

Abstract
Because many peptide and peptide-mimetic drugs are substrates of peptide transporter 1, it is important to evaluate the peptide transporter 1-mediated intestinal absorption of drug candidates in the early phase of drug development. Although intestinal cell lines treated with inhibitors of peptide transporter 1 are widely used to examine whether drug candidates are substrates for peptide transporter 1, these inhibitors are not sufficiently specific for peptide transporter 1. In this study, to generate a more precise evaluation model, we established peptide transporter 1-knockout induced pluripotent stem cells (iPSCs) by using a CRISPR-Cas9 system and differentiated the cells into intestinal epithelial-like cells. The permeability value and uptake capacity of glycylsarcosine (substrate of peptide transporter 1) in peptide transporter 1-knockout intestinal epithelial-like cells were significantly lower than those in wild-type intestinal epithelial-like cells, suggesting that peptide transporter 1 was successfully depleted in the epithelial cells. Taken together, our model can be useful in the development of peptide and peptide-mimetic drugs.
Keywords: human iPS cells, intestinal epithelial-like cells, SLC15A1, PEPT1, genome editing, CRISPR-Cas9, drug transporter, drug absorption
Graphical Abstract
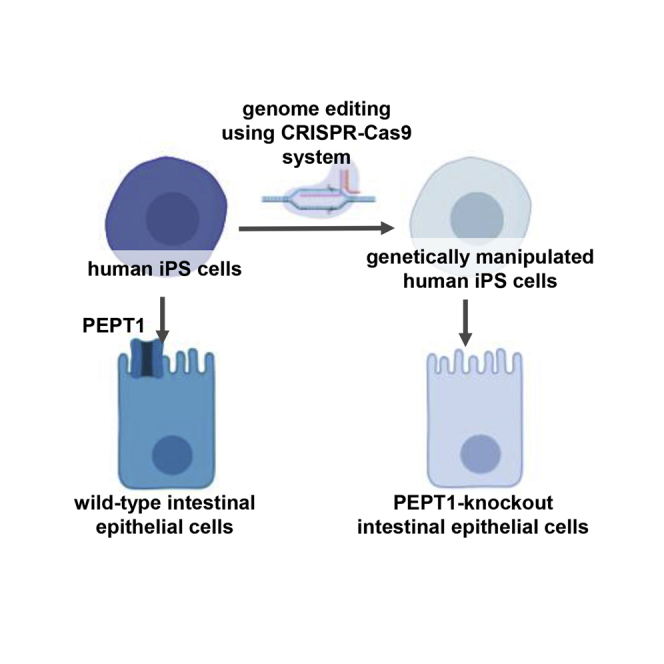
Introduction
Solute carrier family 15 member 1 (SLC15A1; peptide transporter 1 [PEPT1]) is a peptide transporter that is highly expressed in the apical membrane of enterocytes in small intestine.1,2 PEPT1 is classified into the proton-dependent oligopeptide transporter (POT) family and actively transports dipeptides and tripeptides across the apical membrane of enterocytes in small intestine.3,4 In addition, many peptide and peptide-mimetic drugs, such as valaciclovir, captopril, and cephalexin, are substrates of PEPT1.2,5,6 Valaciclovir is an amino acid prodrug of acyclovir. Oral bioavailability of valaciclovir is enhanced by carrier-mediated intestinal absorption via PEPT1 followed by rapid and complete conversion to acyclovir.5,7 An effective strategy to improve the intestinal absorption is to generate peptide-mimetic drugs of low-permeability drugs. Thus, a model that can evaluate PEPT1-mediated intestinal absorption would be useful for discovery of peptide-mimetic drugs. Currently, PEPT1-knockout (KO) mice and PEPT1 inhibitors-treated intestinal cell lines such as Caco-2 cells are widely used to determine whether drug candidates are substrates for PEPT1.8, 9, 10 However, PEPT1-mediated drug absorption differs between humans and mice.11,12 Alternatively, a cell-based transporter assay using PEPT1 inhibitors (such as ibuprofen) can be used to examine whether drug candidates are PEPT1 substrates.13,14 Ibuprofen has been reported to inhibit not only PEPT1 but also solute carrier family 5 member 8 (SLC5A8; sodium-coupled monocarboxylate transporter 1 [SMCT1]).15, 16, 17 Therefore, a novel model that can perform highly specific evaluation of PEPT1-mediated intestinal absorption is required.
Recently, several groups showed that human induced pluripotent stem cell (iPSC)-derived intestinal epithelial-like cells (human iPSC-IECs) can be used for pharmacokinetic testing.18, 19, 20, 21, 22, 23 Our group demonstrated that the PEPT1 expression levels in human iPSC-IECs were similar to those in the human adult small intestine, and that the human iPSC-IECs exhibited PEPT1 activity.23 In addition, we previously developed a method for efficient homologous recombination in human iPSCs using a valproic acid (VPA) and RAD51 recombinase (RAD51)-expression plasmid.24 Using our genome editing method, we succeeded in efficient homologous recombination in transcriptionally inactive loci such as drug-metabolizing enzyme genes.25,26 In this study, therefore, we attempted to generate PEPT1-KO human iPSCs, and then differentiated these cells into IECs that could be used as PEPT1-KO IECs. To our knowledge, no prior study has reported the establishment of a PEPT1-KO human iPSC line. We considered that PEPT1-KO human iPSC-IECs would be useful as a highly specific transporter assay for the evaluation of PEPT1 substrates.
