

Abstract
Background
The serine peptidase inhibitor Kazal type 13 (SPINK13) gene has tumor suppressor activity, but its role in renal cell carcinoma (RCC) remains unknown. This study aimed to investigate mRNA expression of SPINK13 in clear cell renal cell carcinoma (CCRCC) in human tissue and to use bioinformatics data to investigate the role of SPINK13 expression as a clinicopathological and prognostic biomarker for patients with CCRCC.
Material/Methods
Patients with CCRCC (N=533) with available RNA sequence data from The Cancer Genome Atlas (TCGA)-CCRCC database were analyzed with patients who had a tissue diagnosis of CCRCC (N=305) at the Fudan University Shanghai Cancer Center (FUSCC). Differential transcriptional and proteome expression profiles were obtained from the ONCOMINE cancer microarray database, TCGA, and the Human Protein Atlas (HPA) database. Quantitative reverse transcription-polymerase chain reaction (RT-qPCR) measured SPINK13 mRNA expression in 305 samples of CCRCC tissue from the FUSCC. The effects of clinicopathological parameters on progression-free survival (PFS) and overall survival (OS) were analyzed using the Kaplan-Meier and log-rank test.
Results
Transcriptional and proteome expression of SPINK13 were significantly increased CCRCC tissue samples. Increased SPINK13 mRNA expression was significantly associated with reduced PFS and OS in 838 patients with CCRCC patients from the two independent cohorts, the FUSCC and the TCGA-CCRCC cohorts (p<0.01). Gene set enrichment analysis (GSEA) showed that SPINK13 expression was involved in complement, apical junction, epithelial-mesenchymal transition (EMT), glycolysis, hypoxia, and inflammation signaling pathways.
Conclusions
Increased expression of SPINK13 was associated with poor prognosis in patients with CCRCC.
MeSH Keywords: Biological Markers; Carcinoma, Renal Cell; Disease-Free Survival; Prognosis
Background
Worldwide, primary renal cancer is one of the most common urological tumors. In 2019, there were an estimated 73,820 new cases and 14,770 deaths from renal cell carcinoma (RCC) in the United States [1]. The incidence and mortality of renal cancer in China is also increasing [2]. In 2015, the estimated number of new cases was 66,800, and the number of deaths was 23,400 [2]. Clear cell renal cell carcinoma (CCRCC) is a major subtype of renal cancer and the most common subtype of renal cell carcinoma (RCC) in adults. According to the World Health Organisation (WHO), RCC has a poor prognosis with an annual mortality rate of approximately 90,000 worldwide [3].
Although studies have been conducted on the mechanisms of cancer development and progression, the etiology and carcinogenesis of CCRCC remain unclear. Currently, the development and progression of RCC are known to be associated with genetic, cellular, and metabolic factors [4]. Primary renal tumors are small and non-malignant in as many as one-third of the cases, but imaging alone may not accurately identify non-malignant tumors, and potentially harmful overtreatment can occur. Although resection of small RCC is usually effective, the prognosis of metastatic RCC is relatively poor. Surgery and targeted therapy improve patient survival but eventually, most patients die of the disease [5,6]. Considering the high morbidity and mortality of RCC, it is essential to explore molecular biomarkers for early diagnosis, prevention, and targeted therapy by identifying the causes and potential molecular mechanisms.
The serine peptidase inhibitor Kazal type 13 (SPINK13) gene has tumor suppressor activity, but its role in RCC remains unknown. Serine proteinases are a class of proteolytic enzymes that contain a serine residue in their active site. Proteinase inhibitors are responsible for inhibiting and regulating the functions of proteinases [7]. Overexpression of SPINK1 predicts poor outcomes of several cancers [8]. Measurement of serum levels of SPINK1 may be used to identify patients with an increased risk of invasive breast cancer [9]. For example, serum SPINK1 acts as an inhibitor of growth factors and apoptosis in some cancers and has been suggested to be a prognostic marker and therapeutic target for several types of cancer [10,11]. Several studies have shown that the SPINK family is associated with the occurrence and progression of cancer [8,12,13]. Schrödter et al. studied microarray data to profile mRNA expression in malignant renal tumors and adjacent normal renal tissue [13]. Seven upregulated genes were identified in RCC, including SPINK13, SLC6A3, TNFAIP6, NPTX2, NDUFA4L2, ENPP3, and FABP6, and these investigators mainly focused on SLC6A3 as a prognostic biomarker for CCRCC [13]. The SLC6A3 inhibitor, sertraline, had a dose-dependent effect on tumor cell death of CCRCC cells [13]. Transcriptomics, including the use of microarray RNA chips, and other genomics technologies, have developed rapidly with the use of online databases [14]. Understanding the regulation and molecular functions of SPINK13 may facilitate the identification of potential targets for the diagnosis and treatment of CCRCC.
Therefore, this study aimed to investigate mRNA expression of SPINK13 in CCRCC in human tissue from the Fudan University Shanghai Cancer Center (FUSCC) and to use bioinformatics data from The Cancer Genome Atlas (TCGA) to investigate the role of SPINK13 expression as a clinicopathological and prognostic biomarker for patients with CCRCC.
Material and Methods
Patients and transcriptional expression profile
Patients with clear cell renal cell carcinoma (CCRCC) (N=533) were sequentially enrolled in the study who had available level 3 RNA-sequence data from the Cancer Genome Atlas (TCGA)-clear cell renal cell carcinoma (CCRCC) database [15]. The gene expression profiles were downloaded from TCGA database coordination center. The cut-off for SPINK13 mRNA expression was 4.6 after X-tile bioinformatics software analysis. The study also included 305 patients diagnosed with CCRCC after surgery in the Department of Urology, Fudan University Shanghai Cancer Center (FUSCC) (Shanghai, China) from May 2011 to November 2018. Clinicopathological indicators for the CGA-CCRCC and FUSCC cohorts included age at surgery, gender, tumor spread, the TNM stage, American Joint Committee on Cancer (AJCC) stage, and the International Society of Urological Pathology (ISUP) tumor grade. Tumor tissue samples were obtained after surgery or from the FUSCC tissue bank. All clinical protocols and procedures were performed according to the second revision of the Helsinki Declaration.
