibitors of Tubulin Polymerization. Bioorg. Med. Chem 2015, 23 (24), 7497–7520. 10.1016/j.bmc.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tanpure RP; Harkrider AR; Strecker TE; Hamel E; Trawick ML; Pinney KG Application of the McMurry Coupling Reaction in the Synthesis of Tri- and Tetra-Arylethylene Analogues as Potential Cancer Chemotherapeutic Agents. Bioorg. Med. Chem 2009, 17 (19), 6993–7001. 10.1016/j.bmc.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pinney KG; Pettit GR; Trawick ML; Jelinek C; Chaplin DJ The Discovery and Development of the Combretastatins. Anticancer Agents Nat. Prod 2012, 27–63. [Google Scholar]
- (28).Pinney KG Molecular Recognition of the Colchicine Binding Site as a Design Paradigm for the Discovery and Development of Vascular Disrupting Agents. In Vascular-Targeted Therapies in Oncology; Siemann DW, Ed.; Chichester, 2006; pp 95–121. [Google Scholar]
- (29).Siles R; Ackley JF; Hadimani MB; Hall JJ; Mugabe BE; Guddneppanavar R; Monk KA; Chapuis J-C; Pettit GR; Chaplin DJ; Edvardsen K; Trawick ML; Garner GM; Pinney KG Combretastatin Dinitrogen-Substituted Stilbene Analogues as Tubulin-Binding and Vascular-Disrupting Agents. J. Nat. Prod 2008, 71 (3), 313–320. 10.1021/np070377j. [DOI] [PubMed] [Google Scholar]
- (30).Shirali A; Sriram M; Hall JJ; Nguyen BL; Guddneppanavar R; Hadimani MB; Ackley JF; Siles R; Jelinek CJ; Arthasery P; Brown RC; Murrell VL; McMordie A; Sharma S; Chaplin DJ; Pinney KG Development of Synthetic Methodology Suitable for the Radiosynthesis of Combretastatin A-1 (CA1) and Its Corresponding Prodrug CA1P. J. Nat. Prod 2009, 72 (3), 414–421. 10.1021/np800661r. [DOI] [PubMed] [Google Scholar]
- (31).Chaplin DJ; III, C. M. G.; Kane RR; Pinney KG; Prezioso JA; Edvardsen K Functionalized Stilbene Derivatives as Improved Vascular Targeting Agents. US7384925B2, June 10, 2008.
- (32).Chen Z; Mocharla VP; Farmer JM; Pettit GR; Hamel E; Pinney KG Preparation of New Anti-Tubulin Ligands Through a Dual-Mode, Addition−Elimination Reaction to a Bromo-Substituted α,β-Unsaturated Sulfoxide. J. Org. Chem 2000, 65 (25), 8811–8815. 10.1021/jo0004761. [DOI] [PubMed] [Google Scholar]
- (33).Pinney KG; Bounds AD; Dingeman KM; Mocharla VP; Pettit GR; Bai R; Hamel E A New Anti-Tubulin Agent Containing the Benzo[b]Thiophene Ring System. Bioorg. Med. Chem. Lett 1999, 9 (8), 1081–1086. 10.1016/S0960-894X(99)00143-2. [DOI] [PubMed] [Google Scholar]
- (34).Pinney K; Pettit G; Mocharla V; Mejia M. del P.; Shirali A Anti-Mitotic Agents Which Inhibit Tubulin Polymerization. WO/1998/039323, September 12, 1998.
- (35).Pinney K; Sriram M; George C; Tanpure R Efficient Method for Preparing Functionalized Benzosuberenes. WO/2012/068284, May 25, 2012.
- (36).Pinney K; Mocharla V; Chen Z; Garner C; Ghatak A; Hadimani M; Kessler J; Dorsey J; Edvardsen K; Chaplin D; Prezioso J; Ghatak U Tubulin Binding Agents and Corresponding Prodrug Constructs. US20040043969A1, March 4, 2004.