Results
Generation of PEPT1-KO iPS Cells
To generate PEPT1-KO iPSCs, the PEPT1 locus was targeted using our highly efficient genome editing method.24 The schematic overview shows the targeting strategy used for PEPT1 (Figure 1A). PCR analyses were performed to examine whether the human iPSCs were correctly targeted. We confirmed that the EF1α-PuroR-pA (elongation factor 1 alpha promoter followed by the puromycin-resistance gene and polyadenylation sequence) cassette was monoallelically integrated into the PEPT1 locus (Figure 1B). Next, the allele, which does not carry the EF1α-PuroR-pA cassette, was analyzed by Sanger sequencing. Several nucleotide deletions were found in exon 21 of the PEPT1 gene (Figure 1C). We analyzed five clones of puromycin-resistant iPSCs, in all of which the first allele had the EF1α-PuroR-pA cassette, while the second allele had an indel, and thus the establishment efficiency of PEPT1-KO iPSCs was 100% (Figure S1). To examine the pluripotent state of PEPT1-KO iPSCs, the gene expression levels of pluripotent markers (NANOG, POU domain class 5 transcription factor 1 (POU5F1), and sex determining region Y-box 2 [SOX2]) in the PEPT1-KO and wild-type (WT)-iPSCs were examined by real-time RT-PCR analysis. The gene expression levels of pluripotent markers in the PEPT1-KO iPSCs were similar to those in the WT-iPSCs (Figure 1D). Immunocytochemical analysis showed that the PEPT1-KO human iPSCs were positive for POU5F1 (Figure 1E). These results suggest that the pluripotent state was not affected by knockout of PEPT1.
Figure 1.

Generation of PEPT1-KO Human iPSCs
(A) The schematic overview shows the targeting strategy for PEPT1/SLC15A1. The PCR primers that can distinguish WT and mutant alleles are shown with red arrows. Donor plasmid: EF1α, elongation factor 1 alpha promoter; PuroR, puromycin resistant protein; pA, polyadenylation sequence; HDR, homology-directed repair. (B) The genotyping was performed in the PEPT1/SLC15A1 locus. (C) Sequencing analyses were performed to examine whether the PEPT1/SLC15A1-KO iPSC clone was correctly targeted. To confirm the DNA sequence, the PCR products were purified and subjected to sequencing analyses. The single-guide RNA (sgRNA)-targeting sequences are shown in red. (D) The gene expression levels of POU5F1, NANOG, and SOX2 in WT-iPSCs and PEPT1-KO iPSCs were examined by real-time RT-PCR analysis. The gene expression levels in the WT-iPSCs (WT) were taken as 1.0. Data represent the means ± SD (n = 8, technical replicate). (E) Immunostaining analysis of POU5F1 (red) was performed in the WT-iPSCs and PEPT1/SLC15A1-KO cells. Nuclei were stained with DAPI (blue). Scale bars represent 50 μm.
Intestinal Differentiation of PEPT1-KO iPSCs
To examine the intestinal differentiation capacity of PEPT1-KO iPSCs, PEPT1-KO iPSCs were differentiated into the IECs. The schematic overview shows the intestinal differentiation protocol (Figure 2A). The gene expression levels of the intestinal epithelial cell markers (villin 1 [VIL1], sucrase-isomaltase [SI], and intestine-specific homeobox [ISX]) and PEPT1 in the PEPT1-KO iPSC-IECs were compared with those in the WT iPSC-IECs by real-time RT-PCR analysis (Figure 2B). The gene expression levels of VIL1, SI, and ISX were not changed by PEPT1 knockout. Importantly, the gene and protein expression levels of PEPT1 in the PEPT1-KO iPSC-IECs were significantly lower than those of the WT iPSC-IECs (Figures 2B and 2C, respectively). FACS analysis showed that there were no significant differences in the VIL1- and SI-positive cells between the WT iPSC-IECs and PEPT1-KO iPSC-IECs (Figure 2D). Also, immunocytochemical and western blotting analysis showed that both the WT iPSC-IECs and PEPT1-KO iPSC-IECs were positive for VIL1 (Figures 2E and 2F, respectively). These results suggest that the intestinal differentiation capacity was not inhibited by PEPT1 knockout, and that PEPT1 expression was successfully depleted in PEPT1-KO iPSC-IECs.
Figure 2.

Intestinal Differentiation Capacity of PEPT1-KO Human iPSCs
(A) The procedure for intestinal differentiation from human iPSCs is shown. (B) The gene expression levels of intestinal markers (VIL1, ISX, and SI) and PEPT1 in WT-iPSCs and PEPT1-KO iPSC-IECs were examined by real-time RT-PCR analysis. The gene expression levels in the WT iPSC-IECs were taken as 1.0. Data represent the means ± SD. Statistical analyses were performed using the unpaired two-tailed Student’s t test (***p < 0.001). (C) The protein expression levels of PEPT1 and β-actin in WT iPSC-IECs and PEPT1-KO iPSC-IECs were examined by western blotting analysis. (D) The percentages of VIL1-positive cells and SI-positive cells in WT iPSC-IECs and PEPT1-KO iPSC-IECs were measured by FACS analysis. Data are presented as means ± SD (n ≥ 6, technical replicate). (E) Immunostaining analysis of VIL1 (red) was performed in WT iPSC-IECs and SLC15A1-KO iPSC-IECs. Nuclei were stained with DAPI (blue). Scale bars represent 50 μm. (F) The protein expression levels of VIL1 and β-actin in WT iPSC-IECs and PEPT1-KO iPSC-IECs were examined by western blotting analysis.