The ONCOMINE database
In this study, transcriptional expression profiles of SPINK13 in patients with CCRCC were acquired from the ONCOMINE online database (http://www.oncomine.com) [16]. The ONCOMINE database was searched for the expression of SPINK13 in the available datasets based on the following criteria: Cancer type; Gene: SPINK13; Data type: mRNA; Analysis type: Cancer vs. Normal analysis; and threshold setting conditions (p<0.001), fold change >2, gene rank: top 10%. From the ONCOMINE data, the red cells represented gene overexpression, and the blue cells represented gene underexpression. The color intensity represented the top 1%, 5%, or 10% of significantly overexpressed or underexpressed genes [16]. Lenburg renal and Yusenko renal datasets were obtained from the ONCOMINE database.
Human Protein Atlas (HPA)
The Human Pathology Atlas (HPA) project (https://www.proteinatlas.org) included immunohistochemistry data using a tissue microarray-based analysis of 44 different normal tissue types, and proteome analysis of 17 major cancer types [17]. Immunostaining intensity and patient information with the corresponding cancer types were available online. In this study, representative images of protein expression using immunohistochemistry for SPINK13 were captured and compared in CCRCC tissues and normal renal tissues in the HPA.
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR) analysis
Total RNA from harvested cells was isolated by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) from 305 CCRCC tumor tissue samples. Primers were diluted and mixed in RNase-free dH2O in SYBR® Green. The RT-qPCR method (Takara, Minato-ku, Tokyo, Japan) was performed according to the manufacturer’s protocol, using the following primers.
SPINK13, forward: 5′-CGCGGATCCATGGCTGCCTTTCCCCACAAG-3′;
reverse: 5′-CCGCTCGAGTTAATCACATTTTCCATATTTTTCA-3′;
GAPDH, forward: 5′-AGCCACATCGCTCAGACA-3′;
revers: 5′-GCCCAATACGACCAAATCC-3′.
Specific PCR operating cycles conditions for SPINK13 and GAPDH were performed according to SYBR® Green qPCR master mix manufacturer’s protocols (Applied Biosystems, Foster City, CA, USA). Relative SPINK13 mRNA expression was calculated using the ΔCt=Ct(SPINK13)–Ct(GAPDH) method According to cutoff expression value acquired using X-tile software in TCGA cohort, patients with CCRCC were divided into high-expression and low-expression groups.
Protein–protein interaction (PPI) network construction and module analysis
The online database, Search Tool for the Retrieval of Interacting Genes version 10.0 (STRING; http://string-db.org) was used to predict the PPI network of co-regulated hub genes and to analyze the functional interactions between proteins [18]. An interaction with a combined score >0.4 was considered statistically significant. Gene Ontology (GO) was used to identify biological process (BP) and cellular component (CC). The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to detect molecular function (MF), and hub genes in this module were identified using the online Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.8 (http://david.ncifcrf.gov) [19], and then visualized using bubble diagrams. Cytoscape version 3.5 was used to show the relevant internal protein networks and to describe connection specificity of each node [20]. ClueGO, a visualization function enrichment tool, was used to display the non-redundant biological terms based on high-throughput [21]. GO biological process and KEGG pathway analysis of hub genes was performed and visualized using ClueGO version 2.5.3 and CluePedia version 1.5.3 [22].
Data processing using gene set enrichment analysis (GSEA)
Using the R category (version 2.10.1) package, we integrated the TCGA-CCRCC datasets with gene set enrichment analysis (GSEA), or functional enrichment analysis. GSEA used a predefined set of genes, usually from functional annotations or previous experiments, to rank the genes according to the degree of differential expression in the two types of samples. GSEA was used to test whether the set of genes was at the top of the sorted table or at the bottom when enriched. Gene set enrichment analysis detected changes in the expression of a gene set rather than a single gene, and contained subtle expression changes, using adjusted p-values and the false discovery rate (FDR) method to optimize the results [23]. Statistically significant involved hub genes were identified using an adjusted p-value <0.01 and FDR <0.25. Statistical analysis and graphical plotting were conducted using R software (Version 3.3.2).
Statistical analysis
Phenotype and expression profiles of hub genes in 533 patients with CCRCC from the TCGA cohort and 305 patients from the FUSCC cohort were included in the analysis. Patient survival and SPINK13 mRNA expression were compared in the FUSCC and the TCGA-CCRCC cohorts. The primary endpoint was progression-free survival (PFS), and overall survival (OS) was the secondary endpoint. Univariate and multivariate Cox logistic regression models and the chi-squared (χ2) test were used to identify significant variables, including age at diagnosis, age at surgery, gender, T stage, N stage, M stage, AJCC stage, ISUP grade, and SPINK13 expression. A P-value <0.05 was considered to be statistically significant.
Results
Clear cell renal cell carcinoma (CCRCC) cohorts and stages of the study
Patients with clear cell renal cell carcinoma (CCRCC) (N=533) with available RNA sequence data from The Cancer Genome Atlas (TCGA) database were analyzed with patients who had a tissue diagnosis of CCRCC (N=305) at the Fudan University Shanghai Cancer Center (FUSCC) in a study that was conducted in four stages. In the first stage, differentially expressed SPINK13 in was assessed at the transcriptional and protein levels based on datasets hosted on the ONCOMINE and TCGA platforms. In the second stage of the study, survival analysis based on distinct comparison expression of SPINK13 was evaluated in TCGA cohort. In the third stage, the survival implications of SPINK13 were determined in the FUSCC cohort. In the final stage of the study, the significantly involved hub genes were selected, and functional annotation of the hub genes was elaborated.