- (37).Tanpure RP; George CS; Strecker TE; Devkota L; Tidmore JK; Lin C-M; Herdman CA; MacDonough MT; Sriram M; Chaplin DJ; Trawick ML; Pinney KG Synthesis of Structurally Diverse Benzosuberene Analogues and Their Biological Evaluation as Anti-Cancer Agents. Bioorg. Med. Chem 2013, 21 (24), 8019–8032. 10.1016/j.bmc.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tanpure RP; George CS; Sriram M; Strecker TE; Tidmore JK; Hamel E; Charlton-Sevcik AK; Chaplin DJ; Trawick ML; Pinney KG An Amino-Benzosuberene Analogue That Inhibits Tubulin Assembly and Demonstrates Remarkable Cytotoxicity. MedChemComm 2012, 3 (6), 720–724. 10.1039/C2MD00318J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hadimani MB; MacDonough MT; Ghatak A; Strecker TE; Lopez R; Sriram M; Nguyen BL; Hall JJ; Kessler RJ; Shirali AR; Liu L; Garner CM; Pettit GR; Hamel E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG Synthesis of a 2-Aryl-3-Aroyl Indole Salt (OXi8007) Resembling Combretastatin A-4 with Application as a Vascular Disrupting Agent. J. Nat. Prod 2013, 76 (9), 1668–1678. 10.1021/np400374w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hamel E Antimitotic Natural Products and Their Interactions with Tubulin. Med. Res. Rev 1996, 16 (2), 207–231. . [DOI] [PubMed] [Google Scholar]
- (41).Wu X; Wang Q; Li W Recent Advances in Heterocyclic Tubulin Inhibitors Targeting the Colchicine Binding Site. Anticancer Agents Med. Chem 2016, 16 (10), 1325–1338. 10.2174/1871520616666160219161921. [DOI] [PubMed] [Google Scholar]
- (42).Lee RM; Gewirtz DA Colchicine Site Inhibitors of Microtubule Integrity as Vascular Disrupting Agents. Drug Dev. Res 2008, 69 (6), 352–358. 10.1002/ddr.20267. [DOI] [Google Scholar]
- (43).Gigant B; Cormier A; Dorléans A; Ravelli RBG; Knossow M Microtubule-Destabilizing Agents: Structural and Mechanistic Insights from the Interaction of Colchicine and Vinblastine with Tubulin. In Tubulin-Binding Agents: Synthetic, Structural and Mechanistic Insights; Carlomagno T, Ed.; Topics in Current Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 2009; pp 259–278. 10.1007/128_2008_11. [DOI] [PubMed] [Google Scholar]
- (44).Sackett DL Podophyllotoxin, Steganacin and Combretastatin: Natural Products That Bind at the Colchicine Site of Tubulin. Pharmacol. Ther 1993, 59 (2), 163–228. 10.1016/0163-7258(93)90044-E. [DOI] [PubMed] [Google Scholar]
- (45).Wang Y; Zhang H; Gigant B; Yu Y; Wu Y; Chen X; Lai Q; Yang Z; Chen Q; Yang J Structures of a Diverse Set of Colchicine Binding Site Inhibitors in Complex with Tubulin Provide a Rationale for Drug Discovery. FEBS J 2016, 283 (1), 102–111. 10.1111/febs.13555. [DOI] [PubMed] [Google Scholar]
- (46).Ji Y-T; Liu Y-N; Liu Z-P Tubulin Colchicine Binding Site Inhibitors as Vascular Disrupting Agents in Clinical Developments. Curr. Med. Chem 2015, 22 (11), 1348–1360. 10.2174/0929867322666150114163732. [DOI] [PubMed] [Google Scholar]