Next, we examined the global gene expression profile by microarray analysis. The gene expression of PEPT1 was at an almost undetectable level in PEPT1-KO iPSC-IECs (Figure 3A). There was little difference in the gene expression levels of most genes (99.72%) between the WT iPSC-IECs and PEPT1-KO iPSC-IECs (Figure 3A). We observed only 76 genes whose expression levels were upregulated or downregulated more than 4-fold. Moreover, the expression levels of 60 genes that had the highest potential for off-target cleavage were not largely changed (Figure 3B). These results suggest that specific genome editing of the PEPT1 locus was successfully achieved by our genome editing strategy.
Figure 3.

Transcriptomic Analysis of PEPT1-KO iPSC-IECs
DNA microarray analysis was performed in WT iPSC-IECs and PEPT1-KO iPSC-IECs. (A) A scatterplot of the gene expression signals in WT iPSC-IECs and PEPT1-KO iPSC-IECs is shown. Red dots and blue dots indicate the genes whose expression levels were upregulated and downregulated more than 4-fold, respectively. (B) Expression levels of genes with the potential for off-target cleavage. The expression signal values of the 60 genes that had sequences similar (less than four mismatches) to the single-guide RNA (sgRNA) target sequence were extracted from the data generated using the microarray. The relative signal values of the genes in PEPT1-KO iPSC-IECs are shown with WT iPSC-IECs as 1.0. The data were obtained from one differentiation experiment.
The PEPT1-Mediated Drug Absorption Capacities of PEPT1-KO iPSC-IECs
To examine whether the PEPT1-KO iPSC-IECs would be applicable to drug permeability studies, we evaluated the barrier function of the PEPT1-KO iPSC-IEC monolayer by transepithelial electric resistance (TEER) measurements. The TEER value in the WT iPSC-IEC and PEPT1-KO iPSC-IEC monolayer was approximately 400 Ω × cm2 (Figure 4A). In addition, the apparent membrane permeability (Papp) value of lucifer yellow (LY) was not changed by PEPT1 knockout (Figure 4B). These results suggest that barrier function of the human iPSC-IEC monolayer was not affected by PEPT1 knockout. To examine the PEPT1-mediated drug absorption capacity, we performed a glycylsarcosine (a PEPT1 substrate) permeability test. The Papp value of glycylsarcosine in PEPT1-KO iPSC-IECs was significantly lower than that in WT iPSC-IECs (Figure 4C). Moreover, glycylsarcosine uptake capacity in PEPT1-KO iPSC-IECs was much lower than that in WT iPSC-IECs (Figure 4D). These results suggest that PEPT1-KO iPSC-IECs do not retain PEPT1-mediated drug absorption capacity.
Figure 4.

Transport Activity Evaluation of PEPT1/SLC15A1
(A) TEER values of the WT iPSC-IEC and PEPT1-KO iPSC-IEC monolayers were measured by Millicell-ERS2. Data are presented as means ± SD (n ≥ 10, technical replicate). (B) The Papp values of lucifer yellow in the WT iPSC-IEC and PEPT1-KO iPSC-IEC monolayers were measured. Data are presented as means ± SD (n ≥ 3, technical replicate). (C) The apical-to-basolateral permeability of glycylsarcosine across the WT iPSC-IEC and PEPT1-KO iPSC-IEC monolayers was measured. Data are presented as means ± SD (n = 3, technical replicate). (D) The glycylsarcosine uptake capacity in the WT iPSC-IECs and PEPT1-KO iPSC-IECs was examined. Data are presented as means ± SD (n ≥ 3, technical replicate). Statistical analyses were performed using the unpaired two-tailed Student’s t test (*p < 0.05, ***p < 0.001).
Discussion
We have successfully established PEPT1-KO human iPSCs using genome editing technology. The pluripotency and intestinal differentiation capacity were not affected by PEPT1 knockout. We confirmed that the PEPT1-mediated drug transport ability was significantly decreased by PEPT1 knockout.
It was previously reported that PEPT1 loses its transport ability through the introduction of mutations in certain amino acids such as Trp294 or Glu595.27 Thus, in this study, we attempted to deplete PEPT1 transport activity by targeting the glutamate (Glu595) present in exon 21 of PEPT1. As we expected, the PEPT1 transport ability in the PEPT1-KO iPSC-IECs was significantly decreased by deleting exon 21, which includes glutamate (Glu595).