Clinicopathological indicators of patients with clear cell renal cell carcinoma (CCRCC) from the two study cohorts
Analysis using the chi-squared (χ2) test showed that an increased SPINK13 mRNA expression was significantly associated with advanced T-stage (p=0.002), M-stage (p<0.001), American Joint Committee on Cancer (AJCC) stage (p<0.001), and the International Society of Urological Pathology (ISUP) tumor grade (p<0.001) in TCGA cohort (Table 1). The expression of SPINK13 in men was significantly greater than in women (p=0.006). In the 305 patients with CCRCC from the FUSCC cohort, increased expression of SPINK13 was significantly correlated with T-stage (p<0.001), M-stage (p<0.001), AJCC stage (p<0.001), and ISUP grade (p<0.001). Baseline data were balanced for the clinicopathological information, including age, gender, and laterality (p>0.05) (Table 1).
Table 1.
Clinicopathological characteristics at baseline associated with SPINK13 expression status in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts.
| Characteristics | TCGA cohort (N=533) | SPINK13 expression | χ2 | P- value | FUSCC cohort (N=305) | SPINK13 expression | χ2 | P- value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| High (N=230) | Low (N=303) | High (N=138) | Low (N=167) | |||||||
| N (%) | ||||||||||
| Age | 1.392 | 0.238 | 0.224 | 0.636 | ||||||
| <60 years | 245 (46.0) | 99 (43.0) | 146 (48.2) | 199 (65.2) | 92 (66.7) | 107 (64.1) | ||||
| ≥60 years | 288 (54.0) | 131 (57.0) | 157 (51.8) | 106 (34.8) | 46 (33.3) | 60 (35.9) | ||||
| Gender | 7.664 | 0.006 | 0.013 | 0.908 | ||||||
| Male | 345 (64.7) | 164 (71.3) | 181 (59.7) | 189 (62.0) | 86 (62.3) | 103 (61.7) | ||||
| Female | 188 (35.3) | 66 (28.7) | 122 (40.3) | 116 (38.0) | 52 (37.7) | 64 (38.3) | ||||
| Laterality | 2.074 | 0.354 | 0.754 | 0.383 | ||||||
| Left | 251 (47.1) | 113 (49.1) | 138 (45.5) | 152 (49.8) | 65 (47.1) | 87 (52.1) | ||||
| Right | 281 (52.7) | 116 (50.4) | 165 (54.5) | 153 (50.2) | 73 (52.9) | 80 (47.9) | ||||
| Bilateral | 1 (0.2) | 1 (0.4) | 0 | 0 | 0 | 0 | ||||
| pT stage | 14.439 | 0.002 | 20.148 | <0.001 | ||||||
| T1 | 273 (51.2) | 101 (43.9) | 172 (56.8) | 166 (64.4) | 56 (40.6) | 110 (65.8) | ||||
| T2 | 69 (12.9) | 26 (11.3) | 43 (14.2) | 62 (20.3) | 34 (24.6) | 28 (16.8) | ||||
| T3 | 180 (33.8) | 98 (42.6) | 82 (27.1) | 64 (21.0) | 40 (29.0) | 24 (14.4) | ||||
| T4 | 11 (2.1) | 5 (2.2) | 6 (2.0) | 13 (4.3) | 8 (5.8) | 5 (3.0) | ||||
| pN stage | 4.494 | 0.106 | 23.747 | <0.001 | ||||||
| N0 | 240 (45.0) | 100 (43.5) | 140 (46.2) | 264 (86.6) | 105 (76.1) | 159 (95.2) | ||||
| N1 | 16 (3.0) | 11 (4.8) | 5 (1.7) | 41 (13.4) | 33 (23.9) | 8 (4.8) | ||||
| Nx | 277 (52.0) | 119 (51.9) | 158 (52.1) | 0 | 0 | 0 | ||||
| pM stage | 19.473 | <0.001 | 31.740 | <0.001 | ||||||
| M0 | 422 (79.2) | 166 (72.2) | 256 (84.5) | 266 (87.2) | 104 (75.4) | 162 (97.0) | ||||
| M1 | 79 (14.8) | 52 (22.6) | 27 (8.9) | 39 (12.8) | 34 (24.6) | 5 (3.0) | ||||
| Mx | 32 (6.0) | 12 (5.2) | 20 (6.6) | 0 | 0 | 0 | ||||
| AJCC stage* | 20.351 | <0.001 | 30.138 | <0.001 | ||||||
| I | 267 (50.1) | 97 (42.2) | 170 (56.1) | 188 (61.6) | 75 (54.3) | 113 (67.7) | ||||
| II | 57 (10.7) | 21 (9.1) | 36 (11.9) | 60 (19.7) | 18 (13.1) | 42 (25.1) | ||||
| III | 123 (23.1) | 58 (25.2) | 65 (21.5) | 34 (11.1) | 25 (18.1) | 9 (5.4) | ||||
| IV | 86 (16.1) | 54 (23.5) | 32 (10.6) | 23 (7.6) | 20 (14.5) | 3 (1.8) | ||||
| ISUP grade | 32.437 | <0.001 | 11.595 | 0.001 | ||||||
| G1 | 14 (2.6) | 3 (1.3) | 11 (3.6) | 34 (11.1) | 12 (8.7) | 22 (13.2) | ||||
| G2 | 229 (43.0) | 83 (36.1) | 146 (48.2) | 61 (20.0) | 30 (21.7) | 61 (36.5) | ||||
| G3 | 206 (38.6) | 90 (39.1) | 116 (38.3) | 131 (42.9) | 64 (46.4) | 67 (40.1) | ||||
| G4 | 76 (14.3) | 53 (23.0) | 23 (7.6) | 49 (16.0) | 32 (23.2) | 17 (10.2) | ||||
| Gx | 5 (0.9) | 1 (0.4) | 4 (1.3) | 0 | 0 | 0 | ||||
The American Joint Committee on Cancer (AJCC) staging system describes the extent of disease progression in cancer patients, and the tumor size, lymph nodes affected, and metastases (TNM); ISUP, International Society of Urological Pathology tumor grade.