- (47).Nguyen TL; McGrath C; Hermone AR; Burnett JC; Zaharevitz DW; Day BW; Wipf P; Hamel E; Gussio R A Common Pharmacophore for a Diverse Set of Colchicine Site Inhibitors Using a Structure-Based Approach. J. Med. Chem 2005, 48 (19), 6107–6116. 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- (48).Chen J; Liu T; Dong X; Hu Y Recent Development and SAR Analysis of Colchicine Binding Site Inhibitors. Mini. Rev. Med. Chem 2009, 9 (10), 1174–1190. [DOI] [PubMed] [Google Scholar]
- (49).Lu Y; Chen J; Xiao M; Li W; Miller DD An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res 2012, 29 (11), 2943–2971. 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Macdonough MT; Strecker TE; Hamel E; Hall JJ; Chaplin DJ; Trawick ML; Pinney KG Synthesis and Biological Evaluation of Indole-Based, Anti-Cancer Agents Inspired by the Vascular Disrupting Agent 2-(3’-Hydroxy-4’-Methoxyphenyl)-3-(3″,4″,5″-Trimethoxybenzoyl)-6-Methoxyindole (OXi8006). Bioorg. Med. Chem 2013, 21 (21), 6831–6843. 10.1016/j.bmc.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Flynn BL; Gill GS; Grobelny DW; Chaplin JH; Paul D; Leske AF; Lavranos TC; Chalmers DK; Charman SA; Kostewicz E; Shackleford DM; Morizzi J; Hamel E; Jung MK; Kremmidiotis G Discovery of 7-Hydroxy-6-Methoxy-2-Methyl-3-(3,4,5-Trimethoxybenzoyl)Benzo[b]Furan (BNC105), a Tubulin Polymerization Inhibitor with Potent Antiproliferative and Tumor Vascular Disrupting Properties. J. Med. Chem 2011, 54 (17), 6014–6027. 10.1021/jm200454y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kuo C-C; Hsieh H-P; Pan W-Y; Chen C-P; Liou J-P; Lee S-J; Chang Y-L; Chen L-T; Chen C-T; Chang J-Y BPR0L075, a Novel Synthetic Indole Compound with Antimitotic Activity in Human Cancer Cells, Exerts Effective Antitumoral Activity in Vivo. Cancer Res 2004, 64 (13), 4621–4628. 10.1158/0008-5472.CAN-03-3474. [DOI] [PubMed] [Google Scholar]
- (53).Liu L; Beck H; Wang X; Hsieh H-P; Mason RP; Liu X Tubulin-Destabilizing Agent BPR0L075 Induces Vascular-Disruption in Human Breast Cancer Mammary Fat Pad Xenografts. PLOS ONE 2012, 7 (8), e43314 10.1371/journal.pone.0043314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rasolofonjatovo E; Provot O; Hamze A; Rodrigo J; Bignon J; Wdzieczak-Bakala J; Desravines D; Dubois J; Brion J-D; Alami M Conformationally Restricted Naphthalene Derivatives Type Isocombretastatin A-4 and Isoerianin Analogues: Synthesis, Cytotoxicity and Antitubulin Activity. Eur. J. Med. Chem 2012, 52, 22–32. 10.1016/j.ejmech.2012.03.001. [DOI] [PubMed] [Google Scholar]
- (55).Rasolofonjatovo E; Provot O; Hamze A; Rodrigo J; Bignon J; Wdzieczak-Bakala J; Lenoir C; Desravines D; Dubois J; Brion J-D; Alami M Design, Synthesis and Anticancer Properties of 5-Arylbenzoxepins as Conformationally Restricted Isocombretastatin A-4 Analogs. Eur. J. Med. Chem 2013, 62, 28–39. 10.1016/j.ejmech.2012.12.042. [DOI] [PubMed] [Google Scholar]
- (56).Chen Z; O’Donnell CJ; Maderna A Synthesis of 3-Methoxy-9-(3,4,5-Trimethoxyphenyl)-6,7-Dihydro-5H-Benzo[7]Annulen-4-Ol, a Potent Antineoplastic Benzosuberene Derivative for Anti-Cancer Chemotherapy. Tetrahedron Lett 2012, 53 (1), 64–66. 10.1016/j.tetlet.2011.10.145. [DOI] [Google Scholar]
- (57).Chen Z; Maderna A; Sukuru SCK; Wagenaar M; O’Donnell CJ; Lam M-H; Musto S; Loganzo F New Cytotoxic Benzosuberene Analogs. Synthesis, Molecular Modeling and Biological Evaluation. Bioorg. Med. Chem. Lett 2013, 23 (24), 6688–6694. 10.1016/j.bmcl.2013.10.039. [DOI] [PubMed] [Google Scholar]