We successfully obtained iPSCs that are monoallelically targeted at the PEPT1 locus (Figure 1B). Interestingly, indel mutation was frequently observed in the other allele, which does not carry the EF1α-PuroR-pA cassette at the PEPT1 locus. We considered that the indel mutation might have been caused by CRISPR-Cas9 system-mediated non-homologous end-joining (NHEJ) repair, rather than by homology-directed repair. This result suggests that it might be difficult to generate monoallelically targeted iPSCs that have no indel mutation in the other allele, suggesting that heterozygous knockout iPSCs would be difficult to obtain.
The microarray analysis showed that the gene expression levels of carboxylesterase 1 (CES1) were unexpectedly increased by PEPT1 knockout (Figure 3A). It is known that carboxylesterases catalyze the hydrolysis of esters, amides, thioesters, and carbamates.28, 29, 30 In humans, two carboxylesterases, CES1 and CES2, are important mediators of drug metabolism. In the human small intestine, only CES2 is highly expressed.31,32 In the future, it might be necessary to evaluate the CES1 activity. In addition, it would be preferable to use other single-guide RNAs (sgRNAs) to generate PEPT1-KO iPSCs.
It is known that a SNP (P586L) causes a significant decrease in PEPT1 transport capacity.33,34 Although our PEPT1-KO iPSCs would be valuable models for the evaluation of PEPT1 substrates, our models might not faithfully recapitulate the pharmacokinetics of individuals who have an SNP in the PEPT1 locus. We have previously demonstrated that the inter-individual differences in cytochrome P450 (CYP)-metabolism capacity and drug responsiveness, which are prescribed by an SNP in genes encoding CYPs, can be reproduced by generating human iPSC-derived hepatocyte-like cells that have a WT or null allele.35 Therefore, in the future, it might be preferable to generate human iPSC-IECs from individuals who have an SNP in the PEPT1 locus. Recently, it has become possible to introduce single-nucleotide substitutions using genome editing technologies.36, 37, 38, 39 By using these methods, we would like to generate a panel of human iPSC-IECs that have various SNPs observed in drug-metabolizing enzyme and transporter genes.
Our PEPT1-KO iPSCs would be useful for the evaluation of PEPT1 substrates because PEPT1-KO iPSC-IECs enable us to perform highly specific PEPT1 transporter assays without the use of inhibitors. In addition, it is possible to avoid the problem of the species difference in PEPT1 function between humans and experimental animals by using these cells. We think that our model will be successfully applied to a cell-based, PEPT1-mediated drug transporter assay, which is in the early phase of pharmaceutical development.
Materials and Methods
Human iPSCs
YOW-iPSCs generated from primary human hepatocytes were used in this study.35 To passage human iPSCs, near-confluent human iPSC colonies were treated with TrypLE Select Enzyme (Thermo Fisher Scientific) for 5 min at 37°C. Human iPSCs were seeded at an appropriate cell density (5 × 104 cells/cm2) in an uncoated manner using 0.1 μg/cm2 iMatrix-511 (Nippi) with StemFit AK02N medium (Ajinomoto). Human iPSCs were subcultured every 6 days.
Electroporation
The SLC15A1/PEPT1 locus was targeted using the donor plasmids and CRISPR-Cas9 plasmids. The targeting experiments of human iPSCs were performed as described in our previous study.24 Briefly, human iPSCs were treated with 10 μM VPA (Sigma-Aldrich) for 24 h. Human iPSCs (1.0 × 106 cells) were dissociated into single cells by using TrypLE Select Enzyme and resuspended in Opti-MEM (Thermo Fisher Scientific). The electroporation was performed by using NEPA21 (Nepa Gene) according to the manufacturer’s instructions. The ratio of Opti-MEM to the plasmid solution was 90/10 μL (total 100 μL). The plasmid solution consisted of 5 μg of PuroR-expressing donor plasmids, 5 μg of sgRNA CRISPR-Cas9 plasmids (the CRISPR-Cas9 plasmid pX330 was obtained from Addgene, plasmid no. 42230), and 1 μg of RAD51-expressing plasmids (pHMCA-RAD51). After the electroporation, the cells were seeded in an uncoated manner using 0.1 μg/cm2 iMatrix-511 and cultured with StemFit AK02N medium containing 10 μM Y-27632 (FUJIFILM Wako). After culturing for 2 days, the medium was replaced with 10 μM puromycin-containing medium. Then, 48 h after its addition, the puromycin-containing medium was removed and the original medium was added. At 10 days after the electroporation, five individual colonies were picked up and then seeded in an uncoated manner using a 0.1 μg/cm2 iMatrix-511 six-well plate. After most of the wells became nearly confluent, PCR and sequencing analyses were performed to examine whether the clones were correctly targeted. The donor plasmid sequences, sgRNA sequences, genotyping primers, and sequencing primers are described in the Supplemental Materials and Methods section.
DNA Sequencing
Genomic DNA was isolated from human iPSCs using DNAzol (Thermo Fisher Scientific). PCR was performed using Tks Gflex DNA polymerase (Takara Biomedicals) following the manufacturer’s instructions. PCR-amplified DNA was purified using a QIAquick gel extraction kit (QIAGEN) following the manufacturer’s instructions. DNA sequences were confirmed by the FASMAC DNA sequencing service.