Differential SPINK13 expression level in CCRCC tumor tissue and normal renal tissue
SPINK13 mRNA expression level between CCRCC tissue samples and adjacent normal kidney tissues were compared based on RNA-sequence data from TCGA database. Transcriptional levels of SPINK13 expression were upregulated in 533 CCRCC tissues compared with 72 normal tissues (p<0.0001) (Figure 1A). SPINK13 expression in CCRCC primary tumors was significantly higher than adjacent normal tissues in the Lenburg renal dataset [24], (n=18) (p<0.05) (Figure 1B), and the Yusenko renal dataset [25], (n=31) (p<0.01) (Figure 1C). Representative immunohistochemistry images of tissue protein expression showed that SPINK13 proteins were not expressed in normal renal tissues, but significant levels of protein expression were observed in CCRCC tissues (Figure 1D, 1E). These findings showed that transcriptional and protein expression of SPINK13 were highly expressed in patients with CCRCC.
Figure 1.

Transcriptional and protein expression of SPINK13 in clear cell renal cell carcinoma (CCRCC) tumor tissues and adjacent normal renal tissues. (A) Transcriptional level of SPINK13 expression was found highly expressed in 533 CCRCC tissues compared with 72 normal tissues (**** p<0.0001). (B, C) SPINK13 expression in CCRCC primary tumors was significantly higher than adjacent normal tissues in the Lenburg renal dataset (*, p<0.05), and the Yusenko renal dataset (**, p<0.01). (D, E) Representative images of protein expression detected by immunohistochemistry of SPINK13 were detected in CCRCC and normal tissues from the Human Protein Atlas (HPA). SPINK13 proteins were not expressed in normal renal tissues, but significant expression was observed by medium-level immunohistochemical staining in CCRCC tissues using HPA.
Association of SPINK13 mRNA expression with clinicopathological indicators in patients with CCRCC
After integrating clinicopathological and survival data from the TCGA-CCRCC cohort, significantly increased SPINK13 mRNA expression was found in CCRCC samples compared with normal samples. As shown in Figure 2A, SPINK13 mRNA expression was significantly associated with advanced clinical stage, and the highest SPINK13 mRNA expression was found in stage IV CCRCC. Figure 2B shows that when the relationship between SPINK13 mRNA expression and different pathological grades was studied, SPINK13 mRNA expression was significantly correlated with higher (more poorly-differentiated) histopathological tumor grades. Also, the highest expression level of SPINK13 mRNA was found in grade 4 (undifferentiated) CCRCC. Therefore, increased SPINK13 mRNA expression was significantly associated with advanced clinicopathological parameters, in terms of stage and grade, in patients with CCRCC.
Figure 2.

Transcriptional expression of SPINK13 was significantly correlated with advanced clinicopathological parameters and poor survival outcomes in patients with clear cell renal cell carcinoma (CCRCC). (A) Transcriptional expression of SPINK13 was significantly correlated with the American Joint Committee on Cancer (AJCC) stages, as patients who were in more advanced stages tended to express higher mRNA expression of SPINK13. (B) Transcriptional expression of SPINK13 was significantly correlated with the International Society of Urological Pathology (ISUP) tumor grade, patients who were in more advanced grade score tended to express elevated mRNA expression of SPINK13. The highest mRNA expression of SPINK13 was found in stage IV or grade 4 CCRCC. * p<0.05, ** p<0.01, *** p<0.001. (C) Kaplan-Meier survival analysis indicated that SPINK13 was significantly correlated with shorter progression-free survival (PFS) (p=0.002). (D) Survival curves suggested that patients with elevated SPINK13 mRNA levels showed poorer OS in 533 included patients with CCRCC (p<0.001). CCRCC – clear cell renal cell carcinoma; AJCC – American Joint Committee on Cancer.
Cox regression analysis and survival outcome in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts
After multivariate (Tables 2, 3) and univariate (Tables 4, 5) Cox regression analysis, traditional prognostic factors, including the T-stage, the M-stage, the AJCC stage, and the ISUP grade were significantly associated with progression-free survival (PFS) and overall survival (OS) in patients with CCRCC, indicating that the populations studied in the TCGA and FUSCCC cohorts were representative. Multivariate Cox regression analysis indicated that SPINK13 amplification was significantly correlated with poor PFS (HR, 2.506; p=0.029) and OS (HR, 2.244; p<0.001) for patients with CCRCC in the TCGA-CCRCC cohort (Tables 2, 3). Also, the M-stage was significantly associated with a reduced PFS (HR, 2.958; p=0.028) and a reduced OS (HR, 2.330; p=0.001). Kaplan-Meier survival analysis showed that increased expression of SPINK13 mRNA was significantly correlated with a shorter PFS (p=0.002; Figure 2C) and shorter OS (p<0.001) (Figure 2D).
Table 2.