- (58).Galli U; Travelli C; Aprile S; Arrigoni E; Torretta S; Grosa G; Massarotti A; Sorba G; Canonico PL; Genazzani AA; Tron GC Design, Synthesis, and Biological Evaluation of Combretabenzodiazepines: A Novel Class of Anti-Tubulin Agents. J. Med. Chem 2015, 58 (3), 1345–1357. 10.1021/jm5016389. [DOI] [PubMed] [Google Scholar]
- (59).Nikolaus Prileschajew. Oxydation Ungesättigter Verbindungen Mittels Organischer Superoxyde. Berichte Dtsch. Chem. Ges 1909, 42 (4), 4811–4815. 10.1002/cber.190904204100. [DOI] [Google Scholar]
- (60).VanRheenen V; Kelly RC; Cha DY An Improved Catalytic OsO4 Oxidation of Olefins to Cis-1,2-Glycols Using Tertiary Amine Oxides as the Oxidant. Tetrahedron Lett 1976, 17 (23), 1973–1976. 10.1016/S0040-4039(00)78093-2. [DOI] [Google Scholar]
- (61).Gustowski DA; Delgado M; Gatto VJ; Echegoyen L; Gokel GW Electrochemical Switching in Anthraquinone-Substituted Carbon-Pivot Lariat Ethers and Podands: Chain Length Effects in Geometric and Electronic Cooperativity. J. Am. Chem. Soc 1986, 108 (24), 7553–7560. 10.1021/ja00284a019. [DOI] [PubMed] [Google Scholar]
- (62).Traugott Sandmeyer. Ueber Die Ersetzung Der Amidgruppe Durch Chlor in Den Aromatischen Substanzen. Berichte Dtsch. Chem. Ges 2006, 17 (2), 1633–1635. 10.1002/cber.18840170219. [DOI] [Google Scholar]
- (63).Traugott Sandmeyer. Ueber Die Ersetzung Der Amid‐gruppe Durch Chlor, Brom Und Cyan in Den Aromatischen Substanzen. Berichte Dtsch. Chem. Ges 2006, 17 (2), 2650–2653. 10.1002/cber.188401702202. [DOI] [Google Scholar]
- (64).Mori A; Mizusaki T; Miyakawa Y; Ohashi E; Haga T; Maegawa T; Monguchi Y; Sajiki H Chemoselective Hydrogenation Method Catalyzed by Pd/C Using Diphenylsulfide as a Reasonable Catalyst Poison. Tetrahedron 2006, 62 (51), 11925–11932. 10.1016/j.tet.2006.09.094. [DOI] [Google Scholar]
- (65).E. Eaton P; R. Carlson G; T. Lee J Phosphorus Pentoxide-Methanesulfonic Acid. Convenient Alternative to Polyphosphoric Acid. J. Org. Chem 1973, 38, 4071–4073. 10.1021/jo00987a028. [DOI] [Google Scholar]
- (66).Tanis VM; Moya C; Jacobs RS; Little RD Synthesis and Evaluation of the Bioactivity of Simplified Analogs of the Seco-Pseudopterosins; Progress Toward Determining a Pharmacophore. Tetrahedron 2008, 64 (47), 10649–10663. 10.1016/j.tet.2008.09.025. [DOI] [Google Scholar]
- (67).Lin W; Wang Q; Xiao Y; He H; Zheng; Chen X; Chen S; WuXi AppTec. Fast Synthetic Route for 3-Aryl Substituted Propanoic Acid. China, CN 101747171 A, December 17, 2008.
- (68).Andre Benner; Alessandro Bonifazi; Chikako Shirataki; Louisa Temme; Dirk Schepmann; Wilma Quaglia; Osami Shoji; Yoshihito Watanabe; Constantin Daniliuc; Bernhard Wünsch. GluN2B‐Selective N‐Methyl‐d‐aspartate (NMDA) Receptor Antagonists Derived from 3‐Benzazepines: Synthesis and Pharmacological Evaluation of Benzo[7]Annulen‐7‐amines. ChemMedChem 2014, 9 (4), 741–751. 10.1002/cmdc.201300547. [DOI] [PubMed] [Google Scholar]
- (69).Walsh JJ; Sha R; McCormack EM; Hudson GJ; White M; Stack GD; Moran BW; Coogan A; Breen EC Tubulin Binding Agents. US20150018566A1, August 27, 2012.