Real-Time RT-PCR
Total RNA was isolated from human iPSCs and their derivatives using ISOGEN (Nippon Gene). cDNA was synthesized using 500 ng of total RNA with a SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific). Real-time RT-PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems) using a StepOnePlus real-time PCR system (Applied Biosystems). The relative quantitation of target mRNA levels was performed by using the 2−ΔΔCT method. The values were normalized by those of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). PCR primer sequences were obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/).
Immunocytochemistry
To perform the immunocytochemistry, the human iPSCs and their derivatives were fixed with 4% paraformaldehyde (FUJIFILM Wako) in PBS for 10 min. After blocking the cells with PBS containing 2% BSA and 0.2% Triton X-100 for 20 min, the cells were incubated with the anti-OCT4 (POU5F1) antibody (sc5279, Santa Cruz Biotechnology) or anti-villin 1 antibody (ab130751, Abcam) at 4°C overnight, and, finally, with an immunoglobulin (Ig)G secondary antibody, Alexa Fluor 594 conjugate (Thermo Fisher Scientific), at room temperature for 1 h.
In Vitro Differentiation
Human iPSCs were differentiated into IECs according to our previous report with some modifications.23 To perform the definitive endoderm differentiation, human iPSCs were dissociated into single cells by using TrypLE Select Enzyme and plated onto growth factor-reduced BD Matrigel basement membrane matrix (BD Biosciences). These cells were cultured with StemFit AK02N medium until they reached approximately 80% confluence, then cultured with RPMI 1640 medium (Sigma-Aldrich) containing 100 ng/mL activin A (R&D Systems), 1× GlutaMAX (Thermo Fisher Scientific), penicillin-streptomycin (P/S; Nacalai Tesque), 1× B27 supplement minus vitamin A (Thermo Fisher Scientific), and 1 μM CHIR99021 (FUJIFILM Wako) for 1 day, and finally with RPMI 1640 medium containing 100 ng/mL activin A, 1× GlutaMAX, P/S, and 1× B27 supplement minus vitamin A for 2 days. For the induction of intestinal progenitor cells, the definitive endoderm cells were cultured for 4 days in intestinal differentiation medium consisting of DMEM-high glucose medium (FUJIFILM Wako) containing 10% knockout serum replacement (KSR; Thermo Fisher Scientific), 1% non-essential amino acid (NEAA) solution (Thermo Fisher Scientific), P/S, 1× GlutaMAX, and 20 nM LY2090314 (MedChem Express). Then, for the induction of IECs, the intestinal progenitor cells were cultured for 20 days in WNT3A-, R-spondin 3-, and Noggin-expressing cell (ATCC; CRL3276)-conditioned intestinal differentiation medium containing 3 nM LY2090314, 10 μM SB202190 (FUJIFILM Wako), 1 μM dexamethasone (DEX; FUJIFILM Wako), 50 ng/mL epidermal growth factor (EGF; R&D Systems), and 20 ng/mL insulin-like growth factor-1 (IGF-1; R&D Systems).
Flow Cytometry
Single-cell suspensions of the human iPSC-IECs were treated with 1× permeabilization buffer (eBioscience) and then incubated with the anti-villin 1 antibody (ab109516, Abcam) or anti-SI antibody (sc393424, Santa Cruz Biotechnology), followed by an IgG secondary antibody or Alexa Fluor 488 conjugate (Thermo Fisher Scientific). Flow cytometry analysis was performed using a MACSQuant Analyzer (Milteny Biotec).
DNA Microarray
cRNA amplification, labeling, hybridization, and analysis were performed at Takara Bio using the SurePrint G3 Human gene expression 8×60K v3 microarray (Agilent Technologies). The GEO accession number for the microarray analysis is GEO: GSE131550.
TEER Measurements
TEER values of human iPSC-IECs, which were cultured on BD Falcon cell culture inserts (six-well plate, 0.4-μm pore size, 2.0 × 106 pores/cm2, BD Biosciences) from day 0 of differentiation, were measured by a Millicell-ERS volt-ohm meter (Merck Millipore). The raw data were converted to Ω × cm2 based on the culture insert area (4.2 cm2).
Analysis of Papp
Papp in the transport assay was calculated according to the following equation:
where δCr is the final receiver concentration, δt is assay time, Vr is receiver volume, A is the transwell growth area, and C0 is the initial compound concentration in the donor compartment.
PEPT1 Permeability Tests
The human iPSC-IECs were cultured on the cell culture inserts, then washed with Hanks’ balanced salt solution (HBSS). HBSS containing 30 μM glycylsarcosine (Sigma-Aldrich) was added to the apical side, and HBSS was also added to the basolateral side. After 90 min of incubation at 37°C, the solution was collected from the basolateral side. The solutions were mixed with 2-fold vol of acetonitrile. Mixed solutions were centrifuged for 10 min at 20,000 × g. The supernatants were mixed with a 5.6-fold vol of water and centrifuged for 10 min at 20,000 × g, and then the supernatant was analyzed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) to measure the concentration of glycylsarcosine according to a standard curve. UPLC analysis was performed using an Acquity UPLC (Waters), and MS/MS was performed on a Q-Premier XE (Waters). The mass spectrometer was set to the multiple-reaction monitoring (MRM) mode and was operated with the electrospray ionization source in positive ion mode. The MRM transition (m/z of precursor ion/m/z of product ion) for glycylsarcosine was 146.92/89.90. For transition, the cone voltage and collision energy were set at 18 V, 12 eV. The dwell time for each MRM transition was set at 100 ms. LC separations were carried out at 40°C with an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) (Waters). The mobile phase was delivered at a flow rate of 0.5 mL/min using a gradient elution profile consisting of solvent A (0.1% formic acid/distilled water) and solvent B (acetonitrile). The initial composition of the binary solvent was 0% solvent B from 0 to 1.0 min. Solvent B was increased from 0% to 100% during 1.5 min. The composition of solvent remained for 1.0 min at 100% solvent B. Ten microliters of sample solution was injected into the column.