Multivariate Cox logistic regression analysis of progression-free survival (PFS) in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts.
| Covariates | TCGA cohort | FUSCC cohort | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Age | – | – | – | 0.793 | 0.574–1.095 | 0.159 |
| Gender (ref. Male) | 0.506 | 0.138–1.854 | 0.306 | – | – | – |
| T stage (ref. T1–T2) | 0.561 | 0.115–2.734 | 0.475 | 4.004 | 2.514–6.397 | <0.001 |
| N stage (ref. N0) | – | – | – | 1.269 | 0.662–2.433 | 0.474 |
| M stage (ref. M0) | 2.958 | 1.127–7.768 | 0.028 | 2.947 | 1.752–4.958 | <0.001 |
| AJCC stage (ref. I–II) | 3.678 | 0.627–21.589 | 0.149 | 2.182 | 1.653–2.141 | <0.001 |
| ISUP grade (ref. 1–2) | 2.629 | 0.930–7.433 | 0.068 | 1.523 | 1.064–2.181 | 0.022 |
| SPINK13 expression (ref. low) | 2.506 | 1.123–8.199 | 0.029 | 2.295 | 1.627–3.237 | <0.001 |
HR – hazard ratio; CI – confidence interval; TCGA – The Cancer Genome Atlas; FUSCC – Fudan University Shanghai Cancer Center; AJCC – American Joint Committee on Cancer; ISUP – International Society of Urological Pathology.
Table 3.
Multivariate Cox logistic regression analysis of overall survival (OS) in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts.
| Covariates | TCGA cohort | FUSCC cohort | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Age | 1.232 | 0.805–1.885 | 0.337 | 1.108 | 1.003–1.033 | 0.017 |
| Gender (ref. Male) | – | – | – | – | – | – |
| T stage (ref. T1–T2) | 1.692 | 0.740–3.871 | 0.213 | 4.500 | 2.661–7.612 | <0.001 |
| N stage (ref. N0) | 1.455 | 0.726–2.916 | 0.290 | 1.283 | 0.670–2.456 | 0.452 |
| M stage (ref. M0) | 2.330 | 1.383–3.929 | 0.001 | 3.411 | 2.007–5.798 | <0.001 |
| AJCC stage (ref. I–II) | 1.051 | 0.411–2.687 | 0.918 | 3.118 | 2.595–6.099 | <0.001 |
| ISUP grade (ref. 1–2) | 1.416 | 0.854–2.346 | 0.178 | 1.127 | 0.731–1.737 | 0.587 |
| SPINK13 expression (ref. low) | 2.244 | 1.399–3.601 | 0.001 | 1.513 | 1.007–2.272 | 0.046 |
HR – hazard ratio; CI – confidence interval; TCGA – The Cancer Genome Atlas; FUSCC – Fudan University Shanghai Cancer Center; AJCC – American Joint Committee on Cancer; ISUP – International Society of Urological Pathology.
Table 4.
Univariate Cox logistic regression analysis of progression-free survival (PFS) in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts.
| Covariates | TCGA cohort | FUSCC cohort | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Age | 0.971 | 0.428–2.203 | 0.943 | 1.017 | 1.004–1.031 | 0.010 |
| Gender (ref. Male) | 0.243 | 0.072–0.818 | 0.022 | 0.812 | 0.592–1.112 | 0.194 |
| T stage (ref. T1–T2) | 4.737 | 2.006–11.183 | <0.001 | 8.171 | 5.772–11.568 | <0.001 |
| N stage (ref. N0) | 0.048 | 0.000–100 | 0.803 | 6.375 | 4.356–9.332 | <0.001 |
| M stage (ref. M0) | 8.054 | 3.551–18.266 | <0.001 | 9.953 | 6.687–14.814 | <0.001 |
| AJCC stage (ref. I–II) | 6.557 | 2.583–16.644 | <0.001 | 6.294 | 4.477–8.847 | <0.001 |
| ISUP grade (ref. 1–2) | 3.9 | 1.445–10.530 | 0.007 | 2.539 | 1.817–3.549 | <0.001 |
| SPINK13 expression (ref. low) | 4.855 | 1.895–12.443 | 0.001 | 3.557 | 2.584–4.898 | <0.001 |
HR – hazard ratio; CI – confidence interval; TCGA – The Cancer Genome Atlas; FUSCC – Fudan University Shanghai Cancer Center; AJCC – American Joint Committee on Cancer; ISUP, International Society of Urological Pathology.
Table 5.
Univariate Cox logistic regression analysis of overall survival (OS) in The Cancer Genome Atlas (TCGA) and Fudan University Shanghai Cancer Center (FUSCC) cohorts.
| Covariates | TCGA cohort | FUSCC cohort | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Age | 1.795 | 1.311–2.456 | <0.001 | 1.020 | 1.005–1.035 | 0.008 |
| Gender (ref. Male) | 1.054 | 0.775–1.434 | 0.737 | 0.902 | 0.631–1.290 | 0.572 |
| T stage (ref. T1–T2) | 3.138 | 2.320–4.245 | <0.001 | 8.518 | 5.918–12.261 | <0.001 |
| N stage (ref. N0) | 3.380 | 1.795–6.367 | <0.001 | 6.577 | 4.405–9.822 | <0.001 |
| M stage (ref. M0) | 3.589 | 2.636–4.886 | <0.001 | 9.831 | 6.482–14.909 | <0.001 |
| AJCC stage (ref. I–II) | 3.835 | 2.798–5.256 | <0.001 | 6.365 | 4.447–9.111 | <0.001 |
| ISUP grade (ref. 1–2) | 2.651 | 1.887–3.723 | <0.001 | 2.309 | 1.566–3.405 | <0.001 |
| SPINK13 expression (ref. low) | 3.238 | 2.357–4.449 | <0.001 | 2.714 | 1.878–3.922 | <0.001 |
HR – hazard ratio; CI – confidence interval; TCGA – The Cancer Genome Atlas; FUSCC – Fudan University Shanghai Cancer Center; AJCC – American Joint Committee on Cancer; ISUP – International Society of Urological Pathology.