- (70).Gilman H; Jones RG; Woods LA The Preparation of Methylcopper and Some Observations on the Decomposition of Organocopper Compounds. J. Org. Chem 1952, 17 (12), 1630–1634. 10.1021/jo50012a009. [DOI] [Google Scholar]
- (71).Hua J; Sheng Y; Pinney KG; Garner CM; Kane RR; Prezioso JA; Pettit GR; Chaplin DJ; Edvardsen K Oxi4503, a Novel Vascular Targeting Agent: Effects on Blood Flow and Antitumor Activity in Comparison to Combretastatin A-4 Phosphate. Anticancer Res 2003, 23 (2B), 1433–1440. [PubMed] [Google Scholar]
- (72).Benham FJ; Fogh J; Harris H Alkaline Phosphatase Expression in Human Cell Lines Derived from Various Malignancies. Int. J. Cancer 1981, 27 (5), 637–644. [DOI] [PubMed] [Google Scholar]
- (73).Rao SR; Snaith AE; Marino D; Cheng X; Lwin ST; Orriss IR; Hamdy FC; Edwards CM Tumour-Derived Alkaline Phosphatase Regulates Tumour Growth, Epithelial Plasticity and Disease-Free Survival in Metastatic Prostate Cancer. Br. J. Cancer 2017, 116 (2), 227–236. 10.1038/bjc.2016.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Gangjee A; Zhao Y; Lin L; Raghavan S; Roberts EG; Risinger AL; Hamel E; Mooberry SL Synthesis and Discovery of Water Soluble Microtubule Targeting Agents That Bind to the Colchicine Site on Tubulin and Circumvent Pgp Mediated Resistance. J. Med. Chem 2010, 53 (22), 8116–8128. 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Maguire CJ; Chen Z; Mocharla VP; Sriram M; Strecker TE; Hamel E; Zhou H; Lopez R; Wang Y; Mason RP; Chaplin DJ; Trawick ML; Pinney KG Synthesis of Dihydronaphthalene Analogues Inspired by Combretastatin A-4 and Their Biological Evaluation as Anticancer Agents. MedChemComm 2018, 9 (10), 1649–1662. 10.1039/C8MD00322J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Folaron M; Seshadri M Bioluminescence and MR Imaging of the Safety and Efficacy of Vascular Disruption in Gliomas. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2016, 18 (6), 860–869. 10.1007/s11307-016-0963-8. [DOI] [PubMed] [Google Scholar]
- (77).Tumati V; Mathur S; Song K; Hsieh J-T; Zhao D; Takahashi M; Dobin T; Gandee L; Solberg TD; Habib AA; Saha D Development of a Locally Advanced Orthotopic Prostate Tumor Model in Rats for Assessment of Combined Modality Therapy. Int. J. Oncol 2013, 42 (5), 1613–1619. 10.3892/ijo.2013.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Zhao D; Richer E; Antich PP; Mason RP Antivascular Effects of Combretastatin A4 Phosphate in Breast Cancer Xenograft Assessed Using Dynamic Bioluminescence Imaging and Confirmed by MRI. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 2008, 22 (7), 2445–2451. 10.1096/fj.07-103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Zhao D; Jiang L; Hahn EW; Mason RP Tumor Physiologic Response to Combretastatin A4 Phosphate Assessed by MRI. Int. J. Radiat. Oncol. Biol. Phys 2005, 62 (3), 872–880. 10.1016/j.ijrobp.2005.03.009. [DOI] [PubMed] [Google Scholar]
- (80). Zhao D; Chang C-H; Kim JG; Liu H; Mason RP In Vivo Near-Infrared Spectroscopy and Magnetic Resonance Imaging Monitoring of Tumor Response to Combretastatin A-4-Phosphate Correlated with Therapeutic Outcome. Int. J. Radiat. Oncol., Biol., Phys 2011, 80 (2), 574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Galbraith SM; Maxwell RJ; Lodge MA; Tozer GM; Wilson J; Taylor NJ; Stirling JJ; Sena L; Padhani AR; Rustin GJS Combretastatin A4 Phosphate Has Tumor Antivascular Activity in Rat and Man as Demonstrated by Dynamic Magnetic Resonance Imaging. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 2003, 21 (15), 2831–2842. 10.1200/JCO.2003.05.187. [DOI] [PubMed] [Google Scholar]
- (82).Vichai V; Kirtikara K Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc 2006, 1 (3), 1112–1116. 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- (83).Monks A; Scudiero D; Skehan P; Shoemaker R; Paull K; Vistica D; Hose C; Langley J; Cronise P; Vaigro-Wolff A; Gray-Goodrich M; Campbell H; Mayo J; Boyd M Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. JNCI J. Natl. Cancer Inst 1991, 83 (11), 757–766. 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- (84).Hamel E; Lin CM Stabilization of the Colchicine-Binding Activity of Tubulin by Organic Acids. Biochim. Biophys. Acta BBA - Gen. Subj 1981, 675 (2), 226–231. 10.1016/0304-4165(81)90231-2. [DOI] [PubMed] [Google Scholar]
- (85).Hamel E Evaluation of Antimitotic Agents by Quantitative Comparisons of Their Effects on the Polymerization of Purified Tubulin. Cell Biochem. Biophys 2003, 38 (1), 1–22. 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.