PEPT1 Uptake Tests
The human iPSC-IECs were cultured on the cell culture plate and then washed with HBSS. The human iPSC-IECs were cultured with HBSS containing 30 μM glycylsarcosine (Sigma-Aldrich). After 90 min of incubation at 37°C, the cells were washed with HBSS. Accumulated glycylsarcosine in the human iPSC-IECs was extracted by adding 70% methanol. Cell lysis solutions were mixed with 2-fold vol of acetonitrile. Mixed solutions were centrifuged for 10 min at 20,000 × g, and an aliquot of the supernatant was subjected to UPLC-MS/MS. The concentration of glycylsarcosine was measured as described in “PEPT1 Permeability Tests” above.
Western Blotting
The cells were homogenized with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) containing a protease inhibitor mixture (Thermo Fisher Scientific). The homogenates were centrifuged at 15,000 × g at 4°C for 10 min, and the supernatants were collected. The lysates were subjected to SDS-PAGE on 7.5% polyacrylamide gel and then transferred onto polyvinylidene fluoride membranes (Millipore). After the reaction was blocked with 1% skim milk in TBS containing 0.1% Tween 20 at room temperature for 1 h, the membranes were incubated with anti-PEPT1 (sc-373742, Santa Cruz Biotechnology), VIL1 (ab218331, Abcam), and β-actin antibodies (A1978, Sigma) at 4°C overnight, followed by reaction with horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling Technology) at room temperature for 1 h. The band was visualized by Chemi-Lumi One Super (Nakalai Tesque) and the signals were read using an LAS-3000 imaging system (Fujifilm).
LY Permeability Tests
Human iPSC-IECs, which were cultured on the cell culture inserts, were rinsed with HBSS. The 1.5 mL of HBSS containing 100 μM LY CH dipotassium salt (LY, FUJIFILM Wako) with or without 10 mM n-capric acid (C10; Nacalai Tesque) was added to the apical chamber, and 2.6 mL of HBSS was also added to the basolateral chamber. After a 90-min incubation at 37°C, the solution was collected from the basolateral chamber. The LY fluorescent signal was measured with a fluorescence plate reader (TriStar LB 941, Berthold Technologies) using 485-nm excitation and 535-nm emission filters. LY concentrations were calculated using the standard curve generated by serial dilution of LY.
Data and Statistical Analysis
Data are presented as means ± SD. Statistical analysis was performed using Student’s unpaired t test. A value of p < 0.05 was considered statistically significant. All calculations were carried out using Easy R (EZR) software.
Author Contributions
K.T. conceived and supervised the study; K.K., R.N., and K.T. designed the experiments; K.K., R.N., S.D., K. Harada, and K.T. performed the experiments; K.K., R.N., M.I., T.Y., S.D., K.. Harada, K. Hirata, and K.T. analyzed the data; and K.K., R.N., K.T., and H.M. wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Yasuko Hagihara, Natsumi Mimura, and Ayaka Sakamoto for excellent technical support. This research was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (18H05033, 18H05373). This research was also supported by grants from the Japan Agency for Medical Research and Development (AMED) (19be0304320h0003, 19mk0101125h0002). The graphical abstract was created using BioRender.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2019.11.008.
Contributor Information
Kazuo Takayama, Email: takayama@phs.osaka-u.ac.jp.
Hiroyuki Mizuguchi, Email: mizuguch@phs.osaka-u.ac.jp.