In the FUSCC cohort, clinicopathological variables, including age, T-stage, N-stage, M-stage, AJCC stage, ISUP grade, and SPINK13 expression, were included in the multivariate Cox analysis. The multivariate Cox model showed that T-stage, M-stage, and AJCC stage were significantly associated with prognosis, while ISUP grade was correlated with PFS. Also, SPINK13 expression was significantly associated with unfavorable prognosis in terms of PFS (HR, 2.295; p<0.001) and OS (HR, 1.513; p=0.046) for patients with CCRCC. Survival curves indicated that patients with increased expression of SPINK13 had shorter PFS (p<0.001) (Figure 3A) and OS (p<0.001) (Figure 3B).
Figure 3.

Survival analysis of SPINK13 in the Fudan University Shanghai Cancer Center (FUSCC) cohort (N=305). The survival curves show that patients with elevated SPINK13 expression had shorter (A) PFS (p<0.001) and (B) OS (p<0.001). FUSCC – Fudan University Shanghai Cancer Center; PFS – progression-free survival; OS – overall survival.
Functional annotation and signaling pathways
The PPI network significantly co-regulated with SPINK13 included SPINK7, SPINK9, SPINK14, PILRA, FXYD4, NDUFA4L2, OR1E1, UNC45B, SPATA18, and TGFBI, and is shown in Figure 4A. Functional enrichment of 11 involved hub genes were performed and is shown in Figure 4B. Hub genes were significantly involved in polyol transport, the defense response, the immune response, and participated in the plasma membrane components. As shown in Figure 4C, functional annotation of involved nodes using ClueGO suggested functional alterations in biological processes. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were significantly enriched for membrane-bounded organelles, cell components, and regulation of biological processes. After refining the network specificity, SPINK7, SPINK9, SPINK13, and SPINK14 showed the most significant involvement, with negative regulation of serine-type endopeptidase activity, as shown in Figure 5. The Gene Ontology (GO) pathway network connectivity (kappa score) was 0.4, and significant pathways had a p-value ≥0.05.
Figure 4.

Functional annotations and predicted signaling pathways. (A) The protein-protein interaction (PPI) network of SPINK13 was constructed. A network of SPINK13 and its co-expression genes are shown visually. (B) Functional enrichment analysis of a total of 11 involved genes were performed and visualized in a bubble chart. Significant genes were significantly involved in polyol transport, defense response, immune response, and the plasma membrane, which were integral to membrane, intrinsic to the membrane, and part of the plasma membrane. (C) Functional annotation using ClueGO indicated that changes in the biological processes of the SPINK13 were significantly associated with membrane-bound organelles, the cell, and regulation of biological processes.
Figure 5.
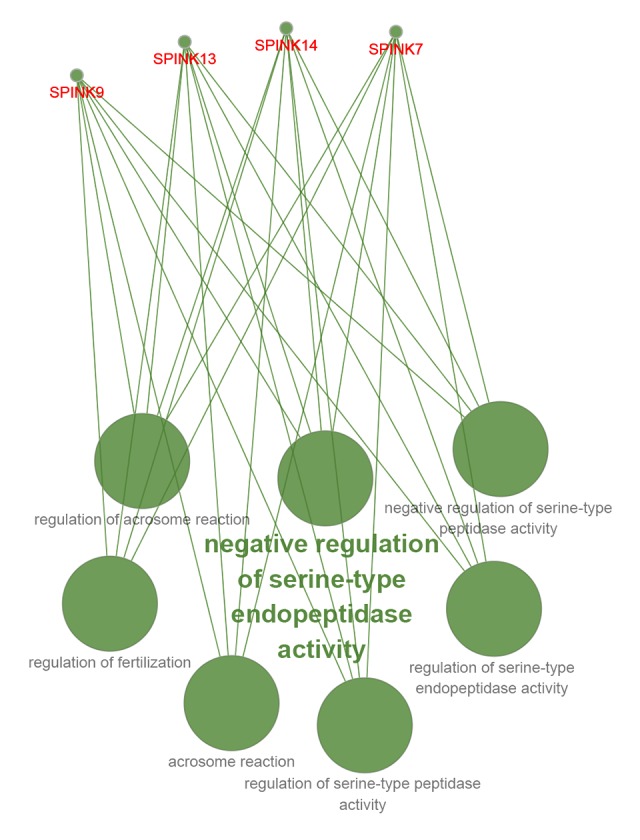
The most significant functional enrichment of hub genes. After refining the network specificity, the most significant functional enrichment of hub genes involved the negative regulation of serine-type endopeptidase activity, which correlated with hub genes, including SPINK7, SPINK9, SPINK13, and SPINK14.
Significant genes and phenotypes obtained by gene set enrichment analysis (GSEA)
Predicted phenotype analysis using GSEA indicated that SPINK13 significantly involved in pathways that included complement, apical junctions, epithelial-mesenchymal transition (EMT), glycolysis, hypoxia, and inflammatory responses, as shown in Figure 6A–6F. Also, transcriptional expression profiles of 100 significantly positive and negative genes are shown from GSEA analysis in Figure 6G.
Figure 6.

Significantly related genes and phenotype pathways in clear cell renal cell carcinoma (CCRCC) obtained by gene set enrichment analysis (GSEA). A total of 100 significant genes were obtained by gene set enrichment analysis (GSEA) with positive and negative correlations. (A–F) The most involved significant pathways included complement, apical junction, epithelial-mesenchymal transition (EMT), glycolysis, hypoxia, and inflammatory response. (G) Transcriptional expression profiles of the 100 most significant genes are shown in a heat map. GSEA, gene set enrichment analysis.