Supplemental Information
References
- 1.Ogihara H., Saito H., Shin B.-C., Terado T., Takenoshita S., Nagamachi Y., Inui K., Takata K. Immuno-localization of H+/peptide cotransporter in rat digestive tract. Biochem. Biophys. Res. Commun. 1996;220:848–852. doi: 10.1006/bbrc.1996.0493. [DOI] [PubMed] [Google Scholar]
- 2.Groneberg D.A., Döring F., Eynott P.R., Fischer A., Daniel H. Intestinal peptide transport: ex vivo uptake studies and localization of peptide carrier PEPT1. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G697–G704. doi: 10.1152/ajpgi.2001.281.3.G697. [DOI] [PubMed] [Google Scholar]
- 3.Ganapathy V., Leibach F.H. Is intestinal peptide transport energized by a proton gradient? Am. J. Physiol. 1985;249:G153–G160. doi: 10.1152/ajpgi.1985.249.2.G153. [DOI] [PubMed] [Google Scholar]
- 4.Inui K., Tomita Y., Katsura T., Okano T., Takano M., Hori R. H+ coupled active transport of bestatin via the dipeptide transport system in rabbit intestinal brush-border membranes. J. Pharmacol. Exp. Ther. 1992;260:482–486. [PubMed] [Google Scholar]
- 5.Han H., de Vrueh R.L.A., Rhie J.K., Covitz K.M., Smith P.L., Lee C.P., Oh D.M., Sadée W., Amidon G.L. 5′-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998;15:1154–1159. doi: 10.1023/a:1011919319810. [DOI] [PubMed] [Google Scholar]
- 6.Brodin B., Nielsen C.U., Steffansen B., Frøkjaer S. Transport of peptidomimetic drugs by the intestinal di/tri-peptide transporter, PepT1. Pharmacol. Toxicol. 2002;90:285–296. doi: 10.1034/j.1600-0773.2002.900601.x. [DOI] [PubMed] [Google Scholar]
- 7.Anand B.S., Katragadda S., Mitra A.K. Pharmacokinetics of novel dipeptide ester prodrugs of acyclovir after oral administration: intestinal absorption and liver metabolism. J. Pharmacol. Exp. Ther. 2004;311:659–667. doi: 10.1124/jpet.104.069997. [DOI] [PubMed] [Google Scholar]
- 8.Herrera-Ruiz D., Wang Q., Gudmundsson O.S., Cook T.J., Smith R.L., Faria T.N., Knipp G.T. Spatial expression patterns of peptide transporters in the human and rat gastrointestinal tracts, Caco-2 in vitro cell culture model, and multiple human tissues. AAPS PharmSci. 2001;3:E9. doi: 10.1208/ps030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamashita S., Konishi K., Yamazaki Y., Taki Y., Sakane T., Sezaki H., Furuyama Y. New and better protocols for a short-term Caco-2 cell culture system. J. Pharm. Sci. 2002;91:669–679. doi: 10.1002/jps.10050. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y., Smith D.E., Ma K., Jappar D., Thomas W., Hillgren K.M. Targeted disruption of peptide transporter Pept1 gene in mice significantly reduces dipeptide absorption in intestine. Mol. Pharm. 2008;5:1122–1130. doi: 10.1021/mp8001655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y., Chen X., Smith D.E. Species-dependent uptake of glycylsarcosine but not oseltamivir in Pichia pastoris expressing the rat, mouse, and human intestinal peptide transporter PEPT1. Drug Metab. Dispos. 2012;40:1328–1335. doi: 10.1124/dmd.111.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Y., Smith D.E. Species differences in the pharmacokinetics of cefadroxil as determined in wildtype and humanized PepT1 mice. Biochem. Pharmacol. 2016;107:81–90. doi: 10.1016/j.bcp.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwao T., Kodama N., Kondo Y., Kabeya T., Nakamura K., Horikawa T., Niwa T., Kurose K., Matsunaga T. Generation of enterocyte-like cells with pharmacokinetic functions from human induced pluripotent stem cells using small-molecule compounds. Drug Metab. Dispos. 2015;43:603–610. doi: 10.1124/dmd.114.062604. [DOI] [PubMed] [Google Scholar]
- 14.Foley D.W., Pathak R.B., Phillips T.R., Wilson G.L., Bailey P.D., Pieri M., Senan A., Meredith D. Thiodipeptides targeting the intestinal oligopeptide transporter as a general approach to improving oral drug delivery. Eur. J. Med. Chem. 2018;156:180–189. doi: 10.1016/j.ejmech.2018.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itagaki S., Gopal E., Zhuang L., Fei Y.-J., Miyauchi S., Prasad P.D., Ganapathy V. Interaction of ibuprofen and other structurally related NSAIDs with the sodium-coupled monocarboxylate transporter SMCT1 (SLC5A8) Pharm. Res. 2006;23:1209–1216. doi: 10.1007/s11095-006-0023-1. [DOI] [PubMed] [Google Scholar]
- 16.Cui D., Morris M.E. The drug of abuse gamma-hydroxybutyrate is a substrate for sodium-coupled monocarboxylate transporter (SMCT) 1 (SLC5A8): characterization of SMCT-mediated uptake and inhibition. Drug Metab. Dispos. 2009;37:1404–1410. doi: 10.1124/dmd.109.027169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaji I., Iwanaga T., Watanabe M., Guth P.H., Engel E., Kaunitz J.D., Akiba Y. SCFA transport in rat duodenum. Am. J. Physiol. Gastrointest. Liver Physiol. 2015;308:G188–G197. doi: 10.1152/ajpgi.00298.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kauffman A.L., Gyurdieva A.V., Mabus J.R., Ferguson C., Yan Z., Hornby P.J. Alternative functional in vitro models of human intestinal epithelia. Front. Pharmacol. 2013;4:79. doi: 10.3389/fphar.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogaki S., Shiraki N., Kume K., Kume S. Wnt and Notch signals guide embryonic stem cell differentiation into the intestinal lineages. Stem Cells. 2013;31:1086–1096. doi: 10.1002/stem.1344. [DOI] [PubMed] [Google Scholar]
- 20.Iwao T., Toyota M., Miyagawa Y., Okita H., Kiyokawa N., Akutsu H., Umezawa A., Nagata K., Matsunaga T. Differentiation of human induced pluripotent stem cells into functional enterocyte-like cells using a simple method. Drug Metab. Pharmacokinet. 2014;29:44–51. doi: 10.2133/dmpk.dmpk-13-rg-005. [DOI] [PubMed] [Google Scholar]
- 21.Ozawa T., Takayama K., Okamoto R., Negoro R., Sakurai F., Tachibana M., Kawabata K., Mizuguchi H. Generation of enterocyte-like cells from human induced pluripotent stem cells for drug absorption and metabolism studies in human small intestine. Sci. Rep. 2015;5:16479. doi: 10.1038/srep16479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takayama K., Negoro R., Yamashita T., Kawai K., Ichikawa M., Mori T., Nakatsu N., Harada K., Ito S., Yamada H. Generation of human iPSC-derived intestinal epithelial cell monolayers by CDX2 transduction. Cell. Mol. Gastroenterol. Hepatol. 2019;8:513–526. doi: 10.1016/j.jcmgh.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negoro R., Takayama K., Kawai K., Harada K., Sakurai F., Hirata K., Mizuguchi H. Efficient generation of small intestinal epithelial-like cells from human iPSCs for drug absorption and metabolism studies. Stem Cell Reports. 2018;11:1539–1550. doi: 10.1016/j.stemcr.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takayama K., Igai K., Hagihara Y., Hashimoto R., Hanawa M., Sakuma T., Tachibana M., Sakurai F., Yamamoto T., Mizuguchi H. Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res. 2017;45:5198–5207. doi: 10.1093/nar/gkx130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takayama K., Hagihara Y., Toba Y., Sekiguchi K., Sakurai F., Mizuguchi H. Enrichment of high-functioning human iPS cell-derived hepatocyte-like cells for pharmaceutical research. Biomaterials. 2018;161:24–32. doi: 10.1016/j.biomaterials.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 26.Deguchi S., Yamashita T., Igai K., Harada K., Toba Y., Hirata K., Takayama K., Mizuguchi H. Modeling of hepatic drug metabolism and responses in CYP2C19 poor metabolizer using genetically manipulated human iPS cells. Drug Metab. Dispos. 2019;47:632–638. doi: 10.1124/dmd.119.086322. [DOI] [PubMed] [Google Scholar]
- 27.Bolger M.B., Haworth I.S., Yeung A.K., Ann D., von Grafenstein H., Hamm-Alvarez S., Okamoto C.T., Kim K.J., Basu S.K., Wu S., Lee V.H. Structure, function, and molecular modeling approaches to the study of the intestinal dipeptide transporter PepT1. J. Pharm. Sci. 1998;87:1286–1291. doi: 10.1021/js980090u. [DOI] [PubMed] [Google Scholar]
- 28.Hosokawa M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules. 2008;13:412–431. doi: 10.3390/molecules13020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vistoli G., Pedretti A., Mazzolari A., Bolchi C., Testa B. Influence of ionization state on the activation of temocapril by hCES1: a molecular-dynamics study. Chem. Biodivers. 2009;6:2092–2100. doi: 10.1002/cbdv.200900174. [DOI] [PubMed] [Google Scholar]
- 30.Ishizaki Y., Furihata T., Oyama Y., Ohura K., Imai T., Hosokawa M., Akita H., Chiba K. Development of a Caco-2 cell line carrying the human intestine-type CES expression profile as a promising tool for ester-containing drug permeability studies. Biol. Pharm. Bull. 2018;41:697–706. doi: 10.1248/bpb.b17-00880. [DOI] [PubMed] [Google Scholar]
- 31.Xie M., Yang D., Liu L., Xue B., Yan B. Human and rodent carboxylesterases: immunorelatedness, overlapping substrate specificity, differential sensitivity to serine enzyme inhibitors, and tumor-related expression. Drug Metab. Dispos. 2002;30:541–547. doi: 10.1124/dmd.30.5.541. [DOI] [PubMed] [Google Scholar]
- 32.Jones R.D., Taylor A.M., Tong E.Y., Repa J.J. Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metab. Dispos. 2013;41:40–49. doi: 10.1124/dmd.112.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang E.Y., Fu D.-J., Pak Y.A., Stewart T., Mukhopadhyay N., Wrighton S.A., Hillgren K.M. Genetic polymorphisms in human proton-dependent dipeptide transporter PEPT1: implications for the functional role of Pro586. J. Pharmacol. Exp. Ther. 2004;310:437–445. doi: 10.1124/jpet.104.065912. [DOI] [PubMed] [Google Scholar]
- 34.Anderle P., Nielsen C.U., Pinsonneault J., Krog P.L., Brodin B., Sadée W. Genetic variants of the human dipeptide transporter PEPT1. J. Pharmacol. Exp. Ther. 2006;316:636–646. doi: 10.1124/jpet.105.094615. [DOI] [PubMed] [Google Scholar]
- 35.Takayama K., Morisaki Y., Kuno S., Nagamoto Y., Harada K., Furukawa N., Ohtaka M., Nishimura K., Imagawa K., Sakurai F. Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes. Proc. Natl. Acad. Sci. USA. 2014;111:16772–16777. doi: 10.1073/pnas.1413481111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 38.Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 39.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.