Discussion
Cancer genetics and abnormal epigenetic regulation, participate in the development and progression of clear cell renal cell carcinoma (CCRCC) [26]. The serine peptidase inhibitor Kazal type (SPINK) family play a major role in the occurrence and progression of tumors such as breast cancer [27], renal cancer [28], and ovarian cancer [29]. Although some members of the SPINK family are carcinogenic, the prognostic role of the expression of SPINK13 in clear cell renal cell carcinoma (CCRCC) has not been previously investigated.
The aim of the present study was to investigate mRNA expression of SPINK13 in CCRCC in human tissue and to use bioinformatics data to investigate the role of SPINK13 expression as a clinicopathological and prognostic biomarker for patients with CCRCC. In this study, the expression levels, prognostic values, and potential functional enrichment of SPINK13 in CCRCC were investigated. Patients with CCRCC (N=533) with available RNA sequence data from The Cancer Genome Atlas (TCGA) database were analyzed with patients who had a tissue diagnosis of CCRCC (N=305) at the Fudan University Shanghai Cancer Center (FUSCC), in a study that included two cohorts. The findings showed that increased expression of SPINK13 in CCRCC tissue samples was significantly associated with reduced progression-free survival (PFS) and overall survival (OS). Increased expression levels of SPINK13 mRNA correlated with a higher risk of tumor recurrence and a reduction in prognosis in patients with CCRCC. Also, functional enrichment and gene set enrichment analysis (GSEA) showed that SPINK13 expression was significantly involved in the expression of phenotypes involved in the inflammatory response, epithelial-mesenchymal transition (EMT), apical junctions, complement, glycolysis, and hypoxia, in CCRCC tumor tissue samples.
Previous studies have shown that the tumor-associated inflammatory response and EMT promote tumor progression. Inflammation is a basic biological behavioral phenotype that has previously been identified in many tumors [30]. Cancer-associated inflammation involves crosstalk between malignant and non-malignant cells in an autocrine and paracrine manner through mediators that include cytokines, chemokines, and prostaglandins [31]. Combined with genetic alterations, the inflammatory tumor environment ultimately leads to tumor progression and metastasis [32]. For example, in the absence of the p53 tumor suppressor gene, the inflammatory response associated with epithelial cell senescence significantly promotes cell transformation and carcinogenesis, which can be inhibited by anti-inflammatory drugs [33]. Treatment with the anti-inflammatory drug dexamethasone also significantly inhibits the spread of tumor cells by inhibiting EMT, a process through which epithelial cells develop the properties of migration and invasion [34]. EMT reduces cell-cell adhesion complexes and confers enhanced migration and invasion properties, which can be used by cancer cells during metastasis. Cancer cells undergoing EMT are more aggressive, exhibiting increased invasiveness, stem-like characteristics, and resistance to apoptosis [35]. The EMT program can also stimulate cancer cells to produce pro-inflammatory factors, and inflammation is a potent inducer of EMT in tumors. Therefore, EMT and cellular and molecular factors of inflammation have a synergistic relationship in tumor biology [36].
Hypoxia can occur during normal development as well as tumorigenesis, and its response at the cellular level is primarily mediated through hypoxia-inducible factors (HIFs) [36]. The general HIF activation pathway is classically associated with reduced prognosis in cancer [37]. HIF pathways in renal cell carcinoma (RCC) are upregulated by inactivation of the von-Hippel-Lindau tumor suppressor [38]. Clinically targeted inhibition of the STAT5 signaling pathway has been proposed as a therapeutic strategy for renal cancer [38]. Currently, the relationship between SPINK13 expression and prognosis in RCC have been unknown. However, the findings from the present study showed that increased SPINK13 expression was significantly associated with more aggressive behavior and poor OS in 838 patients with CCRCC in the TCGA-CCRCC and FUSCC cohorts.
Co-regulatory proteins were included in the protein-protein interaction (PPI) network to investigate the prognostic significance of SPINK13. Functional enrichment analysis was performed using hub gene panels. Data from TCGA database underwent GSEA analysis to screen out the important genomic profile and phenotype pathways. However, this study had several limitations. First, although verification of SPINK13 expression was performed using quantitative reverse transcription-polymerase chain reaction (RT-qPCR) in CCRCC tissue samples from the FUSCC tissue bank, the effects of SPINK13 on tumor cells were not studied. Second, transcriptome expression of SPINK13 was found to be a prognostic biomarker to predict PFS and OS in this study. Although differential proteomic expression of SPINK13 was detected between tumor and normal renal tissues, the prognostic implications were not demonstrated clinically. Third, this study did not explore the underlying mechanisms of the signaling pathways in CCRCC, but a series of functional annotations and enrichment analyses were performed. Further studies are needed to explore the mechanisms involved in the expression of SPINK13 and its role in carcinogenesis in CCRCC, which may provide insights into its effects in other human carcinomas.
Conclusions
The findings from this preliminary study showed that increased expression of SPINK13 was significantly correlated with the progression of clear cell renal cell carcinoma (CCRCC) and with reduced patient survival. Future research should be undertaken to identify the molecular mechanisms for SPINK13 in CCRCC, and controlled clinical studies should be undertaken to investigate its prognostic role.
Footnotes
Conflict of interest
None.
Source of support: This study was supported by Grants from the National Natural Science Foundation of China (No. 81802525), the Natural Science Foundation of Shanghai (No. 16ZR1406400), and the Shanghai Sailing Program of China (No. 17YF1402700)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. Cancer J Clin. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. Cancer J Clin. 2016;66(2):115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Baldewijns MM, van Vlodrop IJ, Schouten LJ, et al. Genetics and epigenetics of renal cell cancer. Biochim Biophys Acta. 2008;1785(2):133–55. doi: 10.1016/j.bbcan.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Linehan WM, Schmidt LS, Crooks DR, et al. The metabolic basis of kidney cancer. Cancer Discov. 2019;9(8):1006–21. doi: 10.1158/2159-8290.CD-18-1354. [DOI] [PubMed] [Google Scholar]
- 5.La Rochelle J, Wood C, Bex A. Refining the use of cytoreductive nephrectomy in metastatic renal cell carcinoma. Semin Oncol. 2013;40(4):429–35. doi: 10.1053/j.seminoncol.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Albiges L, Choueiri T, Escudier B, et al. A systematic review of sequencing and combinations of systemic therapy in metastatic renal cancer. Eur Urol. 2015;67(1):100–10. doi: 10.1016/j.eururo.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Laskowski M., Jr Protein inhibitors of serine proteinases – mechanism and classification. Adv Exp Med Biol. 1986;199:1–17. doi: 10.1007/978-1-4757-0022-0_1. [DOI] [PubMed] [Google Scholar]
- 8.Räsänen K, Itkonen O, Koistinen H, Stenman UH. Emerging roles of SPINK1 in cancer. Clin Chem. 2016;62(3):449–57. doi: 10.1373/clinchem.2015.241513. [DOI] [PubMed] [Google Scholar]
- 9.Soon WW, Miller LD, Black MA, et al. Combined genomic and phenotype screening reveals secretory factor SPINK1 as an invasion and survival factor associated with patient prognosis in breast cancer. EMBO Mol Med. 2011;3(8):451–64. doi: 10.1002/emmm.201100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, Lamontagne J, Lu F, Block TM. Tumor-associated protein SPIK/TATI suppresses serine protease dependent cell apoptosis. Apoptosis. 2008;13(4):483–94. doi: 10.1007/s10495-008-0193-x. [DOI] [PubMed] [Google Scholar]
- 11.Wiksten JP, Lundin J, Nordling S, et al. High tissue expression of tumour-associated trypsin inhibitor (TATI) associates with a more favourable prognosis in gastric cancer. Histopathology. 2005;46(4):380–88. doi: 10.1111/j.1365-2559.2005.02073.x. [DOI] [PubMed] [Google Scholar]
- 12.Chung JS, Wang Y, Henderson J, et al. Circulating tumor cell-based molecular classifier for predicting resistance to abiraterone and enzalutamide in metastatic castration-resistant prostate cancer. Neoplasia. 2019;21(8):802–9. doi: 10.1016/j.neo.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrödter S, Braun M, Syring I, et al. Identification of the dopamine transporter SLC6A3 as a biomarker for patients with renal cell carcinoma. Mol Cancer. 2016;15:10. doi: 10.1186/s12943-016-0495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Xu WH, Wei Y, et al. An integrated score and nomogram combining clinical and immunohistochemistry factors to predict high ISUP grade clear cell renal cell carcinoma. Front Oncol. 2018;8:634. doi: 10.3389/fonc.2018.00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19(1A):A68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asplund A, Edqvist PH, Schwenk JM, Pontén F. Antibodies for profiling the human proteome – The Human Protein Atlas as a resource for cancer research. Proteomics. 2012;12(13):2067–77. doi: 10.1002/pmic.201100504. [DOI] [PubMed] [Google Scholar]
- 18.Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang DW, Sherman BT, Tan Q, et al. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27(3):431–32. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–93. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bindea G, Galon J, Mlecnik B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29(5):661–63. doi: 10.1093/bioinformatics/btt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenburg ME, Liou LS, Gerry NP, et al. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data. BMC Cancer. 2003;3:31. doi: 10.1186/1471-2407-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yusenko MV, Kuiper RP, Boethe T, et al. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152. doi: 10.1186/1471-2407-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao L, Testa JR, Yang H. The roles of chromatin-remodelers and epigenetic modifiers in kidney cancer. Cancer Genet. 2015;208(5):206–14. doi: 10.1016/j.cancergen.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Mezayen HA, Metwally FM, Darwish H. A novel discriminant score based on tumor-associated trypsin inhibitor for accurate diagnosis of metastasis in patients with breast cancer. Tumour Biol. 2014;35(3):2759–67. doi: 10.1007/s13277-013-1366-y. [DOI] [PubMed] [Google Scholar]
- 28.Paju A, Jacobsen J, Rasmuson T, et al. Tumor associated trypsin inhibitor as a prognostic factor in renal cell carcinoma. J Urol. 2001;165(3):959–62. [PubMed] [Google Scholar]
- 29.Nissi R, Talvensaari-Mattila A, Kuvaja P, et al. Claudin-5 is associated with elevated TATI and CA125 levels in mucinous ovarian borderline tumors. Anticancer Res. 2015;35(2):973–76. [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Crusz SM, Balkwill FR. Inflammation and cancer: Advances and new agents. Nat Rev Clin Oncol. 2015;12(10):584–96. doi: 10.1038/nrclinonc.2015.105. [DOI] [PubMed] [Google Scholar]
- 32.Singh R, Mishra MK, Aggarwal H. Inflammation, immunity, and cancer. Mediators Inflamm. 2017;2017 doi: 10.1155/2017/6027305. 6027305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pribluda A, Elyada E, Wiener Z, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24(2):242–56. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J Exp Med. 2019;216(5):1016–26. doi: 10.1084/jem.20181827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017;11(7):805–23. doi: 10.1002/1878-0261.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salama R, Masson N, Simpson P, et al. Heterogeneous effects of direct hypoxia pathway activation in kidney cancer. PLoS One. 2015;10(8):e0134645. doi: 10.1371/journal.pone.0134645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]